

Abstract
Cells lacking the protein kinase ataxia telangiectasia mutated (ATM) have defective responses to DNA double strand breaks (DSBs) including an inability to activate damage response proteins such as p53. However, we previously showed that cells lacking ATM robustly activate p53 in response to DNA strand breaks induced by the radiomimetic enediyne C-1027. To gain insight into the nature of C-1027-induced ATM-independent damage responses to DNA DSBs, we further examined the molecular mechanisms underlying the cellular response to this unique radiomimetic agent. Similar to ionizing radiation (IR) and other radiomimetics, breaks induced by C-1027 efficiently activate ATM by phosphorylation at Ser1981. Yet unlike other radiomimetics and IR, DNA breaks induced by C-1027 results in normal phosphorylation of p53 and the cell cycle checkpoint kinases (Chk1 and Chk2) in the absence of ATM. In the presence of ATM, but under ATM and Rad3-related kinase (ATR) deficient conditions, C-1027 treatment resulted in a decrease of Chk1 phosphorylation but not in p53 and Chk2 phosphorylation. Only when cells were deficient in both ATM and ATR, was there a reduction in phosphorylation of each of these DNA damage response proteins. This reduction was also accompanied by increased cell death in comparison to wild-type cells or cells lacking either ATM or ATR. Our findings demonstrate a unique cellular response to C-1027-induced DNA DSBs in that DNA damage response proteins are unaffected by the absence of ATM, as long as ATR is present.
Keywords: DNA double strand breaks, C-1027, radiomimetic, enediyne, ATM, ATR
MANUSCRIPT TEXT
The successful detection of and response to DNA DSBs, is crucial for cellular maintenance of genomic stability. Therefore, cells utilize a complex network of DNA strand break sensors, signal transmitters and effectors to optimize DNA repair prior to mitosis; helping to avoid the consequences of genomic damage such as apoptosis, mitotic catastrophe and irreversible growth arrest (1). Our understanding of DNA DSBs damage response mechanisms is primarily based upon IR studies. Following IR treatment, cells activate the phosphatidylinositol 3-kinase-like protein kinase (PIKK)1 ATM through an autophosphorylation event (2). Activated ATM initiates a kinase cascade leading to cell cycle checkpoint responses and recruitment of repair proteins to sites of damaged DNA. For example, Chk2, a protein kinase directly phosphorylated by ATM at Thr68, mediates phosphorylation of Cdc25C and Cdk1, resulting in a G2/M checkpoint (3). Another important downstream target of ATM following IR treatment is p53-Ser15 phosphorylation, which mediates the G1/S checkpoint (4-6). Cells without ATM function cannot efficiently activate checkpoint responses to IR-induced DNA DSBs, and are hypersensitive to IR treatment in regard to cell death and growth inhibition (7-9).
A complimentary approach to study PIKK responses to DNA DSBs utilizes radiomimetics such as enediynes. In comparison to IR, which induces multiple types of DNA damage (e.g. thymidine dimers, purine deamination, single-strand breaks (SSBs) and DSBs), enediynes generally induce only DNA strand breaks (both SSBs and DSBs) (10, 11). Despite differences in the nature of the DNA damage, cellular responses to treatment with enediynes such as neocarzinostatin (NCS), share hallmarks of the IR-induced damage response including ATM-dependent activation of p53-Ser15, Nbs1-Ser343 and Chk2-Thr68 (12-14), and cells lacking the ATM kinase are more readily killed by radiomimetic treatment than are ATM wild-type cells (11, 15). Furthermore, enediynes have helped to define the nature of the ATM response to DNA breaks. For example, a study comparing damage induced by IR and calicheamicin, an enediyne known to induce almost exclusively DSBs, has shown that ATM activation strongly correlates with DSBs and not SSBs (16). Enediyne studies have also led to the discovery of DNA DSB response pathways that do not strictly follow the IR model. We have shown that C-1027 treatment induces activation of p53-Ser15 in the absence of ATM and that C-1027-induced cell growth inhibition does not increase in cells lacking ATM nor is radioresistant DNA synthesis observed (17).
Damage responses mechanisms have also been identified that show while ATM is the primary PIKK to respond to DNA DSBs, ATR can play a role albeit the response is typically much more limited. The ATM-Chk2 and ATR-Chk1 pathways both can activate G2 checkpoints by inducing inhibition of Cdc25C phosphatase (3, 18). A recent study has now confirmed that either ATM or ATR can phosphorylate p53-Ser15 as well as the Chk kinases in response to IR, although generally the induced phosphorylation signals were significantly lower when cells are deficient in either of the kinases (8). In contrast, after C-1027 treatment, cells lacking either ATM or ATR can still fully phosphorylate p53-Ser15 (17).
This study elaborates on the nature of C-1027-induced DNA DSB damage responses to identify additional cellular responses to DNA DSBs that are not solely dependent upon ATM. We have found that, similar to the cellular responses to IR or NCS treatment, ATM-Ser1981 is rapidly phosphorylated after C-1027 treatment. Yet, contrary to the IR-mimic NCS, DNA damage induced by C-1027 does not requires ATM to phosphorylate Chk2-Thr68 and Chk1-Ser345. Furthermore, in response to C-1027-induced DNA DSBs, we found that ATR status does not affect Chk2 phosphorylation and only partially affects Chk1 phosphorylation. However, the C-1027-induced damage response requires either ATM or ATR, as simultaneous loss of both kinases results in diminished p53 phosphorylation and nearly a complete loss of Chk1 and Chk2 phosphorylation. Additionally, increased cell death in response to C-1027 treatment is only observed when both PIKKs are compromised.
MATERIALS AND METHODS
Chemicals
C-1027, a gift from Taiho Pharmaceuticals Co. (Saitama, Japan) was stored at 40 μM at 4°C in ddH2O and neocarzinostatin, a gift from Bristol-Myers Squib Co. (Syracuse, NY) was stored at 200 μM at 4°C in ddH2O.
Cells
Isogenic ATM null (AT169A) and ATM restored (YZ510B) human fibroblast cell lines, a gift of Drs. P. Lu and Y. Shiloh, were grown at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum plus 50 μg/ml hygromycin to maintain selective pressure on the transfected constructs (19). Wild-type GM00536 lymphoblast cells2 were grown at 37°C and 5% CO2 in RPMI 1640 medium supplemented with 10% fetal bovine serum.
siRNA
Oligofectamine (Invitrogen, Carlsbad, CA) was incubated at 30°C for 20 min in the presence or absence of siRNA targeted against ATR (Dharmacon Inc., Lafayette, CO) before addition to cells that had been incubated in serum-deficient medium. The siRNA was targeted to CCTCCGTGATGTTGCTTGA, which corresponds to nucleotides 296-314 of the ATR gene (20), and has previously been shown to be effective and selective against ATR in these cells (21). Control siRNA sequences were also examined to ensure that the knockdown was specific for ATR. After 4 h, an equal volume of medium supplemented with 20% fetal bovine serum was added to return serum levels to normal. Fourty-eight h after siRNA treatment, cells were treated for 1 h with C-1027 and used in subsequent assays.
Immunoblotting
Cells with and without siRNA treatment were incubated with drugs at 37 °C for 1 h, harvested and washed in phosphate-buffered saline. Total cellular extracts were prepared by directly adding SDS page buffer or by incubating cells on ice in lysis buffer (50 mM Hepes, pH 7.6, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, 0.5% Triton X-100, 50 mM sodium fluoride, 1 mM sodium o-vanadate, 1 mM β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitors) for 15 min. The cell lysates were cleared by centrifugation, and protein content was determined using the Bradford method (Bio-Rad). Equal amounts of protein were electrophoresed on SDS-polyacrylamide gels and transferred to a PVDF membrane. The membranes were probed with primary antibodies, followed by secondary antibodies conjugated with horseradish peroxidase. The primary antibodies used were anti-phospho-p53-Ser15, anti-phospho-Chk1-Ser345, and anti-phospho-Chk2-Thr68 (Cell Signaling Technology, Beverly, MA), anti-phospho-ATM-Ser1981 (Rockland Immunochemicals, Gilbertsville, PA) anti-ATR (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-β-actin (Sigma, St. Louis, MO). Protein bands were visualized by enhanced chemiluminescence, and phosphorylation levels were measured using a Personal Densitometer SI (Amersham Biosceinces, Piscataway, NJ).
Growth Inhibition Assay
Cells in 6-well dishes were seeded at 2.5*105 cells/well and were drug treated 24 h later. Following a 3-day incubation, dishes were washed and cells were counted by coulter counter (Beckman Coulter Inc., Fullerton, CA). Cell growth inhibition was calculated by comparing the number of treated to non-treated control cells.
Trypan Blue Exclusion
Cells growing in 6-well dishes were seeded at 2.5*105 cells/well. After 24 h, the cells were drug treated. The cells were then incubated for 24 h at which point the medium and cells were collected, spun and incubated with a 0.4% solution of trypan blue for 10 min at RT. Cell death was based on the percentage of trypan stained cells in samples containing 300 or more cells.
Genomic DNA Damage
Following a 30 min drug treatment and harvest, cells were resuspended in PBS at a concentration of 1*107/mL. Cells were then diluted into 0.5% low melting temperature agarose and placed on a frosted slide and covered with a coverslip for 2 h at 4°C. The slide was then immersed in alkaline lysis buffer (2.5 M NaCl. 0.1 M EDTA, 50 mM Tris, pH 10.0, 10% DMSO, 1% Sarcosyl, and 1% Triton X-100) in water overnight at 4°C. The slides were then placed in electrophoresis buffer (30 mM NaOH, 0.5 M EDTA at pH 13.0) and equilibrated for 20 min before electrophoresis at 27 V for 25 min at 4°C. The slides were then placed in 0.4 M Tris, pH 7.5, 3X for 5 min, followed by immersion in 100% methanol, and then ethanol. The slides were dried and stained with 15 μg/mL of ethidium bromide and covered by a coverslip. DNA damage was assessed as described elsewhere (22).
RESULTS
ATM is activated by C-1027-induced DNA breaks
ATM-regulated DNA damage responses after IR and radiomimetic treatment are well-characterized. To test whether the unusual ATM-independent damage responses to C-1027 is related to a diminished ability to activate ATM we tested whether low levels of C-1027 resulted in ATM-Ser1981 autophosphorylation (Fig. 1). Similar to IR-induced responses, ATM was activated within minutes using exceedingly low levels (300 fM) of C-1027 treatment. NCS, another enediyne known to induce IR-like DNA damage responses, also activated ATM at concentrations that induce low levels of DNA breaks, consistent with the findings by others (14).
Fig. 1.

C-1027 treatment induces ATM activation. Wild-type human lymphoblast cells were treated with 0, 0.3 or 3 pM C-1027 or 0.3 and 3 nM NCS for 15 min at 37°C, and the cellular extracts were analyzed by western blotting. Immunoblots were then probed with an antibody specific for phosphorylated ATM-Ser1981.
Verification that C-1027 can induce p53-Ser15 phosphorylation independently of ATM
In our previous study using IR or C-1027 treatment, p53-Ser15 phosphorylation was significantly reduced in an ATM-null line for the former but not the latter, when compared to non-isogenic ATM-wild-type cells (17). Also unlike IR, C-1027 treatment resulted in similar cytotoxicity regardless of ATM status (17). However, since the cell lines were not isogenic, we could not rule out that the observed p53 phosphorylation in the ATM-null cells was affected by the differing genotypes. To confirm that C-1027-induced DNA damage responses could be activated in the absence of ATM, p53-Ser15 phosphorylation was measured in isogenic ATM-wild-type and null cells after C-1027 treatment. We found little difference in p53-Ser15 phosphorylation between the two cell lines in response to C-1027 treatment, while as expected, relative to the ATM-wild-type line, p53-Ser15 phosphorylation in the ATM-null cells was substantially reduced following NCS treatment (Fig. 2). Similarly, we found that growth of the isogenic ATM-null cells when compared to ATM-wild-type cells was more inhibited by NCS but not by C-1027 (17, 19) (Fig 3).
Fig. 2.
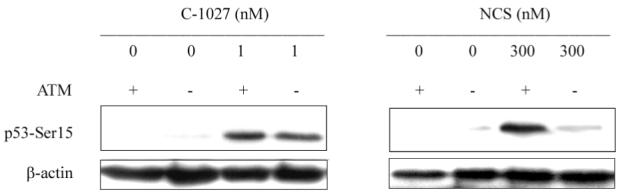
C-1027 induces ATM-independent p53-Ser15 phosphorylation. ATM-null and restored human fibroblast cells were treated with 0 or 1 nM C-1027, or 300 nM NCS for 1 h at 37°C and the cellular extracts were analyzed by western blotting. Immunoblots were then probed with an antibody specific for phosphorylated p53-Ser15.
Fig. 3.

ATM-null cells are not hypersensitive to C-1027 treatment. ATM-null and restored human fibroblast cells were treated with 0 or 30 pM C-1027, or 30 nM NCS for 72 h at 37°C and the cells were harvested. The number of cells were counted and normalized as a percentage of the untreated control from 3 independent experiments. From these percentages, the ratio of growth inhibition between wild-type vs ATM null cells was determined.
C-1027 induces robust p53 phosphorylation in cells deficient in either ATM or ATR, but not in cells deficient in both kinases
Previously we showed that ATM-null cells respond to C-1027 induced damage similarly to ATM-wild-type cells (Fig. 2 & Fig. 3) (17). Prior studies in our laboratory have also revealed that the amount of p53-Ser15 phosphorylation observed after treatment with C-1027 or with IR was unaffected by ATRkd expression (17). We have now extended these results by utilizing ATM-wild-type and null cells in combination with siRNA targeted against ATR (Fig. 4A & 4B). ATR levels averaged 15% of the control level after siRNA knockdown (Fig. 4A), consistent with the findings by others using this siRNA sequence in this and in other mammalian cell lines (20, 21). Reduction of ATR in ATM-wild-type cells had little effect on C-1027-induced phosphorylation of p53-Ser15 (Fig. 4A & 4B). In contrast, ATM-null cells with ATR depleted by siRNA showed an approximate 50% decrease of p53-Ser15 phosphorylation in response to C-1027 treatment (Fig. 4A & 4B). Thus, unlike IR and other radiomimetics such as NCS, C-1027-induced phosphorylation of p53-Ser15 is only decreased in cells deficient in both ATM and ATR.
Fig. 4.
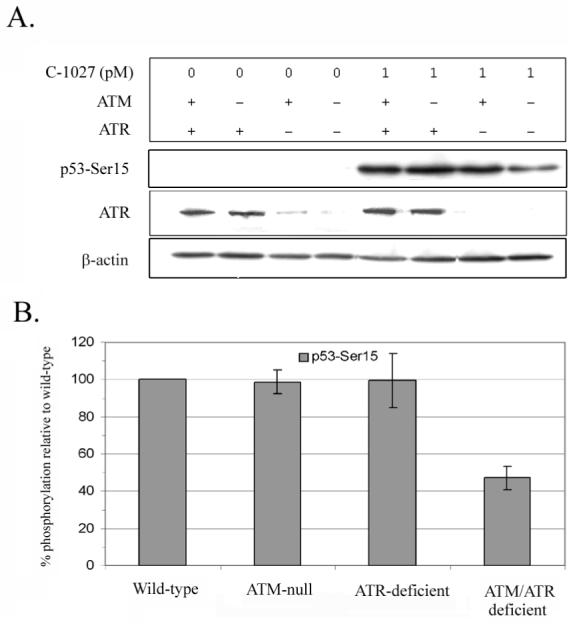
Induction of p53-Ser15 phosphorylation by C-1027 is partially reduced only in the absence of both ATM and ATR. A.) ATM-null and restored human fibroblast cells were pretreated for 48 h with mock siRNA (ATR +) or siRNA directed towards ATR (ATR -) before treatment with 0 or 1 nM C-1027 for 1 h at 37°C. The cellular extracts were analyzed by western blotting. Immunoblots were then probed with an antibody specific for phosphorylated p53-Ser15 and quantitated using image quant software. B.) Each lane was normalize to a loading control (β-actin) and then the normalized value of each untreated signal was subtracted from the C-1027 treated signal under each kinase condition. Since the signal intensities can vary between experiments, the values of 5 independent experiments were converted to a percentage of the wild-type signal to estimate the loss of p53-Ser15 phosphorylation.
C-1027 induces robust Chk2 phosphorylation in cells deficient in either ATM or ATR, but not in cells deficient in both kinases
Chk2 is upstream of p53 and is directly phosphorylated by ATM in response to IR or enediyne-induced DNA breaks (23). The amount of Chk2-Thr68 phosphorylation following 1 h incubation of cells with 1 nM C-1027 was measured by western blotting. Similar to the p53 results and unlike IR, NCS, and calicheamicin treatment, Chk2-Thr68 was similarly phosphorylated in response to C-1027 treatment regardless of ATM status (Fig. 5A & 5B). Also, diminishing ATR kinase activity by siRNA targeting of ATR in ATM-wild-type cells had little effect on Chk2 phosphorylation in response to C-1027-induced DSB (Fig. 5A & 5B), which is also in contrast to the IR model (8). Moreover, diminished phosphorylation of Chk2 was also observed following NCS treatment of ATR reduced ATM-wild-type cells [data not shown]. However, only ATM-null cells with siRNA depleted ATR showed almost a complete loss of Chk2-Thr68 phosphorylation in response to C-1027 (Fig. 5A & 5B). Thus, the ATM independent DNA damage responses induced by C-1027 extends to Chk2-Thr68 phosphorylation.
Fig. 5.

Induction of Chk2-Thr68 phosphorylation by C-1027 is greatly diminished only in the absence of both ATM and ATR. A.) ATM-null and restored human fibroblast cells were pretreated for 48 h with mock siRNA (ATR +) or siRNA directed towards ATR (ATR -) before treatment with 0 or 1 nM C-1027 for 1 h at 37°C. The cellular extracts were analyzed by western blotting. Immunoblots were then probed with an antibody specific for phosphorylated Chk2-Thr68 and quantitated using image quant software. B.) Each lane was normalize to a loading control (β-actin) and then the normalized value of each untreated signal was subtracted from the C-1027 treated signal under each kinase condition. Since the signal intensities can vary between experiments, the values of 5 independent experiments were converted to a percentage of the wild-type signal to estimate the loss of Chk2-Thr68 phosphorylation.
C-1027 induces Chk1 phosphorylation in cells deficient in either ATM or ATR, but not in cells deficient in both kinases
Loss of C-1027-induced activation of DNA damage response proteins seems to only occur when both ATM and ATR are depleted, in contrast to other radiomimetics and IR, where loss of ATM is sufficient to cause a substantial reduction in activation of these proteins. Phosphorylation of Chk1-Ser345 could be a possible exception since it is activated directly by ATR following DNA damage (18). Chk1 phosphorylation is also known to be significantly decreased in the absence of ATM after IR treatment (8), thus Chk1 phosphorylation would be expected to decrease by a loss of either ATM or ATR. In contrast, C-1027 treatment of cells lacking ATM did not result in any decrease of Chk1 phosphorylation (Fig. 6A & 6B). Moreover, in ATM-wild-type cells that are ATR deficient, C-1027 treatment resulted in only a partial loss of Chk1-Ser345 phosphorylation (Fig. 6A & 6B). Only cells deficient in both ATM and ATR showed a nearly complete loss of Chk1-Ser345 phosphorylation after C-1027 treatment (Fig. 6A and 6B).
Fig. 6.

Induction of Chk1-Ser345 phosphorylation requires either ATM or ATR. A.) ATM-null and restored human fibroblast cells were pretreated for 48 h with mock siRNA (ATR +) or siRNA directed towards ATR (ATR -) before treatment with 0 or 1 nM C-1027 for 1 h at 37°C. The cellular extracts were analyzed by western blotting. Immunoblots were then probed with an antibody specific for phosphorylated Chk2-Thr68 and quantitated using image quant software. B.) Each lane was normalize to a loading control (β-actin) and then the normalized value of each untreated signal was subtracted from the C-1027 treated signal under each kinase condition. Since the signal intensities can vary between experiments, the values of 5 independent experiments were converted to a percentage of the wild-type signal to estimate the loss of Chk1-Ser345 phosphorylation.
C-1027-induced DNA breaks are not affected by ATM or ATR status
To test if the amount of C-1027-induced breaks were similar under each kinase deficient condition, DNA breaks were measured by Comet analysis3. SiRNA-treated (either mock or ATR) ATM-wild-type or null cells were incubated with C-1027 under conditions used for western analysis (1 nM C-1027 for 1 h) and then analyzed for induction of DNA breaks (Fig. 7). PIKK status had little effect on the amount of DNA breaks observed after C-1027 treatment; consistent with the idea that difference in DNA damage response signaling is not related to the amount of DNA breaks induced.
Fig. 7.
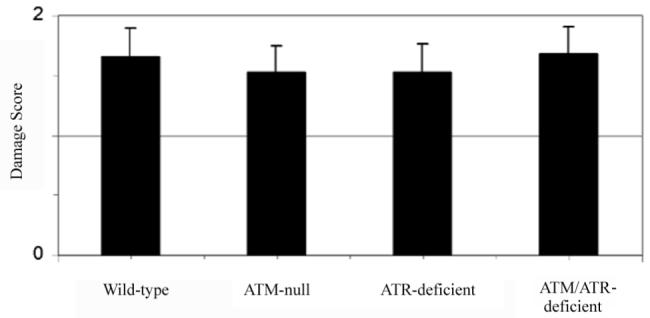
The amount C-1027-induced DNA strand breaks is not affected by PIKK status. Under the conditions used in figures 4, 5, and 6, the amount DNA breaks induced by 1 nM C-1027 was examined under varying PIKK deficient conditions. Cells were harvested and placed on to a microscope slide. Following cell lysis, electrophoresis and the addition of ethidium bromide, the size of the DNA tail was viewed by fluorescence microscopy and DNA breaks were quantitated.
C-1027-induced cell death increases in cells deficient in both ATM and ATR
Cells lacking ATM are unable to induce DNA damage checkpoint responses to DNA DSBs induced by IR and radiomimetics (except C-1027) and as such, are more readily killed by these agents (6). Since reduction of C-1027-induced DNA damage response checkpoints for the most part requires loss of both ATM and ATR, we sought to determine whether hypersensitivity in regard to cell death would vary accordingly. We examined siRNA treated (either mock or ATR) ATM-wild-type or null cells for increased cell death in response to C-1027 treatment. Cells were treated with 0.3 and 1 nM C-1027 for 24 h and examined for their ability to exclude trypan blue. Similar to 3-day growth inhibition (Fig. 3) as well as colony formation assays (17), there was no difference in C-1027-induced cell death in ATM-wild-type and ATM-null cells (Fig. 8). Also consistent with our previous study, there was no change in drug induced cell death between ATM-wild-type cells treated with siRNA targeted toward ATR. In contrast, ATM-null cells that were treated for 48 h with siRNA targeted against ATR showed a substantial increase in cell death following C-1027 treatment (Fig. 8). Thus unlike IR and other radiomimetics, where loss of ATM leads to increased cell death, loss of both ATM and ATR are required for C-1027 to recapitulate this response.
Fig. 8.
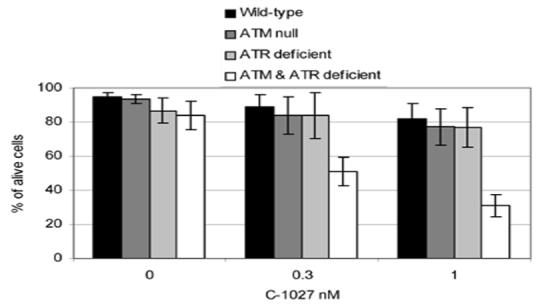
Only cells deficient in both ATM and ATR show increased cell death in response to C-1027 treatment. ATM null and restored human fibroblast cells were pretreated for 48 h with mock siRNA or siRNA directed towards ATR before treatment with 0, 0.3 or 1 nM C-1027 for 24 h at 37°C. Cells were harvested from 3 independent experiments and assessed for the ability to exclude trypan blue. 300 cells from each sample were counted and the percentage of cells excluding trypan blue was determined.
DISCUSSION
Cells lacking ATM are hypersensitive in regards to cytotoxicity induced by IR or radiomimetic treatment consistent with their inability to activate DNA damage response proteins (6). This present study further examined and expanded findings from this laboratory showing that after induction of DNA breaks by the enediyne C-1027, ATM-null cells robustly activated p53-Ser15 and did not display hypersensitive growth inhibition in comparison to ATM-wild-type cells (17). Recent studies have demonstrated that phosphorylation of ATM-Ser1981 occurs within minutes after low levels of IR treatment or treatment with radiomimetics such as NCS and calicheamicin (2, 14, 16). This study revealed that the ATM-independent nature of C-1027-induced DNA damage responses is not due to an inability to activate ATM, as very low levels of C-1027 treatment resulted in ATM-Ser1981 phosphorylation (Fig. 1). These results demonstrate a traditional role for ATM in response to C-1027-induced breaks.
In general, cells lacking ATM show a diminished response to DNA damage induced by IR and a wide variety of radiomimetics including enediynes such as calicheamicin and NCS, as well as glycopeptides such as bleomycin (11). The notable exception to this scenario is C-1027, which induced similar p53 phosphorylation in non-isogenic ATM-wild-type and ATM-null cell lines (17), and now in this study, isogenic ATM-wild-type and null cells, while NCS-induced activation was greatly reduced in the cells lacking ATM (Fig. 2). Similarly, the Chk kinase transducer proteins, located immediately downstream of ATM, are also activated differently after C-1027 treatment when compared to treatment with IR or other radiomimetics. In response to C-1027 treatment, Chk2 and Chk1 are robustly phosphorylated (Fig. 5 & 6), while after treatment with IR or other radiomimetics, cells lacking ATM showed markedly reduced phosphorylation of Chk2 and Chk1 (8).
While all three damage response proteins were fully activated in the absence of ATM after C-1027-induced DNA DSBs, each protein was activated differently in cells deficient in ATR or both ATM and ATR. We found that after C-1027 treatment as long as ATM is present when ATR is suppressed, both p53 and Chk2 were robustly phosphorylated while Chk1 phosphorylation was reduced by about 50% (Fig. 4, 5, & 6). In contrast, ATR activation of DNA damage response proteins in response to IR or other radiomimetics in ATM-null cells is not robust and occurs after longer periods of time and/or at high levels of DSBs when compared against ATM-wild-type cells (16, 24). For example, ATR is thought to be responsible for Chk2 phosphorylation in response to high levels of IR or calicheamicin-induced DSBs in the absence of ATM (16). After IR treatment, phosphorylation of both Chk kinases is diminished in cells deficient in ATR although ATR status did not affect p53-Ser15 phosphorylation (8, 17). We obtained similar results after treating ATR deficient cells with NCS (data not shown). While Chk1 phosphorylation is known to require ATR, the reason Chk2 activation is lost under these conditions is less clear (6). Since ATM mediated phosphorylation of Chk2 in response to IR requires other damage response proteins such as Nbs1 and DNA-PK, perhaps activation of these proteins also involves ATR (21, 25). Taken together, these findings suggest that unlike the observed ATR-dependent activation of the Chk kinases in response to DSBs induced by IR and NCS, ATM can compensate for the loss of ATR when the breaks are induced by C-1027.
The notion that cells can readily use either ATM or ATR to respond to C-1027-induced DSBs is supported by the finding that phosphorylation of p53 Chk2, and Chk1 is drastically or partially reduced respectively only when both PIKKs are lost (Fig. 4, 5, & 6). While there was almost no phosphorylation of either of the Chk kinases, p53-Ser15 phosphorylation levels were still significant, consistent with a previous observation that p53 but not the Chk kinases can be activated by additional PIKK kinases such as DNA-PK and Smg1 (26, 27). Further study is needed to determine the reason for these differences in phosphorylation and whether other PIKKs are involved in the C-1027-induded damage response.
Our findings also revealed that PIKK status dictates not only DNA damage responses but also cell survival in response to DNA breaks. Thus, only cells lacking both ATM and ATR were hypersensitive (regarding cell death) to C-1027 treatment compared to wild-type cells and cells deficient in ATM or ATR (Fig. 8). In contrast, after IR or NCS treatment, deficiency of either ATM or ATR results in greater cell death (17, 19). Thus, activation of cellular damage responses by C-1027 is unique based upon PIKK status, and these responses correlate with cellular hypersensitivity in regard to cell death.
An important question is why do C-1027-induced DNA damage responses differ from those induced by IR and other radiomimetics? Radiomimetics can be classified into two major groups, glycopeptides such as bleomycin, which interact with activated oxygen and a metal cation to damage DNA, and enediynes, which damage DNA by generation of free radicals that undergo a Bergman cycloaromization reaction (11). Furthermore, enediynes can consist of a 10 member ring chromophore such as calicheamicin or a nine member ring chromophore surrounded by a hydrophobic protein such as with NCS and C-1027 (28). However, with the exception of C-1027, these various types of radiomimetics all produce DNA breaks that require ATM to robustly activate DNA damage responses and to avoid increased cell death and growth inhibition. Thus, the particular characteristics of the C-1027 protein-chromophore structure are not likely to be the basis for induction of ATM-independent DNA damage responses.
While the major DNA lesion produced by enediynes are strand breaks, under anaerobic conditions in cell free systems, both interstrand cross-links and monoadducts have also been observed (29). In these studies, C-1027 produced mainly covalent drug-DNA interstrand cross-links, NCS produced mostly monoadducts and calicheamicin was less efficient at introducing either type of lesion (29). Possibly, C-1027 treatment of cells may also result in DNA interstrand cross-links that activate ATR in addition to DSBs, which activate ATM. Recent studies using mitomycin C, a known DNA interstrand cross-linker, have shown that cells utilize ATR to respond to DNA interstrand cross-links (30). There are also studies showing that enediynes can damage proteins or cleave RNA, which could also contribute to C-1027′s uniqueness as an enediyne (31, 32).
It is also possible that the type of DNA strand break (either single or double) produced by IR or radiomimetics influences PIKK regulation of DNA damage responses. Enediyne treatments induce DNA strand breaks by abstracting hydrogen from the C-1′, C-4′ and/or C-5′ carbon atoms of the DNA deoxyribose backbone (33, 34). However, as the efficiency of hydrogen abstraction from each DNA strand differs with the agent, so does the frequency of single to double strand breaks. Even though IR and radiomimetics have ratios of SSBs to DSBs that vary from 100 SSBs for every 1 DSBs for IR to almost exclusively DSBs for calicheamicin, all induce ATM-dependent damage responses (11, 34-36). Thus, the SSB/DSB ratio of 2:1 induced by C-1027 would likely not be a factor in the ATM dependence of the DNA damage response (37).
Could the types of DNA ends generated from the strand scission have an impact on the ATM dependence of the DNA damage response? C-1027-induced DNA DSBs have a 2 base pair 3′ overhang, and produce a combination of 3′-phosphoglycolate and base propenal ends on one strand, and either a nucleoside-5′-aldehyde, 2-deoxyribonolactone or 4′-hydroxylated abasic site on the other (38). Similarly, NCS-induced damage also produces a 2 base pair 3′ overhang, and forms all of the above DNA ends (11). Therefore, it is unlikely that it is solely the products of C-1027-induced damage that can explain the observed responses to DNA DSBs in the absence of ATM. However, more subtle differences in DNA damage can exist between NCS and C-1027. For example, NCS reacts with bulges in the DNA structure associated with mismatch base pairing not seen with C-1027, which might influence how the damaged DNA triggers PIKK-dependent cell cycle checkpoint responses (39, 40).
The ability of radiomimetics to focus DNA damage to particular DNA sequences or regions in the genome might also influence the nature of the DNA damage response. In contrast to IR, radiomimetics such as NCS and bleomycin can preferentially cleave particular subsets of genomic DNA, such as actively transcribing regions (41). However, since these enediynes also induce ATM-dependent damage responses, such regional DNA targeting is also unlikely to be a factor (41). Unlike IR, which induces undirected DNA damage, radiomimetics induce breaks by cleaving DNA at site-specific sequences. Although the highly preferred target sequences vary greatly, all of the agents besides C-1027 follow the IR model of ATM-dependent activation of damage responses (33, 39, 42).
Interestingly, one of C-1027’s preferred target sequences, GTTA is contained in the telomere repeat GGGTTA (37). Telomeres are a repetitive DNA sequence 8-15 kb in length, found at the ends of chromosomes and are responsible for maintaining cell viability and genomic stability (43). We recently showed that at equal levels of DSBs, C-1027 treatment caused telomere erosion accompanied by a significantly higher degree of chromosomal aberrations and genomic instability than did IR treatment (44). Recent studies have also shown that telomere dysfunction activates repair responses in a partially ATM-independent manner (45). While it is unknown if ATR is involved in this telomere dysfunction response, the ATM and ATR yeast homologs are known to work together in the regulation of repair of damaged telomeres (46). Although more study is needed, preferential targeting of telomeres by C-1027 could be contributing to the unique PIKK dependence of its induced damage response.
In conclusion, this study adds to the growing evidence that DNA damage responses to C-1027-induced DNA DSBs are not uniformly regulated by ATM. This study shows for the first time, that loss of both ATM and ATR can be required to diminish activation of DNA damage responses proteins and for an increase in cell death. Future studies will determine if the ATM and ATR kinases have overlapping functions in response to C-1027-induced DNA damage and examine whether preferred targeting of telomere DNA and/or formation of DNA interstand cross-links contributes to the unusual nature of the cellular responses to C-1027-induced breaks.
ACKNOWLEDGMENT
The authors thank Drs. Mary McHugh, Athena Lin, Adam Karpf and Joel Huberman for critical reading of this manuscript. This work was supported in part by National Cancer Institute grants CA 106312 and CA 16056 (to T.A.B.). D.R.K. was supported by National Institute of Health Training grant CA 09072-30.
Footnotes
- ATM
- ataxia-telangiectasia mutated
- ATR
- ATM and Rad3-related
- Chk
- Checkpoint kinase
- DSBs
- double-strand breaks
- IR
- ionizing radiation
- kd
- kinase dead
- NCS
- neocarzinostatin
- PIKK
- phosphatidylinositol 3-kinase-like protein kinase
- SSBs
- single-strand breaks
In testing ATM phosphorylation, we used an intrinsically ATM-wild-type cell line as opposed to the ATM-restored fibroblasts that were used in other experiments, although both cell lines exhibited ATM-Ser1981 phosphorylation in response to C-1027 treatment.
Though Comet detects both SSB and DSB, C-1027 induces almost exclusively DSBs (31). Comet results are consistent with neutral pulse field gel electrophoresis estimates of DNA DSBs from C-1027-treated cells (32).
REFERENCES
- 1.Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer. 2003;3:117–29. doi: 10.1038/nrc992. [DOI] [PubMed] [Google Scholar]
- 2.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 3.Li L, Zou L. Sensing, signaling, and responding to DNA damage: organization of the checkpoint pathways in mammalian cells. J Cell Biochem. 2005;94:298–306. doi: 10.1002/jcb.20355. [DOI] [PubMed] [Google Scholar]
- 4.Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997;11:3471–81. doi: 10.1101/gad.11.24.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–9. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 6.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–68. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 7.Barlow C, Brown KD, Deng CX, Tagle DA, Wynshaw-Boris A. Atm selectively regulates distinct p53-dependent cell-cycle checkpoint and apoptotic pathways. Nat Genet. 1997;17:453–6. [Google Scholar]
- 8.Helt CE, Cliby WA, Keng PC, Bambara RA, O’Reilly MA. Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage. J Biol Chem. 2005;280:1186–92. doi: 10.1074/jbc.M410873200. [DOI] [PubMed] [Google Scholar]
- 9.Ziv Y, Jaspers NG, Etkin S, Danieli T, Trakhtenbrot L, Amiel A, Ravia Y, Shiloh Y. Cellular and molecular characteristics of an immortalized ataxia-telangiectasia (group AB) cell line. Cancer Res. 1989;49:2495–501. [PubMed] [Google Scholar]
- 10.Sutherland BM, Bennett PV, Sidorkina O, Laval J. Clustered damages and total lesions induced in DNA by ionizing radiation: oxidized bases and strand breaks. Biochemistry. 2000;39:8026–31. doi: 10.1021/bi9927989. [DOI] [PubMed] [Google Scholar]
- 11.Povirk LF. DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: bleomycin, neocarzinostatin and other enediynes. Mutat Res. 1996;355:71–89. doi: 10.1016/0027-5107(96)00023-1. [DOI] [PubMed] [Google Scholar]
- 12.Ha L, Ceryak S, Patierno SR. Generation of S phase-dependent DNA double-strand breaks by Cr(VI) exposure: involvement of ATM in Cr(VI) induction of gamma-H2AX. Carcinogenesis. 2004;25:2265–74. doi: 10.1093/carcin/bgh242. [DOI] [PubMed] [Google Scholar]
- 13.Yuan SS, Chang HL, Hou MF, Chan TF, Kao YH, Wu YC, Su JH. Neocarzinostatin induces Mre11 phosphorylation and focus formation through an ATM- and NBS1-dependent mechanism. Toxicology. 2002;177:123–30. doi: 10.1016/s0300-483x(02)00220-2. [DOI] [PubMed] [Google Scholar]
- 14.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. Embo J. 2003;22:5612–21. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shiloh Y, van der Schans GP, Lohman PH, Becker Y. Induction and repair of DNA damage in normal and ataxia-telangiectasia skin fibroblasts treated with neocarzinostatin. Carcinogenesis. 1983;4:917–21. doi: 10.1093/carcin/4.7.917. [DOI] [PubMed] [Google Scholar]
- 16.Ismail IH, Nystrom S, Nygren J, Hammarsten O. Activation of ataxia telangiectasia mutated by DNA strand break-inducing agents correlates closely with the number of DNA double strand breaks. J Biol Chem. 2005;280:4649–55. doi: 10.1074/jbc.M411588200. [DOI] [PubMed] [Google Scholar]
- 17.Dziegielewski J, Beerman TA. Cellular responses to the DNA strand-scission enediyne C-1027 can be independent of ATM, ATR, and DNA-PK kinases. J Biol Chem. 2002;277:20549–54. doi: 10.1074/jbc.M109897200. [DOI] [PubMed] [Google Scholar]
- 18.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- 19.Ziv Y, Bar-Shira A, Pecker I, Russell P, Jorgensen TJ, Tsarfati I, Shiloh Y. Recombinant ATM protein complements the cellular A-T phenotype. Oncogene. 1997;15:159–67. doi: 10.1038/sj.onc.1201319. [DOI] [PubMed] [Google Scholar]
- 20.Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–89. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Stern DF. Regulation of CHK2 by DNA-dependent protein kinase. J Biol Chem. 2005;280:12041–50. doi: 10.1074/jbc.M412445200. [DOI] [PubMed] [Google Scholar]
- 22.Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu JC, Sasaki YF. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen. 2000;35:206–21. doi: 10.1002/(sici)1098-2280(2000)35:3<206::aid-em8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 23.Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–6. [PubMed] [Google Scholar]
- 24.Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–7. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gatei M, Sloper K, Sorensen C, Syljuasen R, Falck J, Hobson K, Savage K, Lukas J, Zhou BB, Bartek J, Khanna KK. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem. 2003;278:14806–11. doi: 10.1074/jbc.M210862200. [DOI] [PubMed] [Google Scholar]
- 26.Woo RA, Jack MT, Xu Y, Burma S, Chen DJ, Lee PW. DNA damage-induced apoptosis requires the DNA-dependent protein kinase, and is mediated by the latent population of p53. Embo J. 2002;21:3000–8. doi: 10.1093/emboj/cdf307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brumbaugh KM, Otterness DM, Geisen C, Oliveira V, Brognard J, Li X, Lejeune F, Tibbetts RS, Maquat LE, Abraham RT. The mRNA surveillance protein hSMG-1 functions in genotoxic stress response pathways in mammalian cells. Mol Cell. 2004;14:585–98. doi: 10.1016/j.molcel.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Smith AL, Nicolaou KC. The enediyne antibiotics. J Med Chem. 1996;39:2103–17. doi: 10.1021/jm9600398. [DOI] [PubMed] [Google Scholar]
- 29.Xu YJ, Xi Z, Zhen YS, Goldberg IH. Mechanism of formation of novel covalent drug.DNA interstrand cross-links and monoadducts by enediyne antitumor antibiotics. Biochemistry. 1997;36:14975–84. doi: 10.1021/bi972101o. [DOI] [PubMed] [Google Scholar]
- 30.Andreassen PR, D’Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004;18:1958–63. doi: 10.1101/gad.1196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zein N, Reiss P, Bernatowicz M, Bolgar M. The proteolytic specificity of the natural enediyne-containing chromoproteins is unique to each chromoprotein. Chem Biol. 1995;2:451–5. doi: 10.1016/1074-5521(95)90262-7. [DOI] [PubMed] [Google Scholar]
- 32.Battigello JM, Cui M, Roshong S, Carter BJ. Enediyne-mediated cleavage of RNA. Bioorg Med Chem. 1995;3:839–49. doi: 10.1016/0968-0896(95)00046-j. [DOI] [PubMed] [Google Scholar]
- 33.Dedon PC, Goldberg IH. Free-radical mechanisms involved in the formation of sequence-dependent bistranded DNA lesions by the antitumor antibiotics bleomycin, neocarzinostatin, and calicheamicin. Chem Res Toxicol. 1992;5:311–32. doi: 10.1021/tx00027a001. [DOI] [PubMed] [Google Scholar]
- 34.Henner WD, Grunberg SM, Haseltine WA. Sites and structure of gamma radiation-induced DNA strand breaks. J Biol Chem. 1982;257:11750–4. [PubMed] [Google Scholar]
- 35.Grimwade JE, Beerman TA. Measurement of bleomycin, neocarzinostatin, and auromomycin cleavage of cell-free and intracellular simian virus 40 DNA and chromatin. Mol Pharmacol. 1986;30:358–63. [PubMed] [Google Scholar]
- 36.Kirk CA, Goodisman J, Beerman TA, Gawron LS, Dabrowiak JC. Kinetics of cleavage of intra- and extracellular simian virus 40 DNA with the enediyne anticancer drug C-1027. Biophys Chem. 1997;63:201–9. doi: 10.1016/s0301-4622(96)02217-x. [DOI] [PubMed] [Google Scholar]
- 37.Xu YJ, Zhen YS, Goldberg IH. C1027 chromophore, a potent new enediyne antitumor antibiotic, induces sequence-specific double-strand DNA cleavage. Biochemistry. 1994;33:5947–54. doi: 10.1021/bi00185a036. [DOI] [PubMed] [Google Scholar]
- 38.Xu YJ, Xi Z, Zhen YS, Goldberg IH. A single binding mode of activated enediyne C1027 generates two types of double-strand DNA lesions: deuterium isotope-induced shuttling between adjacent nucleotide target sites. Biochemistry. 1995;34:12451–60. doi: 10.1021/bi00038a044. [DOI] [PubMed] [Google Scholar]
- 39.Dedon PC, Jiang ZW, Goldberg IH. Neocarzinostatin-mediated DNA damage in a model AGT.ACT site: mechanistic studies of thiol-sensitive partitioning of C4′ DNA damage products. Biochemistry. 1992;31:1917–1927. doi: 10.1021/bi00122a004. [DOI] [PubMed] [Google Scholar]
- 40.Kappen LS, Goldberg IH. Site-specific cleavage at a DNA bulge by neocarzinostatin chromophore via a novel mechanism. Biochemistry. 1993;32:13138–45. doi: 10.1021/bi00211a024. [DOI] [PubMed] [Google Scholar]
- 41.Beckmann RP, Agostino MJ, McHugh MM, Sigmund RD, Beerman TA. Assessment of preferential cleavage of an actively transcribed retroviral hybrid gene in murine cells by deoxyribonuclease I, bleomycin, neocarzinostatin, or ionizing radiation. Biochemistry. 1987;26:5409–15. doi: 10.1021/bi00391a029. [DOI] [PubMed] [Google Scholar]
- 42.Mirabelli CK, Beattie WG, Huang CH, Prestayko AW, Crooke ST. Comparison of the sequences at specific sites on DNA cleaved by the antitumor antibiotics talisomycin and bleomycin. Cancer Res. 1982;42:1399–404. [PubMed] [Google Scholar]
- 43.Collins K. Mammalian telomeres and telomerase. Curr Opin Cell Biol. 2000;12:378–83. doi: 10.1016/s0955-0674(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 44.McHugh MM, Gawron LS, Matsui S, Beerman TA. The antitumor enediyne C-1027 alters cell cycle progression and induces chromosomal aberrations and telomere dysfunction. Cancer Res. 2005;65:5344–51. doi: 10.1158/0008-5472.CAN-05-0015. [DOI] [PubMed] [Google Scholar]
- 45.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–56. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 46.Takata H, Kanoh Y, Gunge N, Shirahige K, Matsuura A. Reciprocal association of the budding yeast ATM-related proteins Tel1 and Mec1 with telomeres in vivo. Mol Cell. 2004;14:515–22. doi: 10.1016/s1097-2765(04)00262-x. [DOI] [PubMed] [Google Scholar]