

Abstract
A conjugate between malelyated albumin and a photosensitizer (PS) shows cell-type specific targeting to macrophages via the scavenger receptor. Administration of this conjugate to a tumor-bearing mouse followed by illumination may allow selective destruction of macrophages within tumors. There is accumulating evidence that tumor-associated macrophages contribute to tumor growth, invasiveness, metastasis and immune suppression. We tested the intravenous injection of a conjugate between maleylated albumin and chlorin(e6) to Balb/c mice bearing three tumor-types with differing proportions of tumor-associated macrophages. The accumulation of PS within the tumors after intravenous (IV) injection and twenty-four hours incubation time was disappointing and we therefore investigated intratumoral (IT) injection. This gave 20−50 times greater concentrations of PS within the tumor compared to IV injection as determined by tissue extraction. Furthermore the amounts of PS in each tumor type correlated well with the numbers of macrophages both as determined by extraction from bulk tumor and fluorescence quantification, and by tissue dissociation to a single cell suspension and two-color flow cytometry with macrophage-specific antibodies. IT injection of non-conjugated PS gave lower tumor accumulation that did not correlate with macrophage content. IT injection of targeted macromolecular delivery systems is an underexplored area and worthy of further study.
Keywords: Tumor-associated macrophages, photosensitizer targeting, scavenger receptors, intratumoral injection, flow cytometry
INTRODUCTION
Photodynamic therapy (PDT) combines a non-toxic photoactivatable dye or photosensitizer (PS) in combination with harmless visible light of the specific wavelength to excite the dye to its high-energy triplet state that will then generate reactive oxygen species (ROS), such as singlet oxygen and free radicals, and kill cells 1-3. Although some PS show intrinsic tumor-localizing properties 4, much effort has been made to increase the targeting of the PS to the tumor by, for example, covalent conjugation of PS to macromolecular vehicles 5, 6 such as antibodies 7, and to polymeric carriers 8. We have previously reported on a method of carrying out highly specific targeting of PS to cells of the monocyte-macrophage lineage via conjugation of a PS to a ligand of the scavenger receptor class A 9, 10.
In contrast to previous theories that tumor-associated macrophages (TAMs) were “fighting a valiant but losing battle against the malignant cells” it is now thought 11 that the relationship between the tumor and the TAMs is more complicated and can be viewed as symbiotic in that both populations help each other to survive and grow in various ways 12, 13. Tumors actively recruit macrophages 14 and encourage their growth by expressing chemotactic molecules and growth factors for macrophages/monocytes. The macrophages in return can help the tumor to grow and spread in four distinct ways: (i) secreting paracrine growth factors for tumor cells 15. (ii) producing a wide range of angiogenic molecules 16, (iii) producing extracellular matrix-degrading enzymes that facilitate tumor invasion and metastatic spread 17, (iv) producing factors that have been shown to de-activate various components of the immune system 18. The consequences of the relationship between tumors and their TAMs are that many (but not all) tumors with a high number of TAMs have an increased tumor growth rate, and more local spread and distant metastasis. The extent of TAM infiltration has been used as an inverse prognostic predictor in breast cancer 19, head and neck cancer 20, prostate 21 and uterine cancer 21, 22. For the above reasons TAMs have been proposed to be a “target for cancer therapy” 21-23. If the concept of targeting TAMs as a cancer therapy is valid then one way of accomplishing this goal in a specific manner is to use scavenger-receptor targeted PDT 10. Since the conjugate is only taken up by macrophage type cells (that express the scavenger receptor), and the illumination can be confined to the tumor, this should ensure that only TAMs will be killed. This may be contrasted to other anti-macrophage therapies that can also kill circulating monocytes and macrophages in other anatomical locations that may have beneficial anti-tumor effects such as destroying micrometastases. This has been demonstrated in practice when two fairly specific macrophage toxins (silica particles <5μm and carrageenan) were administered to mice bearing three different tumors growing subcutaneously. In all cases the growth of the primary tumor was slowed but the number of metastases was increased 24.
In this report we describe the delivery of macrophage-targeted PS to TAMs by intravenous and intratumoral (IT) administration. Free PS was also injected IT for comparison. Three different syngeneic Balb/c mouse tumors were used with differing proportions of TAMs.
MATERIALS AND METHODS
Conjugate preparation and characterization
Preparation of the conjugate was described in detail previously 10. Briefly, chlorin(e6) (ce6) N-hydroxysuccinimide (NHS) ester was made by reacting 1.5 equivalents of dicyclohexylcarbodiimide and 1.5 equivalents of NHS with 1 equivalent of ce6 (Frontier Scientific, Logan, UT) in dry dimethyl sulfoxide. One equivalent of bovine serum albumin (BSA) (Sigma Chemical Co., St Louis, MO) was dissolved in NaHCO3 buffer and 3 equivalents of ce6–NHS ester was added. After the reaction mixture was kept in the dark at room temperature for 6 h, solid maleic anhydride (50 equivalents) was added with vortex mixing and addition of saturated NaHCO3 solution as needed to keep the pH above 7. The reaction mixture was allowed to stand at room temperature in the dark for 3 h. Crude conjugate was purified by adding 5 volumes of acetone (ACS grade) slowly at 4°C, and kept at 4°C for 24 h, followed by centrifugation at 4000 g for 15 min at 4°C. The supernatant was removed, and the pellet was collected. The pellet was redissolved in water and dialyzed against phosphate buffered saline (PBS) to afford the pure conjugate between maleylated BSA and ce6 (BSA-ce6-mal). The concentration of the purified conjugate solution was determined by absorption of ce6 at 400 nm. The conjugates were characterizated by fluorescence imaging SDS-PAGE. Gradients of 4−10% acrylamide were used in a nonreducing gel and ce6 was localized on the gel by fluorescence excitation (400−440 nm bandpass filter, emission 580 longpass filter), (ChemiImager 4000, Alpha Innotech Corp, San Leandro, CA). Protein was localized by Coomassie blue staining. The conjugate had an average of 4 ce6 molecules per BSA molecule and the ε-amino groups of lysine residues were approximately 80% maleylated.
Cell Line and Tissue Culture Conditions
J774 murine Balb/c reticulum cell sarcoma 25 (ATCC TIB-67), mammary sarcoma EMT6 26 (ATCC CRL-2755) and colon adenocarcinoma CT26 27 (ATCC CRL-2638) were obtained from ATCC (Manassas, VA), and cultured in Dulbecco's modified Eagle's medium with 4 mM L-glutamine adjusted to contain 1.5g/L sodium bicarbonate and 4.5 g/L glucose with 10% fetal calf serum (FCS), 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were collected for injection by washing with PBS without Ca2+ and Mg2+, and adding trypsin-EDTA to the plate for 5 minutes at 37°C.
Mouse tumor models
All experiments were carried out with the approval of the Subcommittee on Research Animal Care of Massachusetts General Hospital and were in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Male Balb/c mice (Charles River Laboratories, Inc, Wilmington, MA) (4 weeks old, weighing 20−25 g) were kept in a barrier room under permanent sterile conditions to avoid any infections and had continual access to food (synthetic chlorophyll free mouse diet purified 904606, ICN Pharmaceuticals Inc, Costa Mesa, CA) and water, which were taken ad libitum. Throughout the experiment mice were housed in laminar flow racks under specific pathogen-free conditions, and were monitored daily for general health status. All animals were shaved on the right thigh and depilated with Nair (Carter-Wallace Inc New York, NY). Mice were anesthetized with an i.p. injection of ketamine/xylazine cocktail (90mg/kg ketamine, 10 mg/kg xylazine). One million tumor cells were injected subcutaneously in one mid-thigh area suspended in 100-μL PBS. Tumors grew predictably in all mice and reached a size of 6−7 mm diameter in 8−10 days after injection. The size of the tumor was measured with vernier calipers in three dimensions, from which the volume was calculated using the formula . The average tumor volume used in these experiments was 0.32 ± 0.08 cm3
BSA-ce6-mal conjugate administration
When the tumor reached the appropriate size BSA- ce6-mal conjugate was administered either IV or IT. The injected dose of 3.5 mg ce6 equivalent/kg body weight (approximately 70 μg per mouse) was the same for all the tumors tested and involved the injection (lasting about 3 minutes) of 100 μL solution in sterile PBS. Other groups of mice received same amount of free ce6 injected IT in 100 μL PBS solution. Injections were either carried out in the lateral tail vein or into the center of the tumor with a 26-gauge needle. After IV administration of conjugate in a series of mice with CT26 tumors, blood samples (10−20 μL) were withdrawn from the orbital plexus at the following times: 15, 30 minutes, 1, 2, 4, 6, 12, 24,36 and 48 hours after injection. Blood samples were centrifuged at 1000 g (Eppendorf Centrifuge 5417C; Fisher Scientific, Pittsburg, PA) at 20°C for 5 min and the supernatant was collected by aspiration, weighed and then dissolved in 3 mL 1 M NaOH/0.2% SDS.
Biodistribution
At 6 h and 24 h after injection animals were sacrificed by CO2 inhalation. At necropsy the tumor, liver, blood, skin, muscle, kidney, spleen, large intestine, stomach, bladder, lung, thymus, heart and bone marrow were harvested. Wet tissue samples (about 70 mg) were weighed immediately after resection and placed in 3 mL 1 M NaOH/0.2% SDS for 5 days. The peak height of the fluorescence emission (emission maximum between 655 and 670 nm) was measured with a fluorometer (FluoroMax-3, Jobin Yvon Horiba, Instrument S.A., Inc., NJ) (excitation at 405 nm, emission scanned from 580−720 nm). Quantitation of BSA-ce6-mal conjugate concentration in the tissue extracts was obtained by constructing calibration curves from known amounts of the same conjugate together with weighed specific tissue samples from uninjected mice dissolved in 1 M NaOH/0.2% SDS. A similar set of calibration curves was prepared using free ce6. Results were expressed as mol ce6 equivalent per gram tissue.
Tumor dissociation and photosensitizer and macrophage characterization
Fresh tumor was cut into small pieces, placed in Hanks buffered salt solution (HBSS) containing 2 mM Ca2+ and 100 U/mL collagenase IV (Sigma), and digested at 37°C for 4 hours with continuous shaking. The digests were then diluted with HBSS and filtered through a 70-mm strainer, and the cells pelleted by centrifugation at 400 g for 10 minutes. The pellet was then washed with HBSS. The cells were resuspended in HBSS containing 0.1% BSA. For macrophage analysis, 100 μL cell suspension was stained with 3 μg/mL FITC labeled anti-SRA (clone 2F8) or F4/80 (clone CI:A3−1, both from Serotec Inc, Raleigh, NC) or IgG2b isotype control at 4°C for 1 hour, after blocking with 10% rat serum and 10% mouse serum. The incubation mixture was then washed with 2-mL buffer and the cells resuspended in 200-μL buffer. Fluorescence was then read on a FACScalibur (Becton Dickinson, San Jose, CA). The cell population was gated, and cell-associated fluorescence was analyzed in a histogram of cell number versus fluorescence intensity in log scale. A marker was set to allow 1% of all gated events to be higher than this marker, and samples stained with the FITC-anti-SRA were analyzed with this same marker. Cells with fluorescence higher than this marker were considered macrophages.
For 2 color (fluorescence from both BSA-ce6-mal and FITC-anti-SRA) analysis, the tumor tissue which had been injected with the BSA-ce6-mal was prepared and the cells were stained using the same method as described above. Compensations were made with cells from tumors injected with BSA-ce6-mal without FITC-staining and tumor without BSA-ce6-mal stained with FITC-anti-SRA. Samples were then analyzed with the quadrants set up with the controls.
Histology
Tumors were removed and immediately placed in Tissue-Tek OCT embedding medium (Sakura Finetek USA Inc, Torrance, CA) followed by freezing at −80°C, and sections were cut 5 μm thick on a cryotome. Immunohistochemistry was carried out to characterize endothelial cells and macrophages. Thawed slides were initially fixed in acetone, followed by rinsing in PBS (2X) and in hydrogen peroxide (3%) to block endogenous peroxidase activity. Slides were then incubated in diluted blocking rabbit serum for 30 minutes. Primary antibodies; rat anti-mouse CD31 PECAM IgG2a (clone 390, Serotec) for endothelial cells, and rat anti-mouse F4/80 IgG2b (clone CI:A3-Serotec) for macrophages (both J774 cells and host macrophages), were incubated at 100X dilution in PBS containing 3% FCS overnight. The slides were rinsed with PBS and the secondary antibody (biotinylated rabbit anti-rat IgG, Vector Labs, Burlingame, CA) was applied for 30 minutes. Again the slides were rinsed and preformed avidin: biotinylated peroxidase complex (ABC, Vector Labs) was applied for 30 minutes. Slides were rinsed and NovaRed peroxidase substrate (SK-4800, Vector Labs) was applied according to the manufacturer's instructions until desired intensity was achieved. Slides were rinsed and counterstained in hematoxylin, rinsed and dipped once in lithium carbonate, dehydrated in alcohol and xylene. Slides were visualized on an Axiophot (Carl Zeiss MicroImaging Inc, Thornwood, NY) microscope. Images were captured using a RT Slider camera and Spot Advanced software (both obtained from Diagnostic Instruments Inc, Sterling Heights, MI). Quantitation of brown macrophage staining on F4/80 stained slides was carried out by color segmentation using IP Lab software (Scanalytics Inc, Fairfax, VA), and numbers of vessels were counted on CD31 stained slides and results for both are means of 15−20 fields derived from slides from five separate tumors.
Statistics
Results are presented as means and standard errors. Differences between means were compared with an unpaired 2-tailed Student's t-test assuming equal or non-equal distribution of the standard deviations as appropriate.
RESULTS
Characterization of macrophage content in three Balb/c tumors
In order to determine the cell specificity and efficiency of macrophage-targeted PS delivery in vivo, it was desirable to study mouse tumors with widely differing macrophage contents. The J774 tumor is composed from malignant cells of the monocyte-macrophage lineage and it should therefore have a very high macrophage content comprising both the malignant cells themselves and any host macrophages that are present. After preliminary testing of a number of other mouse tumors we selected EMT6 breast sarcoma and CT26 colon carcinoma as non-macrophage tumors with high and low macrophage content respectively. We carried out immunohistochemistry of frozen sections of the tumors with the macrophage specific antibodies and the ABC-peroxidase method and quantified the resulting brown staining by color segmentation. Because intravenous delivery of the conjugate was disappointing (see next section) we asked whether the tumors differed in the density of blood vessels and whether this could be responsible for poor drug delivery. Therefore we also stained the frozen sections with anti-CD31 (PECAM) to enable the microvascular density to be determined. Figure 1 shows anti-F4/80 and anti-CD31 staining of the three tumor types. Panels 1 A, B, C show representative examples of F4/80 staining. CT26 (Fig 1A) and EMT6 (Fig 1B) tumors show focal staining, with EMT6 demonstrating significantly more macrophage-positive cells than CT26. By contrast J774 tumors (Fig 1C) show a uniformly positive brown staining around the membranes of all the constituent cells of the tumor. The quantification of the F4/80 staining measured on frozen sections is shown in Figure 2 (solid bars). It should be noted that the J774 staining, while visible on virtually all the cells in the section, does not give a numerical value near 100% on color segmentation because the staining is confined to the membrane of the cells and the internal cytoplasm is F4/80 negative. The anti-CD31 staining is shown in panels 1 D,E,F. It can be seen that CT26 (Fig 1D) is the most vascular tumor of the three followed by EMT6 (Fig 1E) and J774 (Fig 1F) is least vascular. The numbers of vessels per high power field for the three tumors (determined from 15−20 fields from 5 slides per tumor) are as follows: CT26 = 73.3 ± 11.7; EMT6 = 62 ± 9.2; J774 42.6 ± 7.6. The difference between J774 and the other two tumors is significant (P < 0.005).
Figure 1.
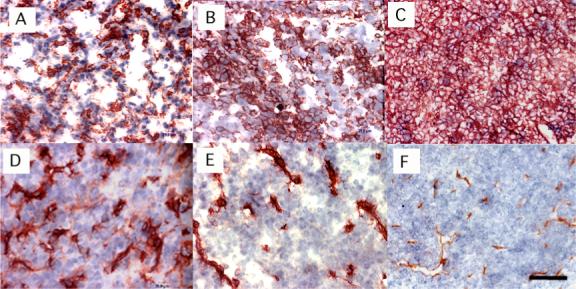
Immunohistochemistry of frozen sections of the three Balb/c tumors. Panels (A)-(C) F4/80 staining; panels (D)-(F) CD31 staining; of (A) and (D) CT26; (B) and (E) EMT6; (C) and (F) J774 tumors. Scale bar is 25 micron.
Figure 2.

Quantification of macrophage content of the three Balb/c tumors. Immunohistochemistry with F4/80 was quantified by color segmentation (15−20 fields from 5 slides for each tumor), flow cytometry with anti-SRA FITC labeled antibody was from triplicate measurements from at least 8 dissociations from each tumor. Bars are SEM. P values are from unpaired 2-tailed Students t-test.
To provide a second independent quantification of macrophage content in these three mouse tumors we also measured actual percentages of macrophage cells out of the total cells by dissociating tumor fragments into cell suspensions and using FITC-labeled rat-anti mouse monoclonal antibodies that recognize macrophage-specific antigens followed by analysis with flow cytometry. We used both rat anti-mouse F4/80 and rat anti-mouse SRA monoclonal antibodies to stain the cell suspensions and the results were very similar. The values reported here are for anti-SRA staining. As can be seen from Figure 2 (open bars) the values of % macrophages are very similar to the values obtained by quantitative immunohistochemistry, with exception of the case of J774 where the value from flow cytometry is higher than that for immunohistochemistry for reasons previously explained (only cell membranes stained with peroxidase on tissue slices).
Pharmacokinetics, tumor targeting and biodistribution of BSA-ce6-mal after intravenous delivery
It is necessary to place mice on a synthetic chlorophyll free diet before carrying out biodistribution experiments involving PS such as chlorins. This is because a metabolite of dietary chlorophyll has identical red fluorescence and accumulates in mouse organs, particularly organs of the digestive tract and skin 28, 29. We injected mice bearing CT26 tumors with BSA- ce6-mal at a dose of 3.5 mg ce6 equivalent dissolved in 100 μL of PBS via the tail vein. Blood samples were removed and analyzed for ce6 concentration, and a pharmacokinetic curve was plotted (Fig 3). When this curve is fitted to a standard two-compartment (biexponential) model it yielded estimated mean alpha and beta half-lives (95% confidence interval) that were 98 minutes (36−256 minutes) and 10 hours (3.2−28 hours), respectively. Although the conjugate is removed fairly rapidly from the bloodstream of mice, we decided to allow twenty-four hours after injection in order to provide sufficient time for the conjugate to be accumulated by the macrophages by the time dependent process of receptor-mediated endocytosis. Figure 4 shows the mean amounts of ce6 equivalent per gram of tissue for tumor and for thirteen normal tissues in mice bearing the three different tumor types (CT26 n=6; EMT6 n=6; J774 n=7). The amounts of ce6 in the tumor increase for the different tumor types in the order CT26 < EMT6 < J774 which is the identical order of increasing macrophage content in the tumors (see Fig 2). However the error bars were sufficiently wide that the only one of these differences that was significant was CT26 < J774 (P<0.05). The largest uptake per gram of tissue in the normal organs was exhibited by the liver. In decreasing order of uptake the bladder was followed by the large intestine, thymus, plasma, kidney, tumor, lung, skin and muscle, stomach and bone marrow, and the lowest uptake was seen in was spleen and heart. In general all these values had large inter-animal variation for the same organ type as shown by the relatively large error bars. There appeared to be no systematic difference in uptake by the same organ type between animals implanted with the different tumor types.
Figure 3.

Plasma pharmacokinetics of BSA-ce6-mal after IV injection into Balb/c mice bearing CT26 tumors. Points are means of values determined from at least six mice per time point. Bars are SD.
Figure 4.

Distribution of ce6 in organs and tumors from Balb/c mice 24 hours after IV injection of BSA-ce6-mal at a dose of 3.5 mg ce6 equivalent/kg. Tumors were dissolved in 1 M NaOH/0.2% SDS and fluorescence measured and converted to mol ce6 equivalent per gram of tissue with calibration curves. Values are means of triplicate tissue samples removed from at least six mice. Bars are SD.
Tumor targeting and biodistribution of BSA-ce6-mal after intratumoral delivery
We wished to compare the intratumoral delivery by direct injection of the macrophage-targeted conjugate into the tumor with that provided by systemic injection into the bloodstream because it appeared that intravenous administration was not highly efficient in delivering the ce6 to the TAMs. In addition the trend of the microvascular density of the three tumor types appeared to be working in opposite direction to the TAM content of the tumor. In other words CT26 had the lowest percentage of TAMs and the highest density of blood vessels, while J774 had the highest percentage of TAMs and the lowest density of blood vessels. The EMT6 tumor was intermediate in both parameters. It was necessary to decide on a dose for intratumoral injection. In the absence of other overriding factors we decided to use the same dose (70 μg ce6 equivalent per mouse) as we used for IV injection. It could be argued that the dose should have been lower for intratumoral injection as compared to IV injection because the total volume of tissue being addressed is much less in the first case than in the second. However there is no obvious way of calculating what precisely the reduced dose should be and the point of intratumoral injection is to maximize the delivery to tumor tissue, while our experiments were designed to test if cell type specific targeting could still be preserved. Figure 5 depicts the accumulation of ce6 in the same tumor types and normal tissues as were measured for IV injection in Figure 4. The amounts in the tumors were 20.4 ± 2.7; 39.5 ± 5.2; and 85.7 ± 34.3 nmol ce6 per g tissue for CT26, EMT6 and J774 tumors respectively twenty-four hours after intratumoral injection. By contrast the comparable values for intravenous injections twenty-four hours before sacrifice were 0.7 ± 0.3; 1.2 ± 0.7; 1.7 ± 0.2 nmol ce6 per g tissue for CT26, EMT6 and J774 tumors respectively. These numbers represent an increase in tumor accumulation of ce6 after intratumoral injection of from 29 times (CT26) to 50 times (J774) the accumulation obtained after intravenous injection. The relative amounts of ce6 in the three tumor types twenty-four hours after IT injection also correlated well with the macrophage contents of the tumors (compare Fig 2). Figure 5 also demonstrates that the ce6 does not accumulate in any of the normal tissues to any appreciable extent after IT injection. The largest concentrations were found in large intestine and plasma but were less than 10% of the tumor concentrations.
Figure 5.

Distribution of ce6 in organs and tumors from Balb/c mice 24 hours after intratumoral injection of BSA-ce6-mal at a dose of 3.5 mg ce6 equivalent/kg. Tumors were dissolved in 1 M NaOH/0.2% SDS and fluorescence measured and converted to mol ce6 equivalent per gram of tissue with calibration curves. Values are means of triplicate tissue samples removed from at least six mice. Bars are SD.
Intratumoral injection of free ce6
In order to study to what extent the tumoral accumulation of BSA-ce6-mal is due to the macrophage-targeting properties of the conjugate, and to what extent it is due to the fact of IT injection, we compared the conjugate with an IT injection of non-conjugated ce6 carried out under the same conditions. The biodistribution of ce6 in mice bearing the three tumor types at 24 h post-injection is shown in Figure 6. The amount of free ce6 in each tumor was less in every case than the amount of BSA-ce6-mal (for CT26 1.17 × 10−8 vs 2.04 × 10−8; for EMT6 2.55 × 10−8 vs 3.95 × 10−8; for J774 1.15 × 10−8 vs 8.57 × 10−8 mol ce6 / g tissue). In addition although there were differences between tumor content of free ce6 within the three tumor types, these did not correlate with the macrophage density although the error bars for free ce6 tumor content were too large to allow statistical comparisons to be made. The highest free ce6 content was in EMT6 followed by CT26 and J774 was the lowest. Another difference observed was the higher amount of free ce6 that was found in distant organs compared with the levels found after IT injection of BSA-ce6-mal. The ce6 was found in the bone marrow, thymus, skin, bladder, large intestine and stomach.
Figure 6.

Distribution of ce6 in organs and tumors from Balb/c mice 24 hours after intratumoral injection of free ce6 at a dose of 3.5 mg ce6 /kg. Tumors were dissolved in 1 M NaOH/0.2% SDS and fluorescence measured and converted to mol ce6 equivalent per gram of tissue with calibration curves. Values are means of triplicate tissue samples removed from at least six mice. Bars are SD.
BSA-ce6-mal is targeted to tumor-associated macrophages as determined by flow cytometry
Although the bulk ce6 content of the three different tumor types correlated well with the macrophage content of the respective tumors, it was still necessary to show that the conjugate was actually recognized and taken up by the macrophages in the tumor. This was done by two-color flow cytometry carried out on dissociated single cell suspensions of tumors removed from mice that had received intratumoral injections with conjugate and were subsequently incubated with FITC labeled antibodies. This technique allowed both the cells that had taken up ce6 (red fluorescence) and the cells labeled as macrophages (green fluorescence) to be quantified simultaneously. Figure 7 depicts the results. The top three scattergrams were obtained with a control isotype IgG labeled with FITC, while the lower three were obtained with FITC-labeled 2F8 anti-SRA antibody. In all three tumors the clear majority of cells that take up ce6 are also labeled with anti-SRA antibody. In CT26 tumors (Fig 7B) it can be seen that a portion of the cells that take up ce6 are not labeled as macrophages, while in EMT6 tumors (Fig 7D) some macrophages do not take up ce6, but in J774 tumors the correlation is almost complete. The reason for the first discrepancy is due to the relative paucity of macrophages in the CT26 tumor. Presumably a small number of another cell type (tumor or endothelial cells) can also take up some conjugate. In the case of EMT6 tumors the discrepancy presumably arises because this is a fibrous tumor and there is more difficulty in the injected conjugate achieving a completely homogeneous distribution within the whole tumor.
Figure 7.

Examples of 2 color flow cytometry analysis of dissociated tumors. FL1 is green fluorescence from FITC-labeled control IgG2b (panels A, C, E) or FITC-labeled anti-SRA (panels D, E, G). FL3 is red fluorescence from cell-associated ce6. Panels A, B are CT26 tumors, C, D are EMT6 tumors, and E, F are J774 tumors.
Comparison of cell-associated and bulk ce6 content in tumors at 6 and 24 hours
We asked whether the time after intratumoral injection affected the tumor content of ce6 both in terms of bulk ce6 extracted from tissue samples and also cell-associated ce6 fluorescence. It is possible that the injected material will diffuse away from the injected tumor site fairly quickly and, at the same time, the uptake of the targeted conjugate by the macrophages in the tumor is likely to be a time dependent process. We selected 6 and 24-hour time points as these appeared to be far enough apart to allow differences in these processes to become apparent. Figures 8A and 8B depict the results. In Fig 8A it can be seen that the respective amounts of ce6 extracted from the three tumor types are remarkably similar at 6 and 24 hours after injection. The ce6 content of CT26 increases somewhat at 24 hours compared to 6 hours, while the ce6 contents of EMT6 and J774 tumors decrease slightly but these differences are not significant. The differences between the three tumor types are significant at both 6 hours and 24 hours. Fig 8B shows the data obtained from measuring the cell associated ce6 fluorescence from the same tumors and time points. The units of cellular fluorescence take into account both the % of red-fluorescent cells and the intensity of the red fluorescence. The overall picture of the various columns in Fig 8B is very similar to that observed in Fig 8A. Again CT26 is somewhat higher at 24 hours than 6 hours and EMT6 is somewhat lower, while J774 is the same. The differences between time points were not significant, while the tumor types were significantly different in most cases. The remarkable similarity of the patterns of the columns in Figs 8A and 8B provides additional evidence that the ce6 extracted from the tumor samples is in reality targeted to the macrophages in the tumor.
Figure 8.



(A) Content of ce6 in tumors from Balb/c mice 6 and 24 hours after intratumoral injection of BSA-ce6-mal at a dose of 3.5 mg ce6 equivalent/kg. Values are means of triplicate determinations from at least 6 tumors. P values are from unpaired 2-tailed Students t-test.
(B) Geometrical means obtained by FACScalibur software of red fluorescence obtained by flow cytometry from Balb/c mice tumors 6 and 24 hours after intratumoral injection described in Fig 7a. Values are means of triplicate determinations from at least 6 tumors. P values are from unpaired 2-tailed Students t-test.
(C) Content of ce6 in tumors from Balb/c mice 6 and 24 hours after intratumoral injection of free ce6-mal at a dose of 3.5 mg ce6/kg. Values are means of triplicate determinations from at least 6 tumors.
We also were able to compare the bulk amounts of ce6 found in the three tumor types at 6 and 24 h after IT injection of free ce6. Fig 8C shows that in all three tumor types the amounts of free ce6 present at 6 h after IT injection are higher than those present at 24 h. This reduction in tumor ce6 content with time is consistent with the non-targeted PS simply diffusing away from the tumor as time progresses. This proposed mechanism is also consistent with the higher amounts of ce6 seen in distant organs in Fig 6 compared to those seen after IT injection of BSA-ce6-mal in Fig 5.
DISCUSSION
Many new cancer therapies rely on the delivery of therapeutic molecules to tumors by an active targeting strategy. This strategy frequently involves their covalent attachment to macromolecules that are recognized by receptors specific to tumor cells or associated cell types. However, it is also well known that large molecules encounter significant barriers to their transport and accumulation in tumors after intravenous injection 30. These barriers include (a) the difficulty faced by large molecules in passing through the “pores” between endothelial cells in the tumor microvasculature 31; (b) raised intratumoral pressure that inhibits convection of large molecules from the blood vessels into the tumor interstitium 32; and (c) rapid clearance of macromolecular conjugates by the reticuloendothelial system in the liver and the spleen leading to short circulation times 33. Many workers have studied ways of overcoming these barriers. Barrier (a) has been tackled by reducing the size of the macromolecular targeting vehicle, for instance by changing from intact IgG antibodies to Fab and scFv antibody fragments 34, 35. Barrier (b) has been tackled by pharmacological approaches to cause vasodilation and hence reduce intratumoral pressure 36, 37. Barrier (c) has been tackled by attaching polyethylene glycol side chains to the macromolecular conjugate (PEGylation) that reduces uptake by cells of the reticuloendothelial system and prolongs circulation time 38. However, despite these multiple novel approaches, the relatively simple approach of directly injecting the targeted conjugate into the tumor has not been much studied.
In our case we wished to test if a PS-conjugate that was recognized by macrophages could be used to deliver PS specifically to TAMs. In order to test the specificity of this delivery we used three Balb/c cell lines that could grow as subcutaneous tumors in syngeneic mice. These were selected to have a range of macrophage contents. CT26 is a colon adenocarcinoma with approximately 20% TAMs, while EMT6 is a mammary sarcoma with about 35% TAMs. J774 however is a reticulum cell sarcoma where all the malignant cells have characteristics of macrophages. In the latter case of J774 tumors there may or may not be additional host macrophages recruited to the tumor. In addition to tumor cells and TAMs the tumors will contain endothelial cells and varying numbers of fibroblasts and non-macrophage leukocytes (lymphocytes, neutrophils and mast cells).
However, since macrophage recognition plays a large role in the accumulation of macromolecular conjugates such as BSA-ce6-mal in organs of the reticuloendothelial sytem, it might be expected that IV injection of these conjugates could be problematic in that the molecules may not survive long enough in the circulation to allow accumulation in TAMs. In addition the microvascular blood supply to the three tumors was different between tumor types. The tumor that had the highest macrophage density (J774) had the lowest blood vessel density and vice versa (CT26). This was an opposite finding to what might have been predicted on the basis that macrophage density is one of the factors that controls the extent of angiogenesis 39, 40. It did however suggest, that the IV route of administration would not be optimal for targeting macrophages in these three Balb/c tumors. The pharmacokinetics of the conjugate in mice displayed serum half-lives that, while not long-lived, were not as short as might be supposed if the conjugate was rapidly cleared by the liver. The biodistribution of the conjugate in organs after 24 hours showed the highest accumulation in the liver as expected. The amounts in spleen were surprisingly low as this organ is reported to accumulate macrophage-targeted molecules. Possible explanations for this observation include difficulties in conjugate molecules exiting the blood vessels, or lack of scavenger receptor expression or low activation state particular to splenic macrophages. The amounts in kidney and bladder were relatively high considering that PS such as ce6 are generally excreted via gall bladder and bile. It is possible that ce6 that is separated from protein by digestion in lysosomes is metabolized and excreted differently. The other organs with relatively high uptakes were the large intestine and the thymus, both of which are relatively rich in macrophages.
The key question to be answered when considering IT injections of receptor-targeted delivery vehicles is; does the receptor recognition apply equally after IT injection as it does after IV injection? It could be possible that the expected high concentrations of the particular molecule of interest in the tumor, would be caused by a non-specific “pooling” within the interstitium of the tumor. There is not a lot of literature information on the diffusion rates of various molecules after IT injection. Our data shows that the amounts of the macrophage-targeted conjugate within the three tumors correlated very well with the actual macrophage content after IT injection. Moreover the majority of this ce6 was associated with target SRA-receptor expressing cells as demonstrated by flow cytometry. The amounts present at 6 and at 24 hours showed little difference between the time points in all three tumor types, suggesting that the conjugate is recognized by its target cells within the first 6 hours and also that unbound conjugate has largely diffused away during this first six hour period. It also appears that the ce6 taken up by the macrophages is retained for some time (at least 24 hours) and not released out of the cell after lysosomal degradation. The data obtained from the IT injection of non-conjugated ce6 corroborates this hypothesis. Free ce6 does not bind to cellular receptors, therefore the amount in the tumor at 6 hours is lower (since some has already diffused away) than that found for scavenger-receptor targeted conjugate. Likewise the amounts of free ce6 are even lower at 24 h than they are at 6 h since the non-bound free PS continues to diffuse away. Consistent with this mechanism are the higher amounts of ce6 found in distant organs 24 h after IT injection of free ce6 compared to those found after 24 h after IT injection of BSA-ce6-mal.
A recent report by Gupta et al 41 compared the intravenous and intratumoral administration of an anti-carcinoembryonic antigen (CEA) monoclonal antibody conjugated to radiolabeled Photosan, to mice bearing subcutaneous CEA expressing tumors. They found that after IV injection the majority of the radioactivity was localized in the liver especially at longer times after injection with relatively small amounts in the tumor. By contrast after IT injection the highest radioactivity remained in the tumor for 24 hours with much smaller amounts appearing in the liver at 6−10 hours post-injection. There have been a few reports about IT injection of non-targeted PS. Lee et al 42 compared IV and IT injection of aluminum phthalocyanine disulfonate into subcutaneous mouse tumors. They found the concentration in the tumor after IT injection reached a peak at 1hour but at 3−6 hours after IV injection. There was more variation of PS concentrations between individual tumors after IV, but more variation of PS concentrations within a single tumor after IT. Hebeda et al 43 injected hematoporphyrin derivative (HpD) IT into intracerebral rat gliosarcomas and found long-lasting retention of PS, and discussed the need for large injection volumes to cover the whole tumor. Amano and coworkers 44 found higher concentrations of HpD after IT injection into subcutaneous mouse bladder tumor compared to IV injection and much higher tumor-skin ratios.
The most important consideration that affects the efficiency of IT injections is the need to obtain homogeneous distribution of PS within the tumor. Here it is clearly important to balance the volume injected with the volume of the tumor. In addition the injection should be carried out slowly to avoid creating traumatic pools of liquid within the tumor. We found that a single injection was preferable to multiple injections as the intratumoral pressure could force liquid out of pre-existing needle puncture holes. It may be possible to use in vivo fluorescence imaging to follow the intratumoral spread of the injected material. It remains to be seen what will be the results when these tumors that have been injected IT with macrophage-targeted PS-conjugate are illuminated with a therapeutic dose of red light. Preliminary experiments suggest that there is low toxicity (either systemically or locally to normal tissue), and that there is positive evidence of tumor response (data not shown). These experiments are currently underway in our laboratory.
In conclusion we have shown that intratumoral injection of a macrophage targeted ce6 conjugate gives high amounts of ce6 in the tumor compared to other organs and compared to intravenous delivery. Moreover the tumor ce6 content correlates with tumor macrophage content, and ce6 is specifically taken up by target macrophage cells at 6 hours and retained for 24 hours. Non-targeted free ce6 gives lower amounts at 6 h that continue to decrease and do not correlate with macrophage content. Intratumoral administration of receptor-targeted therapeutics may be an underexplored technique and worthy of further study.
ACKNOWLEDGEMENTS
This work was supported by the National Cancer Institute (grant R01-CA/AI838801 to MRH). Ana P Castano was supported by a Department of Defense CDMRP Breast Cancer Research Grant (W81XWH-04-1-0676). Emilia Swietlik was supported by UICC grant ICR/05/103/2005. We are grateful to John Demirs for help with histology.
References
- 1.Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part one--photosensitizers, photochemistry and cellular localization. Photodiagn. Photodyn. Ther. 2004;1(4):279–293. doi: 10.1016/S1572-1000(05)00007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part two -cellular signalling, cell metabolism and modes of cell death. Photodiagn. Photodyn. Ther. 2004;2(1):1–23. doi: 10.1016/S1572-1000(05)00030-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dolmans DE, Fukumura D, Jain RK. Photodynamic therapy for cancer. Nat. Rev. Cancer. 2003;3(5):380–387. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- 4.Hamblin MR, Newman EL. On the mechanism of the tumour-localising effect in photodynamic therapy. J. Photochem. Photobiol. B. 1994;23(1):3–8. doi: 10.1016/s1011-1344(94)80018-9. [DOI] [PubMed] [Google Scholar]
- 5.Konan YN, Gurny R, Allemann E. State of the art in the delivery of photosensitizers for photodynamic therapy. J. Photochem. Photobiol. B. 2002;66(2):89–106. doi: 10.1016/s1011-1344(01)00267-6. [DOI] [PubMed] [Google Scholar]
- 6.Sharman WM, van Lier JE, Allen CM. Targeted photodynamic therapy via receptor mediated delivery systems. Adv. Drug Deliv. Rev. 2004;56(1):53–76. doi: 10.1016/j.addr.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 7.Hamblin MR, Governatore MD, Rizvi I, Hasan T. Biodistribution of charged 17.1A photoimmunoconjugates in a murine model of hepatic metastasis of colorectal cancer. Br. J. Cancer. 2000;83(11):1544–1551. doi: 10.1054/bjoc.2000.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamblin MR, Miller JL, Rizvi I, Ortel B, Maytin EV, Hasan T. Pegylation of a chlorin(e6) polymer conjugate increases tumor targeting of photosensitizer. Cancer Res. 2001;61(19):7155–7162. [PubMed] [Google Scholar]
- 9.Demidova TN, Hamblin MR. Macrophage-targeted photodynamic therapy. Int. J. Immunopathol. Pharmacol. 2004;17(2):117–126. doi: 10.1177/039463200401700203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamblin MR, Miller JL, Ortel B. Scavenger-receptor targeted photodynamic therapy. Photochem. Photobiol. 2000;72(4):533–540. doi: 10.1562/0031-8655(2000)072<0533:srtpt>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 11.Mantovani A. Tumor-associated macrophages in neoplastic progression: a paradigm for the in vivo function of chemokines. Lab. Invest. 1994;71(1):5–16. [PubMed] [Google Scholar]
- 12.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J. Pathol. 2002;196(3):254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 13.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer. 2004;4(1):71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 14.Graves DT, Valente AJ. Monocyte chemotactic proteins from human tumor cells. Biochem. Pharmacol. 1991;41(3):333–7. doi: 10.1016/0006-2952(91)90528-d. [DOI] [PubMed] [Google Scholar]
- 15.Wu S, Rodabaugh K, Martinez-Maza O, Watson JM, Silberstein DS, Boyer CM, Peters WP, Weinberg JB, Berek JS, Bast RC., Jr. Stimulation of ovarian tumor cell proliferation with monocyte products including interleukin-1, interleukin-6, and tumor necrosis factor-alpha. Am. J. Obstet. Gynecol. 1992;166(3):997–1007. doi: 10.1016/0002-9378(92)91379-o. [DOI] [PubMed] [Google Scholar]
- 16.Harmey JH, Dimitriadis E, Kay E, Redmond HP, Bouchier-Hayes D. Regulation of macrophage production of vascular endothelial growth factor (VEGF) by hypoxia and transforming growth factor beta-1. Ann. Surg. Oncol. 1998;5(3):271–8. doi: 10.1007/BF02303785. [DOI] [PubMed] [Google Scholar]
- 17.Hagemann T, Robinson SC, Schulz M, Trumper L, Balkwill FR, Binder C. Enhanced invasiveness of breast cancer cell lines upon co-cultivation with macrophages is due to TNF-alpha dependent up-regulation of matrix metalloproteases. Carcinogenesis. 2004;25(8):1543–1549. doi: 10.1093/carcin/bgh146. [DOI] [PubMed] [Google Scholar]
- 18.Elgert KD, Alleva DG, Mullins DW. Tumor-induced immune dysfunction: the macrophage connection. J. Leukoc. Biol. 1998;64(3):275–90. doi: 10.1002/jlb.64.3.275. [DOI] [PubMed] [Google Scholar]
- 19.Leek RD, Landers R, Fox SB, Ng F, Harris AL, Lewis CE. Association of tumour necrosis factor alpha and its receptors with thymidine phosphorylase expression in invasive breast carcinoma. Br. J. Cancer. 1998;77(12):2246–51. doi: 10.1038/bjc.1998.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young MR, Wright MA, Lozano Y, Prechel MM, Benefield J, Leonetti JP, Collins SL, Petruzzelli GJ. Increased recurrence and metastasis in patients whose primary head and neck squamous cell carcinomas secreted granulocyte-macrophage colony-stimulating factor and contained CD34+ natural suppressor cells. Int. J. Cancer. 1997;74(1):69–74. doi: 10.1002/(sici)1097-0215(19970220)74:1<69::aid-ijc12>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 21.Lissbrant IF, Stattin P, Wikstrom P, Damber JE, Egevad L, Bergh A. Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. Int. J. Oncol. 2000;17(3):445–451. doi: 10.3892/ijo.17.3.445. [DOI] [PubMed] [Google Scholar]
- 22.Salvesen HB, Akslen LA. Significance of tumour-associated macrophages, vascular endothelial growth factor and thrombospondin-1 expression for tumour angiogenesis and prognosis in endometrial carcinomas. Int. J. Cancer. 1999;84(5):538–43. doi: 10.1002/(sici)1097-0215(19991022)84:5<538::aid-ijc17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 23.Wahl LM, Kleinman HK. Tumor-associated macrophages as targets for cancer therapy. J. Natl. Cancer Inst. 1998;90(21):1583–4. doi: 10.1093/jnci/90.21.1583. [DOI] [PubMed] [Google Scholar]
- 24.Mantovani A, Giavazzi R, Polentarutti N, Spreafico F, Garattini S. Divergent effects of macrophage toxins on growth of primary tumors and lung metastases in mice. Int. J. Cancer. 1980;25(5):617–20. doi: 10.1002/ijc.2910250511. [DOI] [PubMed] [Google Scholar]
- 25.Ralph P, Prichard J, Cohn M. Reticulum cell sarcoma: an effector cell in antibody-dependent cell-mediated immunity. J. Immunol. 1975;114(2 pt 2):898–905. [PubMed] [Google Scholar]
- 26.Rockwell SC, Kallman RF, Fajardo LF. Characteristics of a serially transplanted mouse mammary tumor and its tissue-culture-adapted derivative. J. Natl. Cancer Inst. 1972;49(3):735–49. [PubMed] [Google Scholar]
- 27.Brattain MG, Strobel-Stevens J, Fine D, Webb M, Sarrif AM. Establishment of mouse colonic carcinoma cell lines with different metastatic properties. Cancer Res. 1980;40:2142–2146. [PubMed] [Google Scholar]
- 28.Gudgin Dickson EF, Holmes H, Jori G, Kennedy JC, Nadeau P, Pottier RH, Rossi F, Russell DA, Weagle GE. On the source of the oscillations observed during in vivo zinc phthalocyanine fluorescence pharmacokinetic measurements in mice. Photochem. Photobiol. 1995;61(5):506–9. doi: 10.1111/j.1751-1097.1995.tb02353.x. [DOI] [PubMed] [Google Scholar]
- 29.Holmes H, Kennedy JC, Pottier R, Rossi R, Weagle G. A recipe for the preparation of a rodent food that eliminates chlorophyll-based tissue fluorescence. J. Photochem. Photobiol. B. 1995;29(2−3) doi: 10.1016/1011-1344(95)90099-3. [DOI] [PubMed] [Google Scholar]
- 30.Jain RK. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev. 1990;9(3):253–66. doi: 10.1007/BF00046364. [DOI] [PubMed] [Google Scholar]
- 31.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752–6. [PubMed] [Google Scholar]
- 32.Jain RK, Baxter LT. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: significance of elevated interstitial pressure. Cancer Res. 1988;48(24 Pt 1):7022–32. [PubMed] [Google Scholar]
- 33.Park YS, Huang L. Distribution within the organs of a reticuloendothelial system of liposomes containing lipid A. J. Drug Target. 1993;1(4):325–330. doi: 10.3109/10611869308996091. [DOI] [PubMed] [Google Scholar]
- 34.Olafsen T, Tan GJ, Cheung CW, Yazaki PJ, Park JM, Shively JE, Williams LE, Raubitschek AA, Press MF, Wu AM. Characterization of engineered anti-p185HER-2 (scFv-CH3)2 antibody fragments (minibodies) for tumor targeting. Protein Eng. Des. Sel. 2004;17(4):315–323. doi: 10.1093/protein/gzh040. [DOI] [PubMed] [Google Scholar]
- 35.Wong JY, Chu DZ, Williams LE, Yamauchi DM, Ikle DN, Kwok CS, Liu A, Wilczynski S, Colcher D, Yazaki PJ, Shively JE, Wu AM, Raubitschek AA. Pilot trial evaluating an 123I-labeled 80-kilodalton engineered anticarcinoembryonic antigen antibody fragment (cT84.66 minibody) in patients with colorectal cancer. Clin. Cancer Res. 2004;10(15):5014–5021. doi: 10.1158/1078-0432.CCR-03-0576. [DOI] [PubMed] [Google Scholar]
- 36.Kristjansen PE, Boucher Y, Jain RK. Dexamethasone reduces the interstitial fluid pressure in a human colon adenocarcinoma xenograft. Cancer Res. 1993;53(20):4764–6. [PubMed] [Google Scholar]
- 37.Lee I, Boucher Y, Jain RK. Nicotinamide can lower tumor interstitial fluid pressure: mechanistic and therapeutic implications. Cancer Res. 1992;52(11):3237–40. [PubMed] [Google Scholar]
- 38.Roberts MJ, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv. Drug Deliv. Rev. 2002;54(4):459–476. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 39.Lackner C, Jukic Z, Tsybrovskyy O, Jatzko G, Wette V, Hoefler G, Klimpfinger M, Denk H, Zatloukal K. Prognostic relevance of tumour-associated macrophages and von Willebrand factor-positive microvessels in colorectal cancer. Virchows Arch. 2004;445(2):160–167. doi: 10.1007/s00428-004-1051-z. [DOI] [PubMed] [Google Scholar]
- 40.Polverini PJ. Role of the macrophage in angiogenesis-dependent diseases. Exs. 1997;79:11–28. doi: 10.1007/978-3-0348-9006-9_2. [DOI] [PubMed] [Google Scholar]
- 41.Gupta S, Mishra AK, Muralidhar K, Jain V. Improved targeting of photosensitizers by intratumoral administration of immunoconjugates. Technol. Cancer Res. Treat. 2004;3(3):295–301. doi: 10.1177/153303460400300307. [DOI] [PubMed] [Google Scholar]
- 42.Lee CC, Pogue BW, O'Hara JA, Wilmot CM, Strawbridge RR, Burke GC, Hoopes PJ. Spatial heterogeneity and temporal kinetics of photosensitizer (AIPcS2) concentration in murine tumors RIF-1 and MTG-B. Photochem. Photobiol. Sci. 2003;2(2):145–150. doi: 10.1039/b207598a. [DOI] [PubMed] [Google Scholar]
- 43.Hebeda KM, Kamphorst W, Sterenborg HJ, Wolbers JG. Damage to tumour and brain by interstitial photodynamic therapy in the 9L rat tumour model comparing intravenous and intratumoral administration of the photosensitiser. Acta Neurochir. 1998;140(5):495–501. doi: 10.1007/s007010050131. [DOI] [PubMed] [Google Scholar]
- 44.Amano T, Prout GR, Jr., Lin CW. Intratumor injection as a more effective means of porphyrin administration for photodynamic therapy. J. Urol. 1988;139(2):392–5. doi: 10.1016/s0022-5347(17)42441-4. [DOI] [PubMed] [Google Scholar]