

Abstract
GPR56 is a member of the family of adhesion G-protein-coupled receptors that have a large extracellular region containing a GPS (G-protein proteolytic site) domain. Loss-of-function mutations in the GPR56 gene cause a specific human brain malformation called bilateral frontoparietal polymicrogyria (BFPP). BFPP is a radiological diagnosis and its histopathology remains unclear. This study demonstrates that loss of the mouse Gpr56 gene leads to neuronal ectopia in the cerebral cortex, a cobblestone-like cortical malformation. There are four crucial events in the development of cobblestone cortex, namely defective pial basement membrane (BM), abnormal anchorage of radial glial endfeet, mislocalized Cajal–Retzius cells, and neuronal overmigration. By detailed time course analysis, we reveal that the leading causal events are likely the breaches in the pial BM. We show further that GPR56 is present in abundance in radial glial endfeet. Furthermore, a putative ligand of GPR56 is localized in the marginal zone or overlying extracellular matrix. These observations provide compelling evidence that GPR56 functions in regulating pial BM integrity during cortical development.
Keywords: GPR56, bilateral frontoparietal polymicrogyria, BFPP, basement membrane, cobblestone cortex, cortical lamination
Introduction
Using positional cloning, we previously demonstrated that mutations in the GPR56 gene cause a specific human brain malformation called bilateral frontoparietal polymicrogyria (BFPP) (Piao et al., 2004). BFPP is a recessively inherited genetic disorder of human cerebral cortical development, characterized by disorganized cortical lamination that is most severe in the frontal and parietal lobes (Piao et al., 2002; Chang et al., 2003). The histopathology of BFPP remains unclear. Our follow-up genotype–phenotype analysis in patients with BFPP and other similar polymicrogyria syndromes have demonstrated that all predicted pathogenic GPR56 sequence alterations are associated with polymicrogyria plus white matter defects and brainstem and cerebellar hypoplasia (Piao et al., 2005; Piao and Walsh, 2007). The findings of impaired myelination, small pons, and cerebellar hypoplasia are common in individuals with congenital muscular dystrophy syndromes; however, they present in very few other disorders (A. J. Barkovich, personal communication).
Cobblestone lissencephaly is defined as aberrant migration of cortical neurons out of the developing brain through breaches in the pial basement membrane (BM), forming neuronal ectopias on the surface of the brain (Olson and Walsh, 2002). Cobblestone lissencephaly is typically seen in three distinct human congenital muscular dystrophy syndromes, muscle–eye–brain disease (MEB), Fukuyama-type muscular dystrophy (FCMD), and Walker–Warburg syndrome (WWS) (Olson and Walsh, 2002). MEB, FCMD, and some WWS cases are caused by aberrant glycosylation of α-dystroglycan, a receptor for laminin (Kobayashi et al., 1998; Yoshida et al., 2001; Michele et al., 2002; Yamamoto et al., 2004). Mutant mice with deletions in some members of the integrin family, downstream associates of integrins, such as FAK (focal adhesion kinase) and Ilk (integrin-linked kinase), or extracellular matrix constituents like the proteoglycan, perlecan, and the nidogen-binding site of laminin γ1, also show cortical migration defects with deficiencies in basal lamina integrity, features that resemble human cobblestone lissencephaly (Georges-Labouesse et al., 1998; Costell et al., 1999; De Arcangelis et al., 1999; Graus-Porta et al., 2001; Halfter et al., 2002; Beggs et al., 2003; Niewmierzycka et al., 2005; Haubst et al., 2006). To date, the majority of cobblestone cortex-causative genes identified encode proteins that are directly or indirectly involved in pial BM organization (Georges-Labouesse et al., 1998; Costell et al., 1999; De Arcangelis et al., 1999; Graus-Porta et al., 2001; Halfter et al., 2002; Beggs et al., 2003; Niewmierzycka et al., 2005; Haubst et al., 2006; Hu et al., 2007).
In this study, we show that loss of GPR56 leads to cortical lamination defects, with overmigration of neurons through defective BM into the pial layer, demonstrating the histopathology of BFPP to be a cobblestone-like brain malformation. Furthermore, by analyzing detailed time courses to evaluate BM integrity and radial glial endfeet anchorage, we demonstrate that breaks in pial BM are likely the leading causal events, which are, in turn, associated with abnormal anchorage of radial glial endfeet. We further reveal that a putative ligand of GPR56 is present in the pial BM. Thus, our findings establish a role for GPR56 in regulating pial BM integrity and corticogenesis.
Materials and Methods
Mice.
The Gpr56 knock-out mice were obtained from Genentech/Lexicon Genetics. The mutant mice were originally created in a 129/BL6 background, but were rederived into the FvB strain and bred to BALB/c at one point. Therefore, the genetic background of the mutant mice is mixed: 129/BL6/FvB/BALB/c. The genotype of the mice was determined by PCR using the following primers: A (5′-CGAGAAGACTTCCGCTTCTG), B (5′-AAAGTAGCTAAGATGCTCTCC), and Neo (5′-GCAGCGCATCGCCTTCTATC).
The time point at 10:00 A.M. on the day of vaginal plug is defined as embryonic day 0.5 (E0.5). Fetal stage was calculated from the day when a vaginal plug was observed. All animals were treated according to the guidelines of the Animal Care and Use Committee of Children's Hospital Boston.
Antibodies.
The C-terminal peptide of mouse GPR56 (KNNSDSAKLPISSGSTSSSRI) was synthesized, conjugated to keyhole limpet hemocyanin, and injected into rabbits for antiserum production (Yenzym Antibodies). Rabbit anti-brain lipid binding protein (BLBP) (Millipore Bioscience Research Reagents; 1:1000), mouse anti-5-bromo-2′-deoxyuridine (BrdU) (Dako; 1:100), mouse anti-calbindin (Swant; 1:1000), goat anti-collagen IV (Southern Biotech; 1:20), rat anti-chicken ovalbumin upstream promoter transcription factor-interacting protein 2 (CTIP2) (Abcam; 1:500), rabbit anti-Cux1 (gift from J. Cunningham, Brigham and Women's Hospital, Harvard Medical School, Boston, MA; 1:200), rabbit anti-dopamine- and cAMP-regulated phosphoprotein-32 (DARPP32) (Millipore Bioscience Research Reagents; 1:1000), guinea pig anti-glutamate-aspartate transporter (GLAST) (Millipore Bioscience Research Reagents; 1:2000), rabbit anti-Engelbreth–Holm–Swarm laminin (Sigma-Aldrich; 1:25), rabbit anti-nidogen (gift from A. Chung, University of Pittsburg, Pittsburgh, PA; 1:250), rabbit anti-Tbr1 (gift from R. Hevner, University of Washington, Seattle, WA; 1:1000), mouse anti-RC2 (hybridoma; 1:200), mouse anti-reelin (Millipore Bioscience Research Reagents; 1:250), and mouse anti-Tuj1 (Covance; 1:1000).
Histology and immunohistochemistry.
Embryos and postnatal mice were fixed or perfused using 4% paraformaldehyde. Brains were either cryoprotected by 30% sucrose, frozen over dry ice and sectioned on a cryostat, or embedded in paraffin and sectioned on a microtome. Sections were stained with 0.1% cresyl violet/0.5% acetic acid, or processed for immunostaining using standard procedures. Primary antibodies were visualized by appropriate fluorophore-conjugated secondary antibodies (Invitrogen; 1:400). Nuclei were stained with Hoechst 33342 (Invitrogen). All images were captured using a confocal LSM 510 NLO system (Carl Zeiss) or a Nikon 80i upright microscope. Representative photographs were obtained with the same exposure setting for control and mutant.
BrdU labeling and quantitative analysis.
Pregnant mice (E12.5, E15.5, and E17.5) were injected intraperitoneally with BrdU (50 mg/g body weight, dissolved in PBS). Brains were removed at postnatal day 1 (P1) and prepared for cryostat sections. Sections for BrdU staining were pretreated for 15 min in 2N HCl at 40°C. Quantification of newborn cells and distribution within cortical layers were analyzed as previously reported (Feng and Walsh, 2004) and are described in detail in the supplemental material (available at www.jneurosci.org).
Electron microscopy.
Embryonic brains were fixed by immersion in mixture of 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 m sodium cacodylate buffer, pH 7.4. After overnight fixation, tissues were postfixed in 1% osmium tetroxide, then dehydrated, and embedded in Epon-Araldite. Ultrathin sections were cut and stained with uranyl acetate and lead citrate. The samples were observed and photographed using the transmission electron microscope (Tecnai G2 Spirit BioTWIN) at the Harvard Medical School EM Facility.
Generation of mouse Fc fusion protein and putative ligand binding assay.
Mouse Fc vector was a kind gift from Dr. H. Lin (Chang Gung University, Tao-Yuan, Taiwan) (Stacey et al., 2003). Mouse IgG Fc-tagged construct was generated by PCR. The PCR primers for mouse GPR56N were as follows: forward, 5′-GGATCCTATGGCTGTCCAGGTGCTGCGGC-3′, and reverse, 5′-GCGGCCGCCAGGTGGTTGCACAGGCAGGATG-3′. Deletion mutagenesis was done by using the QuikChange II XL Site-Directed Mutagenesis kit (Stratagene) according to the protocols suggested by the manufacturer. The primers for GPR56Ndel were as follows: forward, 5′-CCCAGAGCCTAGAGGGCTCTATC-ACTTCTGCCTCTACCAGATTCCCCAGAACACCAGCCTGCCTGGGGCTCCGA-3′, and reverse, 5′-TCGGAGCCCCAGGCAGGCTGGTGTTCTGGGGAATCTGGTAGAGGCAGAAGTG-ATAGAGCCCTCTAGGCTCTGGG-3′. All expression constructs were sequenced to confirm their identity.
HEK-293T cells were transiently transfected with mouse Fc-only construct, mouse Fc-tagged GPR56N or GPR56Ndel constructs. Transfected cells were cultured for 72 h with media changed after the first 24 h. Conditioned media were harvested, centrifuged at 300 × g for 5 min to remove cells and cellular debris, and sterile-filtered through a 0.45 μm pore syringe filter (Whatman). Supernatants were concentrated using Centricon Plus-20 spin columns (Millipore). Equal amounts of concentrated conditioned media from different samples were loaded onto SDS-PAGE gels and transferred to nitrocellulose membranes. The membranes were then probed with HRP-conjugated anti-mouse IgG antibody using standard Western blot protocols. Equivalent amounts of fusion proteins or mouse Fc control were used as probes to examine the distribution of putative ligands of GPR56 in mouse brains of various embryonic stages.
Results
GPR56 is expressed in neural progenitors in murine fetal forebrain
We previously showed that Gpr56 mRNA is detected in the ventricular zone of the mouse developing cortex (Piao et al., 2004). To better localize GPR56 protein in developing forebrain, we stained sections of embryonic mouse brains at E12.5, E14.5, and E16.5 with an affinity-purified rabbit anti-mouse GPR56 antibody. The specificity of the GPR56 antibody was confirmed on cell lines transfected with a GPR56 expression construct, as well as on Gpr56 knock-out mouse brain sections (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). GPR56 immunoreactivity was detected across the entire lateral ventricular wall at E12.5 and E14.5 (Fig. 1A; supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). In the developing cortex at E16.5, GPR56 signals were found in the ventricular zone (VZ), intermediate zone (IZ), and marginal zone (MZ) (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). Double immunohistochemistry (IHC) with anti-GPR56 antibody and a marker for migrating neurons, Tuj1 revealed that GPR56 protein is mostly absent from the migrating neurons (supplemental Fig. 1C,E, available at www.jneurosci.org as supplemental material).
Figure 1.
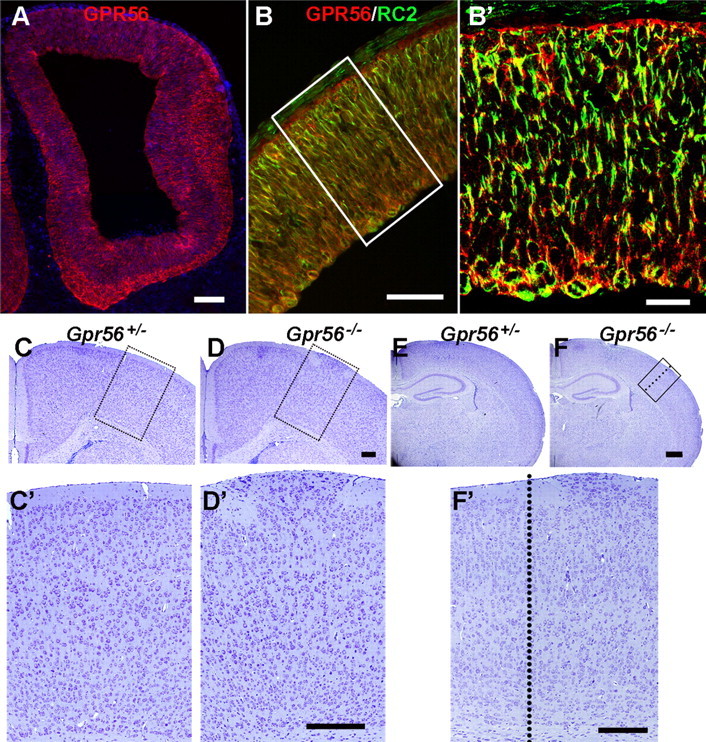
GPR56 is detected in neural progenitor cells and deletion of Gpr56 results in neuronal ectopias. A, B, E12.5 coronal sections through mouse forebrains showing GPR56 as visualized by indirect immunofluorescence. Double-label IHC of GPR56 and RC2 antibody (B) performed on adjacent sections, revealing the expression of GPR56 in radial glial cells. B′, Higher-magnification view of the boxed area in B. C–F, Nissl staining of coronal sections from P14 brains of heterozygous and homozygous mutant mice. No cortical defects were observed in heterozygous brains (C, E), whereas neuronal ectopia was seen in homozygous brains (D, F). E, F, The hippocampus was unaffected. C′, D′, F′, Higher-magnification views of the boxed areas in C, D, and F. Scale bars: A, 100 μm; B, 50 μm; B′, 20 μm; (in D′, F′) C, C′, D, D′, F′, 200 μm; (in F) E, F, 500 μm.
The expression pattern of GPR56 protein is similar to the distribution of radial glial cells. To confirm that radial glia express GPR56, we performed double-label IHC with GPR56 antibody and a radial glial marker, RC2, as well as combined in situ hybridization for Gpr56 and BLBP IHC on mouse developing cortex at various embryonic stages. BLBP, a member of the CRABP (cellular retinoic acid-binding protein)/FABP (fatty acid-binding protein) gene family, is expressed in radial glial cells (Feng et al., 1994). GPR56 protein was predominantly colocalized with radial glial cell markers (Fig. 1B; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This observation is consistent with the expression data generated from Gpr56 BAC transgenic mice (http://www.gensat.org/login.jsp). Together, our data indicate that GPR56 is expressed in radial glia as well as other neural progenitor cells.
Loss of GPR56 leads to regional lamination defects
To study the pathogenesis of BFPP, we obtained Gpr56 knock-out mice from Genentech and Lexicon. The genetic background of the mutant mice is 129/BL6/FvB/BALB/c. The targeting scheme is shown in supplemental Figure 3A (available at www.jneurosci.org as supplemental material). Briefly, Gpr56 exons 2 and 3 are replaced by IRES-lacZ/MCI-neo, which results in the deletion of the starting ATG and a frameshift. The homozygous Gpr56 mutant mice were viable, fertile, and recovered at Mendelian ratios (supplemental Table 1, available at www.jneurosci.org as supplemental material).
GPR56 is cleaved at the GPS (G-protein proteolytic site) domain into an N- and a C-terminal fragments, GPR56N and GPR56C (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material) (Xu et al., 2006; Jin et al., 2007). Western blot analysis with an antibody against the C terminus of GPR56C failed to detect any signal in Gpr56−/− mouse brain, in contrast with wild-type and heterozygous brains, confirming that the targeting strategy results in a true null allele (supplemental Fig. 3C, available at www.jneurosci.org as supplemental material).
Although the overall brain sizes were comparable in mutant and control animals, homozygous mutants revealed frequent occurrence of neuronal ectopias manifested by overmigration of neurons through the marginal zone (Fig. 1D,F). The hippocampus was mostly unaffected in the mutant mice (Fig. 1F). Only homozygous mutant embryos were phenotypically affected, whereas heterozygotes appeared normal and were used as controls in most of the experiments. The phenotype showed full penetrance in a total of 81 homozygous animals from E12.8 to adulthood, whereas there was not a single ectopia observed in a total of 7 wild-type and 62 heterozygous mice (Table 1). Cortical lamination was disorganized in the areas beneath the ectopic outgrowths. In all of the postnatal Gpr56 null mutant animals analyzed, cortical ectopias were only detected in the frontoparietal cortex (Table 2).
Table 1.
Penetrance of cortical dysplasia in Gpr56 knock-out mice
| Stage | Wild-type/heterozygous |
Null |
||
|---|---|---|---|---|
| No. of animals | Ectopias | No. of animals | Ectopias | |
| E12.8 | 3 | No | 8 | Yes |
| E13.5 | 10 | No | 18 | Yes |
| E14.5 | 14 | No | 17 | Yes |
| E15.5 | 1 | No | 3 | Yes |
| E16.5 | 7 | No | 8 | Yes |
| P0.5 | 8 | No | 10 | Yes |
| P6 | 13 | No | 10 | Yes |
| P14 | 3 | No | 3 | Yes |
| 6 week | 10 | No | 4 | Yes |
| Summary | 69 | 81 | ||
There was not a single ectopia detected in a total of 57 Gpr56 wild-type or heterozygous brains, whereas every Gpr56 knock-out mouse brain analyzed displayed ectopias from embryonic day 13.5 to adulthood.
Table 2.
Location of cortical ectopias
| Stage | No. of animals analyzed | Presence of cortical ectopia |
||
|---|---|---|---|---|
| Frontoparietal cortex | Temporal cortex | Occipital cortex | ||
| P0.5 | 10 | Yes | No | No |
| P6 | 10 | Yes | No | No |
| P14 | 3 | Yes | No | No |
| 6 weeks | 4 | Yes | No | No |
Among the 27 Gpr56-null mutant animals analyzed, cortical ectopias were detected only in the frontoparietal cortex.
The ectopic neurons comprise deeper and upper layer neurons
To determine the cellular composition of the ectopias, we performed immunostaining with various layer-specific markers, Cux1 for layers II–IV, Tbr1 for layers II–IV and VI, CTIP2 for layer V, and Darpp32 for layer VI (Nieto et al., 2004; Molyneaux et al., 2005). Neurons positive for Cux1, Tbr1, CTIP2, or Darpp32 were detected in the ectopia, suggesting that the ectopic cells in the Gpr56−/− cortex were neurons from both deep and superficial cortical layers (Fig. 2A). In addition, calbindin-positive interneurons were also detected in the ectopias (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Cortical lamination outside of the regions of the ectopic outgrowths appeared normal.
Figure 2.
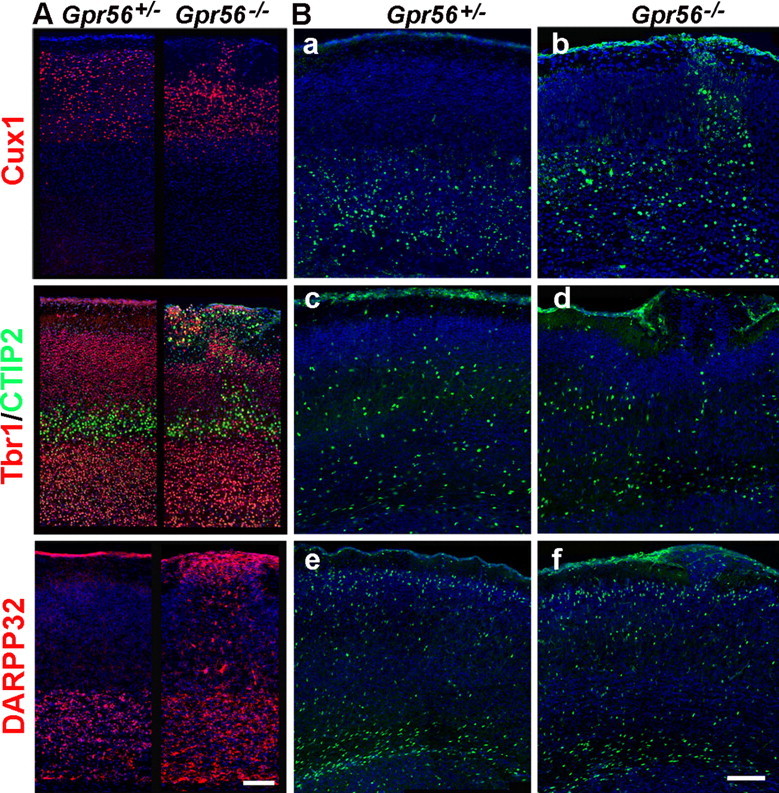
Abnormal cortical lamination in Gpr56−/− mice. A, Immunostaining for cortical layer-specific markers in P6 mouse brains revealed the disorganized lamination in Gpr56−/− mice. Ectopic cluster comprises neurons from both deep and superficial layers. B, In vivo migration assay using BrdU pulse labeling at E12.5 (a, b), E15.5 (c, d), and E17.5 (e, f) revealed that neuronal proliferation and migration were mostly unaffected in Gpr56−/− cortex, except in the ectopic regions. Both deep and upper layer neurons migrated into the marginal zone. Scale bars, 100 μm.
Defects in neuronal migration could be the cause of cortical dysplasia (Anton et al., 1999). To evaluate this possibility, we assessed laminar positioning of cerebral projection neurons by birthdate analysis via injection BrdU at three stages, E12.5, E15.5, and E17.5, according to methods routinely used (Gonzalez et al., 1997; Feng and Walsh, 2004). Animals were killed at P1 and analyzed by immunostaining their brain sections with anti-BrdU antibodies. There were no obvious defects in neuronal migration except in the ectopic regions where postmitotic neurons overmigrated into the marginal zone (Fig. 2B). Again, we detected BrdU+ cells in the ectopic regions from brain sections pulse-labeled at E12.5, E15.5, and E17.5, confirming that the ectopic neurons comprised both upper and deeper layer neurons (Fig. 2B). Furthermore, we find no quantitative difference in the number of BrdU-positive cells located at a range of depths covering all cortical layers of P14 brain between Gpr56−/− and Gpr56+/− mice at all three stages analyzed, suggesting that the neuronal migration is mostly unaffected in the absence of GPR56 (supplemental Fig. 5, available at www.jneurosci.org as supplemental material).
Defective pial basement membrane leads to neuronal overmigration
Defective pial BM, abnormal anchorage of radial glial endfeet, mislocalized Cajal–Retzius (CR) cells, and neuronal overmigration are the four crucial events that are involved in the development of cortical ectopias. To determine the sequence of these four events, we first studied the time course of ectopia formation from E12.5 to E16.5. By Nissl staining, the earliest time point when an ectopia could be detected was E13.5 (supplemental Fig. 6D, arrow; available at www.jneurosci.org as supplemental material). The laminar organization became progressively distorted from E13.5 to E16.5 (supplemental Fig. 6D,F,H, available at www.jneurosci.org as supplemental material).
The pial BM is a specialized extracellular matrix (ECM) that is formed adjacent to the radial glial endfeet. Meningeal fibroblasts contribute to the BM by secreting the majority of basal lamina constituents, including laminin, collage IV, nidogen, and the heparan sulfate proteoglycan (Sievers et al., 1994). Defective pial BM is thought to be the primary cause of cobblestone lissencephaly (Olson and Walsh, 2002). We therefore examined whether there were defects in the pial BM by probing for extracellular matrix constituents, laminin, collagen IV (Col IV), and nidogen on sections of E12.5 and E13.5 mutant and control brains. Interestingly, the BM appeared intact in the mutant brain at E12.5, suggesting that the BM was assembled normally at early developmental stages (Fig. 3C).
Figure 3.

Defective BM leads to neuronal ectopias. A–D, Double-label IHC of Tuj1 and laminin on E12.5 coronal sections revealing intact BM in both control and mutant animals. E–P, Double-label IHC of Tuj1 and various BM constituents on E13.5 coronal sections revealed a continuous thin lining of the BM on the cortical surface in the heterozygous brains (E, I, M) and postmitotic neurons positioned underneath the BM (F, J, N). In contrast, regionally ruptured BM was detected in the homozygous brains (G, K, O, arrows). Tuj1-positive neurons migrated through the defective BM (H, L, P, arrows). Scale bars, 100 μm.
At E13.5, however, we found areas of discontinuity in laminin, Col IV, and nidogen immunoreactivities at the pial surface of Gpr56−/− mouse brains (Fig. 3G,K,O). In contrast, laminin, Col IV, and nidogen immunoreactivities were distributed as a continuous thin lining on the surface of the cortical wall in control animals (Fig. 3E,I,M). Double immunolabeling with Tuj1 and ECM constituents demonstrated that neurons migrated through the defective BM (Fig. 3H,L,P).
It is intriguing that the pial BM was assembled during the early stage of cortical development, and subsequently broke at later stages in the absence of GPR56. One possible explanation is that the ECM polymers are fragile in the absence of GPR56 and cannot sustain the tension generated by the expanding cortical wall. To test this possibility, we performed more detailed time course and EM analyses. By IHC, we could detect a region with disrupted BM and overmigrating neurons as early as E12.8 (i.e., at 6:00 P.M. on day 12 after vaginal plugging) (Fig. 4B). Interestingly, it appeared that Tuj1-positive neurons migrated through a linear break of the pial BM (Fig. 4B, white arrowhead). By EM analysis, we detected a sheet of pial BM together with the attached radial glial endfeet folded over by a herd of overmigrating neurons (Fig. 4D′, red arrows).
Figure 4.
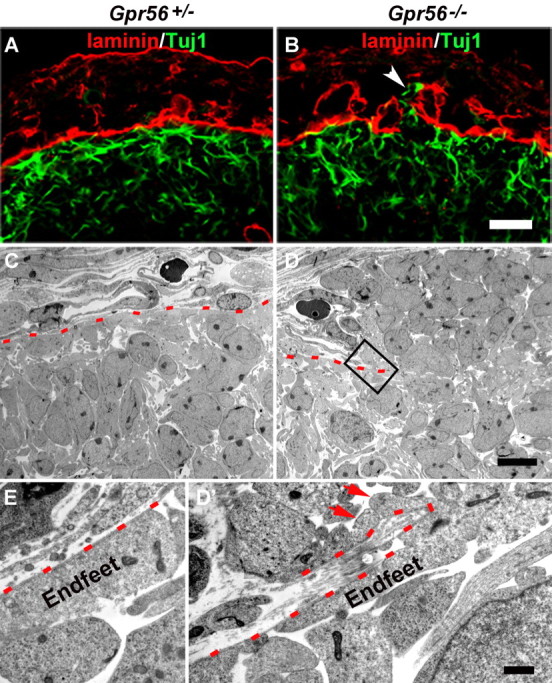
Regional breaks of the pial BM in the absence of GPR56. A, B, Double IHC of laminin (red) and Tuj1 (green) at embryonic stage E12.8 revealing the continuous pial BM and the migrating neurons underneath in a heterozygous brain (A), whereas a linear break of the pial BM and ectopic neurons were detected in a Gpr56 mutant (B, arrow). C–E, Electron-microscopic views at E13.5 showing the continuous pial BM in heterozygous brain (C, red dashed line) and ectopic neurons migrating out through the broken pial BM in Gpr56 knock-out brain (D). D′, Higher-magnification view of the boxed area in D showing radial glial endfeet extending out (arrows) through the broken pial BM (red dashed line). E, Higher-magnification view of the normal attachment of radial glial endfeet to the pial BM in heterozygous brain. Scale bars: (in B) A, B, 20 μm; (in D) C, D, 10 μm; (in D′) D′, E, 1 μm.
GPR56 mutation leads to abnormal position of radial glial endfeet
Radial glial endfeet attach to the BM and form an adhesive lining at the pial surface (Rakic, 2003). Defects in pial BM could lead to abnormal anchorage of radial glial endfeet. To test this hypothesis, we investigated whether loss of GPR56 leads to disorganized/displaced radial glial endfeet by probing for RC2 and BLBP in E12.8 and E15.5 brains, respectively. In control brains, radial glial processes were aligned in parallel arrays throughout the cerebral wall, terminating in well defined endfeet at the pial BM, which was visualized by laminin or collagen IV staining (Fig. 5A,C). In mutant developing cortex, however, the radial glial fibers were disorganized and extended beyond the pia mater into the neuronal ectopia in the areas where the BM was disrupted (Fig. 5B,D,F, arrowheads). Although we could not completely exclude the presence of retracted endfeet, the predominant abnormalities were protruded endfeet that extended through the defective BM.
Figure 5.
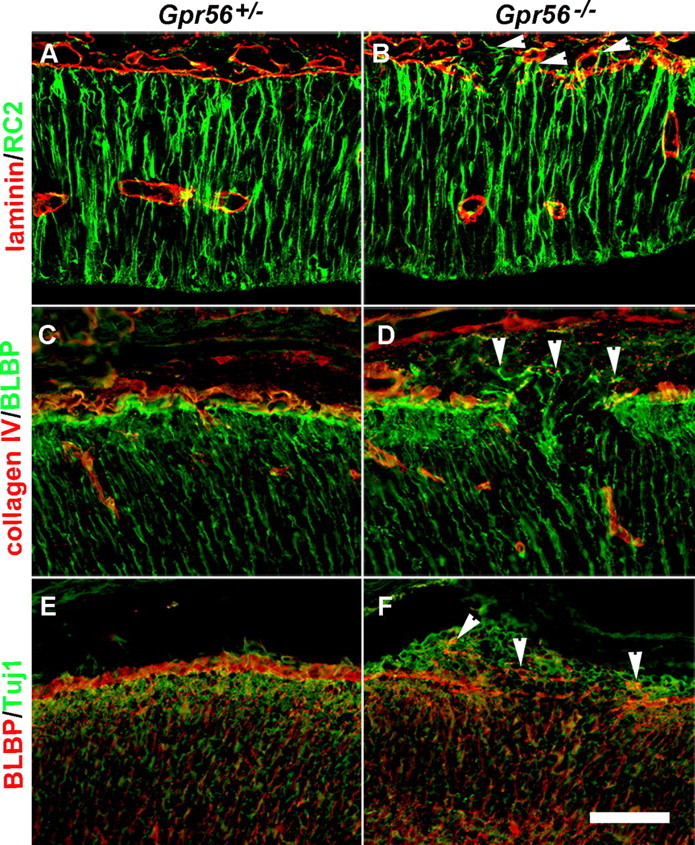
Abnormally positioned radial glial endfeet in Gpr56 mutant mice. A, B, E12.8 coronal section through mouse forebrain showing that radial glial processes (green) extend through the cortical wall and terminate at the continuous BM (red) in Gpr56+/− brain. In contrast, the radial glial processes and endfeet protruded through the defective BM in mutants (B, arrowheads). C, D, Double-label IHC of BLBP (green) and Col IV (red) on E15.5 coronal sections. The endfeet were extended through the defective BM in mutants (D, arrowheads). E, F, Double-label IHC of BLBP (red) and Tuj1 (green) on E15.5 brain sections. The endfeet were extended into the neuronal ectopia in mutants (F, arrowheads). In contrast, a continuous lining of the endfeet on the outer surface of the migrating neurons was found in control animals (E). Scale bar, 50 μm.
Loss of GPR56 leads to CR cell malpositioning
CR cells secrete reelin, an ECM signaling molecule that is critical for proper positioning of postmitotic neurons during cortical development (Tissir and Goffinet, 2003). To determine whether CR cells are abnormally located in developing Gpr56−/− cortex, we compared the distribution of CR cells in brains of Gpr56+/− and Gpr56−/− animals using both reelin and calretinin to identify CR cells. In contrast with the relatively continuous single layer of CR cells at the marginal zone in E12.8 control animals (Fig. 6A,C), we found CR cells located beyond the pial surface on the defective BM and on the peak of the ectopic neuronal clusters (Fig. 6B,D, arrows). In E16.5 brains, in contrast with a single-cell layer of well defined CR neurons at the outer surface of the cortex in control animals (Fig. 6E,G), we observed scattered CR cells within the ectopia (Fig. 6F,H). There was a CR cell gap at the stem of the ectopia where a stream of neurons were migrating through (Fig. 6F,H, between the arrows).
Figure 6.
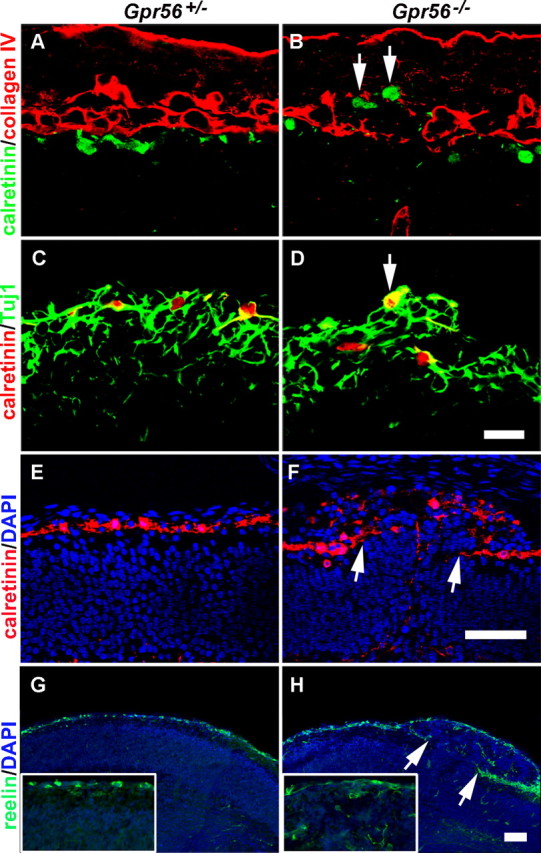
Displaced CR cells in Gpr56 mutant mice. A, B, Calretinin/Col IV double-label IHC on E12.8 brain sections revealed a single layer of CR cells beneath the BM in the heterozygous brain, whereas ectopically placed CR cells were observed in the subarachnoid space at the region of defective BM in the homozygous brain (B, arrows). C, D, Double-label IHC of calretinin (red) and Tuj1 (green) on coronal sections of E12.8 brains. A single layer of CR cells outlined the surface of Tuj1-positive neurons in heterozygous cortex (C), whereas a bump of overmigrated neurons was seen in homozygous neocortex with one CR cell placed on the peak of the ectopia (D, arrow). E–H, Calretinin and reelin IHC on E16.5 coronal sections of heterozygous and mutant brains showing a single layer of CR cells continuously outlining the cortical surface in control animals (E, G), and in contrast, mutant brain showing scattered CR cells within the ectopia (F, H). No CR cells were detected at the stem, where a stream of neurons were migrating through (between the arrows in F and H). Scale bars: (in D) A–D, 20 μm; (in F, H) E–H, 50 μm.
Strong GPR56 expression in radial glial endfeet
Having established a role for GPR56 in BM organization/maintenance, we next investigated whether GPR56 protein is present in the radial glial endfeet. Double immunostaining with antibodies against GPR56 and Col IV revealed GPR56 signals terminating at the Col IV-positive BM at E16.5 (Fig. 7C). We then performed double immunostaining of GPR56 and GLAST, as well as BLBP and GPR56 on adjacent sections because both antibodies are rabbit antisera. We observed strong GPR56 immunoreactivity in the radial glial endfeet (Fig. 7D–H). Thus, our data support the hypothesis that GPR56 protein is trafficked to radial glial endfeet for its potential function of organizing and/or maintaining the BM.
Figure 7.

GPR56 protein is present in radial glial endfeet. A–C, Double-label IHC of GPR56 (green) and Col IV (red) on E16.5 wild-type cortex showing GPR56 immunoreactivities terminate at the pial BM. D–F, Double-label IHC of GPR56 (green) and GLAST (red) on E16.5 wild-type cortex. GPR56 immunoreactivities were colocalized with GLAST in the radial glial endfeet (F). G, H, GPR56 and BLBP IHC on adjacent sections of E16.5 wild-type cortex, revealing that GPR56 immunoreactivity (G) was observed at the pial surface, corresponding to the position of radial glial endfeet outlined by BLBP on the adjacent section (H). Scale bar, 20 μm.
GPR56 binds a putative ligand in pial BM
GPR56 is an orphan G-protein-coupled receptor. To investigate the distribution of putative GPR56 ligands in the developing cortex, we engineered a fusion construct in which mouse IgG Fc tag was fused to the C terminus of GPR56N as illustrated in Figure 8A. The fusion construct was used to transfect HEK-293T cells. Secreted GPR56N-mFc protein in the conditioned medium was collected, concentrated, and verified by Western blot (Fig. 8B). The concentrated GPR56N-mFc protein was used to probe mouse E12.5 and E14.5 brains with the mFc protein as negative control. GPR56N-mFc fusion protein was found to bind the overlying pial surface of the cerebral cortex at all embryonic stages investigated (Fig. 8E,H). This binding pattern was highly specific and could be detected with GPR56N-mFc preparations as low as 40 μg/ml. In contrast, no signal was detected using unfused mFc preparations as high as 500 μg/ml (Fig. 8C,F). The GPR56N-mFc binding pattern likely indicates a local binding partner/GPR56 interaction at the marginal zone or overlying extracellular matrix that is necessary for regulating the pial BM integrity. To confirm the GPR56 binding partner was present in the pial basement membrane, we performed double immunostaining with GPR56N-mFc and laminin, a basement membrane constituent. As expected, the GPR56N-mFc staining was well colocalized with laminin staining in the pial basement membrane (supplemental Fig. 7A, available at www.jneurosci.org as supplemental material).
Figure 8.

GPR56 binds a putative ligand in pial basement membrane. A, Schematic drawing of the fusion constructs. An in-frame deletion mutation was created by deleting amino acids 93–143, GPR56Ndel. Mouse IgG Fc tag was fused to the C terminus of GPR56N or GPR56Ndel to form a fusion construct. B, The fusion constructs were transfected into HEK-293T cells. Secreted proteins in the conditioned media were collected, concentrated, and verified by Western blot. C–H, GPR56N-mFc fusion protein in situ. GPR56N-mFc binds a putative ligand in the ECM at both E12.5 (E) and E14.5 (H) brains, whereas mFc (C, F) and GPR56Ndel-mFc (D, G) controls revealed no binding. Nuclear counterstain was performed by Hoechst 33342 (blue). Scale bars, 100 μm.
To further verify the specificity of above binding pattern, we created a mutant fusion protein by deleting amino acids 93–143, GPR56Ndel-mFc (Fig. 8A). The mutant fusion construct was transfected into HEK-293T cells to produce GPR56Ndel-mFc fusion protein. Western blot analysis revealed a glycosylated and secreted GPR56Ndel-mFc protein that is ∼5 kDa smaller than the wild-type GPR56N-mFc (Fig. 8B). There was no specific staining when GPR56Ndel-mFc fusion proteins were used to probe mouse E12.5 and E14.5 brain sections, thus confirming that the binding pattern revealed by GPR56N-mFc is highly specific (Fig. 8D,G).
To investigate whether the organization of the putative ligand is disrupted in the absence of GPR56, we performed GPR56N-mFc staining in E14.5 Gpr56−/− and Gpr56+/− littermate brain sections. In contrast with the smooth, continuous GPR56N-mFc binding in control animals, we found breakdown and fragmented GPR56N-mFc binding in the regions of ectopias (supplemental Fig. 7B,C, available at www.jneurosci.org as supplemental material). These results supported that GPR56 binds a putative ligand in the pial basement membrane and the organization of this putative ligand is disrupted within the ectopic regions of Gpr56−/− brains.
Discussion
BFPP is a radiological diagnosis. Its pathology is unclear and extremely difficult to study because of the unavailability of brain specimens from affected individuals. Genotype–phenotype analysis in patients with BFPP and other similar polymicrogyria syndromes, however, indicated that BFPP brains have features of cobblestone-like brain malformations (i.e., polymicrogyria, cerebellar hypoplasia, and white matter defect) (Piao et al., 2005; Piao and Walsh, 2007). Here, we demonstrate in a Gpr56 knock-out mouse model that the histopathology of BFPP is indeed a cobblestone-like cortical malformation.
The role of GPR56 in pial BM organization
The pial BM is a specialized ECM that is formed adjacent to the radial glial endfeet. Meningeal fibroblasts contribute to the BM by secreting and organizing the majority of basal lamina constituents (Sievers et al., 1994). The sites of ECM protein synthesis and BM assembly are spatially distinct, suggesting cell-directed mechanisms that can specify the sites of ECM assembly. Indeed, emerging evidence indicates that receptors on the cell surface, such as integrins and dystroglycan, can orchestrate the assembly of the ECM (Schwarzbauer, 1999).
In this study, we establish the role of GPR56 in regulating the pial BM integrity by demonstrating that (1) GPR56 is present in abundance in radial glial endfeet (Fig. 7C,F,G); (2) GPR56 binds a putative ligand in the ECM (Fig. 8E,H); and (3) loss of GPR56 in mice result in defective pial BM and neuronal ectopia, a cobblestone-like cortex. However, it remains to be determined whether GPR56 exerts its function by regulating ECM assembly or remodeling. The presence of intact BM in Gpr56 mutant mice during early embryonic stages suggests that the initial assembly of the pial BM occurs in the absence of GPR56 (Fig. 3C). Regional pial BM breakdown starts at E12.8, concurrent with the beginning of corticogenesis and cortical wall expansion. Therefore, it is possible that the ECM polymers are fragile in the absence of GPR56 and cannot sustain the tension generated by the expanding cortical wall. Indeed, we detected linear breaks in Gpr56−/− cortex at E12.8 (Fig. 4B) and folded pial BM together with the attached radial glial endfeet at E13.5 (Fig. 4D). Alternatively, loss of GPR56 could cause disturbed homeostasis in ECM remodeling. ECM remodeling is critical for many developmental processes as well as tumor metastasis. The establishment of homeostasis in ECM synthesis and proteolytic degradation is the key in ECM maintenance (Larsen et al., 2006).
GPR56 regulates the proper anchorage of radial glial endfeet
In the developing cerebral cortex, radial glial cells serve dual functions as progenitor cells that give rise to neurons as well as “guiding wires” that provide the organizing framework for corticogenesis (Malatesta et al., 2003; Rakic, 2003). Radial glial cells have their somata in the ventricular zone and extend their long radial processes through the entire developing cortex to terminate at the pial BM. The pial BM is formed at the interface between radial glial endfeet and the pial meninges. It is the consensus in the field that the leading pathology of cobblestone cortex is the pial BM disruption (Olson and Walsh, 2002). Although it is inconsistent in the literature whether radial glial endfeet are retracted or protruded at regions of defective BM, it is clear that the normal anchorage of radial glial endfeet is relevant to the presence of intact pial BM (Georges-Labouesse et al., 1998; Graus-Porta et al., 2001; Halfter et al., 2002; Beggs et al., 2003; Niewmierzycka et al., 2005; Haubst et al., 2006; Hu et al., 2007).
In this study, we demonstrate that deletion of the mouse Gpr56 gene predominantly causes protrusion of endfeet beyond the pial surface, coinciding with regions of discontinuous BM (Fig. 5B,D,F). Given the fact that GPR56 is expressed abundantly in radial glial endfeet and that it binds a putative ligand in the ECM (Figs. 7, 8), we postulate that GPR56 regulates the proper anchorage of radial glial endfeet. Consistent with the literature, our data demonstrate that intact pial BM is essential for the existence of normal anchorage of radial glial endfeet and CR cell position (Figs. 5, 6).
A new signaling pathway in pial BM organization
Studies of human cobblestone cortex and mouse models of cobblestone-like malformations have led to the identification of many molecules involved in pial BM assembly/maintenance, including ECM constituents (laminin, nidogen, and perlecan) and laminin receptors (α-dystroglycan and some integrin family members) (Georges-Labouesse et al., 1998; Costell et al., 1999; De Arcangelis et al., 1999; Graus-Porta et al., 2001; Halfter et al., 2002; Beggs et al., 2003; Niewmierzycka et al., 2005; Haubst et al., 2006; Hu et al., 2007). Deletion of the major laminin receptors, α3β1 and α6β1 integrins, results in severe disruption of pial BM and cortical lamination defects, indicating a role for laminin–integrin interactions in the maintenance of the BM and in cortical development (De Arcangelis et al., 1999; Graus-Porta et al., 2001). Here, we introduce a new receptor, GPR56, whose ligand in the pial BM is yet unknown, into this equation. It is to be determined whether GPR56 interacts with α-dystroglycan and/or integrins in regulating the same developmental process.
The GPR56 signaling pathway remains mostly unknown. Recently, GPR56N was shown to bind tissue transglutaminase, TG2 (Xu et al., 2006). However, it is not clear whether TG2 functions as a traditional ligand for GPR56. Moreover, it has been shown that GPR56 associates specifically with Gαq/11 and Gβ as part of a larger complex with tetraspanins CD9 and CD81 (Little et al., 2004). The function of this tetraspanin–GPR56 complex remains unclear. Members of the tetraspanin family of cell surface proteins act as molecular scaffolds with known adhesion proteins such as integrins to facilitate their function (Levy and Shoham, 2005). It is possible that GPR56 functions in a receptor complex with tetraspanins and integrins in regulating/maintaining pial BM integrity.
Footnotes
This work was supported in part by The Richard and Sara Page Mayo Fund (X.P.), National Institute of Neurological Disorders and Stroke (NINDS) Grants K08 NS045762 and R01 NS057536 (X.P.), a Child Health Research Grant from The Charles H. Hood Foundation (X.P.), NINDS Grant R01 NS35884 (G.C.), National Institutes of Health (NIH) Mental Retardation Research Center Grant P30-HD 18655 (G.C.), a Flight Attendant Medical Research Institute Young Clinical Scientist Award (Z.J.), and Fundamental Neurobiology NIH Training Grant T32 NS007484-06 (S.K.). We are grateful to Dr. Anthony Hill for his assistance during the initial phase of this study. We thank Dr. H. Lin for his gift of mFc/pCDNA3.1 vector and mFc/pSecTap2 construct, Dr. J. Cunningham for Cux1 antibody, Dr. A. Chung for nidogen antibody, Dr. R. Hevner for Tbr1 antibody, and the Neurobiology Program Imaging Center for assistance with confocal microscopy. The Gpr56 knock-out mice, kindly provided by Genentech, were produced in collaboration between Genentech and Lexicon Genetics to analyze the function of ∼500 secreted and transmembrane proteins.
References
- Anton ES, Kreidberg JA, Rakic P. Distinct functions of alpha3 and alpha (v) integrin receptors in neuronal migration and laminar organization of the cerebral cortex. Neuron. 1999;22:277–289. doi: 10.1016/s0896-6273(00)81089-2. [DOI] [PubMed] [Google Scholar]
- Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron. 2003;40:501–514. doi: 10.1016/s0896-6273(03)00666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BS, Piao X, Bodell A, Basel-Vanagaite L, Straussberg R, Dobyns WB, Qasrawi B, Winter RM, Innes AM, Voit T, Grant PE, Barkovich AJ, Walsh CA. Bilateral frontoparietal polymicrogyria: clinical and radiological features in 10 families with linkage to chromosome 16. Ann Neurol. 2003;53:596–606. doi: 10.1002/ana.10520. [DOI] [PubMed] [Google Scholar]
- Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, Fassler R. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–1122. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Arcangelis A, Mark M, Kreidberg J, Sorokin L, Georges-Labouesse E. Synergistic activities of alpha3 and alpha6 integrins are required during apical ectodermal ridge formation and organogenesis in the mouse. Development. 1999;126:3957–3968. doi: 10.1242/dev.126.17.3957. [DOI] [PubMed] [Google Scholar]
- Feng L, Hatten ME, Heintz N. Brain lipid-binding protein (BLBP): a novel signaling system in the developing mammalian CNS. Neuron. 1994;12:895–908. doi: 10.1016/0896-6273(94)90341-7. [DOI] [PubMed] [Google Scholar]
- Feng Y, Walsh CA. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron. 2004;44:279–293. doi: 10.1016/j.neuron.2004.09.023. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse E, Mark M, Messaddeq N, Gansmuller A. Essential role of alpha 6 integrins in cortical and retinal lamination. Curr Biol. 1998;8:983–986. doi: 10.1016/s0960-9822(98)70402-6. [DOI] [PubMed] [Google Scholar]
- Gonzalez JL, Russo CJ, Goldowitz D, Sweet HO, Davisson MT, Walsh CA. Birthdate and cell marker analysis of scrambler: a novel mutation affecting cortical development with a reeler-like phenotype. J Neurosci. 1997;17:9204–9211. doi: 10.1523/JNEUROSCI.17-23-09204.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Halfter W, Dong S, Yip YP, Willem M, Mayer U. A critical function of the pial basement membrane in cortical histogenesis. J Neurosci. 2002;22:6029–6040. doi: 10.1523/JNEUROSCI.22-14-06029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubst N, Georges-Labouesse E, De Arcangelis A, Mayer U, Gotz M. Basement membrane attachment is dispensable for radial glial cell fate and for proliferation, but affects positioning of neuronal subtypes. Development. 2006;133:3245–3254. doi: 10.1242/dev.02486. [DOI] [PubMed] [Google Scholar]
- Hu H, Yang Y, Eade A, Xiong Y, Qi Y. Breaches of the pial basement membrane and disappearance of the glia limitans during development underlie the cortical lamination defect in the mouse model of muscle-eye-brain disease. J Comp Neurol. 2007;501:168–183. doi: 10.1002/cne.21238. [DOI] [PubMed] [Google Scholar]
- Jin Z, Tietjen I, Bu L, Liu-Yesucevitz L, Gaur SK, Walsh CA, Piao X. Disease-associated mutations affect GPR56 protein trafficking and cell surface expression. Hum Mol Genet. 2007;16:1972–1985. doi: 10.1093/hmg/ddm144. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K, Osawa M, Hamano K, Sakakihara Y, Nonaka I, Nakagome Y, Kanazawa I, Nakamura Y, Tokunaga K, Toda T. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- Larsen M, Artym VV, Green JA, Yamada KM. The matrix reorganized: extracellular matrix remodeling and integrin signaling. Curr Opin Cell Biol. 2006;18:463–471. doi: 10.1016/j.ceb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Levy S, Shoham T. The tetraspanin web modulates immune-signalling complexes. Nat Rev Immunol. 2005;5:136–148. doi: 10.1038/nri1548. [DOI] [PubMed] [Google Scholar]
- Little KD, Hemler ME, Stipp CS. Dynamic regulation of a GPCR-tetraspanin-G protein complex on intact cells: central role of CD81 in facilitating GPR56-Galpha q/11 association. Mol Biol Cell. 2004;15:2375–2387. doi: 10.1091/mbc.E03-12-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malatesta P, Hack MA, Hartfuss E, Kettenmann H, Klinkert W, Kirchhoff F, Gotz M. Neuronal or glial progeny: regional differences in radial glia fate. Neuron. 2003;37:751–764. doi: 10.1016/s0896-6273(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, Dollar J, Nishino I, Kelley RI, Somer H, Straub V, Mathews KD, Moore SA, Campbell KP. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, Macklis JD. Fezl is required for the birth and specification of corticospinal motor neurons. Neuron. 2005;47:817–831. doi: 10.1016/j.neuron.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Nieto M, Monuki ES, Tang H, Imitola J, Haubst N, Khoury SJ, Cunningham J, Gotz M, Walsh CA. Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II-IV of the cerebral cortex. J Comp Neurol. 2004;479:168–180. doi: 10.1002/cne.20322. [DOI] [PubMed] [Google Scholar]
- Niewmierzycka A, Mills J, St-Arnaud R, Dedhar S, Reichardt LF. Integrin-linked kinase deletion from mouse cortex results in cortical lamination defects resembling cobblestone lissencephaly. J Neurosci. 2005;25:7022–7031. doi: 10.1523/JNEUROSCI.1695-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EC, Walsh CA. Smooth, rough and upside-down neocortical development. Curr Opin Genet Dev. 2002;12:320–327. doi: 10.1016/s0959-437x(02)00305-2. [DOI] [PubMed] [Google Scholar]
- Piao X, Walsh CA. GPR56 and bilateral frontoparietal polymicrogyria. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn errors of development: the molecular basis of clinical disorders of morphogenesis. New York: Oxford UP; 2007. pp. 1309–1312. [Google Scholar]
- Piao X, Basel-Vanagaite L, Straussberg R, Grant PE, Pugh EW, Doheny K, Doan B, Hong SE, Shugart YY, Walsh CA. An autosomal recessive form of bilateral frontoparietal polymicrogyria maps to chromosome 16q12.2–21. Am J Hum Genet. 2002;70:1028–1033. doi: 10.1086/339552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao X, Hill RS, Bodell A, Chang BS, Basel-Vanagaite L, Straussberg R, Dobyns WB, Qasrawi B, Winter RM, Innes AM, Voit T, Ross ME, Michaud JL, Déscarie JC, Barkovich AJ, Walsh CA. G protein-coupled receptor-dependent development of human frontal cortex. Science. 2004;303:2033–2036. doi: 10.1126/science.1092780. [DOI] [PubMed] [Google Scholar]
- Piao X, Chang BS, Bodell A, Woods K, Benzeev B, Topcu M, Guerrini R, Goldberg-Stern H, Sztriha L, Dobyns WB, Barkovich AJ, Walsh CA. Genotype-phenotype analysis of human frontoparietal polymicrogyria syndromes. Ann Neurol. 2005;58:680–687. doi: 10.1002/ana.20616. [DOI] [PubMed] [Google Scholar]
- Rakic P. Elusive radial glial cells: historical and evolutionary perspective. Glia. 2003;43:19–32. doi: 10.1002/glia.10244. [DOI] [PubMed] [Google Scholar]
- Schwarzbauer J. Basement membranes: putting up the barriers. Curr Biol. 1999;9:R242–R244. doi: 10.1016/s0960-9822(99)80153-5. [DOI] [PubMed] [Google Scholar]
- Sievers J, Pehlemann FW, Gude S, Berry M. Meningeal cells organize the superficial glia limitans of the cerebellum and produce components of both the interstitial matrix and the basement membrane. J Neurocytol. 1994;23:135–149. doi: 10.1007/BF01183867. [DOI] [PubMed] [Google Scholar]
- Stacey M, Chang GW, Davies JQ, Kwakkenbos MJ, Sanderson RD, Hamann J, Gordon S, Lin HH. The epidermal growth factor-like domains of the human EMR2 receptor mediate cell attachment through chondroitin sulfate glycosaminoglycans. Blood. 2003;102:2916–2924. doi: 10.1182/blood-2002-11-3540. [DOI] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM. Reelin and brain development. Nat Rev. 2003;4:496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- Xu L, Begum S, Hearn JD, Hynes RO. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc Natl Acad Sci USA. 2006;103:9023–9028. doi: 10.1073/pnas.0602681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Kato Y, Kawaguchi M, Shibata N, Kobayashi M. Expression and localization of fukutin, POMGnT1, and POMT1 in the central nervous system: consideration for functions of fukutin. Med Electron Microsc. 2004;37:200–207. doi: 10.1007/s00795-004-0260-5. [DOI] [PubMed] [Google Scholar]
- Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, Inazu T, Mitsuhashi H, Takahashi S, Takeuchi M, Herrmann R, Straub V, Talim B, Voit T, Topaloglu H, Toda T, Endo T. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1:717–724. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]