

Abstract
A parasite-derived neurotrophic factor (PDNF) produced by the Chagas’ disease parasite Trypanosoma cruzi binds nerve growth factor (NGF) receptor TrkA, increasing receptor autophosphorylation, activating phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK/Erk) pathways, and transcription factor CREB. The end-result is enhanced survival and neuritogenesis of various types of neurons. PDNF also enhances the expression and activity of tyrosine hydroxylase, a rate limiting enzyme in the synthesis of dopamine and other catecholamine neurotransmitters. It remains unknown, however, if PDNF alters expression and metabolism of acetylcholine (ACh), a neurotransmitter thought to play a role in Chagas’ disease progression. Here we demonstrate that PDNF stimulates mRNA and protein expression of choline acetyltransferase (ChAT) and vesicular acetylcholine transporter (VAChT), which are critical for synthesis and storage of ACh. Stimulation requires functional TrkA because it did not occur in cell mutants that lack the receptor and in TrkA-expressing wild-type cells treated with K252a, an inhibitor of TrkA kinase activity. It also requires TrkA-dependent PI3K and MAPK/Erk signaling pathways because PDNF stimulation of cholinergic transcripts is abolished by specific pharmacological inhibitors. Furthermore, the cholinergic actions of PDNF were reproduced by PDNF-expressing extracellular T. cruzi trypomastigotes at the start of host cell invasion. In contrast, host cells bearing intracellular T. cruzi showed decreased, rather than increased, cholinergic gene expression. These results suggest that T. cruzi invasion of the nervous system alters cholinergic gene expression and that could play a role in neuropathology, and/or lack thereof, in Chagas’ disease patients.
Keywords: choline acetyltransferase, vesicular acetylcholine transporter, Trypanosoma cruzi, TrkA
Introduction
Acetylcholine (ACh) mediates neurotransmission throughout the central and peripheral nervous systems. It is essential for learning and memory (Robbins, 2005), somatic nervous system function (Madhavan and Peng, 2003), and fast excitatory synaptic neurotransmission in the autonomic nervous system (Galligan et al., 2000). Perturbed ACh signaling resulting from selective degradation of cholinergic neurons in basal forebrain and from deficits in neuromuscular junction are associated with Alzheimer disease (Lahiri et al., 2005) and myasthenia gravis (Nizri et al., 2007) pathogenesis, respectively. ACh is also an important component of a recently described vagus nerve-mediated mechanism termed cholinergic anti-inflammatory pathway, where the cholinergic neurons are believed to interact with the innate immune system to repress potentially damaging immune responses (Pavlov and Tracey, 2005).
Many aspects of cholinergic neuron morphology and function are closely associated with nerve growth factor (NGF). NGF promotes survival of cholinergic neurons after damage, maintains the cholinergic phenotype of uninjured neurons, and alters neuronal excitability by remodeling neurotransmitter receptors (Yeh et al., 2001). NGF also directly regulates cholinergic neurotransmission by increasing levels of choline acetyltransferase (ChAT), ACh release, and choline uptake in synaptosomes (Auld et al., 2001; Ekstrom and Reinhold, 2004; Heisenberg et al., 1994; Knipper et al., 1994; Sofroniew et al., 1993). Reduction in NGF supply to basal forebrain causes atrophy of cholinergic cortical projection neurons reminiscent of neuronal changes in Alzheimer’s disease (Tuszynski et al., 1990). Similarly, the selective loss of NGF-sensitive enteric neurons in experimental colitis correlates with a decrease in cholinergic excitatory neurons (Lin et al., 2005a), further emphasizing the role of NGF in the maintenance of cholinergic phenotype.
NGF signaling is critically dependent on membrane expression of two cognate receptors – TrkA and p75NTR – on target cells. Signaling through high-affinity TrkA receptor, a member of the tyrosine receptor kinase superfamily, underlies most of the biological activities of NGF (Counts and Mufson, 2005; Miller and Kaplan, 2001). TrkA is expressed in all cholinergic neurons in the brain (Holtzman et al., 1995), and in the peripheral nervous system, in most sympathetic and sensory nociceptive neurons (Snider and McMahon, 1998) and in more than 75% of enteric cholinergic neurons (Lin et al., 2005b). The widespread expression of TrkA in cholinergic neurons, and the particular dependence of these neurons on NGF for survival and maintenance of cholinergic phenotype, strongly indicates a direct relationship between the functional integrity of the cholinergic system and NGF/TrkA signaling.
PDNF, a neurotrophin-like factor produced by the Chagas’ disease parasite Trypanosoma cruzi, the agent of Chagas’ disease, binds TrkA and starts a complex cascade of intracellular signaling, including activation of MAPK and PI3K kinetic pathways and cAMP-response element CRE-dependent (CREB) gene transcription (Chuenkova and Pereira, 2000; Chuenkova and Pereira, 2003; Chuenkova and PereiraPerrin, 2004; Chuenkova and Pereiraperrin, 2006). This triggers multiple TrkA-dependent neurotrophic responses in neuronal cells, including enhanced survival and differentiation, and upregulation of catecholaminergic response (Chuenkova and Pereira, 2000; Chuenkova and Pereira, 2001; Chuenkova and PereiraPerrin, 2005; Chuenkova and Pereiraperrin, 2006).
Given the specific effects of NGF/TrkA signaling on the cholinergic phenotype, and the altered ACh metabolism in experimental Chagas’ disease (Machado et al., 1979; Machado et al., 1987), we sought to determine whether PDNF and T. cruzi invoke cholinergic responses by investigating whether PDNF regulates expression of two key proteins that largely define cholinergic phenotype in neurons: choline acetyltransferase (ChAT) and vesicular ACh transporter (VAChT).
ChAT synthesizes ACh from choline and acetyl-CoA, while VAChT is the proton-ACh antiporter that shuttles ACh into synaptic vesicles. The genes encoding ChAT and VAChT are colocalized in the same “cholinergic” locus, where VAChT gene is clustered within the first intron of the ChAT gene (De Gois et al., 2000; Eiden, 1998); thus their products are co-expressed and known to be coordinately regulated by NGF through TrkA binding (Berse and Blusztajn, 1995; Eiden, 1998; Oosawa et al., 1999; Pongrac and Rylett, 1998a). The signaling pathways through which the cholinergic locus is activated by NGF are not well understood, although there is evidence that protein kinase pathways of PI3K and PKA/CREB and the immediate-early gene c-fos play a role in NGF-induced ChAT expression (Castell et al., 2003; Madziar et al., 2005; Pongrac and Rylett, 1998b; Toliver-Kinsky et al., 2000).
We show here that PDNF and invasive extracellular T. cruzi trypomastigotes increase the expression of cholinergic genes in PC12 neuronal cells via activation of TrkA, while the intracellular T. cruzi infection has the opposite effect. The findings suggest that T. cruzi invasion of the nervous system deregulates ACh metabolism, which could be important in the neuropathology that characterizes Chagas’ disease progression.
Results
PDNF induces ChAT and VAChT expression in PC12 cells
PC12 is a cholinergic cell line that expresses tyrosine kinase TrkA, but not TrkB or TrkC (Segal and Greenberg, 1996), and responds to NGF by acquiring sympathetic neuron phenotype, including an increase in cholinergic gene expression (Greene and Tischler, 1976; Pongrac and Rylett, 1998a). We previously demonstrated that PDNF induces TrkA-dependent PC12 cell survival and sympathetic neuron-like differentiation (Chuenkova and Pereira, 2000).
To test whether PDNF regulates cholinergic gene expression, PC12 cells were treated with various concentrations of PDNF in 1% FCS followed by analysis of ChAT and VAChT transcripts by RT-PCR. The results showed that after 48 hrs mRNA of both ChAT and VAChT increased in the PDNF-treated cells in a dose-dependent manner (Fig. 1A). The PDNF-induced increase in the transcripts at 200 ng/ml (3.5 nM) was similar to that of mammalian NGF, 100ng/ml (0.8 nM) (Fig. 1A). Such PDNF-dependent stimulation was confirmed by quantitative real-time PCR, which revealed a statistically significant dose-dependent increase in cholinergic gene expression (Fig. 1B). As with the RT-PCR results, the stimulatory effect of NGF at 100 ng/ml (1.87 ± 0.30 and 2.19 ± 0.52 fold-increase in ChAT and VAChT mRNA, respectively) was close to that of PDNF at 250 ng/ml (ChAT and VAChT mRNA increase by 1.69 ± 0.17 fold and 1.81 ± 0.33 fold, respectively) (Figure 1B).
Figure 1. PDNF stimulates ChAT and VAChT transcripts in PC12 cells.
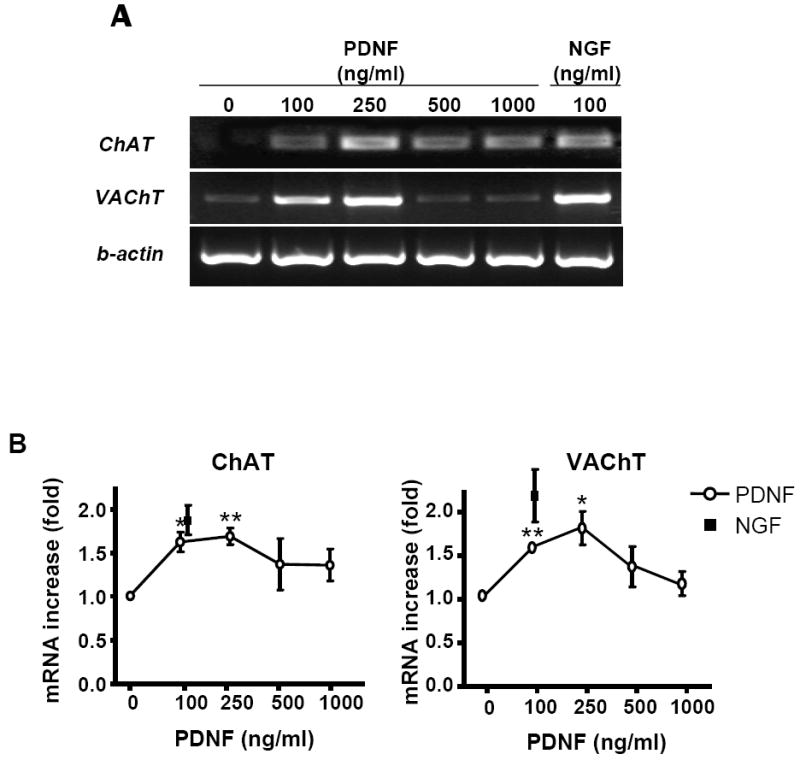
Cells plated in 6-well plates were treated with the indicated concentrations of PDNF or NGF for 48 h in DMEM/FCS (1%).
(A) ChAT and VAChT expression analyzed by RT-PCR and by
(B) q PCR. β-actin expression was used as an internal control.
Values given are fold difference between treated samples and untreated controls. Error bars represent SEM (n=3), * - P < 0.05, ** - P < 0.01.
The PDNF-induced increase in ChAT transcript was reflected in the production of the corresponding protein determined by western blotting and immunofluorescence. PDNF augmented ChAT protein levels in PC12 cells in a dose-dependent manner, with the fourfold increase at 250 ng/ml comparable to that of NGF at 100 ng/ml (Fig. 2A). Similarly, enhanced ChAT production by both PDNF and NGF was visualized by immunofluorescence (Figure 3).
Figure 2. PDNF increases ChAT protein levels in PC12 cells.
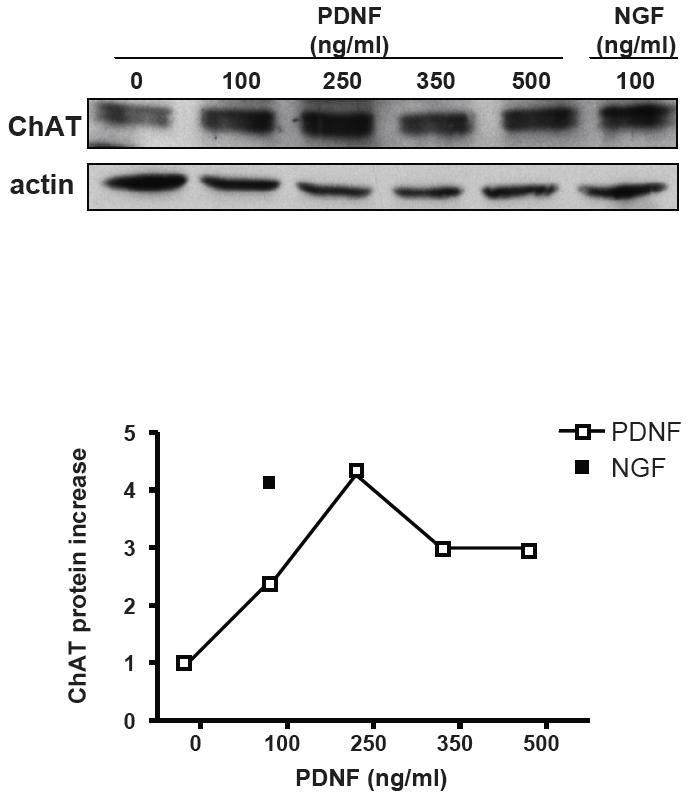
PC12 cells were incubated without or with indicated concentrations of PDNF or NGF for 48 h, and ChAT protein expression was tested by western blot using ChAT (H-95) antibody as a probe. Graph represents densitometry analysis of ChAT expression compared with untreated control after normalization to actin (average of four experiments).
Figure 3. PDNF increases number of ChAT-expressing PC12 cells.
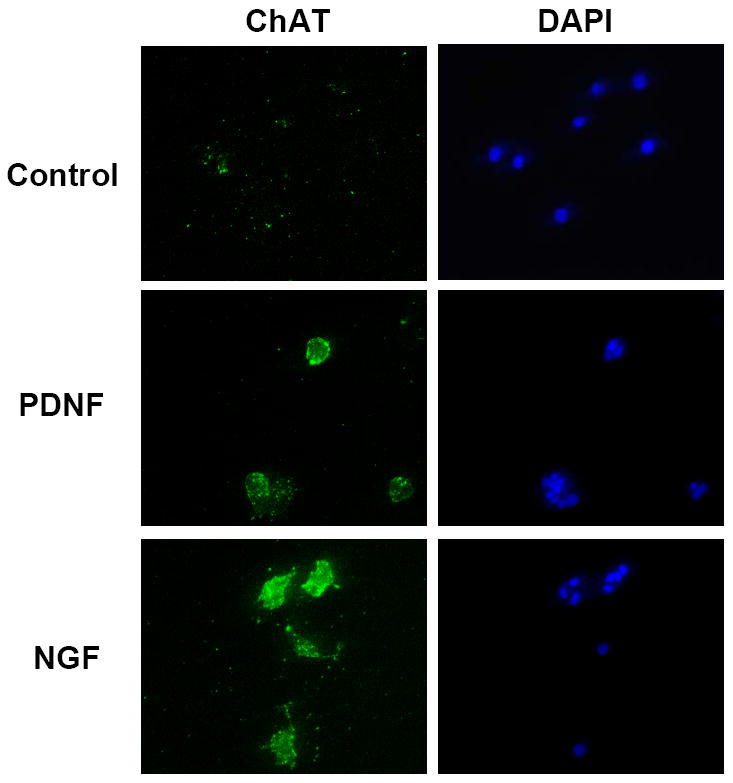
Cells were treated as in Fig. 1 with or without 250 ng/ml PDNF or 100 ng/ml NGF, fixed in paraformaldehyde, permeabilized with a non-ionic detergent, ChAT visualized with specific antibody (AB144P) followed by a fluorescent-labeled secondary antibody (green), and nuclear DNA with DAPI (blue).
PDNF-regulated cholinergic gene expression via TrkA and downstream PI3K and MAPK/Erk1/2 signaling
To determine if PDNF-induced cholinergic gene expression is TrkA-dependent, we tested whether the T. cruzi ligand would be active or not in TrkA-deficient PC12nnr5 cells in a protocol similar to that used for wild-type PC12 cells. We found that, in contrast to the stimulatory action on parental PC12 cells, PDNF and NGF did not enhance expression of ChAT and VAChT in PC12nnr5 cells (Fig. 4A). Consistent with this result, K252a, an inhibitor of TrkA tyrosine kinase autophosphorylation, blocked PDNF-induced increase in ChAT mRNA, as it did with NGF activated cells (Fig. 4B). However, K252a did not alter PDNF- and NGF-induced expression of VAChT mRNA, in agreement with the notion that ChAT and VAChT, coordinately regulated in general, also have distinct trancriptional control mechanisms (Castell et al., 2003; Holler et al., 1996).
Figure 4. PDNF-induced expression of cholinergic gene depends on functional TrkA, and PI3K and MAPK/Erk signaling.
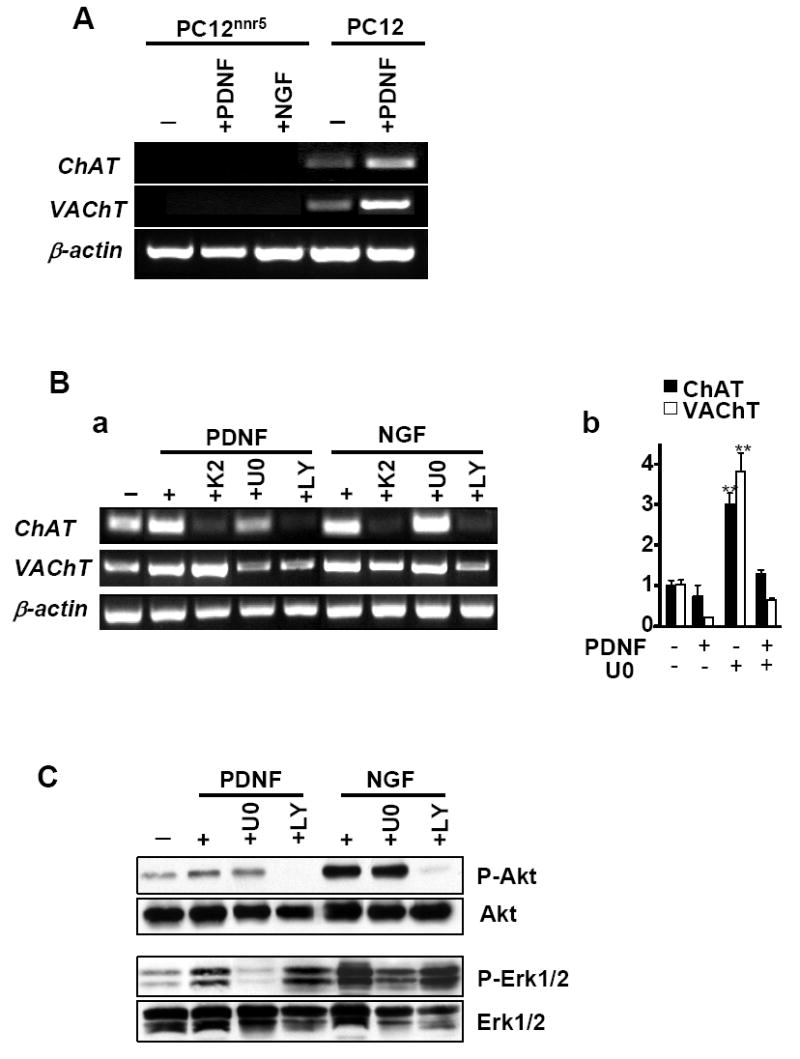
(A) TrkA-deficient (PC12nnr5) and wild-type (PC12) cells were treated with PDNF or NGF for 48 h in 1% FCS/DMEM. (B) PC12 cells were pretreated for 1 h with K252a (K2, 100 nM), U0126 (U0, 10 mM), LY294002 (LY, 10 mM) prior to addition of PDNF or NGF (250 ng/ml and 100 ng/ml, respectively) for 48 h in 1% FCS/DMEM. b. Cells were treated with U0126 and PDNF as described above. ChAT, VAChT, and β-actin expression was analyzed by RT-PCR (A and B,a) and by quantitative PCR (B, b). Error bars represent SEM (n=3), ** - P < 0.01.
(C) Cells were treated as described for B and total cell lysates were analyzed by western blot with phospho-Akt (Ser473), phospho-ERK1/2 (Thr202/Tyr204), Akt, or ERK1/2 antibodies.
To verify whether the PDNF actions on cholinergic gene expression depended on PI3K and MAPK/Erk pathways, which are stimulated following TrkA activation (Chuenkova and Pereira, 2000; Chuenkova and Pereira, 2001), we determined whether PDNF would still be active in PC12 cells treated with LY294002 (a selective inhibitor of PI3K (Vlahos et al., 1994)) or U0126 (a specific inhibitor of MEK, a kinase that exclusively phosphorylates Erk1/2 (Dudley et al., 1995)). We found that upregulation of ChAT mRNA was completely reversed by the PI3K inhibitor LY294002 and inhibited by the MEK inhibitor U0126 (Figure 4B). Both PI3K and MAPK inhibitors also blocked PDNF-induced VAChT expression. In contrast to PDNF, NGF-dependent increase in ChAT expression was sensitive only to the PI3K but not to MEK inhibitor (Figure 4B), in agreement with previous results (Madziar et al., 2005).
To confirm the conclusion that PDNF induced cholinergic gene via PI3K and MAPK, the state of respective signaling was analyzed by western blot. LY294002 completely blocked PDNF-induced phosphorylation of Akt kinase, an enzyme that works downstream of PI3K; and the MAPK kinase inhibitor U0126 also abrogated PDNF-induced Erk1/2 phosphorylation (Fig. 4C), in accordance with our earlier results (Chuenkova and Pereira, 2000; Chuenkova and Pereira, 2001).
The data demonstrate that PDNF-induced increase in ChAT and VAChT mRNA expression required activation of PI3K/Akt, as well as MAPK/Erk1/2 signaling pathways, and underscore that PDNF-elicited TrkA activation is not identical to that of NGF.
Intracellular T. cruzi infection decreases and extracellular parasites increases cholinergic gene expression in PC12 cells
PDNF is expressed on the outer surface of T. cruzi trypomastigotes, the infectious form, and allows parasites to directly activate TrkA receptor (Chuenkova and PereiraPerrin, 2004) as well as utilize TrkA for invasion of host cells (de Melo-Jorge and PereiraPerrin, 2007). After adhering and entering cells by receptor-mediated endocytosis, trypomastigotes differentiate into a replicative stage - amastigotes - that after several division cycles re-differentiate into trypomastigotes and exit infected cells to adhere and invade other cells through the circulation. To investigate whether T. cruzi invasion alters cholinergic gene expression, PC12 cells were infected with 106× trypomastigotes/ml for 3 hrs, extracellular parasites were removed, and cells were incubated for additional 24 or 48 hrs to develop the intracellular infection. Quantitative PCR analysis revealed that transcription of ChAT and VAChT was reduced in infected cells (Figure 5A). However, cells that were incubated with T. cruzi for only 3 hrs showed an increase in ChAT mRNA (Fig. 5A). Therefore, we sought to verify that a surface-mediated host-parasite interaction, presumably via T. cruzi-expressed PDNF, rather than intracellular infection, is responsible for cholinergic gene stimulation. To eliminate the influence of cell invasion, T. cruzi trypomastigotes were heat-inactivated at 50°C for 10 minutes, then assayed for PDNF trans-sialidase (TS) activity to ensure native conformation of the protein and to approximate PDNF concentration, 300 ng PDNF per 1 × 106 parasites. This amount of inactivated parasites stimulated expression of ChAT and VAChT in PC12 cells after 48 h of treatment, while higher concentrations produced less effect (Figure 5B), consistent with the results obtained with purified PDNF (Figs. 1, 2). Thus the data suggest that the stimulation of cholinergic gene expression is a result of surface -mediated interactions between T. cruzi and host cells rather than intracellular infection.
Figure 5. Cholinergic gene expression is down-regulated in PC12 cells with intracellular T. cruzi infection and up-regulated by extracellular T. cruzi trypomastigotes.
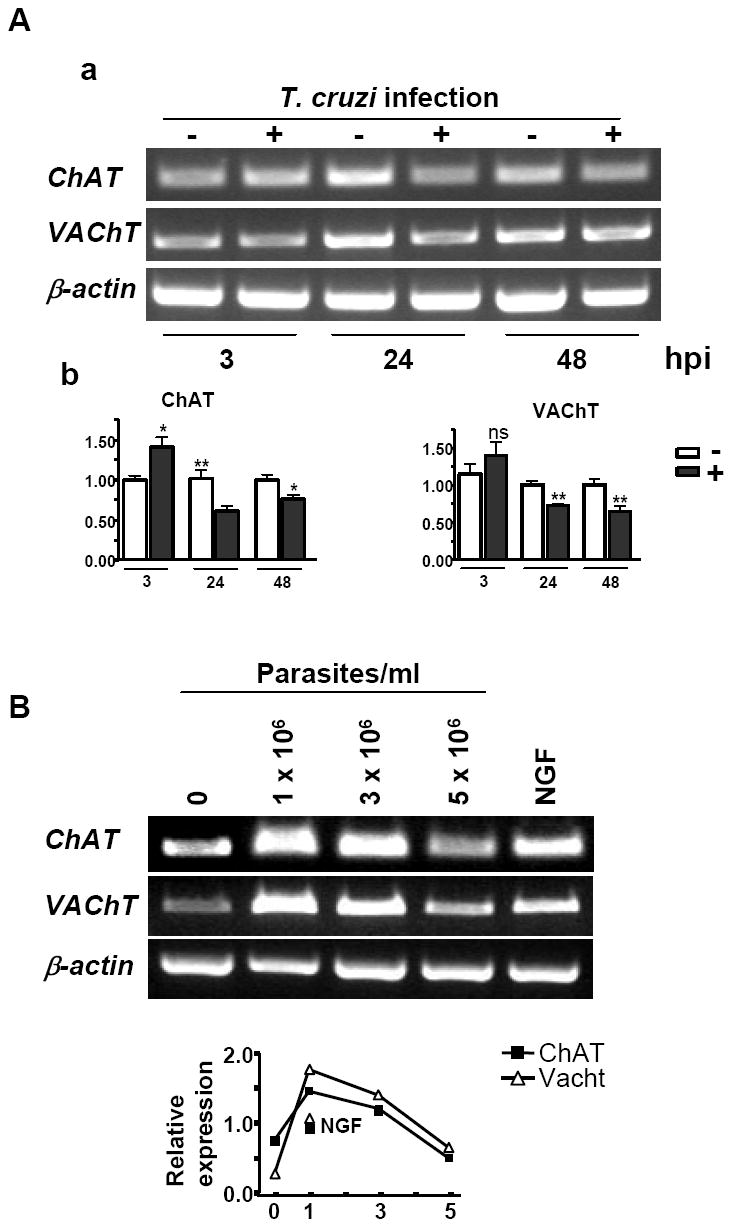
(A) PC12 cells were infected with 1.0 × 106 trypomastigotes for 3, 24 and 48 h, cells harvested and analyzed for ChAT, VAChT, and β-actin expression by RT-PCR (a) and qPCR (b). Error bars represent SEM (n=4), * - P < 0.05, ** - P < 0.01, ns – non-significant.
(B) PC12 cells were exposed to heat-treated trypomastigotes (106/ml) or NGF (100 ng/ml) for 48 h, and ChAT, VAChT transcripts determined by RT-PCR. Graph represents quantitation of the response determined by scanning densitometry.
Discussion
PDNF, first discovered as an enzyme with neuraminidase or trans-sialidase activity (Schenkman et al., 1994), is located in the outer membrane of trypomastigotes through a glycosylphosphatidylinisotol (GPI) anchor (Prioli et al., 1991) and as a soluble protein in the extracellular environment (Prioli et al., 1990) where it can act on substrates on cell surfaces. Subsequently, it was discovered that it can function as a parasite-derived neurotrophic factor (PDNF) by stimulating neurite outgrowth and neuron survival through the activation of NGF receptor TrkA in a way that does not require neuraminidase or trans-sialidase activities (Chuenkova and Pereira, 2000; Chuenkova and PereiraPerrin, 2004); it was also shown that PDNF targets TrkB receptor (Woronowicz et al., 2007). Recognition of TrkA by PDNF on the surface of trypomastigotes also promotes T. cruzi invasion (de Melo-Jorge and PereiraPerrin, 2007). PDNF activity in triggering trophic responses in neuronal cells and in promoting trypanosome invasion of host cells is an important cue for understanding progression of Chagas’ disease pathogenesis.
Given that PDNF functions described so far depend on NGF-like binding and activation of TrkA, it may be that T. cruzi could have other neurotrophin-like biological actions of possible relevance to Chagas’ disease pathogenesis. Here we show that soluble PDNF potently up-regulates expression of ChAT and VAChT in PC12 cells at the mRNA and protein levels (Figs. 1-3), and that the actions of soluble PDNF is reproduced by trypomastigote, the extracellular stage of T. cruzi that expresses PDNF on the surface and that uses PDNF to invade cells (Fig. 5A). In contrast, cells bearing intracellular T. cruzi amastigotes decreased, rather than increased, cholinergic gene expression (Fig. 5B).
As with neurotrophic activity and cellular invasion, the cholinergic actions of PDNF were strictly dependant on the expression of functional TrkA on the target cells because TrkA-deficient mutant of PC12 cells (PC12nnr5) did not respond to PDNF and T. cruzi, nor did wild-type PC12 cells treated with a pharmacological inhibitor of TrkA autophosphorylation (Fig. 4). Accordingly, blocking TrkA-dependent PI3K/Akt and MAPK/Erk signaling with pharmacological inhibitors prevented the cholinergic actions of PDNF (Fig. 4). Additionally, stimulation of ChAT and VAChT expression by both PDNF and parasites increased with the concentration of the stimulants to decrease at high concentrations (Figs 1, 5), an effect reminiscent of that of bona fide TrkA ligand NGF (Conti et al., 2004).
The cholinergic actions of T. cruzi in general and of PDNF in particular could relate to the pathogenesis of Chagas’ disease, especially concerning TrkA-expressing neurons in brain and PNS. In acute Chagas’ disease, when parasites multiply avidly inside cells, ChAT activity and ACh content are reduced (Machado et al., 1979; Machado et al., 1987). Accordingly cholinergic gene expression is down-regulated in T. cruzi-infected PC12 cells (Fig. 5A), as well as in fibroblasts and cardiomyocytes (Imai et al., 2005; Mukherjee et al., 2003). In vivo such decrease could relate not only to ChAT activity (Machado et al., 1979), but also to the degeneration of cholinergic neurons during acute T. cruzi infection (Rodrigues et al., 2002; Tafuri, 1970).
Extracellular T. cruzi could activate TrkA by adhering to neurons as a prelude to cellular entry or by releasing PDNF that could diffuse in the nervous tissue, act on un-invaded cells and restore cholinergic phenotype, ChAT activity and ACh levels as observed in the heart, sub-mandibular gland, and esophagus of rats chronically infected with T. cruzi (Machado et al., 1979; Machado et al., 1987). The increase in ACh concentration could be a factor in the regeneration of sympathetic neurons observed in many Chagasic patients (Koberle, 1968), because ACh stimulation of nicotinic receptors not only promotes neuron survival in ANS (Garrido et al., 2001; O’Neill et al., 2002), it also elicits potent anti-inflammatory responses in regions surrounding the vagus nerve - a major nerve fiber involved with autonomic innervation to the heart and gut (Pavlov and Tracey, 2005). Since the gastro-intestinal and cardiac ganglia are affected in Chagas’ disease, it is possible that this cholinergic anti-inflammatory pathway plays a role in recovery of the damaged tissues via parasite-induced ACh.
It may be that PDNF stimulation of cholinergic gene expression facilitates recovery of parasympathetic function in Chagas’ disease. It would be consistent with our earlier results showing that parasite invasion and PDNF promoted survival of neuronal cells (Chuenkova and Pereira, 2000; Chuenkova et al., 2001; Chuenkova and Pereira, 2003; Chuenkova and PereiraPerrin, 2005) and activated tyrosine hydroxylase, a rate-limiting enzyme in the synthesis of catecholamine neurotransmitters (Chuenkova and Pereiraperrin, 2006), reflecting a corresponding increase in T. cruzi-infected mammals (Machado et al., 1978). Such help is in keeping with the chronic nature of T. cruzi infection, which can last for decades in humans without causing significant pathology.
Finally, our results raise the possibility that PDNF, as a positive regulator of neuronal survival and cholinergic gene expression, may be beneficial in restoration of lost cholinergic phenotype, and may have therapeutic potential for Chagas’ disease.
Experimental Procedures
Materials
Dulbecco’s Modified Eagle’s Medium, fetal calf serum (FCS), and penicillin/streptomycin cocktail were from Gibco-Invitrogen (Carlsbad, CA). Mouse recombinant nerve growth factor (NGF, 7S) and MEK inhibitor U0126 were purchased from Sigma (St. Louis, MI); TrkA-tyrosine kinase inhibitor K252a and PI3-kinase inhibitor LY294002 from Calbiochem (La Jolla, CA); Trizol reagent, chloroform, SuperScript III reverse transcription kit and Platinum Taq DNA polymerase High Fidelity Kit from Invitrogen (Carlsbad, CA); and Quantitect SYBR green from Qiagen (Valencia, CA). Antibodies to phospho-Akt (Ser473), phospho-p44/42 MAPK (Thr202/Tyr204), Akt, and p44/42 MAPK were from Cell Signaling (Beverly, MA); ChAT and actin from Santa Cruz Biotechnology (Santa Cruz, CA) and Chemicon (Temecula, CA). Recombinant PDNF was expressed in E. coli and purified by Ni-affinity chromatography and Fast Protein Liquid Chromatography (FPLC) as described earlier (Chuenkova et al., 1999).
PC12 and Parasite Cultures
Rat pheochromocytoma cells PC12 wt and PC12nnr5 mutant, a generous gift of Dr. Lloyd Greene (College of Physicians and Surgeons, Columbia University, New York, NY), were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively) at 37 °C in 5% CO2. For all experiments, PC12 cells were plated in 35 mm plates in DMEM containing10% FCS at a density of 2.0 – 3.0 × 105 cells/ml. After 24h, medium was replaced with DMEM containing low serum (~1% FCS), and cells were incubated with PDNF and/or other reagents for additional 24 or 48 h. For experiments with pharmacological inhibitors of protein kinases, cells were treated with inhibitors (K252a, 100 nM; LY294002, 10 μM; U0126, 10 μM) for 1 h prior to the addition of growth factors, and the protocol repeated at 24 h intervals. T. cruzi parasites, Silvio strain, were maintained in Vero cell cultures (Prioli et al., 1990), and swimming trypomastigotes obtained after 3-5 days post-infection, (1 × 106 /ml) were used to infect PC12 cells. After 3 h, unattached parasites were removed; the monolayers washed with DMEM, and incubated with DMEM/1% FCS for 24 or 48 h. For experiments with heat-inactivated T. cruzi, trypomastigotes in DMEM were heated at 50°C for 10 min, a condition that blocks parasite invasion (Yoshida, 1983); heat-killed parasites and control viable parasites were used at various concentrations to treat PC12 cells for 48 h, which were subsequently analyzed for ChAT and VAChT expression by RT-PCR.
RT-PCR
Total RNA was isolated from PC12 cultures with Trizol reagent and chloroform. cDNA was synthesized from 500 ng total RNA with Superscript III reverse transcriptase using manufacturers specifications for priming with random hexamers. The conditions for PCR reactions performed with 1-2 μl cDNA using Platinum Taq DNA polymerase High Fidelity were: 94°C for 2 min for initial denaturation, then 30-35 cycles 94 °C for 30 s, 53°C for 30 s, and 68°C for 30 sec, followed by a final extension of 68 °C for 3 min. ChAT primers common for all three ChAT isoforms were FWD: 5′~GCC TCA TCT CTG GTG TGC TTA G~3′; REV: 5′~CCC TCA CTG AGA CGG CGG AA~3′ (Madziar et al., 2005). VAChT primers were FWD: 5′~AGC GGG CCT TTC ATT GAT CG~3′; REV: 5′~GGC GCA CGT CCA CCA GAA AGG~3′ and β-actin primers were FWD: 5′~GTG GGC CGC CCT AGG CAC CAG~3′; REV: 5′~CTC TTT GAT GTC ACG CAC GAT TTC~3′. PCR products were analyzed by electrophoresis in 2% agarose gels with 5 ug/ml ethidium bromide.
Quantitative Real-time PCR (qPCR)
RNA was isolated and cDNA prepared as described above. Quantitative PCR reactions were performed with QuantiTect SYBR Green PCR kit (Qiagen) using Applied Biosystems 7300 Real-Time PCR system (Applied Biosystems, Foster City, CA). Conditions for the PCR reaction were: 95 °C for 15 min, followed by 40 cycles of 94 °C for 15 s, 56 °C for 30 s, and 72 °C for 30 s, concluded with a dissociation stage to measure primer specificity. ChAT, VAChT and β-actin real-time primers amplify within the same regions as the RT-PCR primers. Chat primers were FWD: 5′~AGC CCT GCT GTG ATC TTT GCT CG~3′; REV: 5′~CCT TGG CCC AGT CAG TGG GAA~3′; VAChT primers - FWD: 5′~CCC TTA AGC GGG CCT TTC ATT GAT~3′; REV: 5′~AAA GGC AAA CAT GAC TGT GGA GGC~3′ and β-actin primers - 5′~GTG GGC CGC CCT AGG CAC CAG~3′; REV: 5′~GGA TAC CTC TCT TGC TCT GGG CC~3′. Experiments were performed in triplicates, and fold difference was calculated using the 2ΔΔCt method [ΔΔCt = (Cttarget gene − Ctβ-actin)treated - (Cttarget gene − Ctβ-actin)untreated] (Livak and Schmittgen, 2001).
Immunoblotting
Total cell proteins were extracted with a buffer containing 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 2.5 mM sodium pyrophosphate, 1 mM glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin and 1 mM PMSF on ice for 10min and used immediately or kept frozen at -80°C. Protein concentration was measured by the Bio-Rad DC protein assay (Richmond, CA). Samples (50-70 μg of protein in each) were resolved by SDS-PAGE, transferred to nitrocellulose, blocked with 5% nonfat milk, and probed overnight at 4°C with antibody to ChAT (1:200), phospho-Akt (1:1000) or phospho-Erk1/2 (1:1000). Antibody reaction was detected with horseradish peroxidase (HRP)-conjugated secondary antibody and Western Lightning Chemiluminescence Reagent Plus (PerkinElmer, Boston, MA). Some blots were stripped in appropriate buffer (50mMTris–HCl, pH 7.8, 2% SDS, 1% β-mercaptoethanol) at 50 °C for 60 min and re-probed with Akt (1:1000), Erk1/2 (1:1000) or actin (1:500) antibodies to ensure equal loading of proteins.
Immunofluorescence
Cells were plated on collagen-coated slides overnight in 10% FCS DMEM, which was replaced with serum-free medium alone or with PDNF (250 ng/ml) or NGF (100 ng/ml) for 48 h. Fresh PDNF and NGF were added after 24 h. Cells were washed with PBS, fixed with 4% paraformaldehyde in PBS for 15 min at RT and permeabilized with 0.1% Triton X in PBS for 15 min at RT. After blocking with 10% goat serum in PBS, slides were incubated overnight at 4°C with anti-ChAT antibody (1:100) in 5% goat serum followed by Alexa Flour 488-conjugated secondary antibodies (30 min) (Molecular Probes). Cells were stained with DAPI (250 ng/ml) for 30 sec to visualize DNA, and examined by fluorescent microscopy. Micrographs were taken by SPOT camera (Diagnostic Instruments, Sterling Heights, MI).
Statistical Analysis
For statistical analysis of real-time data, unpaired Student’s t test was performed using GraphPad Prism version 4.0 (GraphPad software, San Diego, CA). The statistical significance level was set at P < 0.05. Data from three or more independent experiments were plotted as mean ± SEM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Auld DS, Mennicken F, Day JC, Quirion R. Neurotrophins differentially enhance acetylcholine release, acetylcholine content and choline acetyltransferase activity in basal forebrain neurons. J Neurochem. 2001;77:253–62. doi: 10.1046/j.1471-4159.2001.t01-1-00234.x. [DOI] [PubMed] [Google Scholar]
- Berse B, Blusztajn JK. Coordinated up-regulation of choline acetyltransferase and vesicular acetylcholine transporter gene expression by the retinoic acid receptor alpha, cAMP, and leukemia inhibitory factor/ciliary neurotrophic factor signaling pathways in a murine septal cell line. J Biol Chem. 1995;270:22101–4. doi: 10.1074/jbc.270.38.22101. [DOI] [PubMed] [Google Scholar]
- Castell X, Cheviron N, Barnier JV, Diebler MF. Exploring the regulation of the expression of ChAT and VAChT genes in NG108-15 cells: implication of PKA and PI3K signaling pathways. Neurochem Res. 2003;28:557–64. doi: 10.1023/a:1022829608540. [DOI] [PubMed] [Google Scholar]
- Chuenkova M, Pereira M, Taylor G. trans-sialidase of Trypanosoma cruzi: location of galactose-binding site(s) Biochem Biophys Res Commun. 1999;262:549–56. doi: 10.1006/bbrc.1999.1154. [DOI] [PubMed] [Google Scholar]
- Chuenkova MV, Pereira MA. A trypanosomal protein synergizes with the cytokines ciliary neurotrophic factor and leukemia inhibitory factor to prevent apoptosis of neuronal cells. Mol Biol Cell. 2000;11:1487–98. doi: 10.1091/mbc.11.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuenkova MV, Furnari FB, Cavenee WK, Pereira MA. Trypanosoma cruzi trans-sialidase: a potent and specific survival factor for human Schwann cells by means of phosphatidylinositol 3-kinase/Akt signaling. Proc Natl Acad Sci U S A. 2001;98:9936–41. doi: 10.1073/pnas.161298398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuenkova MV, Pereira MA. The T. cruzi trans-sialidase induces PC12 cell differentiation via MAPK/ERK pathway. Neuroreport. 2001;12:3715–8. doi: 10.1097/00001756-200112040-00022. [DOI] [PubMed] [Google Scholar]
- Chuenkova MV, Pereira MA. PDNF, a human parasite-derived mimic of neurotrophic factors, prevents caspase activation, free radical formation, and death of dopaminergic cells exposed to the Parkinsonism-inducing neurotoxin MPP+ Brain Res Mol Brain Res. 2003;119:50–61. doi: 10.1016/j.molbrainres.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Chuenkova MV, PereiraPerrin M. Chagas’ disease parasite promotes neuron survival and differentiation through TrkA nerve growth factor receptor. J Neurochem. 2004;91:385–94. doi: 10.1111/j.1471-4159.2004.02724.x. [DOI] [PubMed] [Google Scholar]
- Chuenkova MV, PereiraPerrin M. A synthetic peptide modeled on PDNF, Chagas’ disease parasite neurotrophic factor, promotes survival and differentiation of neuronal cells through TrkA receptor. Biochemistry. 2005;44:15685–94. doi: 10.1021/bi0512039. [DOI] [PubMed] [Google Scholar]
- Chuenkova MV, Pereiraperrin M. Enhancement of tyrosine hydroxylase expression and activity by Trypanosoma cruzi parasite-derived neurotrophic factor. Brain Res. 2006;1099:167–75. doi: 10.1016/j.brainres.2006.04.128. [DOI] [PubMed] [Google Scholar]
- Conti AM, Brimijoin S, Miller LJ, Windebank AJ. Suppression of neurite outgrowth by high-dose nerve growth factor is independent of functional p75NTR receptors. Neurobiol Dis. 2004;15:106–14. doi: 10.1016/j.nbd.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Counts SE, Mufson EJ. The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. J Neuropathol Exp Neurol. 2005;64:263–72. doi: 10.1093/jnen/64.4.263. [DOI] [PubMed] [Google Scholar]
- De Gois S, Houhou L, Oda Y, Corbex M, Pajak F, Thevenot E, Vodjdani G, Mallet J, Berrard S. Is RE1/NRSE a common cis-regulatory sequence for ChAT and VAChT genes? J Biol Chem. 2000;275:36683–90. doi: 10.1074/jbc.M006895200. [DOI] [PubMed] [Google Scholar]
- de Melo-Jorge M, PereiraPerrin M. The Chagas’ disease parasite Trypanosoma cruzi exploits nerve growth factor receptor TrkA to infect mammalian hosts. Cell Host Microbe. 2007;1:251–61. doi: 10.1016/j.chom.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1995;92:7686–9. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiden LE. The cholinergic gene locus. J Neurochem. 1998;70:2227–40. doi: 10.1046/j.1471-4159.1998.70062227.x. [DOI] [PubMed] [Google Scholar]
- Ekstrom J, Reinhold AC. Increases in nerve growth factor immunoreactivity in the submandibular gland, but not in the parotid gland, of the rat following sympathetic denervation. Arch Oral Biol. 2004;49:3–9. doi: 10.1016/s0003-9969(03)00181-x. [DOI] [PubMed] [Google Scholar]
- Galligan JJ, LePard KJ, Schneider DA, Zhou X. Multiple mechanisms of fast excitatory synaptic transmission in the enteric nervous system. J Auton Nerv Syst. 2000;81:97–103. doi: 10.1016/s0165-1838(00)00130-2. [DOI] [PubMed] [Google Scholar]
- Garrido R, Mattson MP, Hennig B, Toborek M. Nicotine protects against arachidonic-acid-induced caspase activation, cytochrome c release and apoptosis of cultured spinal cord neurons. J Neurochem. 2001;76:1395–403. doi: 10.1046/j.1471-4159.2001.00135.x. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–8. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisenberg CP, Cooper JD, Berke J, Sofroniew MV. NMDA potentiates NGF-induced sprouting of septal cholinergic fibres. Neuroreport. 1994;5:413–6. doi: 10.1097/00001756-199401120-00010. [DOI] [PubMed] [Google Scholar]
- Holler T, Berse B, Cermak JM, Diebler MF, Blusztajn JK. Differences in the developmental expression of the vesicular acetylcholine transporter and choline acetyltransferase in the rat brain. Neurosci Lett. 1996;212:107–10. doi: 10.1016/0304-3940(96)12808-1. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Kilbridge J, Li Y, Cunningham ET, Jr, Lenn NJ, Clary DO, Reichardt LF, Mobley WC. TrkA expression in the CNS: evidence for the existence of several novel NGF-responsive CNS neurons. J Neurosci. 1995;15:1567–76. doi: 10.1523/JNEUROSCI.15-02-01567.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Mimori T, Kawai M, Koga H. Microarray analysis of host gene-expression during intracellular nests formation of Trypanosoma cruzi amastigotes. Microbiol Immunol. 2005;49:623–31. doi: 10.1111/j.1348-0421.2005.tb03654.x. [DOI] [PubMed] [Google Scholar]
- Knipper M, da Penha Berzaghi M, Blochl A, Breer H, Thoenen H, Lindholm D. Positive feedback between acetylcholine and the neurotrophins nerve growth factor and brain-derived neurotrophic factor in the rat hippocampus. Eur J Neurosci. 1994;6:668–71. doi: 10.1111/j.1460-9568.1994.tb00312.x. [DOI] [PubMed] [Google Scholar]
- Koberle F. Chagas’ disease and Chagas’ syndromes: the pathology of American trypanosomiasis. Adv Parasitol. 1968;6:63–116. doi: 10.1016/s0065-308x(08)60472-8. [DOI] [PubMed] [Google Scholar]
- Lahiri DK, Chen DM, Lahiri P, Bondy S, Greig NH. Amyloid, cholinesterase, melatonin, and metals and their roles in aging and neurodegenerative diseases. Ann N Y Acad Sci. 2005;1056:430–49. doi: 10.1196/annals.1352.008. [DOI] [PubMed] [Google Scholar]
- Lin A, Lourenssen S, Stanzel RD, Blennerhassett MG. Selective loss of NGF-sensitive neurons following experimental colitis. Exp Neurol. 2005a;191:337–43. doi: 10.1016/j.expneurol.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Lin A, Lourenssen S, Stanzel RD, Blennerhassett MG. Nerve growth factor sensitivity is broadly distributed among myenteric neurons of the rat colon. J Comp Neurol. 2005b;490:194–206. doi: 10.1002/cne.20654. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Machado AB, Machado CR, Gomez MV. Trypanosoma cruzi: acetylcholine content and cholinergic innervation of the heart in rats. Exp Parasitol. 1979;47:107–15. doi: 10.1016/0014-4894(79)90012-2. [DOI] [PubMed] [Google Scholar]
- Machado CR, Machado AB, Chiari CA. Recovery from heart norepinephrine depletion in experimental Chagas’ disease. Am J Trop Med Hyg. 1978;27:20–4. doi: 10.4269/ajtmh.1978.27.20. [DOI] [PubMed] [Google Scholar]
- Machado CR, Gomez MV, Machado AB. Changes in choline acetyltransferase activity of rat tissues during Chagas’ disease. Braz J Med Biol Res. 1987;20:697–702. [PubMed] [Google Scholar]
- Madhavan R, Peng HB. A synaptic balancing act: local and global signaling in the clustering of ACh receptors at vertebrate neuromuscular junctions. J Neurocytol. 2003;32:685–96. doi: 10.1023/B:NEUR.0000020617.05656.68. [DOI] [PubMed] [Google Scholar]
- Madziar B, Lopez-Coviella I, Zemelko V, Berse B. Regulation of cholinergic gene expression by nerve growth factor depends on the phosphatidylinositol-3’-kinase pathway. J Neurochem. 2005;92:767–79. doi: 10.1111/j.1471-4159.2004.02908.x. [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR. On Trk for retrograde signaling. Neuron. 2001;32:767–70. doi: 10.1016/s0896-6273(01)00529-3. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Belbin TJ, Spray DC, Iacobas DA, Weiss LM, Kitsis RN, Wittner M, Jelicks LA, Scherer PE, Ding A, Tanowitz HB. Microarray analysis of changes in gene expression in a murine model of chronic chagasic cardiomyopathy. Parasitol Res. 2003;91:187–96. doi: 10.1007/s00436-003-0937-z. [DOI] [PubMed] [Google Scholar]
- Nizri E, Wirguin I, Brenner T. The role of cholinergic balance perturbation in neurological diseases. Drug News Perspect. 2007;20:421–9. doi: 10.1358/dnp.2007.20.7.1149629. [DOI] [PubMed] [Google Scholar]
- O’Neill MJ, Murray TK, Lakics V, Visanji NP, Duty S. The role of neuronal nicotinic acetylcholine receptors in acute and chronic neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2002;1:399–411. doi: 10.2174/1568007023339166. [DOI] [PubMed] [Google Scholar]
- Oosawa H, Fujii T, Kawashima K. Nerve growth factor increases the synthesis and release of acetylcholine and the expression of vesicular acetylcholine transporter in primary cultured rat embryonic septal cells. J Neurosci Res. 1999;57:381–7. [PubMed] [Google Scholar]
- Pavlov VA, Tracey KJ. The cholinergic anti-inflammatory pathway. Brain Behav Immun. 2005;19:493–9. doi: 10.1016/j.bbi.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Pongrac JL, Rylett RJ. NGF-induction of the expression of ChAT mRNA in PC12 cells and primary cultures of embryonic rat basal forebrain. Brain Res Mol Brain Res. 1998a;62:25–34. doi: 10.1016/s0169-328x(98)00215-0. [DOI] [PubMed] [Google Scholar]
- Pongrac JL, Rylett RJ. Molecular mechanisms regulating NGF-mediated enhancement of cholinergic neuronal phenotype: c-fos trans-activation of the choline acetyltransferase gene. J Mol Neurosci. 1998b;11:79–93. doi: 10.1385/jmn:11:1:79. [DOI] [PubMed] [Google Scholar]
- Prioli RP, Mejia JS, Pereira ME. Monoclonal antibodies against Trypanosoma cruzi neuraminidase reveal enzyme polymorphism, recognize a subset of trypomastigotes, and enhance infection in vitro. J Immunol. 1990;144:4384–91. [PubMed] [Google Scholar]
- Prioli RP, Mejia JS, Aji T, Aikawa M, Pereira ME. Trypanosoma cruzi: localization of neuraminidase on the surface of trypomastigotes. Trop Med Parasitol. 1991;42:146–50. [PubMed] [Google Scholar]
- Robbins TW. Chemistry of the mind: neurochemical modulation of prefrontal cortical function. J Comp Neurol. 2005;493:140–6. doi: 10.1002/cne.20717. [DOI] [PubMed] [Google Scholar]
- Rodrigues E, Liberti EA, Maifrino LB, de Souza RR. Cardiac denervation in mice infected with Trypanosoma cruzi. Ann Trop Med Parasitol. 2002;96:125–30. doi: 10.1179/000349802125000583. [DOI] [PubMed] [Google Scholar]
- Schenkman S, Eichinger D, Pereira ME, Nussenzweig V. Structural and functional properties of Trypanosoma trans-sialidase. Annu Rev Microbiol. 1994;48:499–523. doi: 10.1146/annurev.mi.48.100194.002435. [DOI] [PubMed] [Google Scholar]
- Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–89. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- Snider WD, McMahon SB. Tackling pain at the source: new ideas about nociceptors. Neuron. 1998;20:629–32. doi: 10.1016/s0896-6273(00)81003-x. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Cooper JD, Svendsen CN, Crossman P, Ip NY, Lindsay RM, Zafra F, Lindholm D. Atrophy but not death of adult septal cholinergic neurons after ablation of target capacity to produce mRNAs for NGF, BDNF, and NT3. J Neurosci. 1993;13:5263–76. doi: 10.1523/JNEUROSCI.13-12-05263.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafuri WL. Pathogenesis of lesions of the autonomic nervous system of the mouse in experimental acute Chagas’ disease. Light and electron microscope studies. Am J Trop Med Hyg. 1970;19:405–17. doi: 10.4269/ajtmh.1970.19.405. [DOI] [PubMed] [Google Scholar]
- Toliver-Kinsky T, Wood T, Perez-Polo JR. Nuclear factor kappaB/p49 is a negative regulatory factor in nerve growth factor-induced choline acetyltransferase promoter activity in PC12 cells. J Neurochem. 2000;75:2241–51. doi: 10.1046/j.1471-4159.2000.0752241.x. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, U HS, Amaral DG, Gage FH. Nerve growth factor infusion in the primate brain reduces lesion-induced cholinergic neuronal degeneration. J Neurosci. 1990;10:3604–14. doi: 10.1523/JNEUROSCI.10-11-03604.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)- 8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–8. [PubMed] [Google Scholar]
- Woronowicz A, Amith SR, Davis VW, Jayanth P, De Vusser K, Laroy W, Contreras R, Meakin SO, Szewczuk MR. Trypanosome trans-sialidase mediates neuroprotection against oxidative stress, serum/glucose deprivation, and hypoxia-induced neurite retraction in Trk-expressing PC12 cells. Glycobiology. 2007;17:725–34. doi: 10.1093/glycob/cwm034. [DOI] [PubMed] [Google Scholar]
- Yeh J, Ferreira M, Ebert S, Yasuda RP, Kellar KJ, Wolfe BB. Axotomy and nerve growth factor regulate levels of neuronal nicotinic acetylcholine receptor alpha3 subunit protein in the rat superior cervical ganglion. J Neurochem. 2001;79:258–65. doi: 10.1046/j.1471-4159.2001.00545.x. [DOI] [PubMed] [Google Scholar]
- Yoshida N. Surface antigens of metacyclic trypomastigotes of Trypanosoma cruzi. Infect Immun. 1983;40:836–9. doi: 10.1128/iai.40.2.836-839.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]