

Abstract
Oligomerization of numerous G protein-coupled receptors has been documented, including the prototypic family B secretin receptor. The clinical significance of oligomerization of this receptor became clear with the recent observation that a misspliced form present in pancreatic cancer could associate with the wild type receptor and act as a dominant negative inhibitor of its normal growth inhibitory function. Our goal was to explore the molecular mechanism of this interaction using bioluminescence (BRET) and fluorescence (FRET) resonance energy transfer and fluorescence microscopy with a variety of receptor constructs tagged with luciferase or cyan or yellow fluorescent proteins. BRET signals comparable to those obtained from cells coexpressing differentially-tagged wild type receptors were observed for similarly-tagged secretin receptors in which all or part of the amino-terminal domain was deleted. As expected, neither of these constructs bound secretin, and only the partially truncated construct sorted to the plasma membrane. Receptors lacking the majority of the carboxyl-terminal domain, including that important for phosphorylation-mediated desensitization, also produced BRET signals above background. These findings suggested that the receptor's membrane-spanning core is responsible for secretin receptor oligomerization. Interestingly, alanine substitutions for a –GxxxG– helix interaction motif in transmembrane segment seven created non-functional receptors that were capable of forming oligomers. Furthermore, treatment of receptor-expressing cells with brefeldin A did not eliminate the BRET signals, and morphologic FRET experiments confirmed the expected subcellular localizations of receptor oligomers. We conclude that secretin receptor oligomerization occurs through –GxxxG– motif-independent interactions of transmembrane segments during the maturation of nascent molecules.
Quaternary assemblies of G protein-coupled, plasma membrane-bound heptahelical receptors (GPCRs)1 appear to be a common structural feature of these functionally diverse, pharmacologically important molecules. In most cases, however, it has been particularly difficult to determine the stoichiometry of association, such that the general term “oligomerization” is most appropriate for describing quaternary assemblies that have been characterized only partially. Such assemblies have been documented for numerous family A, family B and family C GPCRs as homomeric associations of receptors with themselves or as heteromeric associations of receptors with structurally-related family members (1-3). Functional characterizations of known receptor oligomers have demonstrated that these assemblies are important modulators of receptor maturation, ligand-binding specificity, and downstream signaling processes (4, 5). Interestingly, heteromer formation is required in some cases for the maturation of receptor complexes that may exhibit pharmacological, signaling, and turnover properties that distinguish them from their homomeric equivalents (6-8). Although these exciting observations have illuminated novel opportunities for therapeutic manipulation (9), much remains to be learned of how GPCR interactions are regulated both structurally and functionally.
Several recent studies have identified specific receptor domains and/or residues that were necessary for the formation of receptor oligomers. For example, one of the earliest studies described a synthetic peptide corresponding to TM6 of the β2-adrenergic receptor that disrupted homodimers of this receptor and prevented stimulation of adenylate cyclase activity (10). Similar effects have been reported for TM1 and TM4 of the chemokine CCR5 receptor (11), TM5 of the adenosine A2A receptor (12), and TM6 and TM7 of the cholecystokinin receptor (Harikumar et al., submitted). Mutations incorporated into TM and other receptor domains (11-14), including the extracellular domains of family C receptors (15) and the intracellular domains of certain family A receptors (16), also have been shown in various energy transfer, biochemical and functional analyses to disrupt homomeric and heteromeric interactions. However, these data have yet to reveal consistent structural motifs for predicting GPCR oligomerization. One enticing candidate found in various GPCRs is the –GxxxG– pattern, a face-specific helix-helix interaction motif first identified in studies that addressed the assembly of glycophorin A dimers (17). Although this motif provides a potential mechanism for GPCR associations (10, 13), its location varies among family members that often show poor conservation within a given TM segment. Interestingly, a pairwise glycine motif was not present in the aforementioned chemokine, adenosine or cholecystokinin receptor TM domains, and in some membrane proteins its presence was not related to oligomer formation (18, 19). Nonetheless, rigorous comparisons of sequence and structural information are beginning to reveal further candidate domains and/or residues, including hydrophobic residues juxtaposed with the membrane interface (20, 21).
The emerging picture for GPCR oligomerization is that amino acids within receptor core domains, especially the TM segments, provide the interfaces for receptor associations that occur early during the biosynthetic maturation of nascent receptors. With respect to the latter, a series of studies that have asked when and where in the cell newly-synthesized receptor monomers associate has led to the conclusion that oligomerization is a prerequisite for receptor maturation and plasma membrane sorting (14, 22, 23). These findings pertain mostly to the more well-studied family A GPCRs, and even in those cases only the most interesting or functionally relevant receptor domains have been evaluated for their effects on oligomerization and sorting. Moreover, the specific core domain residues that are responsible for oligomerization thus far appear to vary greatly from one receptor to another. We addressed these concerns with a systematic approach to understanding the mechanism of secretin receptor oligomerization, the prototypic family B GPCR that regulates the secretion of pancreatic and biliary bicarbonate (24, 25). Secretin receptor is characterized by a large extracellular amino-terminal domain, a typical receptor core domain of seven TM and intervening loop segments, and an intracellular carboxyl-terminal domain. Previous studies have shown that binding of the peptide agonist secretin at the receptor's amino-terminal domain has no effect on the demonstrated ability of the receptor to form homomers with itself (26) and heteromers with structurally-related vasoactive intestinal polypeptide receptors (27).
A detailed understanding of secretin receptor oligomerization recently was given clinical relevance when it was found that a splicing defect in human pancreatic cancer cells created an inactive, oligomerization-competent secretin receptor (26, 28). Presence of the misspliced variant resulted in a dominant negative phenotype that was observed as a marked decrease in cAMP production by secretin-stimulated cancer cells coexpressing misspliced and wild type secretin receptors. This functional defect was explained in part by the demonstrated association of misspliced and wild type receptors residing in the plasma membrane, an increasingly common theme that illustrates the importance of oligomerization processes in the establishment of functional receptor for normal cell growth (29-32). Considering the implications of secretin receptor interactions for potential advancements in cancer therapy and detection, we aimed to determine the molecular and cellular bases of these interactions, and how they relate to the regulation of secretin receptor function. To this end, we present a series of resonance energy transfer studies that employed secretin receptor mutants designed to ask which receptor domains may be involved in forming the interaction surfaces necessary for monomer assembly. Associations monitored by BRET demonstrated clearly that residues within the amino- and carboxyl-terminal domains were not required for homomeric interactions or heteromeric interactions with wild type receptors. As a first look at similar analyses of established oligomerization motifs within the receptor core domain, we found that mutational disruptions of a pairwise glycine motif in TM7 also did not alter oligomer formation. Functional characterizations of all mutants also provided opportunities for understanding the role that oligomerization plays in secretin receptor maturation, where it was concluded that like family A GPCRs, the family B secretin receptor can form oligomeric associations early during receptor maturation and plasma membrane sorting. Coupled with morphological FRET studies that confirmed the presence of cell surface and intracellular associations, these data enhance our understanding of the mechanisms that drive oligomeric assembly of the clinically important secretin receptor.
EXPERIMENTAL PROCEDURES
Materials
Reagents for recombinant DNA work were purchased from New England Biolabs (Beverly, MA), Stratagene (La Jolla, CA), Bio-Rad Laboratories (Hercules, CA), Eppendorf (Hamburg, Germany), and Qiagen (Valencia, CA). Invitrogen (San Diego, CA) supplied the cell culture growth media and antibiotics, HyClone Laboratories (Logan, UT) the Fetal Clone II serum supplements, and Sigma-Aldrich (St. Louis, MO) the non-enzymatic cell dissociation solution. BFA, formaldehyde and coelenterazine h were purchased from Molecular Probes (Eugene, OR), Ted Pella (Redding, CA) and Biotium (Hayward, CA), respectively. The natural rat secretin and [Tyr10]secretin peptides used in binding assays were synthesized in our laboratory (33) and have been shown to be functional at human secretin receptors in intact cells (34). Oxidative radioiodination of the Tyr10 residue to form the 125I-[Tyr10]secretin radioligand has been described elsewhere (33). All other reagents were of the highest quality appropriate for the given experiment.
Receptor mutagenesis
The molecular mechanism of SecR oligomerization was examined by systematically deleting or mutating receptor residues that comprise key extracellular, receptor core, or intracellular domains (Figure 1). These modifications were incorporated into a native human SecR open reading frame (ORF) that was subcloned through an EcoRV/SmaI blunt-end ligation from pBKCMV/SecR (28) into the cytomegalovirus-driven eukaryotic expression vector pcDNA3 (Invitrogen). The same mutations also were incorporated into Rlu and YFP carboxyl-terminally tagged versions of the same SecR coding sequence, minus the 5' and 3' untranslated regions (35). Construction of pcDNA3/SecR-CFP (27) and pcDNA3/CCKBR-YFP (36) were described previously.
FIGURE 1.

Structural representation of human secretin receptor. Single letter abbreviations denote primary amino acid sequences within and adjacent to the amino- and carboxyl-terminal domains, as well as the two glycine residues of the –GxxxG– motif in TM7, that are pertinent to this study (enlarged, shaded residues). Solid arrowheads mark the positions of truncation points within the amino and carboxyl termini.
The Δ(1−102) and Δ(1−121) (numbering starts with the first amino acid after the signal sequence) amino-terminal truncations were created by first inserting with QuikChange site-directed mutagenesis (Stratagene) two NheI sites positioned after codons representing the last signal sequence residue and either Cys102 or Leu121. Sequences between these sites were excised with NheI, and then the resulting vector fragments were re-ligated to yield SecR mutants that retained signal sequence residues but lacked sequences coding for the first 102 or 121 amino acids. The Δ(376−419) and Δ(387−419) carboxyl-terminal truncations were created in much the same way, except that an XhoI site was inserted before codons representing Leu376 or Leu387, respectively, to remove the sequences spanning these sites and the XhoI linker that was used to incorporate the Rlu or YFP tags. For untagged SecR, the corresponding carboxyl-terminal mutants were created by QuikChange insertion of a TGA stop codon immediately before the codons representing Leu376 or Leu387. Alanine substitutions for either or both of the glycine residues within the receptor core domain's conserved –GxxxG– motif in TM7 were introduced in a single QuikChange reaction that included a mixture of mutagenic primers that changed Gly356 (G356) and/or Gly360 (G360) to alanine. All receptor mutations were confirmed by automated dye-terminator cycle sequencing.
Cell cultures and transfections
African green monkey kidney (COS) cells from the American Type Culture Collection (Manassas, VA) were propagated in Dulbecco's modified Eagle's growth medium (DMEM) supplemented with 5% (v/v) Fetal Clone II serum. The cells were maintained in a humidified chamber with 5% (v/v) CO2 at 37 °C and were passaged twice per week on Corning (Acton, MA) tissue culture flasks. Cells inoculated to 10-cm Petri dishes were transfected according to a modified diethylaminoethyl (DEAE)-dextran method (35, 37, 38), and then were cultured for 24 h in DMEM with serum before lifting with 0.05% (w/v) trypsin and inoculating well plates or flasks for subsequent analyses. All transfections received the same amount of plasmid DNA regardless of the downstream experiment. For experiments designed to test SecR oligomerization under conditions in which protein sorting to the plasma membrane was inhibited, cells were cultured post-transfection in DMEM containing 10 μg/ml BFA that was added directly to the culture medium from a 10 mg/ml stock solution in 95% (v/v) ethanol. Control cultures received an equivalent amount of ethanol without BFA.
Receptor binding assays
To test the ability of each of the secretin receptor mutants to bind a secretin agonist, transfected COS cells were transferred to 24-well plates and then incubated an additional 48 h prior to use in intact cell binding assays that were carried out as described previously (38). Briefly, cells were incubated for 1 h at room temperature in an agonist competition mixture that included increasing concentrations (from 0 to 1 μM) of secretin and a constant, saturating amount of 125I-[Tyr10]secretin radioligand diluted in Krebs-Ringers-HEPES (KRH) (25 mM HEPES, pH 7.4, 104 mM NaCl, 5 mM KCl, 1 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2) containing 0.2% (w/v) bovine serum albumin and 0.01% (w/v) soybean trypsin inhibitor. Bound radioactivity was quantified with a γ-spectrometer after washing and lysing cells with KRH and 0.5 M NaOH, respectively. Secretin dissociation constants (Ki values) were calculated with LIGAND software (39), and in all cases data represent the means ± S.E.M. of duplicate assays from at least three independent experiments. Receptors reported as having Ki and percent wild type values of >1000 nM and <5%, respectively, were those for which inadequate evidence for saturable binding was observed, with 1 μM unlabeled secretin displacing less than 70% of bound radioligand.
Fluorescence microscopy
To determine whether each of the mutant secretin receptors could sort to the plasma membrane, COS cells expressing YFP-tagged SecR variants were seeded on UV-sterilized 25-mm round coverslips in 6-well plates. Roughly 48 to 72 h post-transfection, the cells were fixed immediately after removal from the growth incubator with fresh 2% (w/v) formaldehyde in PBS for 15 min at room temperature. Coverslips then were immersed in three 10-min washes in PBS before mounting on microscope slides in 70% (v/v) glycerol in PBS containing 4% (w/v) n-propyl gallate (Sigma) to prevent photobleaching. A Zeiss (Thornwood, NY) LSM 510 confocal microscope was used to excite (514-nm argon laser) and collect (LP530 long-pass filter) YFP emissions from fluorescent cells that represented in routine experiments ∼50−75% of the transfected cell population. Images of single cells and cell groups were collected from photomultiplier tube signals with a Plan-Apochromat 63X/1.4NA oil objective and a pinhole diameter of one airy unit. Cells coexpressing Rlu- and YFP-tagged receptors in the presence of BFA were fixed and mounted 48 h after transfection, as described above. YFP epifluorescence signals from these cells were recorded with a Diagnostic Instruments (Sterling Heights, MI) SPOT 1.3.0 digital camera connected to a Leica (Heidelberg, Germany) DMRB microscope (PL Fluotar 40X/0.7NA objective, G/R filter cube). All micrographs were adjusted for contrast and assembled into figures using Adobe Photoshop 7.0 (Mountain View, CA).
BRET assays
Protein associations were measured in living cells transiently (co)expressing Rlu- and/or YFP-tagged versions of the various SecR mutants created for the present study. COS cells in 162 cm2 culture flasks were lifted 48 h after transfection with a non-enzymatic dissociation solution (Sigma) and then washed twice with KRH containing 0.2% (w/v) bovine serum albumin and 0.01% (w/v) soybean trypsin inhibitor. The cells then were resuspended in KRH to roughly 1 million cells/ml and held at room temperature until analysis. BRET assays were performed by mixing with 1 ml resuspended cells in a quartz cuvette the cell-permeant Renilla luciferase substrate coelenterazine h (5 μM final concentration). Rlu and YFP emissions were recorded immediately with a Jobin Yvon (Edison, NJ) FluoroMax-3 spectrofluorometer set to acquire data from 400 to 600 nm in 2-nm increments with an integration time of 2 s per data point. Acceptor fluorescence was confirmed for all YFP-tagged mutants by direct excitation at 480 nm. BRET ratios (35) were calculated from baseline-corrected peak integrations as follows: [Em510−590–(Em440−500*Cf)]/Em440−500. The correction factor Cf = Em510−590/Em440−500 defined the amount of signal in the acceptor portion of the spectrum that was attributable to donor bioluminescence, and was calculated separately for each experiment from cells expressing the appropriate Rlu-tagged SecR mutant alone.
Morphologic FRET microscopy
The specific subcellular compartment(s) that possessed receptor oligomers were identified by first seeding, culturing, and fixing on coverslips COS cells (co)expressing CFP- and/or YFP-tagged receptors. After three washes in PBS, the cells were mounted in Vectashield (Vector Laboratories, Burlingame, CA) and then observed and photographed with the FRET epifluorescence microscope setup described in (27). Images for the dedicated CFP, YFP, and FRET filter channels were acquired at equivalent exposure times and gain settings that were adjusted for each sample to provide a maximum range of intensity values with a minimum amount of pixel saturation. These acquisition parameters ensured that the subsequent image corrections addressed the spectral limitations of the filter sets and not the saturation limits of the digital camera. FRET images were corrected for bleed-through with intensity-based pixel-by-pixel calculations in MetaMorph 6.3 (Molecular Devices, Sunnyvale, CA) according to the sensitized emission method (27). The acceptor and donor bleed-through coefficients used were A = 0.055 ± 0.004 and B = 0.435 ± 0.008 (means ± S.E.M. for at least eight separate data sets), respectively, and were calculated from background-corrected images of cells expressing CFP- or YFP-tagged receptors alone. Micrographs were assembled into figures without significant contrast adjustments using Adobe Photoshop 7.0.
RESULTS
Creation and functional characterization of domain-specific secretin receptor mutants
Several oligomerization studies have established that the extracellular and intramembranous domains of some receptors may be functionally independent in providing the structural requirements necessary for quaternary assembly (40, 41). Thus, the primary sequence of SecR was divided logically into three domains based upon the known orientation of the membrane-bound receptor, namely an amino-terminal extracellular domain, an intramembranous receptor body domain, and a carboxyl-terminal intracellular domain. Figure 1 shows a schematic representation of how the amino- and carboxyl-terminal domains were truncated in whole or in part, and the receptor body domain was substituted in site-directed fashion, in order to determine whether such mutations were capable of disrupting SecR oligomerization. Complete truncations of the amino and carboxyl termini were made within five amino acids of the extracellular side of TM1 and the intracellular side of TM7, respectively (Figure 1). Partial truncations removed known regions of functional interest, such as the six conserved cysteine residues that are required for forming the ligand binding structure of the amino-terminal domain (38), and the multiple serine and threonine residues that are known phosphorylation sites for receptor regulation within the carboxyl-terminal domain (24). Finally, site-directed mutations within the receptor body domain targeted the glycine residues of a conserved –GxxxG– oligomerization motif within TM7 as alanine substitutions either singly or in tandem (Figure 1).
Each of the seven domain-specific mutations were incorporated into carboxyl-terminal Rlu- or YFP-tagged SecRs that were characterized in intact cells for their ability to bind secretin competitively in the presence of a related secretin radioligand. Table 1 compares the results of these analyses to the binding affinities exhibited by Rlu-, YFP- or CFP-tagged receptors that like wild type, untagged SecR bound secretin with nanomolar affinities (28). None of the tagged, amino-terminally truncated receptor mutants bound ligand at concentrations up to 1 μM secretin. Likewise, alanine substitutions of the –GxxxG– motif glycines G360 or both G356 and G360 created receptor mutants that were unable to bind secretin, although substitution of G356 alone yielded receptors that bound secretin saturably with five to twelve-fold lower affinity than comparably-tagged wild type receptors. Complete truncation of the SecR carboxyl terminus also eliminated secretin binding, whereas removal of only the 33 most carboxyl-terminal residues produced tagged receptors that bound ligand with nanomolar or near nanomolar affinities. It should be noted that the two mutants that exhibited high-affinity secretin binding did so at levels that were one-fourth to one-half of those demonstrated with tagged wild type receptors (Table 1).
Table 1.
Secretin Binding and Plasma Membrane Sorting of Various Human Secretin Receptor Mutants
| Receptor constructs | Secretin binding |
Plasma membrane sortingb | |
|---|---|---|---|
| Ki (nM) | % WTa | ||
| SecR-Rlu | 7.1 ± 2.7 | 100. ± 0.0 | |
| SecR-YFP | 4.9 ± 0.1 | 100. ± 0.0 | +++ |
| SecR-CFP | 5.3 ± 0.7 | 100. ± 0.0 | +++ |
| Δ(1−102)-SecR-Rlu | >1000 | <5 | |
| Δ(1−102)-SecR-YFP | >1000 | <5 | +++ |
| Δ(1−121)-SecR-Rlu | >1000 | <5 | |
| Δ(1−121)-SecR-YFP | >1000 | <5 | − |
| G356A-SecR-Rlu | 90 ± 29 | 29.9 ± 1.4 | |
| G356A-SecR-YFP | 23 ± 6 | 27.1 ± 0.2 | +++ |
| G360A-SecR-Rlu | >1000 | <5 | |
| G360A-SecR-YFP | >1000 | <5 | + |
| G356A-G360A-SecR-Rlu | >1000 | <5 | |
| G356A-G360A-SecR-YFP | >1000 | <5 | + |
| SecR-Δ(376−419)-Rlu | >1000 | <5 | |
| SecR-Δ(376−419)-YFP | >1000 | <5 | − |
| SecR-Δ(387−419)-Rlu | 18 ± 12 | 38.6 ± 3.2 | |
| SecR-Δ(387−419)-YFP | 8.1 ± 3.3 | 56.9 ± 3.7 | +++ |
Percent bound in absence of competitor with respect to corresponding Rlu- or YFP-tagged wild type receptors.
Substantial (+++), partial (+), or no (−) sorting of CFP- or YFP-tagged receptors visualized in COS cells via confocal fluorescence microscopy.
COS cells expressing transiently the YFP-tagged versions of each of the SecR oligomerization mutants were imaged via confocal fluorescence microscopy to determine if the variant receptors had retained the ability to sort to the plasma membrane. Figure 2 shows representative micrographs of individual cells that each possess YFP fluorescence that is distributed differentially among intracellular endoplasmic reticulum and Golgi compartments of the biosynthetic pathway, as well as at the plasma membrane. Receptors having partial truncations of either the amino or carboxyl terminus, or having a G356A substitution within the receptor body's –GxxxG– motif, were observed at the cell surface similar to wild type receptors. The G360A and paired G356A-G360A mutant receptors were localized mostly within intracellular compartments, indicating that these variants sorted to the plasma membrane less efficiently than the related G356A single mutant. In contrast, neither of the fully-truncated amino- or carboxyl-terminal receptor mutants were detected at the cell surface and instead were localized almost exclusively within tubules and/or lamellar sheets of endoplasmic reticulum. Fluorescence signals were absent from control cells that had been transfected with an empty expression vector (Figure 2). Summarized in Table 1, these data demonstrate clearly that the secretin binding defects of all but the Δ(1−102) amino terminus partial truncation mutant were due at least in part by coincident defects in cell surface targeting. However, these data do not preclude the notion that the close membrane proximity of the full amino- and carboxyl-terminal truncations may have resulted in improperly folded receptors that retained an intact, fluorescent YFP domain.
FIGURE 2.
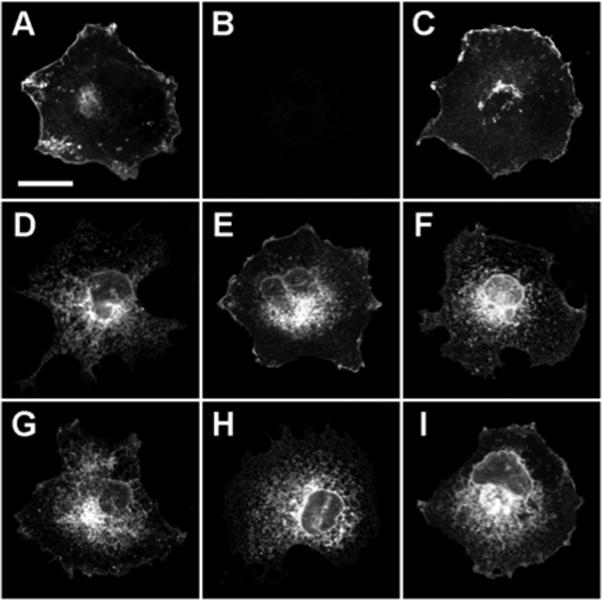
Fluorescence localizations of secretin receptor oligomerization mutants. Representative confocal micrographs (single optical sections) of formaldehyde-fixed COS cells exhibiting plasma membrane and/or intracellular fluorescence attributable to the following YFP-tagged secretin receptors: (A) SecR-YFP, (B) pcDNA3 expression vector, (C) Δ(1−102)-SecR-YFP, (D) Δ(1−121)-SecR-YFP, (E) G356A-SecR-YFP, (F) G360A-SecR-YFP, (G) G356A-G360A-SecR-YFP, (H) SecR-Δ(376−419)-YFP, and (I) SecR-Δ(387−419)-YFP. Bar in panel A = 25 μm.
Bioluminescence resonance energy transfer analyses of domain-specific secretin receptor mutants
Luminescence-stimulated YFP emissions were recorded from cells (co)expressing various combinations of SecR truncation and –GxxxG– mutants to identify which, if any, of these mutations could disrupt the demonstrated ability of secretin receptors to form oligomers. Figure 3A shows the BRET ratios calculated from a series of control experiments in which cells expressing differentially-tagged wild type SecRs yielded typical responses of 0.06 ± 0.01 that were well above the background responses achieved from those expressing Rlu-tagged receptors alone. These values represented about one-sixth of the signal achieved from an Rlu-YFP fusion protein (0.38 ± 0.01), and routinely were at least twice the signal obtained from cells coexpressing an Rlu-tagged SecR and a fully functional, YFP-tagged version of the structurally unrelated family A CCKBR (0.03 ± 0.01) (36). These important controls defined maximum and minimum BRET ratios that represented ideal and non-specific interactions, respectively, and thus the range within which signals such as those from wild type SecRs could be interpreted to indicate specific protein-protein interactions. CCKBR provided an appropriate test for the specificity of the BRET signals because the helical bundles of family A and B receptors are structurally dissimilar and thus may prevent oligomeric interactions (24), and because meaningful interactions between secretin and CCK receptors were not detected in previous BRET and coimmunoprecipitation analyses (26, 35). Our procedures mimicked the optimal transfection protocols established in those studies, which resulted in a roughly consistent ∼1−3 pmol/mg of membrane protein for each membrane-bound secretin and CCK receptor construct (35).
FIGURE 3.

BRET analyses of secretin receptor oligomerization mutants. Each bar indicates the BRET ratio calculated from integrated emissions of the indicated Rlu- and YFP-tagged receptors (co)expressed in COS cells. The four panels describe series of experiments that under controlled conditions (A) evaluated the abilities of amino-terminally truncated (B), receptor body modified (C), or carboxyl-terminally truncated (D) receptor mutants to form homomers with themselves or heteromers with wild type receptors. Horizontal lines group the experiments according to the receptor mutant specified above the line. In panel D, the fourth and fifth bars from the left represent the signals derived from SecR-Δ(376−419)-Rlu/SecR-Δ(376−419)-YFP homomers and SecR-Δ(376−419)-Rlu/SecR-Δ(387−419)-YFP heteromers, respectively, for the Δ(376−419) grouping of carboxyl-terminal truncation mutants. All BRET ratios are expressed as the means ± S.E.M. of at least five observations collected from two or more independent experiments.
As shown in Figure 3B, cells coexpressing Rlu-tagged amino-terminal truncation mutants either with their YFP-tagged counterparts (homomeric conditions) or with YFP-tagged wild type receptors (heteromeric conditions) exhibited BRET ratios that were equal to or greater than those achieved from cells coexpressing differentially-tagged wild type receptors. Homomers of both the Δ(1−102) and the Δ(1−121) mutants exhibited larger ratios than did heteromers between these mutants and wild type receptors, e.g., signals from Δ(1−121) homomers were twice those achieved from Δ(1−121)/wild type heteromers. Figure 3C and 3D show that analogous results were obtained respectively for each of the receptor body and carboxyl-terminal domain mutants. Like the Δ(1−121) mutant, the G360A, paired G356A-G360A and Δ(387−419) mutants displayed BRET ratios as homomers that were twice those detected from the corresponding mutant/wild type heteromers. Interestingly, homomers of the Δ(376−419) carboxyl-terminal truncation mutants yielded the largest BRET signals detected from any receptor construct (Figure 3D). These signals were equal to those obtained from the Rlu-YFP fusion protein, were twice those achieved from Δ(376−419)/Δ(387−419) heteromers, and were at least five times those exhibited by Δ(376−419)/wild type heteromers. Many of these increased BRET signals were significant statistically (data not shown), particularly with respect to the trend of greater signals achieved from mutant homomers. However, their relevance was difficult to establish because the spatial requirements for resonance energy transfer make it impossible to correlate the differences with specific changes in protomer interactions and/or changes in the distances or orientations of the Rlu and YFP tags. Nonetheless, these data were not due to notable differences in expression levels because secretin binding was comparable for either member of a donor/acceptor pair when expressed alone (Table 1). All YFP signals were compared routinely to same-day acquisitions from cells expressing the corresponding Rlu-tagged receptor alone to minimize the potential effects of expression level variations between experiments. Likewise, expression-related differences within an experiment were nullified by normalizing all donor-corrected signals to the total bioluminescence of each sample. In all cases, the resulting BRET ratios fell within the range of signals considered to indicate specific interactions. Cumulatively, these results indicate that residues within the amino terminus, receptor body –GxxxG– motif, and carboxyl terminus are not required for SecR oligomerization, but that certain mutations within these domains can alter the BRET signals detected under various homomeric and heteromeric expression conditions.
Dynamics of secretin receptor oligomerization
The finding that the Δ(1−121) and Δ(376−419) truncation mutants retained the ability to form receptor oligomers despite being localized exclusively within the endoplasmic reticulum and Golgi prompted us to ask whether quaternary interactions could occur intracellularly during receptor maturation. Figure 4 shows the results of experiments in which cells were made to express wild type and/or amino-terminally truncated mutant receptors in the presence of brefeldin A (BFA), a fungal toxin known to inhibit sorting via the secretory pathway by blocking anterograde vesicular traffic from the endoplasmic reticulum. The Δ(1−121) mutant was chosen as a representative example of one of the two non-sorting receptors that were created for the present study. Analogous experiments with the other domain-specific mutants were not performed because those receptor variants behaved similarly to wild type receptors with respect to plasma membrane sorting and oligomerization.
FIGURE 4.
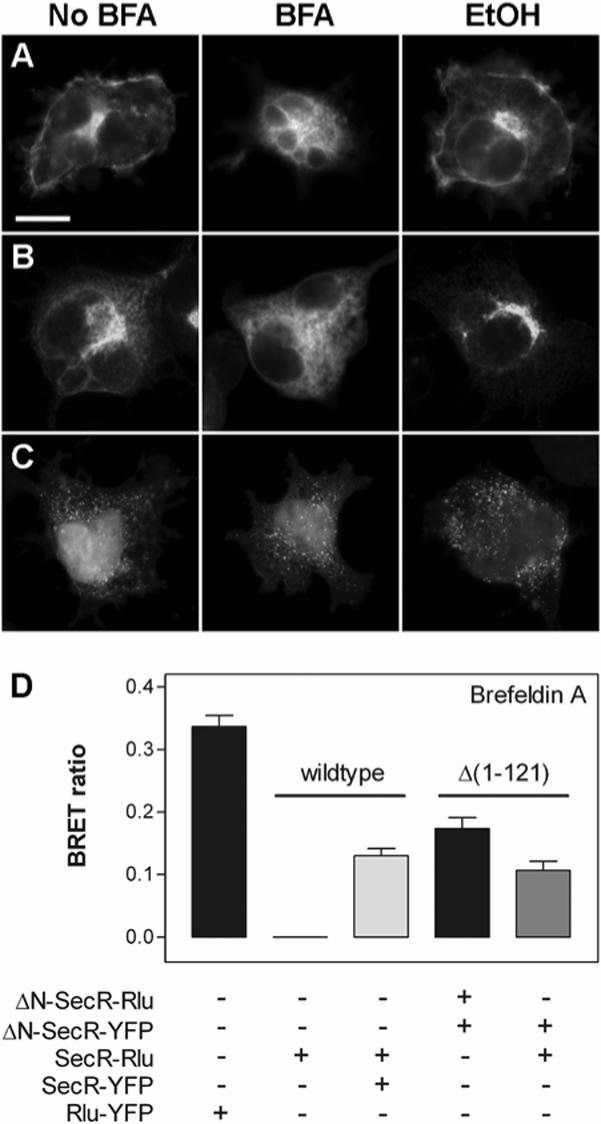
Effects of BFA-induced disruptions of protein sorting on secretin receptor oligomerization. The top panel shows representative epifluorescence micrographs of YFP signals detected in individual COS cells expressing wild type SecR-YFP (A), amino-terminally truncated Δ(1−121)-SecR-YFP (B) or the BRET fusion protein control Rlu-YFP (C) in the absence (left column) or presence (middle column) of BFA, or in the presence of the ethanol diluent used in the BFA experiments (right column). Bar in panel A = 25 μm. The bottom panel (D) shows the BRET ratios calculated from integrated emissions of cells (co)expressing the indicated wild type and Δ(1−121) amino-terminally truncated receptors. Horizontal lines group the experiments according to the abilities of the receptors specified above the line to form homo- or heteromers in the presence of BFA. All BRET ratios are expressed as the means ± S.E.M. from three observations collected from at least two independent experiments.
The COS cells in Figure 4A show that YFP-tagged wild type receptors that typically localized to the plasma membrane instead were found after BFA treatment within diffuse intracellular compartments, namely endoplasmic reticulum, that largely were absent from diluent-only control cells. BFA-treated cells expressing YFP-tagged Δ(1−121) amino-terminal truncation mutants exhibited less dramatic differences in receptor localization, but did show a clear redistribution of fluorescence from the Golgi to the endoplasmic reticulum (Figure 4B). In contrast, BFA treatment had no apparent effect on the cytosolic and nuclear localizations of the Rlu-YFP fusion proteins shown in Figure 4C. We concluded from these epifluorescence results that BFA treatments had the intended effect of blocking protein sorting to the plasma membrane via the biosynthetic secretory pathway, thus causing newly-synthesized wild type or mutant secretin receptors to become trapped within the endoplasmic reticulum.
The data in Figure 4D show in part that BFA-treated cells expressing the Rlu-YFP fusion protein control gave robust BRET signals that resembled the signals detected in untreated cells. This important control indicated that BFA treatments did not have a deleterious effect on the synthesis or stability of Rlu-YFP in the cytosol. Coexpression of Rlu- and YFP-tagged wild type receptors in the presence of BFA also produced BRET responses attributable to receptor homomers that were well above the background responses detected from Rlu-tagged receptors expressed alone. Likewise, amino-terminally truncated receptors were shown to interact after BFA treatments both with themselves as homomers and with oppositely-tagged wild type receptors as heteromers that displayed BRET ratios equal to or greater than wild type homomers (Figure 4D). All of the BFA-induced BRET responses from receptors were similar to analogous responses detected without BFA (Figure 3), indicating that the drug also did not affect the synthesis and/or folding of membrane-bound receptors. Furthermore, these findings demonstrate that potential changes in the local concentration of receptors in the endoplasmic reticulum did not lead to artifactual increases in BRET, probably because the proportion of receptors redirected to and/or impeded from leaving the endoplasmic reticulum was small relative to the large amount typically detected throughout the biosynthetic sorting machinery. In summary, these data demonstrate that plasma membrane trafficking is not required for secretin receptor associations, and may be interpreted to indicate that newly-synthesized receptors oligomerize first in the endoplasmic reticulum during maturation and are trafficked to the cell surface as dimeric or multimeric receptor assemblies.
Subcellular morphologic localizations of secretin receptor oligomers
The observed abilities of secretin receptors to associate intracellularly under conditions that restricted plasma membrane sorting, e.g., amino-terminal truncations and/or drug treatments, prompted us to examine in more detail the subcellular localizations of these complexes. These efforts also were motivated by the concern that the observed BRET results from receptors sequestered within the endoplasmic reticulum could be explained by protein associations that were induced artifactually by overexpression conditions and/or drug treatments. Thus, we employed CFP- and YFP-tagged wild type receptors in morphologic FRET microscopy experiments in an attempt to validate or refute the hypothesis that secretin receptor oligomers are formed during biosynthetic maturation and persist through cell surface sorting. If this is the case, cells coexpressing differentially-tagged receptors would be expected to exhibit under carefully controlled conditions FRET signals that are specific for secretin receptor interactions occurring within at least portions of the endoplasmic reticulum, Golgi and the plasma membrane.
The results in Figure 5 show that cells expressing CFP- (Figure 5A) or YFP-tagged (Figure 5B) secretin receptors alone exhibited respective fluorescence signals attributable to receptors that were localized in organelles of the biosynthetic pathway and the plasma membrane. These distributions matched the localizations of the YFP-tagged receptors shown in Figure 2A and 4A, and together provided the controls necessary for determining levels of background fluorescence and donor and acceptor bleed-through into the FRET channel. The corrected FRET image in Figure 5C depicts under coexpression conditions a representative cell that possessed sensitized YFP fluorescence within the same compartments observed in cells expressing either of the tagged secretin receptors alone. In particular, well-resolved FRET signals were noted separately in tubular endoplasmic reticulum, Golgi clusters and plasma membrane. These signals were absent completely in cells coexpressing CFP-tagged secretin receptor and a YFP-tagged version of the structurally dissimilar CCKBR (Figure 5D), despite the apparent subcellular and plasma membrane colocalizations of these molecules in the separate CFP and YFP channels. It should be noted that although these results represent the distributions of receptors at only a single snapshot in time, the observations of FRET in several subcellular compartments provide convincing evidence for the presence of secretin receptor oligomers throughout the biosynthetic cascade. Moreover, this approach provides an important independent assessment of receptor associations that validates the specificity of oligomerization demonstrated by BRET, and for the first time pinpoints the localization of secretin receptor oligomers within cultured cells.
FIGURE 5.

Morphological FRET analyses of wild type secretin receptor oligomers. Three-channel epifluorescence micrographs of individual COS cells expressing SecR-CFP (A) or SecR-YFP (B), or coexpressing SecR-CFP and SecR-YFP (C) or SecR-CFP and CCKBR-YFP (D). Each panel shows a representative background-corrected image of CFP donor (left column), YFP acceptor (middle column) or bleed-through corrected FRET (right column) emissions that were collected from fluorescence filter sets specific for each signal. Arrowheads denote plasma membrane-localized receptors and receptor oligomers. Bar in panel A = 25 μm.
DISCUSSION
Understanding the structural and functional consequences of GPCR oligomerization clearly holds promise for identifying new ways of manipulating cell functions through physiologically relevant signaling molecules. Our previous observation that a splicing defect in cancer cells may alleviate secretin receptor's normal growth inhibitory effect, and thus promote tumor growth, justified more detailed analyses of the mechanisms of secretin receptor association. Unfortunately, the general lack of precise structural information for most GPCRs, especially family B receptors, has made accumulation of these types of data difficult. Nonetheless, the current report's utilization of two resonance energy transfer methods to monitor associations of rationally mutated receptors provides important contributions to our understanding of both secretin receptor oligomerization and maturation.
The various secretin receptor mutants that were created for the present study addressed the contribution of each receptor domain to oligomerization based upon our current understanding of secretin receptor tertiary structure and function. For example, we found that the Δ(1−102) partial and Δ(1−121) complete amino-terminal truncation mutants were able to form oligomers that were detected by BRET. The former were designed specifically to test whether this process required any of the seven cysteine residues within the distal amino-terminal tail (38), similar to the oligomerization responsibilities of the conserved cysteines of many family C receptors (15). However, our results suggested that amino-terminal residues, including cysteines, were not involved in the constitutive oligomerization of secretin receptors, consistent with the previous finding that a Δ(44−79) splicing defect also did not alter oligomerization (26). The lack of secretin binding by all of the amino-terminal truncation mutants was consistent with the role of this domain for ligand recognition (24), although the binding responses of the Δ(1−121) mutant (and probably the Δ(376−419) carboxyl-terminal truncation mutant) at least in part were the result of its inability to sort to the plasma membrane. The data also support the current 1:1 ligand:receptor model of binding and activation for family B receptors in which the amino terminus of the peptide agonist works together with portions of the receptor's amino-terminal domain to tether the propagation of conformational changes to the TM helices within the receptor core (42, 43). Thus, this model and our results describe a ligand-induced activation mechanism that is independent of potential associations between amino-terminal domains, unlike the single-pass growth hormone receptor in which one ligand is bound by a dimer of two receptor protomers (44, 45). It should be noted, however, that these ideas do not preclude the notion that the protomers of a secretin receptor dimer or multimer may function cooperatively via conformational changes that cross-activate non-bound members of the oligomer.
Similar arguments can be made with respect to the importance of the secretin receptor carboxyl-terminal domain for down-regulation of receptor signaling. In general, desensitization is achieved by phosphorylation and internalization machineries that for secretin receptor have been shown to act in a coordinated, but mutually exclusive, manner (46, 47). These functions are mediated in part by several serine and threonine residues within the distal carboxyl-terminal tail (47); the Δ(387−419) partial carboxyl-terminal truncation was designed specifically to remove these known sites for desensitization. Partial truncation of the carboxyl-terminal domain had only minor effects on secretin binding and plasma membrane sorting, and all of the carboxyl-terminal truncation mutants exhibited BRET signals that were comparable to or better than those of intact receptors. It should be noted that because it was impossible to distinguish changes in tag orientations from actual changes in receptor associations, the observed differences in the BRET signals, particularly for experiments that employed the Δ(376−419) mutant, could not be interpreted as differences in protomer affinity or function. Nonetheless, the data are consistent with ligand binding having no effect on secretin receptor associations (27), and were supported by the finding that the Δ(387−419) partial truncation mutant stimulated adenylate cyclase and internalized similarly to wild type receptor (data not shown). Cumulatively, these findings suggest that the regulatory steps that occur after receptor activation and G protein coupling act upon secretin receptor oligomers rather than dissociated receptor monomers.
The foregoing discussion points to a responsibility of the receptor core domain for providing the contacts necessary for secretin receptor quaternary structure. As a first look at which residues may be involved, we mutated the –GxxxG– pairwise glycine motif in TM7 of the receptor core domain. This motif was the most logical choice because disruptions of a similar motif in yeast α-factor receptor (13) and β2-adrenergic receptor (14) impaired oligomerization of those family A GPCRs. The –GxxxG– motif is conserved in TM7 among secretin, vasoactive intestinal polypeptide and growth hormone-releasing factor receptors from various organisms, but not among all members of family B GPCRs (13, 48). Alanine substitutions for one or both of secretin receptor's –GxxxG– glycine residues led to poor or nonexistant binding responses that could not be explained by misfolding or maturation, because all three mutant receptors sorted to the plasma membrane and formed homomers with themselves or heteromers with wild type receptors. These results differed from those obtained with similar mutants of the α-factor and β2-adrenergic receptors, many of which exhibited biosynthetic defects that resulted in abnormal targeting of the receptors to the cell surface (13, 14). In those studies, the combined sorting and oligomerization defects were interpreted to indicate that quaternary assembly was a pre-requisite for receptor maturation. Overton et al. (13) concluded that the assembly and maturation of α-factor receptor required the –GxxxG– motif because its position on the lipid-exposed face of TM1 favored the helix-helix interactions necessary for oligomerization. Interestingly, in a helical packing model for family B receptor core domains, TM7 is situated such that G356 and G360 of the secretin receptor motif form a portion of the helical face that is directed towards TM2 and buried within the lipid-protected core (48). Although this orientation has not been confirmed experimentally, Gardella et al. (49) has described a functional relationship between specific residues within TM2 and TM7 of the parathyroid hormone receptor. Thus, we conclude that instead of mediating interhelical interactions between protomers, secretin receptor's – GxxxG– motif likely dictates intrahelical packing interactions that stabilize the receptor core domain and/or establish the conformation required for ligand binding and activation.
Cumulatively, the current data indicate that secretin receptor oligomerization occurs through as yet unidentified interactions of core domain residues that happen early during the biosynthetic processes of receptor maturation. In support of the latter conclusion, truncated receptors that did not sort to the plasma membrane were found to form oligomers, even when trapped in the endoplasmic reticulum of cells that had been treated with BFA. Several groups have noted that various family A GPCRs also assemble higher order structures in biosynthetic compartments, thus indicating that receptor oligomerization is required for cell surface targeting (13, 14, 23, 50, 51). Perhaps more interesting are the findings that physiologically-relevant misspliced or mutated receptors cause sorting defects that through heteromerization result in the trapping of wild type receptors intracellularly (30, 31, 50). These interactions create dominant negative phenotypes that serve as important justification for oligomerization's impact on both biosynthetic trafficking and receptor function. However, defects in plasma membrane sorting are not an inherent characteristic of oligomerization-related phenotypes, because both misspliced and wild type secretin receptors sorted to and colocalized on the surfaces of COS and Panc-1 cells (26, 28). Consistent with our previous hypotheses, oligomerization thus provides a structural mechanism for how misspliced secretin receptor inhibits wild type receptor function, perhaps by changing the ability of wild type receptors to compete for G protein coupling at the cell surface (32). In conclusion, these initial assessments, and this study's findings in general, are important contributions to our understanding of the mechanism of secretin receptor association and its implications for receptor function and disease progression.
ACKNOWLEDGMENT
We thank Renee Happs for constructing several SecR mutants and Laura Bruins for managing cell culture activities. Gratitude is also extended to Delia Pinon and Keiko Hosohata for creating and radioiodinating, respectively, the secretin probes used in binding assays. Furthermore, we appreciate greatly the helpful discussions and fluorescence expertise provided by Kaleeckal Harikumar.
Footnotes
This work was supported by grants from the National Institutes of Health (DK46577) and the Fiterman Foundation.
Abbreviations: BFA, brefeldin A; BRET, bioluminescence resonance energy transfer; CCKBR, type B cholecystokinin receptor; CFP, cyan fluorescent protein; FRET, fluorescence resonance energy transfer; GPCR, G protein-coupled receptor; PBS, phosphate-buffered saline; Rlu, Renilla luciferase; SecR, secretin receptor; TM, transmembrane; YFP, yellow fluorescent protein.
REFERENCES
- 1.Gomes I, Jordan BA, Gupta A, Rios C, Trapaidze N, Devi LA. G protein coupled receptor dimerization: implications in modulating receptor function. J. Mol. Med. 2001;79:226–42. doi: 10.1007/s001090100219. [DOI] [PubMed] [Google Scholar]
- 2.Angers S, Salahpour A, Bouvier M. Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu. Rev. Pharmacol. Toxicol. 2002;42:409–35. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- 3.Park PS, Filipek S, Wells JW, Palczewski K. Oligomerization of G protein-coupled receptors: past, present, and future. Biochemistry. 2004;43:15643–56. doi: 10.1021/bi047907k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Terrillon S, Bouvier M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004;5:30–4. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hansen JL, Sheikh SP. Functional consequences of 7TM receptor dimerization. Eur. J. Pharm. Sci. 2004;23:301–17. doi: 10.1016/j.ejps.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Maggio R, Novi F, Scarselli M, Corsini GU. The impact of G-protein-coupled receptor hetero-oligomerization on function and pharmacology. FEBS J. 2005;272:2939–46. doi: 10.1111/j.1742-4658.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- 7.Prinster SC, Hague C, Hall RA. Heterodimerization of G protein-coupled receptors: specificity and functional significance. Pharmacol. Rev. 2005;57:289–98. doi: 10.1124/pr.57.3.1. [DOI] [PubMed] [Google Scholar]
- 8.Devi LA. Heterodimerization of G-protein-coupled receptors: pharmacology, signaling and trafficking. Trends Pharmacol. Sci. 2001;22:532–7. doi: 10.1016/s0165-6147(00)01799-5. [DOI] [PubMed] [Google Scholar]
- 9.George SR, O'Dowd BF, Lee SP. G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat. Rev. Drug Discov. 2002;1:808–20. doi: 10.1038/nrd913. [DOI] [PubMed] [Google Scholar]
- 10.Hebert TE, Moffett S, Morello JP, Loisel TP, Bichet DG, Barret C, Bouvier M. A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J. Biol. Chem. 1996;271:16384–92. doi: 10.1074/jbc.271.27.16384. [DOI] [PubMed] [Google Scholar]
- 11.Hernanz-Falcon P, Rodriguez-Frade JM, Serrano A, Juan D, del Sol A, Soriano SF, Roncal F, Gomez L, Valencia A, Martinez AC, Mellado M. Identification of amino acid residues crucial for chemokine receptor dimerization. Nat. Immunol. 2004;5:216–23. doi: 10.1038/ni1027. [DOI] [PubMed] [Google Scholar]
- 12.Thevenin D, Lazarova T, Roberts MF, Robinson CR. Oligomerization of the fifth transmembrane domain from the adenosine A2A receptor. Protein Sci. 2005;14:2177–86. doi: 10.1110/ps.051409205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Overton MC, Chinault SL, Blumer KJ. Oligomerization, biogenesis, and signaling is promoted by a glycophorin A-like dimerization motif in transmembrane domain 1 of a yeast G protein-coupled receptor. J. Biol. Chem. 2003;278:49369–77. doi: 10.1074/jbc.M308654200. [DOI] [PubMed] [Google Scholar]
- 14.Salahpour A, Angers S, Mercier JF, Lagace M, Marullo S, Bouvier M. Homodimerization of the beta2-adrenergic receptor as a prerequisite for cell surface targeting. J. Biol. Chem. 2004;279:33390–7. doi: 10.1074/jbc.M403363200. [DOI] [PubMed] [Google Scholar]
- 15.Pin JP, Kniazeff J, Liu J, Binet V, Goudet C, Rondard P, Prezeau L. Allosteric functioning of dimeric class C G-protein-coupled receptors. FEBS J. 2005;272:2947–55. doi: 10.1111/j.1742-4658.2005.04728.x. [DOI] [PubMed] [Google Scholar]
- 16.Fan T, Varghese G, Nguyen T, Tse R, O'Dowd BF, George SR. A role for the distal carboxyl tails in generating the novel pharmacology and G protein activation profile of mu and delta opioid receptor hetero-oligomers. J. Biol. Chem. 2005;280:38478–88. doi: 10.1074/jbc.M505644200. [DOI] [PubMed] [Google Scholar]
- 17.Lemmon MA, Treutlein HR, Adams PD, Brunger AT, Engelman DM. A dimerization motif for transmembrane alpha-helices. Nat. Struct. Biol. 1994;1:157–63. doi: 10.1038/nsb0394-157. [DOI] [PubMed] [Google Scholar]
- 18.Kobus FJ, Fleming KG. The GxxxG-containing transmembrane domain of the CCK4 oncogene does not encode preferential self-interactions. Biochemistry. 2005;44:1464–70. doi: 10.1021/bi048076l. [DOI] [PubMed] [Google Scholar]
- 19.Schneider D, Engelman DM. Motifs of two small residues can assist but are not sufficient to mediate transmembrane helix interactions. J. Mol. Biol. 2004;343:799–804. doi: 10.1016/j.jmb.2004.08.083. [DOI] [PubMed] [Google Scholar]
- 20.Filizola M, Weinstein H. The study of G-protein coupled receptor oligomerization with computational modeling and bioinformatics. FEBS J. 2005;272:2926–38. doi: 10.1111/j.1742-4658.2005.04730.x. [DOI] [PubMed] [Google Scholar]
- 21.Arluison V, Seguin J, Le Caer JP, Sturgis JN, Robert B. Hydrophobic pockets at the membrane interface: an original mechanism for membrane protein interactions. Biochemistry. 2004;43:1276–82. doi: 10.1021/bi030205v. [DOI] [PubMed] [Google Scholar]
- 22.Bulenger S, Marullo S, Bouvier M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol. Sci. 2005;26:131–7. doi: 10.1016/j.tips.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Wilson S, Wilkinson G, Milligan G. The CXCR1 and CXCR2 receptors form constitutive homo- and heterodimers selectively and with equal apparent affinities. J. Biol. Chem. 2005;280:28663–74. doi: 10.1074/jbc.M413475200. [DOI] [PubMed] [Google Scholar]
- 24.Dong M, Miller LJ. Molecular pharmacology of the secretin receptor. Receptors Channels. 2002;8:189–200. [PubMed] [Google Scholar]
- 25.Chey WY, Chang TM. Secretin, 100 years later. J. Gastroenterol. 2003;38:1025–35. doi: 10.1007/s00535-003-1235-3. [DOI] [PubMed] [Google Scholar]
- 26.Ding WQ, Cheng ZJ, McElhiney J, Kuntz SM, Miller LJ. Silencing of secretin receptor function by dimerization with a misspliced variant secretin receptor in ductal pancreatic adenocarcinoma. Cancer Res. 2002;62:5223–9. [PubMed] [Google Scholar]
- 27.Harikumar KG, Morfis MM, Lisenbee CS, Sexton PM, Miller LJ. Constitutive Formation of Oligomeric Complexes between Family B G Protein-Coupled Vasoactive Intestinal Polypeptide and Secretin Receptors. Mol. Pharmacol. 2006;69:363–73. doi: 10.1124/mol.105.015776. [DOI] [PubMed] [Google Scholar]
- 28.Ding WQ, Kuntz S, Bohmig M, Wiedenmann B, Miller LJ. Dominant negative action of an abnormal secretin receptor arising from mRNA missplicing in a gastrinoma. Gastroenterology. 2002;122:500–11. doi: 10.1053/gast.2002.31039. [DOI] [PubMed] [Google Scholar]
- 29.Herrick-Davis K, Grinde E, Harrigan TJ, Mazurkiewicz JE. Inhibition of serotonin 5-hydroxytryptamine2c receptor function through heterodimerization: receptor dimers bind two molecules of ligand and one G-protein. J. Biol. Chem. 2005;280:40144–51. doi: 10.1074/jbc.M507396200. [DOI] [PubMed] [Google Scholar]
- 30.Lee SP, O'Dowd BF, Ng GY, Varghese G, Akil H, Mansour A, Nguyen T, George SR. Inhibition of cell surface expression by mutant receptors demonstrates that D2 dopamine receptors exist as oligomers in the cell. Mol. Pharmacol. 2000;58:120–8. doi: 10.1124/mol.58.1.120. [DOI] [PubMed] [Google Scholar]
- 31.Zhou F, Filipeanu CM, Duvernay MT, Wu G. Cell-surface targeting of alpha(2)-adrenergic receptors - Inhibition by a transport deficient mutant through dimerization. Cell. Signal. 2006;18:318–27. doi: 10.1016/j.cellsig.2005.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dosil M, Giot L, Davis C, Konopka JB. Dominant-negative mutations in the G-protein-coupled alpha-factor receptor map to the extracellular ends of the transmembrane segments. Mol. Cell. Biol. 1998;18:5981–91. doi: 10.1128/mcb.18.10.5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Powers SP, Pinon DI, Miller LJ. Use of N,O-bis-Fmoc-D-Tyr-ONSu for introduction of an oxidative iodination site into cholecystokinin family peptides. Int. J. Pept. Protein Res. 1988;31:429–34. doi: 10.1111/j.1399-3011.1988.tb00899.x. [DOI] [PubMed] [Google Scholar]
- 34.Dong M, Asmann YW, Zang M, Pinon DI, Miller LJ. Identification of two pairs of spatially approximated residues within the carboxyl terminus of secretin and its receptor. J. Biol. Chem. 2000;275:26032–9. doi: 10.1074/jbc.M000612200. [DOI] [PubMed] [Google Scholar]
- 35.Cheng ZJ, Miller LJ. Agonist-dependent dissociation of oligomeric complexes of G protein-coupled cholecystokinin receptors demonstrated in living cells using bioluminescence resonance energy transfer. J. Biol. Chem. 2001;276:48040–7. doi: 10.1074/jbc.M105668200. [DOI] [PubMed] [Google Scholar]
- 36.Cheng ZJ, Harikumar KG, Holicky EL, Miller LJ. Heterodimerization of type A and B cholecystokinin receptors enhance signaling and promote cell growth. J. Biol. Chem. 2003;278:52972–9. doi: 10.1074/jbc.M310090200. [DOI] [PubMed] [Google Scholar]
- 37.Holtmann MH, Hadac EM, Miller LJ. Critical contributions of amino-terminal extracellular domains in agonist binding and activation of secretin and vasoactive intestinal polypeptide receptors. Studies of chimeric receptors. J. Biol. Chem. 1995;270:14394–8. doi: 10.1074/jbc.270.24.14394. [DOI] [PubMed] [Google Scholar]
- 38.Lisenbee CS, Dong M, Miller LJ. Paired cysteine mutagenesis to establish the pattern of disulfide bonds in the functional intact secretin receptor. J. Biol. Chem. 2005;280:12330–8. doi: 10.1074/jbc.M414016200. [DOI] [PubMed] [Google Scholar]
- 39.Munson PJ, Rodbard D. Ligand: a versatile computerized approach for characterization of ligand-binding systems. Anal. Biochem. 1980;107:220–39. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- 40.Milligan G, Ramsay D, Pascal G, Carrillo JJ. GPCR dimerisation. Life Sci. 2003;74:181–8. doi: 10.1016/j.lfs.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 41.Giese B, Roderburg C, Sommerauer M, Wortmann SB, Metz S, Heinrich PC, Muller-Newen G. Dimerization of the cytokine receptors gp130 and LIFR analysed in single cells. J. Cell Sci. 2005;118:5129–40. doi: 10.1242/jcs.02628. [DOI] [PubMed] [Google Scholar]
- 42.Dong M, Li Z, Pinon DI, Lybrand TP, Miller LJ. Spatial approximation between the amino terminus of a peptide agonist and the top of the sixth transmembrane segment of the secretin receptor. J. Biol. Chem. 2004;279:2894–903. doi: 10.1074/jbc.M310407200. [DOI] [PubMed] [Google Scholar]
- 43.Grace CR, Perrin MH, DiGruccio MR, Miller CL, Rivier JE, Vale WW, Riek R. NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc. Natl. Acad. Sci. U. S. A. 2004;101:12836–41. doi: 10.1073/pnas.0404702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Vos AM, Ultsch M, Kossiakoff AA. Human growth hormone and extracellular domain of its receptor: crystal structure of the complex. Science. 1992;255:306–12. doi: 10.1126/science.1549776. [DOI] [PubMed] [Google Scholar]
- 45.Cunningham BC, Ultsch M, De Vos AM, Mulkerrin MG, Clauser KR, Wells JA. Dimerization of the extracellular domain of the human growth hormone receptor by a single hormone molecule. Science. 1991;254:821–5. doi: 10.1126/science.1948064. [DOI] [PubMed] [Google Scholar]
- 46.Holtmann MH, Roettger BF, Pinon DI, Miller LJ. Role of receptor phosphorylation in desensitization and internalization of the secretin receptor. J. Biol. Chem. 1996;271:23566–71. doi: 10.1074/jbc.271.38.23566. [DOI] [PubMed] [Google Scholar]
- 47.Ozcelebi F, Holtmann MH, Rentsch RU, Rao R, Miller LJ. Agonist-stimulated phosphorylation of the carboxyl-terminal tail of the secretin receptor. Mol. Pharmacol. 1995;48:818–24. [PubMed] [Google Scholar]
- 48.Donnelly D. The arrangement of the transmembrane helices in the secretin receptor family of G-protein-coupled receptors. FEBS Lett. 1997;409:431–6. doi: 10.1016/s0014-5793(97)00546-2. [DOI] [PubMed] [Google Scholar]
- 49.Gardella TJ, Luck MD, Fan MH, Lee C. Transmembrane residues of the parathyroid hormone (PTH)/PTH-related peptide receptor that specifically affect binding and signaling by agonist ligands. J. Biol. Chem. 1996;271:12820–5. doi: 10.1074/jbc.271.22.12820. [DOI] [PubMed] [Google Scholar]
- 50.Calebiro D, de Filippis T, Lucchi S, Covino C, Panigone S, Beck-Peccoz P, Dunlap D, Persani L. Intracellular entrapment of wild-type TSH receptor by oligomerization with mutants linked to dominant TSH resistance. Hum. Mol. Genet. 2005;14:2991–3002. doi: 10.1093/hmg/ddi329. [DOI] [PubMed] [Google Scholar]
- 51.Terrillon S, Durroux T, Mouillac B, Breit A, Ayoub MA, Taulan M, Jockers R, Barberis C, Bouvier M. Oxytocin and vasopressin V1a and V2 receptors form constitutive homo- and heterodimers during biosynthesis. Mol. Endocrinol. 2003;17:677–91. doi: 10.1210/me.2002-0222. [DOI] [PubMed] [Google Scholar]