

Abstract
The lateral membrane of the cochlear outer hair cell (OHC) is the site of a membrane-based motor that powers OHC electromotility, enabling amplification and fine-tuning of auditory signals. The OHC membrane protein prestin plays a central role in this process. We have previously shown that membrane cholesterol modulates the peak voltage of prestin-associated nonlinear capacitance in vivo and in vitro. The present study explores the effects of membrane cholesterol and docosahexaenoic acid content on the peak and magnitude of prestin-associated charge movement in a human embryonic kidney (HEK 293) cell model. Increasing membrane cholesterol results in a hyperpolarizing shift in the peak voltage of the nonlinear capacitance (Vpkc) and a decrease in the total charge movement. Both measures depend linearly on membrane cholesterol concentration. Incubation of cholesterol-loaded cells in cholesterol-free media partially restores the Vpkc toward normal values but does not have a compensatory effect on the total charge movement. Decreasing membrane cholesterol results in a depolarizing shift in Vpkc that is restored toward normal values upon incubation in cholesterol-free media. However, cholesterol depletion does not alter the magnitude of charge movement. In contrast, increasing membrane docosahexaenoic acid results in a hyperpolarizing shift in Vpkc that is accompanied by an increase in total charge movement. Our results quantify the relation between membrane cholesterol concentration and prestin-associated charge movement and enhance our understanding of how membrane composition modulates prestin function.
The mammalian cochlea is a fluid-filled organ in which viscous damping could diminish hearing. This obstacle is overcome by a membrane-based motor in the lateral membrane of the cochlear outer hair cell (OHC)4 (1–6). Here, energy stored in the transmembrane electric field is converted to mechanical energy in the form of length changes in the OHC at frequencies approaching 100 kHz (7). The OHC transfers this mechanical energy into the basilar membrane, augmenting the vibrations of the cochlear partition and counteracting the effects of viscous damping.
Prestin (SLC26A5) is a polytopic integral membrane protein located in the OHC lateral wall (8–10) and to a lesser extent in the basal plasma membranes (10–12). Its distinct role in OHC electromotility is characterized by its propensity to greatly increase charge movement into and out of the lateral wall plasma membrane (8, 9, 13). Consistent with the prestin membership in the SLC26A family of anion transporters (8), chloride and bicarbonate have been identified as charge carriers in this process (13). The effect of prestin on charge movement results in a voltage-dependent nonlinear capacitance (NLC), which has been demonstrated in several mammalian cell lines (9, 13–16).
Multiple studies have investigated OHC mechanosensitive properties (17–27). Depolarizing shifts in the peak voltage of the NLC (Vpkc) have been demonstrated in OHCs with increased turgor pressure due to positive pipette pressure or hypo-osmotic extracellular solutions (17, 19, 20) as well as in OHCs subjected to longitudinal mechanical stretching (18). Reducing cell turgor using negative pipette pressure or hyperosmotic extracellular solutions induced hyperpolarizing shifts in Vpkc (19, 20, 24). After the identification of the role of prestin in OHC electromotility (8), Santos-Sacchi et al. (28) demonstrated similar Vpkc shifts in prestin-transfected human embryonic kidney (HEK) cells with increased turgor pressure. Depolarizing shifts in Vpkc have also been shown in OHCs upon exposure to exogenous fructose (23), chlorpromazine (25), and furosemide (26), and hyperpolarizing shifts have been demonstrated upon exposure to the lipophilic anion, tetraphenylborate (TPB–) (22) and chloroform (27). Finally, temperature changes were shown to induce shifts in Vpkc in both OHCs (21) and HEK cells (29), with higher temperatures causing depolarizing shifts. Each of these manipulations alters the material properties of the plasma membrane, suggesting that membrane composition itself may be an important factor in prestin function.
Cholesterol comprises a significant portion of the plasma membrane of most eukaryotic cells, where the number of cholesterol molecules is nearly the same as the total number of phospholipid molecules (30, 31). The OHC lateral wall plasma membrane contains significantly less cholesterol than the basal or apical membranes of the OHC (14, 32–36). Although prestin is present in both the lateral wall and the basal membranes (8, 11, 12, 37), the motor only functions in the lateral wall membrane (1, 3, 38). Our recent findings indicate that membrane cholesterol content affects prestin-associated charge movement in both OHCs (14) and HEK 293 cells expressing prestin (14, 16). Cholesterol was shown to influence prestin distribution, self-association, and the voltage dependence of prestin-associated charge movement. Additionally, membrane cholesterol content was found to decrease during OHC maturation, concurrent with the onset and increase in OHC electromotility (14).
The effect of cholesterol on prestin function parallels its effect on rhodopsin, a membrane protein that initiates the light cascade in the rod outer segment of the eye (39, 40). Conversion of rhodopsin to its active form in the stacked disk membranes of the rod outer segment occurs in a gradient that corresponds to the increase in age and decrease in cholesterol content of the disk membranes (41). Rhodopsin functions optimally in older disks in which membrane cholesterol content is significantly lower (39, 42). The gradient of increased rhodopsin function also correlates with the presence of docosahexaenoic acid (DHA) (43), a polyunsaturated, omega-3 fatty acid (44). DHA content increases with disk age, concomitant with the increase in rhodopsin function (45, 46). The exact role of membrane composition in rhodopsin function is a topic of active investigation. In the case of prestin, the precise composition of the OHC plasma membrane and its effect on prestin function and hearing remains unknown. In the present study we investigate the effect of changes in membrane cholesterol and DHA content on the magnitude and peak of prestin-associated charge movement in prestin-expressing HEK 293 cells.
EXPERIMENTAL PROCEDURES
Prestin Construct and Transfection—Gerbil prestin was cloned into the pIRES-hrGFP vector (Stratagene, La Jolla, CA) as a HA-tag fusion protein (HA-prestin) as previously described (15, 16, 47). HEK 293 cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum and antibiotics (Mediatech, Herndon, VA). At ∼50–70% confluence, cells were transfected with HA prestin at a 3:2 ratio of FuGENE 6 (Roche Applied Science) to DNA. Plates containing transfected cells were incubated for 16–20 h then split into coated microwell Petri dishes (MatTek, Ashland, MA) and incubated for an additional 24–34 h before treatment with lipid-altering agents and performance of electrophysiological measurements.
Cholesterol Manipulations—Cholesterol manipulations produce qualitatively similar results in OHCs and prestin-expressing HEK 293 cells, validating the use of HEK 293 cells to study the effects of membrane composition on prestin function (14). Steady-state electrophysiological measurements were performed on HA prestin-transfected HEK 293 cells at 40–54 h post-transfection directly after incubation for 30–40 min at 37 °C in DMEM containing either 4 mm methyl-β-cyclodextrin (MβCD) (Sigma) or water-soluble cholesterol with MβCD as a vehicle (49 mg of cholesterol per gram of cholesterol + MβCD; Sigma). The series of cholesterol concentrations used corresponded to 1, 2, 3, 4, and 8 mm MβCD concentrations. Additional treatments of cells with altered cholesterol content involved cholesterol manipulation as described above followed by incubation in bathing media composed of cholesterol-free, MβCD-free DMEM. Incubation took place at 37 °C for 60–75 min directly before steady-state electrophysiological measurements.
DHA Manipulation—Steady-state electrophysiological measurements were performed on HA prestin-transfected HEK 293 cells at 40–54 h post-transfection directly after incubation for 60–75 min at 37 °C in DMEM containing 15 μm DHA (Sigma 6217-54-5) complexed with 5 μm human serum albumin (HSA; Sigma 70024-90-7, St. Louis, MO) as a vehicle. The effects of DHA were followed kinetically by adding DHA to the external solution after the establishment of a whole-cell patch until a final concentration of 75 μm DHA was reached. A higher final concentration of DHA was used to compensate for the lower temperature (room temperature, 23 °C) and shorter incubation timescales of the kinetic experiment. For steady-state treatment with HSA alone, cells were incubated for 60–75 min at 37 °C in DMEM containing 5 μm HSA. Kinetic readings were taken with HSA added to the external solution to a 5 μm final concentration after the establishment of a whole cell patch.
Electrophysiological Measurements—Electrophysiological data were obtained from cells using the whole-cell voltage clamp technique. The recording techniques have been fully described previously (15), and a brief description follows. Coated microwell Petri dishes containing transfected cells were extensively perfused with an extracellular solution containing Ca2+ and K+ channel blockers (100 mm NaCl, 20 mm CsCl, 20 mm tetraethylammonium chloride, 10 mm HEPES, 2 mm CoCl2, 1.47 mm MgCl2, and 2 mm CaCl2) before recording. The dishes were placed on the stage of an inverted microscope (Carl Zeiss, Gottingen, Germany) under 20× magnification, and all recordings were conducted at room temperature (23 ± 1 °C). Quartz glass patch pipettes with resistances in the range of 1.5–7 megaohms were constructed using a CO2 laser-based micropipette puller (P-2000, Sutter Instrument Co., Novato, CA). Patch pipettes were filled with an intracellular solution containing channel blockers (140 mm CsCl, 2 mm MgCl2, 10 mm EGTA, and 10 mm HEPES) before patching. The pH and osmolality of the external and internal solutions were adjusted to 7.2 ± 0.02 and 300 ± 2 mosm/kg using CsOH or HCl and glucose, respectively.
The cell membrane admittance was measured utilizing the patch clamp technique in whole-cell mode. First, an electrical seal >1 gigaohm was formed between the pipette and cell membrane by drawing the two into contact and rapidly applying ∼60 mm Hg negative pressure through the pipette. Next, the compensation circuitry of the amplifier (Axon 200B; Molecular Devices, Union City, CA) was used to correct the pipette capacitance, and whole-cell mode was achieved by gradually increasing the negative pressure through the pipette. Then the cell membrane admittance was measured during a direct current (DC) voltage ramp. The holding potential was 0 mV before and after the ramp and increased at a rate of 300 mV/s during the ramp. The range of the voltage ramp was either –140 to 140 mV or –180 to 140 mV depending on treatment. Voltages were measured relative to an Ag+/AgCl reference electrode in contact with the external solution. The admittance was probed using a dual-frequency stimulus (48, 49) formed by the sum of two 10-mV (amplitude) sine waves with frequencies of 390.625 Hz and 781.250 Hz. Cell parameters were calculated from the admittance as described previously (50).
The conductance was also experimentally determined using a DC protocol (15, 50). Briefly, a square wave pulse with 10-mV amplitude was applied to the cell through the pipette. At each voltage the current was measured every 10 or 100 μs for a total of 1000 readings (400 points before the pulse, 500 points during the pulse, and 100 points after the pulse), with the longer time intervals used to determine the conductance of cells with high resistances (>4 gigaohms). Conductance was calculated from the change in the steady-state component of measured current relative to the change in voltage. The relationship between current and voltage was also determined using a voltage ramp from –140 to 140 mV in 10-mV steps with 1000 readings taken at 100-μs intervals for each step.
A total of 941 HEK 293 cells were patched, and data from 371 cells were used for subsequent analysis. Isolated, non-mobile cells were patched within 60 min of initial exposure to external blocking solution. Data were included or excluded from the final pool based on a combination of parameters; assessment of the patch quality at the time of patching was based on the transient current plot, and the magnitude of the membrane resistance was calculated from the admittance measurements as well as measured directly using the DC protocol. Additional data inclusion criteria included the noise level of the capacitance versus voltage curve, the magnitude of the average membrane resistance (greater than 200 megaohms), the average series resistance (between 2 and 10 megaohms), and the reversal potential (less than –5 mV or greater than +5 mV). The linear capacitance of the cells ranged from 4.8 to 41.6 pF, which based on the specific membrane capacitance for inflated cells (51), represents a range of surface areas from ∼960 to 8300 μm2. Charge density data were excluded if the corresponding Boltzmann fit had an R2 value less than 0.97. For kinetics data, the reversal potential was not used to evaluate the quality of the patch, and assessment was made on a point-by-point rather than cell-by-cell basis, although entire cells were excluded if sufficient data were not present beyond 6 min after patching.
For all representations the linear capacitance was subtracted from the calculated capacitance to yield the nonlinear component. This nonlinear component of the capacitance was then normalized relative to the peak capacitance as indicated in the equation below. The linear capacitance was taken as the average capacitance at 5 consecutive voltages centered around 126 mV, where the nonlinear component is negligible. The peak capacitance varied for each cell,
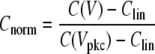 |
(Eq. 1) |
where C(V) is the calculated capacitance at voltage V, C(Vpkc) is the calculated capacitance at peak voltage Vpkc, and Clin is the linear capacitance. Cells in all treatment groups exhibited bell-shaped capacitance versus voltage curves. A Boltzmann function was fit to the NLC curves as described previously (24), and the charge density was calculated by dividing the maximum charge transferred (calculated from the area under the Boltzmann curve) by the linear capacitance (15).
Quantification of Cholesterol in HEK 293 Cell Membrane Fractions—HEK 293 cells were cultured in 60-mm poly-d-lysine-coated plates until they were at or close to 100% confluence. Cells were then treated for 30 min at 37 °C with either 10 mm MβCD or 1–10 mm MβCD-cholesterol in DMEM containing 10% fetal bovine serum and antibiotics or left untreated. Cells were washed twice with cold phosphate-buffered saline for 5 min and then washed with cell lysis buffer (50 mm HEPES, 10 mm EDTA, 200 mm NaCl, pH 8.0). Cells were harvested by scraping in 300 μl of lysis buffer, incubated for 10 min on ice, and lysed by syringe extrusion (20 times through a 25½ gauge needle). Lysed cells were centrifuged at 1000 × g for 10 min at 4 °C to pellet the nuclear fraction and cell debris. The supernatant (membrane fraction) was collected and stored.
Protein content in the membrane fraction was estimated using the Bio-Rad protein assay based on the Bradford method. After this, the membrane fractions were subjected to the Amplex Red Cholesterol Assay (Molecular Probes, Eugene, OR). Briefly, this colorimetric assay is based on the reaction of cholesterol with cholesterol oxidase to yield H2O2, which can then be detected using the Amplex Red reagent. A standard curve was created using the supplied cholesterol reference standards diluted to final concentrations of 0–8 μg/ml. 50 μl of each of the standards and samples (undiluted and 2× diluted) were added to the wells of a 96-well plate in duplicate and 50 μl of the Amplex Red Reagent working solution containing 300 μm Amplex Red reagent, 2 units/ml horseradish peroxidase, 2 units/ml cholesterol oxidase, and 0.2 units/ml cholesterol esterase was added to each well. The plate was incubated for 30 min at 37 °C to allow product to form, and a plate reader was used to measure the absorption at 570 nm. The cholesterol content of each sample was quantified based on the standard curve generated and normalized to total protein content. Final cholesterol content was expressed in terms of pmol/μg of protein in the sample.
Statistical Analysis—Vpkc and charge density data for cells subjected to different treatments were compared using two-tailed Student t tests.
RESULTS
We have previously explored the qualitative relationship between membrane cholesterol and prestin function (14, 16). To gain a quantitative understanding of this relationship, we measured prestin function over a series of specific membrane cholesterol concentrations. The membrane cholesterol concentration was determined in cells depleted of cholesterol using 10 mm MβCD, left untreated, or loaded with cholesterol using MβCD-cholesterol, which was varied from 1 to 10 mm. MβCD-treated, and untreated cells had membrane cholesterol concentrations of 2.8 and 7.4 pmol/μg of protein, respectively. Cholesterol-loaded cells had membrane cholesterol concentrations that ranged from 14.4 to 30.4 pmol/μg of protein. The relative change in membrane cholesterol concentration that we observed is consistent with what has been previously reported (52).
The Effect of Cholesterol Depletion on Vpkc Is Partially Restored over Time—Untreated HEK 293 cells expressing prestin exhibited a bell-shaped NLC curve with a mean Vpkc of approximately –72 mV, similar to previous reports (14, 38). To explore the effect of reduced membrane cholesterol on Vpkc, prestin-expressing HEK 293 cells were depleted to a membrane cholesterol concentration of ∼3 pmol/μg of protein using MβCD. Past studies using MβCD to deplete membrane cholesterol have shown a large depolarizing shift of Vpkc (14). Our data also demonstrate a large depolarizing shift of Vpkc (p < 1 × 10–21) to a mean of –17 mV (Fig. 1A).
FIGURE 1.

Steady-state cholesterol depletion- and loading-induced shifts in Vpkc return toward control values upon incubation in bathing media. A, the mean Vpkc in untreated cells (▪, n = 39), which had a membrane cholesterol concentration of 7.4 pmol/μg protein, was approximately –72 mV. Cells treated with MβCD (⋄, n = 25) to a membrane cholesterol concentration of 2.8 pmol/μg of protein exhibited a large and significant depolarizing shift to a mean Vpkc of –17 mV (*, p < 1 × 10–21). Cells treated with MβCD and incubated in bathing media (♦, n = 24) had a mean Vpkc of –29 mV. Although this is a significant depolarizing shift relative to the control Vpkc (p < 1 × 10–17), it is also a significant reduction in the shift seen for MβCD treatment alone (**, p < 0.002). The traces shown here and in subsequent NLC plots are representative of cells within a treatment group, and each capacitance trace was normalized with respect to peak and base-line capacitance values. B, the box plots represent the distribution of steady-state Vpkc values for each treatment. C, cholesterol-loaded cells with a membrane cholesterol concentration of 23.5 pmol/μg of protein (♦, n = 19) exhibited a large and significant hyperpolarizing shift to a mean Vpkc of –139 mV (*, p < 1 × 10–13). Cells exposed to the same cholesterol treatment followed by incubation in bathing media (♦, n = 14) had a mean Vpkc of –99 mV. Although this is a significant hyperpolarizing shift relative to the control Vpkc (p < 1 × 10–11), it is also a significant reduction in the shift seen for cholesterol treatment alone (**, p < 1 × 10–8). D, the box plots represent the distribution of steady-state Vpkc values for each treatment. Ticks in these and subsequent box plots mark the maximum and minimum values, boxes contain values falling between the first and third quartiles, and bars mark the median value for each group.
We investigated the dynamics of cholesterol depletion by incubating depleted cells in bathing media composed of cholesterol-free, MβCD-free DMEM for 60–75 min. These cells had a mean Vpkc of –29 mV (Fig. 1B). Although the shift is still significant relative to untreated cells (p < 1 × 10–17), it is also significant when compared with cholesterol depletion alone (p < 0.002). This recovery occurs despite the lack of an exogenous source of cholesterol in the bathing media.
The Effect of Cholesterol Loading on Vpkc Is Partially Restored over Time—To determine the effects of increased membrane cholesterol on Vpkc, cells were loaded with water-soluble cholesterol to a membrane cholesterol concentration of ∼24 pmol/μg of protein. The treatment resulted in a bell-shaped NLC curve with a mean Vpkc of –139 mV (Fig. 1C). This large and significant hyperpolarizing shift (p < 1 × 10–13) closely corresponds to our previous cholesterol-loading NLC data (14).
We next determined if recovery from cholesterol loading would occur by incubating loaded cells in bathing media. These cells exhibited a mean Vpkc of –99 mV (Fig. 1D), which is a significant hyperpolarizing shift relative to the control Vpkc (p < 1 × 10–11) but represents a significantly smaller shift than that observed for cholesterol loading alone (p < 1 × 10–8). The compensatory shifts of Vpkc seen upon recovery from cholesterol depletion or loading indicate the presence of a cholesterol homeostasis mechanism operating in the cell.
Cholesterol Loading Alters Charge Density—We then determined the effect of cholesterol manipulations on the magnitude of prestin-associated charge movement (Fig. 2). The mean charge density for untreated cells was 16.1 fC/pF. Cholesterol depletion did not significantly alter charge density relative to untreated cells, having a mean of 17.3 fC/pF (p > 0.65; Fig. 2A). However, cholesterol-loaded cells exhibited a significantly lower mean charge density at 6.8 fC/pF (p < 1 × 10–6; Fig. 2B), indicating that cholesterol loading results in a decrease in the magnitude of prestin-associated charge movement.
FIGURE 2.

Cholesterol depletion does not significantly alter charge density, whereas cholesterol loading causes a decrease in charge density that does not return toward control values upon incubation in bathing media. A, the mean charge density in untreated cells (striped, membrane cholesterol 7.4 pmol/μg of protein; n = 39) was 16.1 fC/pF. Cells treated with MβCD (white, membrane cholesterol 2.8 pmol/μg of protein; n = 25) had a mean charge density of 17.3 fC/pF which was not significantly altered relative to untreated cells (p > 0.65). Cells treated with MβCD and incubated in bathing media (light gray, n = 24) had a mean charge density of 19.6 fC/pF which was not significantly altered relative to untreated (p > 0.21) or MβCD-treated cells (p > 0.48). B, the histogram represents the mean charge density for untreated cells (striped), cells loaded to a membrane cholesterol concentration of 23.5 pmol/μg protein (black, n = 19), and cells loaded to a membrane cholesterol concentration of 23.5 pmol/μg protein and incubated in bathing media (dark gray, n = 14). The mean charge density was significantly reduced in both cholesterol-loaded cells (6.8 fC/pF, p < 1 × 10–6) and cholesterol-loaded cells incubated in bathing media (6.8 fC/pF; *, p < 1 × 10–6), with no significant difference between the two (p > 0.92). Error bars in both graphs represent 2 S.E. in either direction relative to the mean.
We investigated the reversibility of cholesterol-induced charge density changes by incubating loaded cells in bathing media. Unlike Vpkc values, the charge density did not move toward control values, nor was it significantly different from that of cholesterol loading alone (p > 0.92; Fig. 2B). Likewise, incubation of cholesterol-depleted cells in bathing media did not significantly alter the charge density relative to cholesterol-depleted or untreated cells (Fig. 2A).
Cholesterol Loading Alters Vpkc and Charge Density in a Concentration-dependent Manner—We evaluated the relationship between membrane cholesterol concentration and Vpkc by determining Vpkc values at different membrane cholesterol concentrations. Untreated cells with a membrane cholesterol concentration of ∼7 pmol/μg of protein had a mean Vpkc of –72 mV. Cholesterol-loaded cells, with a range of membrane cholesterol concentrations from 14 to 30 pmol/μg protein, exhibited Vpkc values ranging from –103 to –143 mV (Fig. 3A). Increasing concentrations exhibited a linear relationship with hyperpolarization (R2 = 0.923), indicating a direct correlation between membrane cholesterol concentration and Vpkc.
FIGURE 3.

Cholesterol loading alters both Vpkc and charge density in a concentration-dependent manner. A, shown is a plot of the mean Vpkc (mV) versus the membrane cholesterol concentration (pmol/μg of protein). In untreated cells (▪, membrane cholesterol 7.4 pmol/μg of protein; n = 39), the mean Vpkc was approximately –72 mV. In MβCD-treated cells (⋄, membrane cholesterol 2.8 pmol/μg of protein; n = 25), the mean Vpkc was –17 mV. The series of cholesterol-treated cell groups (♦, n = 10, 7, 11, 19, and 8) had membrane cholesterol concentrations ranging from 14.4 to 30.4 pmol/μg protein and Vpkc values ranging from –103 to –143 mV. A significant hyperpolarizing shift was seen in Vpkc for each group of cholesterol-loaded cells relative to the control (p < 0.0001), and the shifts became larger as membrane cholesterol concentration increased. A linear regression function has been fit to the control and cholesterol-loading data (y = –3.26x – 52.7, R2 = 0.923). B, the curves represent the mean charge moved (pC) versus voltage (mV) for cholesterol-loaded cells at membrane cholesterol concentrations of 14.4 (solid line, n = 10), 20.7 (dashed line, n = 7), 23.5 (dotted line, n = 19) and 30.4 pmol/μg of protein (dash-dotted line, n = 7). The charge at Vpkc is labeled for each treatment (♦, ▪, ▴, and •, respectively). The total charge movement, represented by the height of the curve, decreased as membrane cholesterol concentration increased. C, as the membrane cholesterol concentration was increased, the mean charge density (fC/pF) decreased. The mean charge densities for untreated cells (▪, n = 39) and MβCD-treated cells (⋄, n = 25) were 16.1 and 17.3 fC/pF, respectively. The series of cholesterol-treated cell groups (♦, n = 10, 7, 11, 19, and 7) had mean charge densities ranging from 5.2 to 12.9 fC/pF. A linear regression function has been fit to the control and cholesterol-loading data (y = –0.486x + 19.8, R2 = 0.952). In A and C, horizontal and vertical error bars represent 2 S.E. in either direction relative to the mean.
The charge movement as a function of voltage was determined for cholesterol-loaded cells at several membrane cholesterol concentrations (Fig. 3B). The voltage at which the charge increase is most rapid (the point of inflection) marks the mean Vpkc, whereas the maximum charge value indicates the mean total charge moved at each respective membrane cholesterol concentration. The total charge movement decreased with increasing membrane cholesterol concentration.
Charge density was also analyzed (Fig. 3C). The mean charge density decreased linearly with increasing membrane cholesterol concentration (R2 = 0.952), from 16.1 fC/pF in untreated cells to 5.2 fC/pF in the maximally loaded cells. The charge density, therefore, exhibits a strong inverse correlation to the membrane cholesterol concentration.
Alterations in DHA Content Modulate Vpkc—We investigated the effect of DHA manipulation on Vpkc by loading cells with DHA using HSA as a vehicle. To account for the potential influence of HSA, cells were also treated with HSA alone. This produced a bell-shaped NLC curve with a mean Vpkc of –74 mV that was not significantly different from the control Vpkc of –72 mV (p > 0.46). By contrast, DHA loading shifted the Vpkc to approximately –85 mV (Fig. 4, A and B). This hyperpolarizing shift was statistically significant compared with both untreated (p < 1 × 10–6) and HSA-treated cells (p < 0.0002).
FIGURE 4.

DHA shifts Vpkc. A, the mean Vpkc in untreated cells (▪, n = 39) was approximately –72 mV. Cells treated with HSA alone (▵, n = 30) exhibited no significant change in Vpkc relative to untreated cells (p > 0.46). In cells loaded with DHA (▴, n = 41), the mean Vpkc shifted in the hyperpolarizing direction to approximately –85 mV. This shift was statistically significant with respect to both untreated and HSA-treated cells (*, p < 0.0002). B, the box plots represent the distribution of steady-state Vpkc values for each treatment. C, shown is the change in Vpkc as a function of time after the establishment of a whole-cell patch. HSA (▵, n = 10–13) and DHA (▴, n = 17–18) were added 3 min after the patch was established, as indicated by the arrow. Vpkc changes rapidly upon the addition of DHA. Untreated (▪, n = 13–21) and HSA-treated cells have similar kinetic values, indicating that HSA does not contribute to the shift seen with DHA loading. The traces shown are averages from each respective group of the change in Vpkc of individual cells relative to their initial values.
Kinetic studies of DHA loading revealed a similar hyperpolarizing shift of Vpkc which occurred within minutes of the addition of DHA (Fig. 4C). Both the steady-state and kinetic data demonstrate a direct and dynamic correlation between DHA content and Vpkc.
Alterations in DHA Content Modulate Charge Density—We also investigated the effect of DHA on the magnitude of prestin-associated charge movement (Fig. 5). The mean charge density of 16.9 fC/pF observed for HSA-treated cells was not significantly different from the control value of 16.1 fC/pF (p > 0.75). However, the mean charge density for DHA-loaded cells was significantly higher than both untreated (p < 0.007) and HSA-treated cells (p < 0.05) at 23.0 fC/pF. DHA loading, thus, induced a marked increase in the magnitude of prestin-associated charge movement.
FIGURE 5.

DHA increases charge density. The mean charge density in untreated cells (striped, n = 39) was 16.1 fC/pF. Cells treated with HSA alone (white, n = 30) exhibited no significant change in charge density relative to untreated cells (16.9 fC/pF, p > 0.75). In cells loaded with DHA (black, n = 41), the mean charge density increased to 23.0 fC/pF. This shift was statistically significant with respect to untreated (p < 0.007) and HSA-treated cells (*, p < 0.044). Error bars represent 2 S.E. in either direction relative to the mean.
DISCUSSION
Membrane properties are known to modulate prestin function and electromotility in OHCs (19, 25, 27, 28, 53–55); however, the concentration dependence and specific physical and biological effects of membrane components have not been addressed. Cholesterol exhibits a concentration-dependent effect on Vpkc and the total charge movement. Both Vpkc and charge density display a strong linear correlation to increasing membrane cholesterol concentration. An interpretation based on the physical properties of membranes is that increasing membrane cholesterol increases membrane rigidity (56), thereby decreasing the conformational flexibility of prestin. This would result in decreased prestin-associated charge movement. Increased prestin rigidity with elevated membrane cholesterol is also consistent with the hyperpolarizing shift in Vpkc observed for cholesterol loading. A stronger electric field might be needed to induce conformational changes in an increasingly rigid prestin protein. The extent of conformational change might also be reduced, which could contribute to the decrease in total charge movement observed. On the other hand, cholesterol depletion would be expected to increase prestin-associated charge movement by increasing membrane fluidity and the conformational flexibility of prestin. The reason this is not observed might be due to biological effects.
The effect of cholesterol on Vpkc was previously shown to be fully reversible with cyclodextrin-mediated depletion and repletion of membrane cholesterol (14). We now demonstrate that cholesterol-loaded and -depleted cells also exhibit partial reversal of the Vpkc shift upon incubation in bathing media. The fact that cholesterol-depleted and -loaded cells partially recover in the absence of an exogenous source of cholesterol or depleting agent suggests that internal trafficking and homeostasis of membrane lipids plays a role in this process.
Unlike Vpkc, the decrease in charge density with cholesterol loading was not restored upon incubation in bathing media. Increasing membrane cholesterol is known to increase lipid raft formation, a requisite step in cholesterol-dependent endocytosis. Increased endocytosis of prestin could account for the decrease in charge density with cholesterol loading and the lack of change with cholesterol depletion. These findings are also consistent with the immediate non-reversibility of charge density reduction upon recovery from cholesterol loading; a recovery of charge density would require either endocytic recycling to the plasma membrane or de novo synthesis to replenish the membrane prestin population. These observations suggest that the Vpkc is largely modulated by physical properties of the membrane, whereas the charge density is affected by both physical and biological events.
Vpkc and charge density have been shown to change in OHCs during postnatal development (24, 37). Oliver et al. (24) reported a depolarizing shift in Vpkc of nearly 50 mV from birth to day 14 and a large increase in charge density which displayed saturation at day 12 in rats. A later study showed that the abundance of prestin transcripts increased rapidly from birth to day 9 in rats but then leveled at mature values (57). Abe et al. (37) showed that prestin expression in mice peaked at day 10 although OHC motor activity did not reach adult levels until day 18. This suggests that prestin expression itself is not wholly responsible for the NLC changes seen during postnatal development. Rajagopalan et al. (14) demonstrated that OHC membrane cholesterol decreases in mice during OHC maturation, and we have shown that both Vpkc and charge density increase linearly as membrane cholesterol decreases. The changes seen during OHC maturation could be explained by the decrease in membrane cholesterol described by Rajagopalan et al. (14). This mechanism could also explain the delay Abe et al. (37) reported between peak prestin mRNA expression and OHC maturation. Alteration of membrane cholesterol is the only mechanism to cause shifts of Vpkc of the magnitude seen during OHC maturation. Although it is possible that many factors contribute to this change in maturing OHCs, it is likely that cholesterol plays a key role. It might even be possible to predict membrane cholesterol composition during OHC maturation based on the electrophysiological properties of the cell.
A dynamic correlation also exists between membrane DHA and prestin-associated charge movement. We demonstrated a steady-state hyperpolarizing Vpkc shift in DHA-loaded cells and a dynamic hyperpolarizing Vpkc shift upon the addition of DHA to patched cells. The magnitude of the shift is important because it determines the relationship between the Vpkc and the resting membrane potential in OHCs. The Vpkc in normal OHCs lies around –50 mV (14, 38), ∼20 mV to the depolarizing side of the resting membrane potential. Previous studies from our group showed a transient increase in prestin function by measuring distortion product otoacoustic emissions after injection of cholesterol into the cochlea of mice (14). This increase is consistent with the Vpkc briefly aligning with the resting membrane potential as the large cholesterol-induced shift occurs (14). In prestin-expressing HEK 293 cells, DHA induced a Vpkc shift of 10–15 mV in the hyperpolarizing direction. In OHCs, a shift of this magnitude would bring the Vpkc into closer alignment with the resting membrane potential; in other words, DHA could effectively shift Vpkc closer to the optimal operating voltage range of the cell.
DHA loading also caused an increase in total charge movement. This could be attributed to increased membrane fluidity enhancing prestin conformational flexibility because DHA is a highly unsaturated and flexible omega-3 fatty acid (58–60) that readily incorporates into phospholipid membranes (44). Its multiple double bonds give it a coiled, bulky structure that hinders membrane packing leading to increased membrane fluidity, decreased bending rigidity (61), and altered membrane thickness and curvature.
DHA, unlike cholesterol, contains a carboxyl group which gives it a negative charge. The lipophilic anion tetraphenylborate, TPB–, has previously been shown to have an effect similar to that of DHA on Vpkc and to cause an increase in the peak capacitance in OHCs (22). The unbalanced charge that DHA loading brings to the plasma membrane may also contribute to the increase in total charge movement and the hyperpolarizing shift in Vpkc. The increase in charge density seen upon DHA loading is important because its ability to increase total charge movement suggests that increasing membrane DHA content could potentially enhance OHC function and hearing.
The present study demonstrates the importance of membrane lipid composition as a modulator of prestin function. Our results provide new insight into the mechanisms by which membrane composition modulates prestin function and quantifies the relation between membrane cholesterol concentration and prestin-associated charge movement.
Acknowledgments
We thank Brenda Farrell for manuscript review and consultation regarding experimental design, Haiying Liu and Dowin Boatright for technical assistance, and Mary Wiese for manuscript review.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DC00354 (NIDCD; to W. E. B. and F. A. P.). This work was also supported by NSF Grant BES-0522862 (to F. A. P.) and by the Deafness Research Foundation (to L. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: OHC, outer hair cell; NLC, nonlinear capacitance; Vpkc, voltage at peak nonlinear capacitance; HEK, human embryonic kidney; DHA, docosahexaenoic acid; DMEM, Dulbecco's modified Eagle's medium; MβCD, methyl-β-cyclodextrin; HSA, human serum albumin; DC, direct current; fC, femtocoulomb; pF, picofarads.
References
- 1.Brownell, W. E., Bader, C. R., Bertrand, D., and de Ribaupierre, Y. (1985) Science 227 194–196 [DOI] [PubMed] [Google Scholar]
- 2.Dallos, P., Evans, B. N., and Hallworth, R. (1991) Nature 350 155–157 [DOI] [PubMed] [Google Scholar]
- 3.Kalinec, F., Holley, M. C., Iwasa, K. H., Lim, D. J., and Kachar, B. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 8671–8675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang, G., and Santos-Sacchi, J. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 12268–12272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brownell, W. E., Spector, A. A., Raphael, R. M., and Popel, A. S. (2001) Annu. Rev. Biomed. Eng. 3 169–194 [DOI] [PubMed] [Google Scholar]
- 6.Brownell, W. E. (2006) in Vertebrate Hair Cells (Eatock, R. A., Fay, R. R., and Popper, A. N., eds) pp. 313–347, Springer-Verlag New York Inc., New York
- 7.Frank, G., Hemmert, W., and Gummer, A. W. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 4420–4425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng, J., Shen, W., He, D. Z., Long, K. B., Madison, L. D., and Dallos, P. (2000) Nature 405 149–155 [DOI] [PubMed] [Google Scholar]
- 9.Ludwig, J., Oliver, D., Frank, G., Klocker, N., Gummer, A. W., and Fakler, B. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 4178–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adler, H. J., Belyantseva, I. A., Merritt, R. C., Jr., Frolenkov, G. I., Dougherty, G. W., and Kachar, B. (2003) Hear. Res. 184 27–40 [DOI] [PubMed] [Google Scholar]
- 11.Belyantseva, I. A., Adler, H. J., Curi, R., Frolenkov, G. I., and Kachar, B. (2000) J. Neurosci. 20 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu, N., Zhu, M. L., and Zhao, H. B. (2006) Brain Res. 1095 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oliver, D., He, D. Z., Klocker, N., Ludwig, J., Schulte, U., Waldegger, S., Ruppersberg, J. P., Dallos, P., and Fakler, B. (2001) Science 292 2340–2343 [DOI] [PubMed] [Google Scholar]
- 14.Rajagopalan, L., Greeson, J. N., Xia, A., Liu, H., Sturm, A., Raphael, R. M., Davidson, A. L., Oghalai, J. S., Pereira, F. A., and Brownell, W. E. (2007) J. Biol. Chem 282 36659–36670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajagopalan, L., Patel, N., Madabushi, S., Goddard, J. A., Anjan, V., Lin, F., Shope, C., Farrell, B., Lichtarge, O., Davidson, A. L., Brownell, W. E., and Pereira, F. A. (2006) J. Neurosci. 26 12727–12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sturm, A. K., Rajagopalan, L., Yoo, D., Brownell, W. E., and Pereira, F. A. (2007) Otolaryngol. Head Neck Surg. 136 434–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwasa, K. H. (1993) Biophys. J. 65 492–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gale, J. E., and Ashmore, J. F. (1994) Proc. Biol. Sci. 255 243–249 [DOI] [PubMed] [Google Scholar]
- 19.Kakehata, S., and Santos-Sacchi, J. (1995) Biophys. J. 68 2190–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gale, J. E., and Ashmore, J. F. (1997) Pfluegers Arch. Eur. J. Physiol. 434 267–271 [DOI] [PubMed] [Google Scholar]
- 21.Santos-Sacchi, J., and Huang, G. (1998) Hear Res. 116 99–106 [DOI] [PubMed] [Google Scholar]
- 22.Wu, M., and Santos-Sacchi, J. (1998) J. Membr. Biol. 166 111–118 [DOI] [PubMed] [Google Scholar]
- 23.Geleoc, G. S., Casalotti, S. O., Forge, A., and Ashmore, J. F. (1999) Nat. Neurosci. 2 713–719 [DOI] [PubMed] [Google Scholar]
- 24.Oliver, D., and Fakler, B. (1999) J. Physiol. 519 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lue, A. J., Zhao, H. B., and Brownell, W. E. (2001) Otolaryngol Head Neck Surg. 125 71–76 [DOI] [PubMed] [Google Scholar]
- 26.Santos-Sacchi, J., Wu, M., and Kakehata, S. (2001) Hear Res. 159 69–73 [DOI] [PubMed] [Google Scholar]
- 27.Santos-Sacchi, J., and Wu, M. (2004) J. Membr. Biol. 200 83–92 [DOI] [PubMed] [Google Scholar]
- 28.Santos-Sacchi, J., Shen, W., Zheng, J., and Dallos, P. (2001) J. Physiol. (Lond.) 531 661–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meltzer, J., and Santos-Sacchi, J. (2001) Neurosci. Lett. 313 141–144 [DOI] [PubMed] [Google Scholar]
- 30.Cooper, G. M. (2000) The Cell: A Molecular Approach, 2nd Ed. p. 469, American Society for Microbiology, Washington, D. C. and Sinauer Associates, Inc., Sunderland, MA
- 31.Alberts, R., Johnson, A., Lewis, J., Raff, M., Roberts, K., and Walter, P. (2002) Molecular Biology of the Cell, 4th Ed. pp. 588–589, Garland Science, New York
- 32.Forge, A. (1991) Cell Tissue Res. 265 473–483 [DOI] [PubMed] [Google Scholar]
- 33.Santi, P. A., Mancini, P., and Barnes, C. (1994) J. Histochem. Cytochem. 42 705–716 [DOI] [PubMed] [Google Scholar]
- 34.Nguyen, T. V., and Brownell, W. E. (1998) Otolaryngol. Head Neck Surg. 119 14–20 [DOI] [PubMed] [Google Scholar]
- 35.Oghalai, J. S., Patel, A. A., Nakagawa, T., and Brownell, W. E. (1998) J. Neurosci. 18 48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brownell, W. E., and Oghalai, J. S. (2000) Cell and Molecular Biology of the Ear (Lim, D. J., ed) pp. 69–83, Kluwer Academic/Plenum Publishers, New York
- 37.Abe, T., Kakehata, S., Kitani, R., Maruya, S., Navaratnam, D., Santos-Sacchi, J., and Shinkawa, H. (2007) J. Membr. Biol. 215 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santos-Sacchi, J. (1991) J. Neurosci. 11 3096–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boesze-Battaglia, K., and Albert, A. D. (1990) J. Biol. Chem. 265 20727–20730 [PubMed] [Google Scholar]
- 40.Albert, A. D., and Boesze-Battaglia, K. (2005) Prog. Lipid Res. 44 99–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Albert, A. D., Boesze-Battaglia, K., Paw, Z., Watts, A., and Epand, R. M. (1996) Biochim. Biophys. Acta 1297 77–82 [DOI] [PubMed] [Google Scholar]
- 42.Boesze-Battaglia, K., Hennessey, T., and Albert, A. D. (1989) J. Biol. Chem. 264 8151–8155 [PMC free article] [PubMed] [Google Scholar]
- 43.Mitchell, D. C., Straume, M., and Litman, B. J. (1992) Biochemistry 31 662–670 [DOI] [PubMed] [Google Scholar]
- 44.Zerouga, M., Stillwell, W., Stone, J., Powner, A., and Jenski, L. J. (1996) Anticancer Res. 16 2863–2868 [PubMed] [Google Scholar]
- 45.Boesze-Battaglia, K., and Albert, A. D. (1989) Exp. Eye Res. 49 699–701 [DOI] [PubMed] [Google Scholar]
- 46.Albert, A. D., Young, J. E., and Paw, Z. (1998) Biochim. Biophys. Acta 1368 52–60 [DOI] [PubMed] [Google Scholar]
- 47.Greeson, J. N., Organ, L. E., Pereira, F. A., and Raphael, R. M. (2006) Brain Res. 1091 140–150 [DOI] [PubMed] [Google Scholar]
- 48.Barnett, D. W., and Misler, S. (1997) Biophys. J. 72 1641–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santos-Sacchi, J., Kakehata, S., and Takahashi, S. (1998) J. Physiol. (Lond.) 510 225–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farrell, B., Do Shope, C., and Brownell, W. E. (2006) Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 73 041930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Solsona, C., Innocenti, B., and Fernandez, J. M. (1998) Biophys. J. 74 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reversi, A., Rimoldi, V., Brambillasca, S., and Chini, B. (2006) Am. J. Physiol. Regul. Integr. Comp. Physiol. 291 861–869 [DOI] [PubMed] [Google Scholar]
- 53.Rajendran, L., Masilamani, M., Solomon, S., Tikkanen, R., Stuermer, C. A., Plattner, H., and Illges, H. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 8241–8246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao, Z., Popel, A. S., Brownell, W. E., and Spector, A. A. (2005) J. Acoust. Soc. Am. 118 3737–3746 [DOI] [PubMed] [Google Scholar]
- 55.Murdock, D. R., Ermilov, S. A., Spector, A. A., Popel, A. S., Brownell, W. E., and Anvari, B. (2005) Biophys. J. 89 4090–4095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Needham, D., and Nunn, R. S. (1990) Biophys. J. 58 997–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reisinger, E., Zimmermann, U., Knipper, M., Ludwig, J., Klocker, N., Fakler, B., and Oliver, D. (2005) Mol. Cell. Neurosci. 28 106–117 [DOI] [PubMed] [Google Scholar]
- 58.Saiz, L., and Klein, M. L. (2001) Biophys. J. 81 204–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feller, S. E., Gawrisch, K., and MacKerell, A. D., Jr. (2002) J. Am. Chem. Soc. 124 318–326 [DOI] [PubMed] [Google Scholar]
- 60.Huber, T., Rajamoorthi, K., Kurze, V. F., Beyer, K., and Brown, M. F. (2002) J. Am. Chem. Soc. 124 298–309 [DOI] [PubMed] [Google Scholar]
- 61.Rawicz, W., Olbrich, K. C., McIntosh, T., Needham, D., and Evans, E. (2000) Biophys. J. 79 328–339 [DOI] [PMC free article] [PubMed] [Google Scholar]