

Abstract
We have shown previously that when postsynaptic NMDA receptors are blocked, the frequency, but not amplitude, of spontaneous EPSCs (sEPSCs) at synapses in the entorhinal cortex is reduced by NMDA receptor antagonists, demonstrating that glutamate release is tonically facilitated by presynaptic NMDA autoreceptors. In the present study, we recorded sEPSCs using whole-cell voltage clamp in neurons in layer V in slices of the rat entorhinal cortex. Using specific antagonists for NR2A [(R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl]-phosphonic acid] and NR2B [(αR, βS)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidinepropanol hydrochloride (Ro 25-6981)] subunit-containing receptors, we confirmed that in slices from juvenile rats (4–6 weeks of age), the autoreceptor is predominantly of the NR1–NR2B subtype. In older (4–6 months of age) control animals, the effect of the NR2B antagonist was less marked, suggesting a decline in autoreceptor function with development. In slices from rats (aged 4–6 months) exhibiting spontaneous recurrent seizures induced with a lithium-pilocarpine protocol, Ro 25-6981 again robustly reduced sEPSC frequency. The effect was equal to or greater than that seen in the juvenile slices and much more pronounced than that seen in the age-matched control animals. In all three groups, the NR2A antagonist was without effect on sEPSCs. These results suggest that there is a developmental decrease in NMDA autoreceptor function, which is reversed in a chronic epileptic condition. The enhanced autoreceptor function may contribute to seizure susceptibility and epileptogenesis in temporal lobe structures.
Keywords: NMDA, NR2B receptors, presynaptic, glutamate, entorhinal, chronic epilepsy
Introduction
The release of glutamate at cortical synapses is controlled by an array of presynaptic receptors, providing a powerful means of dynamically regulating excitatory transmission at both the network level and in a synapse-delimited way. For example, heteroreceptor control of release is effected by GABAB, adenosine, and muscarinic receptors, whereas both ionotropic (NMDA, kainate) and metabotropic glutamate receptors act as autoreceptors. Work in this laboratory has focused on presynaptic receptor control of glutamate release at synapses in the rat entorhinal cortex (EC). We provided one of the earliest physiological demonstrations that NMDA autoreceptors tonically facilitate glutamate release at excitatory synapses (Berretta and Jones, 1996). Subsequently, we have shown that these receptors are likely to be predominantly of the NR1–NR2B subtype and that they mediate frequency-dependent facilitation of glutamate transmission, probably by increasing intraterminal Ca via the receptor ionophore (Woodhall et al., 2001). A number of other studies have also demonstrated functional presynaptic NMDA receptors in different brain areas (Breukel et al., 1998; Hamada et al., 1998; Cochilla and Alford, 1999; Glitsch and Marty, 1999; Casado et al., 2000; Dalby and Mody, 2003; Sjostrom et al., 2003; Duguid and Smart, 2004; Samson and Pare, 2005).
There is evidence to suggest that NMDA receptor function is altered in chronically epileptic states. In particular, an increase in receptor number and/or function has often been observed (Dalby and Mody, 2001; Avanzini and Franceschetti, 2003; Morimoto et al., 2004). These studies have concentrated on NMDA receptors at postsynaptic sites, and we do not know yet whether chronic epileptic conditions are associated with adaptive changes in NMDA autoreceptors or heteroreceptors. However, a recent study showed that the density of (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) binding sites is increased in synaptosomes isolated from temporal cortical tissue from epileptic patients (Steffens et al., 2005), suggesting that presynaptic NMDA function may be increased. In the present study, we examined the functional physiological effect of NMDA autoreceptors in EC slices from chronically epileptic rats. We found that facilitation of glutamate release by the NR2B subtype of receptor declined with age in normal animals but became functionally upregulated in the chronic epileptic condition.
Materials and Methods
Combined entorhinal–hippocampal slices were prepared from male Wistar rats, as described previously (Jones and Heinemann, 1988). Rats were anesthetized with an intramuscular injection of ketamine (120 mg/kg) plus xylazine (8 mg/kg) and decapitated. The brain was removed rapidly and immersed in oxygenated artificial CSF (ACSF) chilled to 4°C. Slices (350 μm) were cut using a Vibroslice and stored in ACSF bubbled with 95% O2/5% CO2 at room temperature. After recovery for at least 1 h, individual slices were transferred to a recording chamber mounted on the stage of a Zeiss (Oberkochen, Germany) Axioskop FS microscope. The chamber was perfused (2 ml/min) with oxygenated ACSF, pH 7.4, at 30–32°C. The ACSF contained the following (in mm): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 2 MgSO4, 2.5 CaCl2, and 10 d-glucose. Neurons were visualized using differential interference contrast optics and an infrared video camera.
Patch pipettes (1–4 MΩ) were pulled from borosilicate glass on a Flaming/Brown microelectrode puller. Pipettes were filled with a Cs-gluconate-based solution containing the following (in mm): 100 d-gluconate, 40 HEPES, 1 N-ethyl bromide quaternary salt, 0.6 EGTA, 4 NaCl, 5 MgCl2, 1 tetraethylammonium-Cl, 4 ATP-Na, 0.3 GTP-Na, and 1 MK-801. The solution was adjusted to 275 mOsm by dilution and set to pH 7.3 with CsOH. Whole-cell voltage-clamp recordings were made from neurons in layer V of the medial division of the EC, using an Axopatch 200B amplifier. Using this pipette solution and with membrane potential voltage clamped at –60 mV, neurons displayed spontaneous EPSCs (sEPSCs). The open channel blocker MK-801 was included in the patch pipette to block postsynaptic NMDA receptor in the recorded neuron (Berretta and Jones, 1996). To facilitate this blockade, neurons were depolarized to –10 mV for 10 s at intervals during a 10 min period after breakthrough to whole-cell access. Thus, it should be stressed that all of the recordings presented here were conducted with postsynaptic NMDA receptors blocked, and under these experimental conditions, EPSCs were mediated by spontaneous release of glutamate acting at AMPA/kainate receptors. Series resistance compensation was not used, but access resistance (10–30 MΩ) was monitored at regular intervals throughout each recording, and cells were discarded from analysis if it changed by more than ±10%. Liquid junction potentials were estimated using the calculator of pClamp 8 software and compensated for in the holding potentials.
Data were recorded to computer hard disk using Axoscope software. Minianalysis (Synaptosoft, Decatur, GA) was used for analysis of sEPSCs off-line. sEPSCs were detected automatically using a threshold-crossing algorithm. Two hundred sEPSCs were sampled during a continuous recording period for each neuron under each condition. To compare pooled data under control and drug conditions, we determined median values for interevent interval (IEI) and amplitude for events in each cell and calculated the mean median values for the population, using a paired t test for statistical comparison. All error values stated in the text refer to SEM. We also assessed the significance of shifts in cumulative probability distributions of IEI using the nonparametric Kolmogorov–Smirnoff (KS) test.
Previously, we used the relatively selective antagonist ifenprodil to assess whether presynaptic NMDA receptors contained the NR2B subunit (Woodhall et al., 2001). In the present study, we examined the effect of a more specific antagonist at NR2B receptors, (αR, βS)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidinepropanol hydrochloride (Ro 25-6981; Tocris Cookson, Bristol, UK) (Fischer et al., 1997). This was compared with the specific NR2A antagonist (R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl]-phosphonic acid (NVP-AAM077; a kind gift from Dr. Yves Auberson, Novartis Pharmaceuticals, Basel, Switzerland) (Auberson et al., 2002).
Initial experiments to determine the effects of the NR2A- and NR2B-selective agonists were performed in slices taken from juvenile animals (4–6 weeks of age). In studies comparing chronically epileptic rats with age-matched controls, the animals were aged 4–6 months at the time of electrophysiological experiments.
Rats were rendered chronically epileptic using a modified version of the pilocarpine model described by Glien et al. (2001). They were pretreated with LiCl (300 mg/kg, i.p.). Twenty-four hours later, they received α-methyl scopolamine (1 mg/kg, i.p.) to block the peripheral cholinergic effects of pilocarpine, and 15 min later received pilocarpine, administered in very low doses (5 mg/kg), repeated as necessary at 30 min intervals until an acute status epilepticus (SE) was induced. After induction of SE, animals were administered the central muscle relaxant xylazine (4 mg/kg, i.m.), which reduced the severity of clonic contractions without preventing electrical seizure activity. After 2 h in SE, seizures were terminated with diazepam (10 mg/kg, i.p.) and animals monitored to full recovery of consciousness. When necessary, they were given normal saline subcutaneously on the day with SE and on the following day for rehydration. Using this modified approach, we achieved a mortality rate of ∼5–10% and development of spontaneous recurrent seizures in 100% of animals. Age-matched control animals received the initial injection of LiCl but nothing else. This procedure was performed under a UK Home Office project license (PPL30/1877).
After the acute SE, animals developed evidence of epileptic seizure activity during a latent period of 6–10 weeks. This included the appearance of occasional head twitches, wet dog shakes, forepaw clonus, orofacial movements, and whole-body jerks. Eventually, all animals began to display spontaneous recurrent seizures, characterized by rearing plus loss of balance and accompanied by generalized clonic seizures. Animals were used for electrophysiology 2–4 weeks after first developing spontaneous recurrent seizures.
Results
Differences in sEPSCs between groups
There were some differences in sEPSC characteristics in the three groups that were evident even in the relatively small sample of neurons reported on here. These differences, and their significance, will be described in detail for a much larger population of neurons in a report being prepared for submission (G. Ayman, G. L. Woodhall, and R. S. G. Jones, unpublished observations). Although there are quantitative differences in the wider population, qualitatively the differences are the same. sEPSC amplitudes were slightly larger in the cells from epileptic animals compared with both age-matched controls and juvenile animals. Rise and decay times were slower in older animals compared with younger but are very similar in both groups (epileptic and controls). The biggest difference was in the frequency of events, which was similar in young animals (3.4 ± 0.6 Hz; n = 6) and old controls (2.7 ± 1.1 Hz; n = 6) but higher and more variable in slices from epileptic animals (12 ± 8.4 Hz; n = 7).
NR2B but not NR2A receptor blockade depresses sEPSC frequency
In juvenile animals, we have shown previously that the nonspecific antagonist 2-AP-5 and the relatively selective NR2B antagonist ifenprodil both decrease the frequency of sEPSCs in EC neurons by blocking tonically active presynaptic NMDA receptors (Berretta and Jones, 1996; Woodhall et al., 2001). We now report a similar effect with the highly specific NR2B antagonist Ro 25-6981. In six cells, the mean IEI under control conditions was 334.6 ± 49.2 ms with a mean median of 183.8 ± 38.2 ms. During perfusion with Ro 25-6981 (1 μm), the mean IEI increased to 600.0 ± 136.6 ms with a mean median of 360.6 ± 75.9 ms (p > 0.05). Mean median amplitudes were unaltered (10.7 ± 0.8 vs 10.4 ± 0.6 pA). Rise and decay times of sEPSCs were also unaltered (data not shown). Figure 1A shows an example of one neuron in which Ro 25-6981 clearly decreased the frequency of sEPSCs. The cumulative probability distributions for IEI for pooled data from six neurons in Figure 1B shows that in the presence of the drug the distribution is shifted to the right (KS, p > 0.01), reflecting the decreased frequency of sEPSCs.
Figure 1.
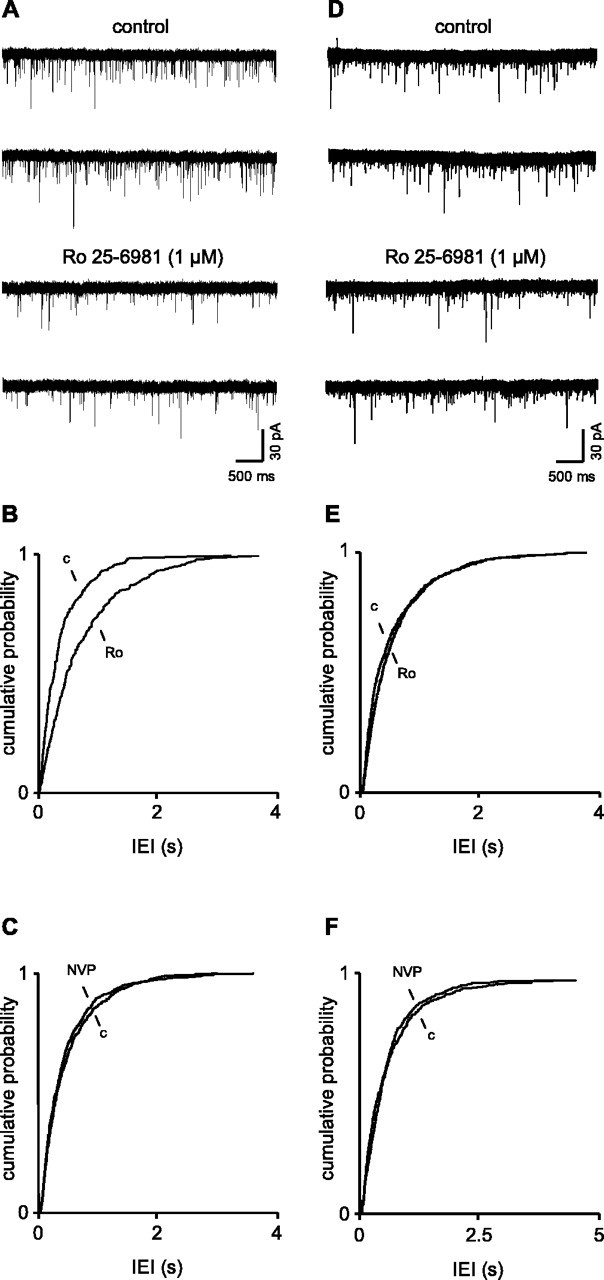
Presynaptic NR2B-containing receptors facilitate glutamate release more in young animals than in old. A, Voltage-clamp recordings of sEPSCs in a layer V neuron in a slice from a 4-week-old animal. With postsynaptic receptors blocked by intracellular MK-801, blockade of NR2B receptors decreases the frequency of sEPSCs. B, Pooled data for an interevent interval from six neurons shows a shift to the right of the cumulative probability distribution in the presence of Ro 25-6981 compared with the control. C, In contrast, in the presence of the NR2A antagonist NVP-AAM077 (1 μm), there was a small, nonsignificant shift to the right of the cumulative probability distribution, indicating that these receptors were not controlling glutamate release in juvenile animals. D, Voltage-clamp recordings from a neuron in a slice from a 5-month-old rat. The NR2B antagonist reduced sEPSC frequency, but the effect was much less robust than in the juvenile animal. This is reflected in the pooled data for an interevent interval in adult control neurons shown in E. F, Pooled data for the effects of NVP-AAM077 in adult neurons. Again there was a small, nonsignificant increase in frequency. Ro, Ro 25-6981; c, control; NVP, NVP-AAM077.
In contrast to the effect of the NR2B antagonist, the NR2A antagonist NVP-AAM077 (3 μm) had no significant effect on sEPSCs in five neurons in slices from young animals. Thus, mean median IEI was 305.3 ± 68.4 ms in control conditions. If anything, there was a slight increase in frequency of sEPSCs in the presence of the drug, but the mean median IEI (279.5 ± 52.3 ms) was not significantly different. Likewise, mean median amplitude (11.5 ± 2.8 vs 11.3 ± 2.0 pA) rise and decay times (data not shown) were also unaffected. Cumulative probability distributions of IEI for pooled data in the presence and absence of NVP-AAM077 are shown in Figure 1C. KS analysis showed that the slight shift to the left was not significant.
NR2B receptor blockade is more effective in young versus adult rats
In adult animals, the effect of Ro 25-6981 on sEPSC frequency was considerably less robust than in the young animals (Fig. 1D). Thus, the mean IEI in control conditions was 543.7 ± 77.4 ms (n = 6) with a mean median of 367.0 ± 74.8 ms. In the presence of Ro 25-6981, the corresponding values were 606.9 ± 83.8 and 451.3 ± 69.4 ms. Neither change was significant when assessed with a paired t test. However, KS analysis of the cumulative probability distributions of pooled IEIs (Fig. 1E) showed that the small rightward shift in the presence of the antagonist was significant (p > 0.05). Concurrently, there was no significant change in amplitude (mean median, 9.9 ± 0.5 vs 9.9 ± 0.9 pA) or rise and decay times (data not shown). Thus, the influence of presynaptic NMDA receptors on glutamate release in adult control animals was reduced compared with that seen in juveniles. On average, the frequency of sEPSCs decreased by ∼40% in neurons in young slices but only 15% in the adult group. The reduction in effect of the NR2B antagonist was not accompanied by an increased effect of blocking NR2A receptors in slices (n = 4) from older animals. Thus, there was no significant effect of NVP-AAM077 on mean median IEI (414.3 ± 50.5 vs 362 ± 31.3 ms) or amplitude (8.8 ± 0.9 vs 8.4 ± 0.9 pA) of sEPSCs, although again, there was a slight increase in overall frequency in the presence of the drug. Pooled data showing the effect of NVP-AAM077 on IEI can be seen in Figure 1F.
Increased effect of presynaptic NR2B receptors in epileptic rats
As mentioned above, the frequency of sEPSCs in neurons from epileptic animals was higher than that in controls and quite variable from neuron to neuron. Thus, the mean IEI in seven neurons was 222.3 ± 104.0 ms with a mean median of 145 ± 61.0 ms. In the presence of Ro 25-6981, these values increased to 337.4 ± 207.7 and 341 ± 131.8 ms, respectively. Thus, the NR2B antagonist, on average, induced a reduction in frequency of ∼55%. The reduction in one neuron is shown in Figure 2A. Despite the variability, the difference in IEI was significantly different when assessed either by a paired t test (p > 0.05) or by KS analysis (p > 0.001) of cumulative probability distributions (Fig. 2B). The decreased frequency was not accompanied by a significant change in amplitude. In the control situation, the mean amplitude was 15.4 ± 1.3 pA (with a mean median of 12.7 ± 0.9) compared with 14.3 ± 1.1 pA (12.0 ± 1.0 pA) in the presence of the drug. Neither rise time nor decay was altered (data not shown).
Figure 2.
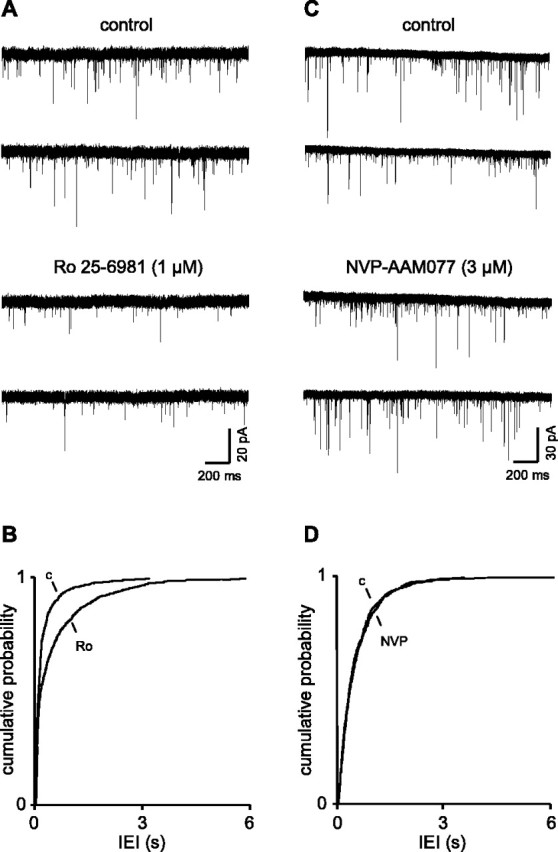
Presynaptic NR2B receptors strongly facilitate glutamate release in epileptic animals.A,B, Voltage-clamp recordings show a robust decrease in sEPSC frequency in the presence of the NR2B antagonist in a neuron recorded in a slice from a 5-month-old epileptic rat, which contrasts with its weak effect in age-matched control animals (Fig. 1 D, E) but compares favorably with the juvenile animals (Fig. 1 A, B). C, D, The NR2B antagonist failed to alter sEPSC frequency in epileptic animals, as in young and age-matched control animals. Ro, Ro 25-6981; c, control; NVP, NVP-AAM077.
There was no evidence for change in subunit composition of the presynaptic receptors in the epileptic rats. Thus, the powerful effect of the NR2B antagonist in the epileptic animals was not matched by the NR2A receptor blocker (Fig. 2C,D). Thus, as in the neurons in juvenile and age-matched older control groups, NVP-NAM077 had no discernible effect on mean median IEI (220 ± 70 vs 239 ± 83.7 ms) or amplitude (10.6 ± 1.1 vs 10.3 ± 1.6 pA) of sEPSCs in five neurons recorded in slices from epileptic animals.
Discussion
We have shown previously that presynaptic NMDA receptors tonically facilitate glutamate release at synapses in the EC (Berretta and Jones, 1996; Woodhall et al., 2001) and suggested that the autoreceptor is likely to be predominantly of the NR1–NR2B subtype. The latter observation was strengthened in the current study, because the frequency of sEPSCs at EC synapses was reduced by the NR2B-selective antagonist Ro 25-6981 but unaltered by the NR2A antagonist NVP-AAM077. Investigations of postsynaptic NMDA receptors have shown that cortical expression of NR2B receptors is present at birth, rises through early development [up to around postnatal day 20 (P20)], and then decreases slightly or is sustained throughout adulthood. NR2A expression is almost undetectable at P0 but increases rapidly up to P20–P28 and then also remains stable throughout adulthood (Sheng et al., 1994; Zhong et al., 1995; Wenzel et al., 1997; Liu et al., 2004). A similar profile of changes has been demonstrated in human hippocampal formation (Law et al., 2003). Interestingly, the latter study found the biggest decline in NR2B expression between neonates and adult to occur in the subiculum and EC and the largest change in NR2A:NR2B ratio in the EC.
These changes in subunit expression have led to the suggestion that there is a developmental switch from NR2B-containing receptors to NR2A at glutamate synapses, with important consequences for postsynaptic responsiveness and synaptic plasticity (Quinlan et al., 1999; Liu et al., 2004). Our studies show a developmental change in the tonic facilitation of glutamate release by NMDA autoreceptors, with a reduced effectiveness in 4- to 5-month-old animals. This is unlikely to be the result of a switch from NR2B- to NR2A-containing receptors at presynaptic sites, because the specific NR2A antagonist was ineffective in reducing glutamate release in either young or old animals. It is possible that the loss of the autoreceptor function in the older animals may reflect a simple loss of NR2B receptors from presynaptic sites, or it could also reflect displacement of the NR2B receptors to sites at which they may be relatively inaccessible to ambient glutamate. Certainly, at postsynaptic sites, there appears to be a developmental shift of the NR2B receptors to extrasynaptic locations (Tovar and Westbrook, 1999; Liu et al., 2004), with an increased contribution from subsynaptic NR2A receptors. However, it would not appear that the presynaptic receptors switch to NR2A in the adult, because the antagonist for the latter did not become effective in these animals. Nevertheless, our data do suggest that NR2B-containing autoreceptors may play a role in development at excitatory synapses in the EC.
The reduced autoreceptor function in the older animals was reversed in animals exhibiting chronic recurrent seizures. Thus, blockade of NR2B receptors in slices from these animals resulted in a reduction in sEPSC frequency that was more robust than that seen in the juvenile age group. Again, NVP-AAM077 had no effect. There is a substantial literature on changes in NMDA receptors associated with the development and maintenance of cortical seizures, much of it suggesting an enhanced activation, function, or expression of the receptors (Dalby and Mody, 2001; Avanzini and Franceschetti, 2003; Morimoto et al., 2004). None of the studies published to date have shown that changes in NMDA receptor function apply to presynaptic sites. However, a recent investigation has shown an increased Bmax of MK-801 binding, with no change in Kd, in synaptosomes prepared from temporal cortex resected during surgery for epilepsy, suggesting an increased density of presynaptic NMDA receptors with no change in affinity (Steffens et al., 2005). There is increasing evidence for enhanced expression of the NR2B-containing receptor in both human epilepsy and animal models (Al-Ghoul et al., 1997; Mathern et al., 1999; DeFazio and Hablitz, 2000; Ying et al., 2004). Such studies do not distinguish between presynaptic and postsynaptic receptors, so it is possible that an increased expression of presynaptic NR2B-containing receptors may arise in epileptic conditions. Our data support this possibility. However, we cannot say whether the increased autoreceptor function reflects an increased density of receptors on the terminals. Other possibilities include a redistribution of receptors to sites more accessible to ambient glutamate or altered properties or composition of existing receptors.
We cannot say at present whether the change in autoreceptor function in the EC is causally or casually related to epileptogenesis. It is interesting that dysplastic neocortical tissue resected from patients with epilepsy shows enhanced NR2B binding, an increased severity of epileptiform discharges provoked by lowering extracellular Mg2+, and an increased sensitivity of such discharges to the NR2B antagonist ifenprodil (Moddel et al., 2005). An increase in sensitivity of bicuculline-induced epileptiform activity was seen in slices from rats with freeze lesion-induced cortical dysplasia (DeFazio and Hablitz, 2000). Such effects could reflect a contribution from increased NMDA autoreceptor function. It will be interesting to conduct similar experiments in EC slices from control and epileptic rats, although it should be stressed that such studies cannot discriminate between blockade of receptors at presynaptic and postsynaptic sites.
Despite the fact that we cannot yet provide a functional link between increased presynaptic NMDA autoreceptors and epileptogenesis, the enhanced level of background glutamate release is certainly partly attributable to the increased autoreceptor function, and the enhanced background excitation will contribute to elevated network excitability. It is interesting that the autoreceptor function appears to be diminished in normal adult animals but is restored in the epileptic adults, essentially recapitulating an earlier phase of development. There is evidence for recapitulation of other aspects of cortical synaptic transmission in temporal lobe epilepsy (e.g., re-emergence of excitatory actions of GABA) (Cohen et al., 2003; Cossart et al., 2005), and it has been suggested that the condition reflects a “pathological replay of developmental mechanisms” (Cohen et al., 2003). Thus, properties of cortical networks in epilepsy may bear resemblance to those during early development, a stage of heightened susceptibility to epileptogenesis (Holmes et al., 2002).
Footnotes
We thank the Epilepsy Research Foundation and the Wellcome Trust for financial support and the University of Bristol for a postgraduate research scholarship to J.Y.
Correspondence should be addressed to Gavin L. Woodhall, Department of Pharmacy and Pharmacology, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: g.l.woodhall@aston.ac.uk.
DOI:10.1523/JNEUROSCI.4413-05.2006
Copyright © 2006 Society for Neuroscience 0270-6474/06/260406-05$15.00/0
References
- Al-Ghoul WM, Meeker RB, Greenwood RS (1997) Amygdala kindling alters N-methyl-d-aspartate receptor subunit messenger RNA expression in the rat supraoptic nucleus. Neuroscience 77: 985–992. [DOI] [PubMed] [Google Scholar]
- Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M (2002) 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett 12: 1099–1102. [DOI] [PubMed] [Google Scholar]
- Avanzini G, Franceschetti S (2003) Cellular biology of epileptogenesis. Lancet Neurol 2: 233–242. [DOI] [PubMed] [Google Scholar]
- Berretta N, Jones RSG (1996) Tonic facilitation of glutamate release by presynaptic N-methyl-d-aspartate autoreceptors in the entorhinal cortex. Neuroscience 75: 339–344. [DOI] [PubMed] [Google Scholar]
- Breukel AI, Besselsen E, Lopes da Silva FH, Ghijsen WE (1998) A presynaptic N-methyl-d-aspartate autoreceptor in rat hippocampus modulating amino acid release from a cytoplasmic pool. Eur J Neurosci 10: 106–114. [DOI] [PubMed] [Google Scholar]
- Casado M, Dieudonne S, Ascher P (2000) Presynaptic N-methyl-d-aspartate receptors at the parallel fiber-Purkinje cell synapse. Proc Natl Acad Sci USA 97: 11593–11597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochilla AJ, Alford S (1999) NMDA receptor-mediated control of presynaptic calcium and neurotransmitter release. J Neurosci 19: 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Le Duigou C, Miles R (2003) Mesial temporal lobe epilepsy: a pathological replay of developmental mechanisms? Biol Cell 95: 329–333. [DOI] [PubMed] [Google Scholar]
- Cossart R, Bernard C, Ben-Ari Y (2005) Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci 28: 108–115. [DOI] [PubMed] [Google Scholar]
- Dalby NO, Mody I (2001) The process of epileptogenesis: a pathophysiological approach. Curr Opin Neurol 14: 87–92. [DOI] [PubMed] [Google Scholar]
- Dalby NO, Mody I (2003) Activation of NMDA receptors in rat dentate gyrus granule cells by spontaneous and evoked transmitter release. J Neurophysiol 90: 786–797. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Hablitz JJ (2000) Alterations in NMDA receptors in a rat model of cortical dysplasia. J Neurophysiol 83: 315–321. [DOI] [PubMed] [Google Scholar]
- Duguid IC, Smart TG (2004) Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat Neurosci 7: 525–533. [DOI] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA (1997) Ro 25-6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterization in vitro. J Pharmacol Exp Ther 283: 1285–1292. [PubMed] [Google Scholar]
- Glien M, Brandt C, Potschka H, Voigt H, Ebert U, Loscher W (2001) Repeated low-dose treatment of rats with pilocarpine: low mortality but high proportion of rats developing epilepsy. Epilepsy Res 46: 111–119. [DOI] [PubMed] [Google Scholar]
- Glitsch M, Marty A (1999) Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci 19: 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada T, Sonoda R, Watanabe A, Ono M, Shibata S, Watanabe S (1998) NMDA induced glutamate release from the suprachiasmatic nucleus: an in vitro study in the rat. Neurosci Lett 256: 93–96. [DOI] [PubMed] [Google Scholar]
- Holmes GL, Khazipov R, Ben-Ari Y (2002) New concepts in neonatal seizures. NeuroReport 13: A3–A8. [DOI] [PubMed] [Google Scholar]
- Jones RSG, Heinemann U (1988) Synaptic and intrinsic responses of medical entorhinal cortical cells in normal and magnesium-free medium in vitro. J Neurophysiol 59: 1476–1496. [DOI] [PubMed] [Google Scholar]
- Law AJ, Weickert CS, Webster MJ, Herman MM, Kleinman JE, Harrison PJ (2003) Expression of NMDA receptor NR1, NR2A and NR2B subunit mRNAs during development of the human hippocampal formation. Eur J Neurosci 18: 1197–1205. [DOI] [PubMed] [Google Scholar]
- Liu XB, Murray KD, Jones EG (2004) Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J Neurosci 24: 8885–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathern GW, Pretorius JK, Mendoza D, Leite JP, Chimelli L, Born DE, Fried I, Assirati JA, Ojemann GA, Adelson PD, Cahan LD, Kornblum HI (1999) Hippocampal N-methyl-d-aspartate receptor subunit mRNA levels in temporal lobe epilepsy patients. Ann Neurol 46: 343–358. [DOI] [PubMed] [Google Scholar]
- Moddel G, Jacobson B, Ying Z, Janigro D, Bingaman W, Gonzalez-Martinez J, Kellinghaus C, Prayson RA, Najm IM (2005) The NMDA receptor NR2B subunit contributes to epileptogenesis in human cortical dysplasia. Brain Res 1046: 10–23. [DOI] [PubMed] [Google Scholar]
- Morimoto K, Fahnestock M, Racine RJ (2004) Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol 73: 1–60. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF (1999) Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci 2: 352–357. [DOI] [PubMed] [Google Scholar]
- Samson RD, Pare D (2005) Activity-dependent synaptic plasticity in the central nucleus of the amygdala. J Neurosci 25: 1847–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY (1994) Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368: 144–147. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB (2003) Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39: 641–654. [DOI] [PubMed] [Google Scholar]
- Steffens M, Huppertz HJ, Zentner J, Chauzit E, Feuerstein TJ (2005) Unchanged glutamine synthetase activity and increased NMDA receptor density in epileptic human neocortex: implications for the pathophysiology of epilepsy. Neurochem Int 47: 379–384. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL (1999) The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro J Neurosci 19: 4180–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel A, Fritschy JM, Mohler H, Benke D (1997) NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J Neurochem 68: 469–478. [DOI] [PubMed] [Google Scholar]
- Woodhall GL, Evans DIP, Cunningham MO, Jones RSG (2001) NR2B containing NMDA auto- and heteroreceptors on entorhinal cortical neurones. J Neurophysiol 86: 1644–1651. [DOI] [PubMed] [Google Scholar]
- Ying Z, Bingaman W, Najm IM (2004) Increased numbers of coassembled PSD-95 to NMDA-receptor subunits NR2B and NR1 in human epileptic cortical dysplasia. Epilepsia 45: 314–321. [DOI] [PubMed] [Google Scholar]
- Zhong J, Carrozza DP, Williams K, Pritchett DB, Molinoff PB (1995) Expression of mRNAs encoding subunits of the NMDA receptor in developing rat brain. J Neurochem 64: 531–539. [DOI] [PubMed] [Google Scholar]