

Abstract
A novel approach to the intracellular delivery of aryl phosphates has been developed that utilizes a phosphoramidate-based prodrug approach. The prodrugs contain an ester group that undergoes reductive activation intracellularly with concomitant expulsion of a phosphoramidate anion. This anion undergoes intramolecular cyclization and hydrolysis to generate aryl phosphate exclusively with a t1/2 = ∼ 20 min. Phosphoramidate prodrugs (8-10) of phosphate-containing peptidomimetics that target the SH2 domain were synthesized. Evaluation of these peptidomimetic prodrugs in a growth inhibition assay and, in a cell-based transcriptional assay, demonstrated that the prodrugs had IC50 values in the low micromolar range. Synthesis of phosphorodiamidate analogs containing a P-NH-Ar linker (16 – 18) was also carried out in the hope that the phosphoramidates released might be phosphatase-resistant. Comparable activation rates and cell-based activities were observed for these prodrugs, but the intermediate phosphoramidate dianion underwent spontaneous hydrolysis with a t1/2 = ∼ 30 min.
Introduction
Protein tyrosine phosphorylation is a common intracellular signaling event that controls numerous signaling pathways. Specific sequences, contained within a protein that undergoes phosphorylation on tyrosine, can serve as recognition sites for other proteins. Src homology 2 (SH2) domains mediate these protein-protein interactions. SH2 domains are non-catalytic motifs of about 100 amino acids that are contained in a variety of proteins. 1,2 The ability of these SH2 domains to bind to phosphotyrosine-containing sequences is governed by the immediately adjacent C-terminal sequence to the phosphotyrosine residue. In particular, it has been found that the SH2 domains of the cytoplasmic tyrosine kinases Src and Lck bind with micromolar affinity to the peptide sequence pY-E-E-I. 3,4 Src and Lck have been implicated in a variety of disorders that include breast cancer, colon cancer, autoimmune diseases and osteoporosis, and therefore constitute good targets to develop new therapeutic agents. 5-8
Several peptidomimetics that target the SH2 domain of Lck have been developed. 9-11 However, the presence of a highly ionizable phosphate group in these compounds compromises their cell membrane permeability. To overcome this problem, several less anionic phosphotyrosine isosteres have been developed and included in the design of SH2 domain inhibitors. 12-14 Another approach that has been used is the generation of phosphotyrosine prodrugs in the form of phosphoesters that are simply activated by enzymatic hydrolysis. 15 However, due to the ubiquitous presence of esterases, this type of approach cannot be considered selective. We have developed previously a series of cell membrane permeable nucleoside phosphoramidate prodrugs that deliver nucleotides intracellularly. 16-18 These phosphoramidates are composed of a phosphoester delivery group and a masking phosphoramide group. The release of the active nucleotide is triggered by the enzymatic transformation and elimination of the delivery group, which is followed by a spontaneous intramolecular cyclization and hydrolysis. Herein we report the application of this prodrug technology to the development of aryl phosphoramidates and apply it to the generation of cell membrane permeable phosphotyrosine mimetic prodrugs.
Results and Discussion
a) Model compounds
In an effort to simplify the reaction and experimental conditions, initial studies were carried out on model p-cresol phosphoramidates. Two different phosphoramidates were synthesized (Scheme 1). Phosphoramidate 1 contained a 2-bromoethyl masking group19 and a benzyl ester as a conditional activating group. 20 Phosphoramidate 2 contained an extended 4-bromobutyl masking group and a nitrofurfuryl ester as an activating group. 17 The syntheses of phosphoramidates 1 and 2 are illustrated in Scheme 1. Phosphorus oxychloride (POCl3) was consecutively reacted with p-cresol and benzyl alcohol in the presence of triethylamine followed by reaction with N-methyl-N-(2-bromoethylamine) hydrobromide to generate phosphoramidate 1. For the synthesis of phosphoramidate 2, POCl3 was reacted with N-methyl-N-(4-bromobutylamine) hydrobromide in the presence of diisopropylethylamine to generate the phosphoramidic chloride which was then consecutively reacted with the lithium alkoxides of p-cresol and nitrofurfuryl alcohol.
Scheme 1a.

aReagents: a) p-cresol, Et3N, CH2Cl2, -10 ° - 0 °C, 30 min; b) benzyl alcohol, Et3N, 0 °C, 1h; c) N-methyl-N-(2-bromo-ethyl)amine hydrobromide, Et3N, THF, 20 min, rt;d) N-methyl-N-(4-bromobutyl)amine hydrobromide, i-Pr2NEt, CH2Cl2, -20 °C to rt, 4 hr; e) p-cresol, LiHMDS, THF; f) nitrofurfuryl alcohol, LiHMDS, THF
Based on results obtained from the study of nucleotide phosphoramidate prodrugs, it was expected that the aryl phosphoramidates would behave in a similar manner, i.e., following delivery group activation and elimination, a phosphoramidate anion A would be formed (Scheme 2). The inductive effect of the oxygen anion would drive the intramolecular cyclization to yield the formation of the reactive intermediate B. This intermediate could react with water following pathway a and/or b to generate solvolysis product C and/or the hydrolysis product D, respectively.
Scheme 2.

To test this hypothesis, phosphoramidate 1 was hydrogenolyzed and its subsequent transformation was monitored by 31P NMR. The analysis showed that when n = 2 the phosphoramidate anion A disappeared with a half-life of 60 min (0.4 M cacodylate buffer, pH ∼ 7.4, 22 °C), and that the desired aryl phosphate D represented approximately 25% of the product. Attack of water at the carbon of the aziridinium ring clearly predominates in this system. In contrast, phosphoramidate A (n = 4) (generated by reduction of 2 with sodium dithionite) was converted to the aryl monophosphate D quantitatively with a half-life of < 3 min (0.4 M cacodylate buffer, pH ∼ 7.4, 22 °C). It is interesting to note that cyclization to the 5-membered ring is much faster than to the 3-membered ring in this phosphoramidate.
b) Peptidomimetic prodrugs
The results obtained from the model compounds prompted the application of this strategy to the synthesis of phosphotyrosine peptidomimetic prodrugs. Peptidomimetics targeted to the SH2 domain were selected to demonstrate the feasibility of the approach. However, it was discovered subsequently that the bromobutyl compound 2 was unstable during storage and appeared to decompose via a reaction initiated by intramolecular cyclization. In an attempt to enhance the stability of the phosphoramidate esters while retaining facile cyclization of the phosphoramidate anion intermediate, the bromine in the masking group was replaced with chlorine. Compound 11 (Scheme 4), which has low micromolar binding affinity to pp60src, 21 was selected as the initial peptidomimetic for the prodrug approach (e.g., 8). By analogy, two additional phosphoramidate prodrugs 9 and 10 that differ in the spacer between the P and P+3 sites were synthesized (Scheme 4). To accomplish the synthesis of phosphoramidates 8-10, a convergent approach was used involving preparation of two building blocks: 1) a carboxy-containing aryl phosphoramidate and 2) a benzylamine. The synthesis of the carboxy-containing building blocks is illustrated in Scheme 3. Reaction of POCl3 and N-methyl-N-(4-chlorobutylamine) hydrochloride produced phosphoramidic dichloride 3, which could be isolated and purified. We have shown previously that allyl esters can be used to protect carboxylic acids in the presence of phosphoramidates, 22 so allyl esters 6a-c were prepared by refluxing the respective free acids in allyl bromide in the presence of diisopropylethylamine. 23 These allyl esters were then reacted with 3 in the presence of diisopropylethylamine to generate phosphoramidic monochlorides 4a-c, respectively. Addition of pre-formed nitrofurfuryl lithium alkoxide to 4a-c generated the corresponding phosphoramidates which were de-allylated to give the carboxylic acids 5a-c. These aryl phosphoramidates were then coupled to benzylamine 7 (synthesized according to the published procedure) 21 in the presence of PyBOP and diisopropylethylamine to generate phosphoramidate prodrugs 8-10 (Scheme 4).
Scheme 4.

Scheme 3a.

aReagents: a) N-methyl-4-chlorobutylamine hydrochloride, i-Pr2NEt, CH2Cl2, 0 °C - rt, 4h; b) 6a-c, i-Pr2NEt, CH2Cl2; c) LiHMDS, nitrofurfuryl alcohol, THF, -30 °C, 5h; d) Pd(PPh3)4, TolSO2Na, THF/H2O.
To verify that the free phosphates could be released from their corresponding prodrugs, phosphoramidate 8 was subjected to reductive activation using sodium dithionite, the residue was dissolved in 0.4 M cacodylate buffer, final pH 7.2 and its transformation studied by 31P NMR kinetics. The phosphoramidate anion derived from 8 generated phosphate 11 with a half-life of 20 minutes at 37°C and 2.5 h at room temperature (Scheme 4).
c) Phosphorodiamidates
Because both phosphate ionization and stability to phosphatase degradation can be modulated by replacement of the linking oxygen in phosphotyrosine mimetics, 24,25 the replacement of oxygen by NH was explored in these peptidomimetics. p-Toluidine phosphate 12b (Scheme 5) was generated in situ from basic hydrolysis of phosphoramidic dichloride 12a and explored as a simple model. The pKa2 for this compound was determined using the pH-dependent chemical shift change of the 31P NMR resonance and found to be 6.9 (Figure 1). The overall 31P NMR chemical shift change of about 1.25 ppm suggests that protonation of 12b occurs on oxygen rather than nitrogen. 26,27
Scheme 5a.

aReagents: a) POCl3, i-Pr2NEt, CH2Cl2, 0 °C - rt; b) Na2CO3 or NaOH, CH3CN/H2O; c) Allyl bromide, DMF; d) Allyl alcohol, cat. H2SO4, reflux; e) Ac2O, NaOH, H2O/acetone; f) H2, Pd/C.
Figure 1.
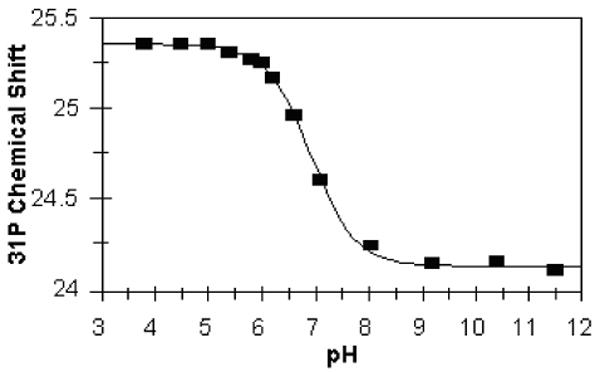
Chemical shift titration curve of p-toluidine phosphoramidate 12b
Synthesis of the analogous phosphorodiamidate peptidomimetic prodrugs 16 – 18 is outlined in Schemes 5 and 6. Allyl ester 13a was prepared by reaction of p-aminobenzoic acid potassium salt with allyl bromide in DMF. Attempts to prepare 13b and 13c28 under similar conditions, however, resulted in allylation of the amino group. These compounds were prepared successfully using Fischer esterification. Phosphorylation of 13a was slow and generated 14a in poor yield even after reflux with phosphoramidic dichloride 3 for 10 days. Intermediates 14b and 14c were prepared in modest yield using a two-step procedure in which 13b or 13c was reacted with POCl3 followed by reaction with N-methyl-N-(4-chlorobutylamine) hydrochloride. Addition of 5-nitrofurfuryl lithium alkoxide to the three different phosphorodiamidic chlorides 14a-c generated the corresponding phosphorodiamidates which were cleaved to the corresponding acids 15a-c by treatment with p-TolSo2Na and Pd(PPh3). Finally, the peptidomimetics 16 - 18 were prepared by coupling 15a-c with benzylamine 7 in the presence of PyBOP and diisopropylethylamine.
Scheme 6a.

aReagents: a) 3, i-Pr2NEt, (ClCH2)2, reflux; b) POCl3, i-Pr2NEt, CH2Cl2, -5 °C; c) N-methyl-4-chlorobutylamine hydrochloride, i-Pr2NEt, CH2Cl2, -5 °C; d) LiHMDS, nitrofuryl alcohol, THF, -20 °C; e) Pd(PPh3)4, TolSO2Na, THF/H2O; f) PyBOP, i-Pr2NEt, CH2Cl2.
In order to demonstrate that the phosphorodiamidates would undergo activation and release of the analogous phosphoramidate dianion, prodrug 16 was activated by sodium dithionite reduction and the subsequent reactions (Scheme 7) studied by 31P NMR; the kinetic profile is shown in Figure 2. Although conversion of the phosphorodiamidate 19 (round symbols) to the phosphate analog 20 (square symbols) is clearly evident at early time points, this product is unstable and proceeds to hydrolyze to the corresponding aniline 21 and inorganic phosphate (triangle symbols). Kinetic analysis (0.4M cacodylate buffer, pH 7.2, 37 °C) gives rate constants k1 = 0.052 and k2 = 0.022 min-1, corresponding to half-lives of 13 min and 32 min, respectively. The pH dependence of the phosphoramide hydrolysis reaction was investigated using p-toluidine phosphate 12b; the pH – rate profile (Figure 3) clearly demonstrates that this is an acid-catalyzed reaction, and that the rate of hydrolysis is near half-maximal at physiologic pH.
Scheme 7.

Figure 2.
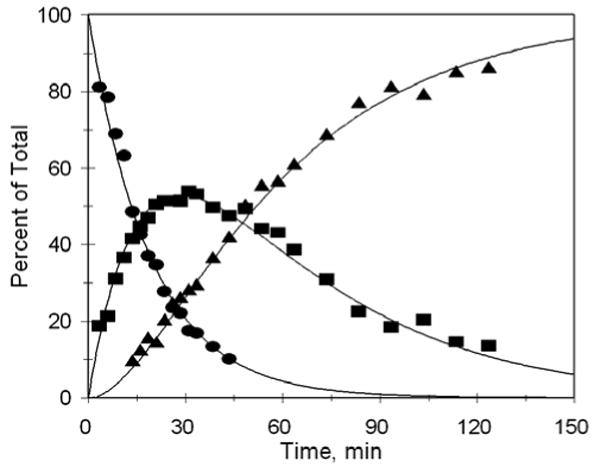
Formation and hydrolysis of NH-phosphate from phosphoramidate 19 at 37 °C, pH = 7.2. (●), phosphoramidate 19; (■), NH-phosphate 20; (▲), inorganic phosphate.
Figure 3.
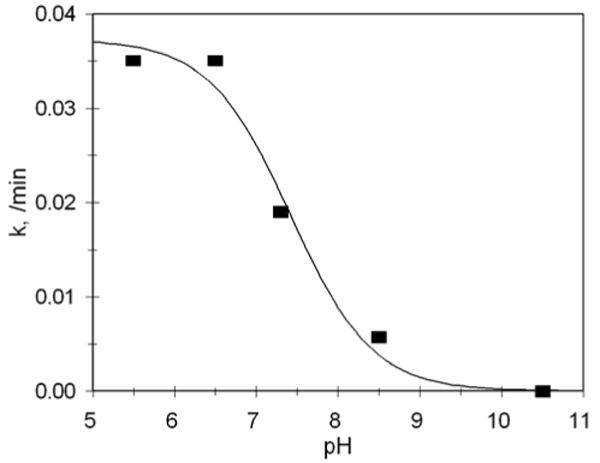
pH – rate profile for the hydrolysis of phosphoramidate 12b, 37 °C
Biological Evaluation
Although the chemical activation and 31P analysis had provided insight into the ability of these prodrugs to generate aryl phosphates, there was the need to evaluate the feasibility of this approach in cell-based systems. Therefore, the phosphoramidate prodrugs 8-10 and 16-18 were evaluated in a cell-based assay that consists of a human leukemia Jurkat J77 T cell line transiently transfected with a luciferase reporter gene driven by four adjacent binding sites for the NFAT (nuclear factor of activated T cells) transcription factor. The activation of NFAT occurs in response to the mobilization of calcium following crosslinking of the T cell antigen receptor and is dependent on the expression of Lck with a functional SH2 domain. 8,29 Thus, inhibitors directed against the SH2 domain of Lck block both the mobilization of calcium and the subsequent activation of gene transcription. 12,30
Transiently transfected Jurkat cells were incubated with phosphoramidates 8-10 and 16-18 for 1 h at concentrations from 0.1-1000 μM and then activated with a combination of anti-CD3 antibody and PMA. At the end of the 6-h incubation period the cells were lysed, supernatant was collected and the luciferase activity was measured. The Src family-selective tyrosine kinase inhibitor PP231 was also studied as a positive control; it showed an IC50 = 0.9 μM. The growth inhibitory activity of the compounds was also evaluated in the NCI tumor cell panel. The results from the cell-based assays are shown in Table 1. The prodrugs showed modest and similar activity in both the luciferase and growth inhibition assays. The cell-based IC50 values for prodrug 8 are in the same range as the published binding affinity (6 μM) for the free phosphate 11, 21 consistent with intracellular prodrug activation and phosphate release. These results are also consistent with the targeting of phosphate 11 to the Lck SH2 domain, but it is also possible that binding of this phosphate to other sites could contribute to the reduced levels of luciferase expression. Although it was anticipated that the difference in length of the spacer between the P and P+3 groups would affect the activity of the prodrugs, it appears that all three structures can be accommodated at the binding site. It is surprising that the activities of prodrugs 16 – 18 are comparable to those of 8 – 10, considering that the phosphorodiamidates released from the former compounds are hydrolytically unstable. However, it is likely that the phosphoramidates released from 8 – 10 will be substrates for intracellular phosphatases, so the intracellular lifetimes of the enzymatically labile phosphoramidates and hydrolytically labile phosphorodiamidates may in fact be comparable.
Table 1.
Response of Jurkat cells and the NCI tumor cell panel to prodrugs. NFAT assay, cells exposed to drug for 6 hr; NCI panel, cells exposed to drug for 48 hr. See Experimental section for details.
| Cpd | NFAT IC50, μMa | NCI Panel GI50, μMb |
|---|---|---|
| 8 | 31 ± 3 | 12 ± 7 |
| 9 | 9 ± 5 | 3 ± 1 |
| 10 | 21 ± 4 | 6 ± 5 |
| 16 | 16 ± 5 | 18 ± 10 |
| 17 | 20 ± 4 | N.D.c |
| 18 | 28 ± 3 | N.D. |
Mean ± S.E., n = 4.
Mean ± S.D. for 62 cell lines in the panel.
Not determined.
Conclusions
A novel approach for the intracellular delivery of aryl phosphate peptidomimetics has been developed. This approach consists of cell membrane permeable phosphoramidate prodrugs that undergo intracellular enzymatic activation followed by intramolecular cyclization and hydrolysis to generate the dianionic phosphate. Although the half-life for activation (cyclization) of the phosphoramidate is slower than that for aliphatic phosphoramidate prodrugs (20 min vs. < 5 min, respectively), the slower activation rate is still adequate for the generation of a biological response. Attempts to utilize a phosphorodiamidate (by replacing the P(O)-O-Ar bond with P(O)-NH-Ar) that generates a presumed phosphatase-resistant ligand resulted in a prodrug of comparable activity, although NMR studies showed that the phosphoramide dianion undergoes spontaneous hydrolysis with a half-life of ∼ 30 min. Efforts to extend this work to phosphatase-resistant difluoromethylphosphonate analogs are underway and will be reported in due course.
Experimental Section
Materials and Methods
NMR spectra were recorded using a 250 MHz Bruker spectrometer equipped with a 5-mm multinuclear probe. 1H chemical shifts are reported in parts per million using tetramethylsilane as an internal reference. 31P NMR spectra were obtained using broadband 1H decoupling, and chemical shifts are reported in parts per million using 1% triphenylphosphine oxide in benzene-d6 as a coaxial insert. 31P NMR kinetics carried out at 37 °C were conducted using a Bruker variable temperature unit. Silica gel grade 60 was used to carry out all chromatographic purifications. HPLC analysis was done using a Beckman System Gold equipped with a 168 detector set to 250 nm, a 126 solvent module and an econosphere C18 column (5 μM, 4×250mm) from Alltech Associates. Mass spectral analyses were obtained from the Mass Spectrometry Laboratory at Purdue University, West Lafayette, IN. All anhydrous reactions were carried out under argon, using flamed dried flasks, and all organic solvents were distilled prior to use.
31P NMR Studies
Kinetics experiments were carried out as described previously.17 Briefly, the compound (∼20 mg) was dissolved in acetonitrile (80 μL); a solution of the activating agent (sodium dithionite; 3 eq) in cacodylate buffer (500 μL, 0.4 M, pH=7.4) was added. The reaction mixture was transferred to a 5-mm NMR tube and data acquisition was started with the probe maintained at 37 °C. Spectra were acquired every 2.5 min for 30 min and when necessary every 10 min for additional time. Time points were assigned to each data acquisition from the start of the reaction. The integration of the peak areas was used to determine the relative concentration of the intermediates and products.
Kinetic Analysis
The methodology has been described. 20 The 31P peak areas were measured, and the product composition was determined at each time point as a percentage of the total material. For simple first order reactions, the natural log of the peak area vs. time was analyzed by linear regression and the rate constant obtained from the slope of the line. For multi-step reactions, rate expressions were derived for each reaction, and the rate constants were determined by minimization of the least-squares difference between observed and calculated product composition at each time point using the Quattro Pro optimization routine.
Luciferase Assay
J77 Jurkat cells (2 × 107 cells in 500 μL of FBS-free RPMI media) were transiently transfected with 15 μg of NF-AT-luciferase plasmid by electroporation (300 V, 800 μF), transferred to a flask with 10 mL of FBS-containing RPMI media and incubated for 24 h. Cells were then harvested, resuspended in 12 mL of FBS-containing RPMI media and divided into a 6-well plate (2 mL/well). Drug stock solutions were prepared in DMSO, and each well was treated with a different concentration and incubated for 1 h. Each well was then treated with 10 μL of a solution of 1.25 μg of PMA (Calbiochem) in 115 μL of serum-free RPMI media, followed by the addition of 2 μg/well of anti-CD3 antibody (Pharmingen). The cells were incubated for 6 h, spun down, washed with PBS, lysed (1× Promega lysis buffer) for 15 min and centrifuged. The supernatant was collected and stored at −78 °C overnight. Luciferase activity was measured, using a Lumat LB 9501 luminometer, from a mixture of 50 μL of luciferase substrate (Promega) and 10 μL of supernatant. The data obtained were analyzed by sigmoidal curve fit of relative luciferase units (RLU) vs log of drug concentration. The results are expressed as the drug concentration that generates a 50% decrease in RLU (IC50) of the control value (DMSO).
NCI Human Tumor Cell Line Screen
Details of the methodology are described at http://dtp.nci.nih.gov/branches/btb/ivclsp.html. Briefly, cells are grown in supplemented RPMI 1640 medium for 24 h, then incubated with drug for 48 h at 5 concentrations from 10-8 to 10-4 M. The assay is terminated by addition of cold trichloroacetic acid, and the cells are fixed and stained with sulforhodamine B. Bound stain is solubilized and the absorbance is read on an automated plate reader. Percentage growth inhibition is calculated from time zero, control growth, and the 5 concentration level absorbances.
N-Methyl-N-(4-bromobutyl)amine hydrobromide
Hydrobromic acid (20 mL, 48% by wt) was added slowly with stirring to N-methyl-N-4-hydroxybutylamine (4.0 g, 0.39 mmol) at 0 °C, and the reaction mixture was heated to reflux for 2 h. A distillation apparatus was attached, 10 mL of distillate was collected, and an additional 10 mL of 48% HBr was added. After refluxing for 4 h, the procedure was repeated and the reaction mixture was refluxed overnight. Approximately 20 mL of distillate was removed from the reaction mixture and the still pot residue was poured into acetone that was cooled to -78 °C. A white precipitate formed that was collected by filtration (4.24 g, 42%), mp 86-88 °C. 1H NMR (D2O): δ 2.84 (t, 2H), 2.39 (t, 2H), 2.04 (s, 3H), 1.23 (m, 4H).
N-Methyl-N-(2-bromoethyl) O-benzyl O-(4-methyl)phenyl phosphoramidate (1). Triethylamine (0.37 mL, 2.6 mmol) was diluted in dry CH2CL2 (1 mL) and added slowly to a pre-cooled solution of p-cresol (0.25 mL, 2.4 mmol) and POCl3 (0.22 mL, 2.4 mmol) in dry CH2CL2 (6 mL) at −10 °C. The reaction mixture was stirred and warmed to 0 °C over 30 min. Benzyl alcohol (0.25 mL, 2.4 mmol) was added, followed by triethylamine (0.37 mL, 2.6 mmol) diluted in dry CH2CL2 (1 mL). The reaction was stirred for 1 h at 0 °C, quenched with saturated ammonium chloride and extracted with CH2CL2 (3 × 5 mL). The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to yield an oil. The crude product was subjected to column chromatography (3:1 Hexanes/EtOAc) to give the phosphoryl monochloride (0.35 g, 50%) as an oil, which was used directly in the next step. Rf = 0.49 (3:1 Hexanes/EtOAc). 1H NMR (CDCl3): δ 7.38 (s, 5H), 7.10 (m, 4H), 5.29 (m, 2H), 2.32 (s, 3H). 31P NMR (CDCl3): δ -24.0.
Triethylamine (0.090 mL, 0.78 mmol) was added dropwise to a solution of N-Methyl-N-2-bromoethylamine hydrobromide (0.070 g, 0.34 mmol) and the phosphoryl monochloride prepared above (0.10 g, 0.34 mmol) in dry THF (1.2 mL) at room temperature. The reaction mixture was stirred for 80 min, filtered through a plug of cotton, and the filtrate was evaporated to give an oil. The crude product was purified using column chromatography (10:1 CHCl3/EtOAc) to yield 1 (60 mg, 47%) as an oil. Rf = 0.56 (10:1 CHCl3/EtOAc). 1H NMR (CDCl3): δ 7.35 (s, 5H), 7.10 (m, 4H), 5.10 (d, 2H), 4.48-3.21 (m, 4H), 2.70 (d, 3H), 2.29 (s, 3H). 31P NMR (CDCl3): δ -19.7. MS (ESI) m/z 398/400 (M+H)+.
N-Methyl-N-(4-bromobutyl) O-(5-nitrofuryl-2-methyl) O-(4-methyl)phenyl phosphoramidate (2). Methyl bromobutyl phosphoramidic dichloride (1.0 g, 3.6 mmol) (prepared as described for the chlorobutyl compound 3) was dissolved in dry THF (5 mL), and cannulated to a pre-cooled solution of p-cresol (0.38 g, 3.6 mmol) and LiHMDS (0.1 M solution in THF, 3.9 mL, 3.9 mmol) in dry THF (18 mL) at −78 °C. The reaction mixture was stirred and warmed to room temperature over 3 h. This mixture was then cannulated to a pre-cooled solution of nitrofurfuryl alcohol (0.51 g, 3.6 mmol) and LiHMDS (3.9 mL, 3.9 mmol) in dry THF (18 mL) at −78 °C. The reaction mixture was then warmed to −15 °C and stirred for 2.5 h. The reaction mixture was quenched with saturated ammonium chloride and extracted with EtOAc (2 × 10 mL). The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to yield a brown oil. The residue was purified using column chromatography to yield 2 (0.75 g, 46%) as an orange oil. Rf = 0.5 (100:10:0.5, CHCl3/EtOAc/MeOH). 1H NMR (CDCl3): δ 7.26 (d, 1H), 7.09 (m, 4H,), 6.621 (d, 1H), 5.06 (d, 2H), 3.38 (t, 2H), 3.09 (m, 2H), 2.71 (d, 3H), 2.31 (s, 3H), 1.76 (m, 2H), 1.63 (m, 2H). 31P NMR (CDCl3): δ -19.0. MS (ESI) m/z 461/463 (M+H)+.
N-Methyl-N-(4-chlorobutyl) phosphoramidic dichloride (3). POCl3 (2.4 mL, 25 mmol) was added to a pre-cooled solution of N-Methyl-N-(4-chlorobutyl)amine hydrochloride (4.0 g, 25 mmol) in dry CH2CL2 (40 mL) at 0 °C, followed by the addition of triethylamine (7.1 mL, 51 mmol) diluted in dry CH2CL2 (20 mL). The reaction mixture was removed from the cooling bath and stirred overnight at room temperature, then quenched with saturated ammonium chloride and extracted with CH2CL2 (3 × 10 mL). The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to dryness to yield an oil. The crude product was purified using column chromatography (3:1 Hexanes/EtOAc) to yield 12 (4.8 g, 80%) as a clear oil. Rf = 0.45 (3:1 Hex/ EtOAc). 1H NMR (CDCl3): δ 3.58 (t, 2H), 3.29 (m, 2H), 2.84 (d, 3H), 1.79 (m, 4H). 31P NMR (CDCl3): δ -6.6.
Allyl 4-Hydroxybenzoate (6a). Diisopropylethylamine (6.3 mL, 36 mmol) was added to a suspension of 4-hydroxybenzoic acid (5.0 g, 36 mmol) in allyl bromide (86 mL). The reaction mixture was refluxed for 2 h and cooled to room temperature. Excess allyl bromide was evaporated under reduced pressure, the remaining oil was dissolved in EtOAc (50 mL) and washed with water (3 × 10 mL). The organic extract was washed with brine, dried over MgSO4, filtered and evaporated to yield a yellow oil. The crude product was purified using column chromatography (10:1 CHCl3/EtOAc) to yield 6a (5.0 g, 78%) as a white solid, mp 100-102 °C. Rf = 0.44 (10:1 CHCl3/EtOAc). 1H NMR (CDCl3): δ 7.99 (d, 2H), 6.86 (d, 2H), 6.02 (m, 1H), 5.30 (m, 2H), 4.80 (d, 2H). MS (ESI) m/z 179 (M+H)+.
Allyl 4-Hydroxyphenylacetate (6b). Allyl ester 6b was synthesized from phenylacetic acid (2.0 g, 13 mmol) as described for 6a and obtained as an oil (2.3 g, 89%). Rf = 0.31 (10:1 CHCl3/EtOAc). 1H NMR (CDCl3): δ 7.16 (d, 2H), 6.76 (d, 2H), 5.92 (m, 1H), 5.28 (m, 2H), 4.60 (d, 2H), 3.64 (s, 2H).
N-Acetyl-L-tyrosine allyl ester (6c). Allyl ester 6c was synthesized from Ac-Tyr-OH (10 g, 45 mmol) as described for 6a and obtained as an oil (9.2 g, 80%). 1H NMR (CDCl3): δ 7.00 (d, 2H), 6.77 (d, 2H), 5.97 (m, 2H), 5.31 (m, 2H), 4.89 (m, 1H), 4.61 (d, 2H,), 3.13 (m, 2H), 2.00 (s, 3H).
N-Methyl-N-(4-chlorobutyl) O-4-carboallyloxyphenyl phosphoramidic chloride (4a). Diisopropylethylamine (0.73 mL, 4.2 mmol) was added neat to a pre-cooled solution of phosphoramidic dichloride 3 (0.50 g, 2.1 mmol) and allyl ester 6a (0.37 g, 2.1 mmol) in dry CH2CL2 (8 mL) at −15 °C. The reaction mixture was stirred for 1 h at -15 °C and 3 h at -5 °C, then quenched with saturated ammonium chloride and extracted with CH2CL2 (2 × 10 mL). The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to give an oil. The crude product was purified using column chromatography (10:1 CHCl3/EtOAc) to yield 4a (0.50 g, 63%) as a clear oil. Rf = 0.60 (10:1 CHCl3/EtOAc). 1H NMR (CDCl3): δ 8.10 (d, 2H,), 7.34 (d, 2H), 6.09-5.98 (m, 1H), 5.45-5.28 (m, 2H), 4.84-4.81 (m, 2H), 3.57 (t, 2H), 3.35-3.17 (m, 2H), 2.84 (d, 3H), 1.82-1.79 (m, 4H). 31P NMR (CDCl3): δ -13.1. MS (ESI) m/z 380/382 (M+H)+.
N-Methyl-N-(4-chlorobutyl) O-(4-carboallyloxymethyl)phenyl phosphoramidic chloride (4b). Phosphoramidic chloride 4b was synthesized from allyl ester 6b (0.20 g, 10 mmol) as described for 4a. The crude product was purified using column chromatography (10:1 CHCl3/EtOAc) to yield 4b a clear oil (0.13 g, 32%). Rf = 0.71 (10:1 CHCl3/EtOAc). 1H NMR (CDCl3): δ 7.32-7.19 (m, 4H), 5.98-5.82 (m, 1H), 5.31-5.20 (m, 2H), 4.60 (d, 2H), 3.64 (s, 2H), 3.58 (t, 2H), 3.25-3.18 (m, 2H), 2.83 (d, 3H), 1.80-1.78 (m, 4H). 31P NMR (CDCl3): δ -12.7. MS (ESI) m/z 394/396 (M+H)+.
N-Methyl-N-(4-chlorobutyl) O-(4-((S)-2-acetylamino-2-carboallyloxyethyl))phenyl phosphoramidic chloride (4c). Phosphoramidic chloride 4c was synthesized from 6c (0.50 g, 1.9 mmol) as described for 4a, with the exception that the reaction mixture was stirred overnight. The crude product was purified using column chromatography (5% MeOH in CH2CL2) to yield 4c (0.49 g, 55%) as an oil. Rf = 0.57 (5% MeOH in CH2CL2). 1H NMR (CDCl3:) δ 7.20-7.09 (m, 4H), 5.99-5.78 (m, 2H), 5.35-5.25 (m, 2H), 4.93-4.85 (m, 1H), 4.61 (d, 2H), 3.58 (t, 2H), 3.26-3.13 (m, 4H), 2.83 (d, 3H), 2.00 (s, 3H), 1.96-1.75 (m, 4H). 31P NMR (CDCl3): δ -12.7. MS (ESI) m/z 465/467 (M+H)+.
N-Methyl-N-(4-chlorobutyl) O-(5-nitrofuryl-2-methyl) O-(4-carboxy)phenyl phosphoramidate (5a). Phosphoramidic monochloride 4a (0.63g, 1.2 mmol) was dissolved in dry THF (2 mL) and cannulated to a pre-cooled solution of nitrofurfuryl alcohol (0.19 g, 1.3 mmol) and LiHMDS (1.0 M solution in THF, 1.5 mL, 1.5 mmol) in dry THF (2 mL) at -78°C. The reaction was brought to -40 °C and stirred for 5.5 h. Saturated ammonium chloride was added and the mixture was extracted with ethyl acetate (3 × 5 mL). The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to yield a dark oil. The crude product was purified by column chromatography (10:1 CHCl3/EtOAc) to yield the allyl ester of phosphoramidate 5a (0.30g, 52%) as an orange oil. Rf = 0.25 (10:1 CHCl3/EtOAc). 1H NMR (CDCl3): δ 8.05 (m, 2H), 7.26 (m, 3H), 6.64 (d, 1H), 6.05 (m, 1H), 5.31 (m, 2H), 5.09 (d, 2H), 4.82 (d, 2H), 3.53 (t, 2H), 3.1 (m, 2H), 2.72 (d, 3H), 1.67 (m, 4H). 31P NMR (CDCl3): δ -21.7.
Sodium p-toluenesulfinate (0.29 g, 1.6 mmol) was dissolved in water (2.5 mL) and added to a solution of the allyl ester (0.71 g, 1.5 mmol) and Pd(PPh3)4 (80 mg, 73 μmol) in THF (6 mL). The reaction mixture was stirred for 30 min at room temperature, diluted with diethyl ether and washed with water (3 × 5 mL). The aqueous extracts were combined, washed with ether (1 × 5 mL), acidified to pH 3 with 2% HCl and extracted with EtOAc. The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to yield phosphoramidate 5a (0.51 g, 78%) as an orange foam. 1H NMR (CDCl3): δ 8.08 (d, 2H), 7.31 (m, 3H), 6.66 (d, 1H), 5.11 (d, 2H), 3.53 (t, 2H), 3.13 (m, 2H), 2.74 (d, 3H), 1.71 (m, 4H). 31P NMR (CDCl3): δ -20.4.
N-Methyl-N-(4-chlorobutyl) O-(-5-nitrofuryl-2-methyl) O-(4-carboxymethyl)-phenyl phosphoramidate (5b). Phosphoramidate 5b (0.12 g, 36%) was synthesized in two steps from 4b (0.20 g, 0.51 mmol) as described for 5a. 1H NMR (CDCl3): δ 7.26-7.04 (m, 5H), 6.60 (d, 1H), 5.43-5.19 (m, 2H), 5.06 (d, 2H), 3.62 (s, 2H), 3.51 (t, 2H), 3.18-2.96 (m, 2H), 2.71 (d, 3H), 1.77-1.55 (m, 4H). 31P NMR (CDCl3): δ -19.0.
N-Methyl-N-(4-chlorobutyl) O-(5-nitrofuryl-2-methyl) O-(4-((S)-2-acetylamino-2-carboxyethyl))phenyl phosphoramidate (5c). Phosphoramidate 5c (0.15g, 27%) was synthesized in two steps from 4c (0.48 g, 1.0 mmol) as described for 5a. Rf = 0.41 (10:1 CHCl3/EtOAc). 1H NMR (CDCl3): δ 7.30-7.25 (m, 1H), 7.23-6.83 (m, 4H), 6.80-6.47 (m, 1H), 5.09 (d, 2H), 5.01-4.59 (m, 2H), 3.76-3.33 (m, 2H), 3.36-2.98 (m, 4H), 2.98-2.55 (m, 3H), 2.14-1.77 (d, 3H), 1.89-1.55 (m, 4H). 31P NMR (CDCl3): δ -19.3, -19.4 (1:1 mixture of diastereomers). MS (ESI) m/z 532/534 (M+H)+.
5-(1-Aminoethyl)-2-cyclohexylmethoxybenzamide (7) was prepared from 5-acetylsalicylamide according to a 3-step procedure described by Lunney et al. 21
N-Methyl-N-(4-chlorobutyl) O-(5-nitrofuryl-2-methyl) O-(4-(1-(3-carbamoyl-4-cyclohexylmethoxyphenyl)ethylcarbamoyl))phenyl phosphoramidate (8). Diisopropylethylamine (0.11 mL, 0.62 mmol) was added neat to a pre-cooled solution of phosphoramidate 5a (0.13 g, 0.28 mmol), amine 7 (0.090 g, 0.31 mmol) and PyBOP (0.16 g, 0.31 mmol) in dry CH2CL2 (2 mL) at 0 °C. The reaction was stirred for 10 min at 0 °C and 30 min at room temperature, then quenched with saturated ammonium chloride and extracted with CH2CL2 (3 × 5 mL). The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to yield an oil. The crude product was purified using column chromatography (10% MeOH in EtOAc) to yield 8 (13 mg, 67%) as a white foam. 1H NMR (CDCl3): δ 8.25 (d, 1H), 7.72 (d, 2H), 7.50 (d, 1H), 7.24 (m, 3H), 6.95 (d, 2H), 6.63 (d, 1H), 5.28 (t, 1H), 5.07 (d, 2H), 3.93 (d, 2H), 3.52 (t, 2H), 3.09 (m, 2H), 2.70 (d, 3H), 1.87-1.58 (m, 14H), 1.48-0.85 (m, 4H). 31P NMR (CDCl3): δ -21.2. HRMS (ESI) C33H42ClN4O9P calculated 705.2456 (M+H)+, found 705.2432. Anal. (C33H42ClN4O9P.H2O) C, H, N.
N-Methyl-N-(4-chlorobutyl) O-(5-nitrofuryl-2-methyl) O-(1-(3-carbamoyl-4-cyclohexylmethoxyphenyl)methyl)phenyl phosphoramidate (9) was obtained from 5b (0.12 g, 0.25 mmol) as described for the preparation of 8. The crude product was purified using column chromatography (5% MeOH in EtOAc) to yield 9 (0.10 g, 55%) as a brown foam. Rf = 0.44 (5% MeOH in EtOAc). 1H NMR (CDCl3): δ 8.10-8.00 (m, 1H), 7.99-7.86 (m, 1H), 7.45-7.09 (m, 7H), 6.98-6.85 (m, 1H), 6.69-6.56 (m, 1H), 6.05-5.89 (m, 1H), 5.87-5.69 (m, 1H), 5.19-4.95 (m, 3H), 4.02-3.81 (m, 2H), 3.64-3.29 (m, 4H), 3.26-2.92 (m, 2H), 2.87-2.52 (m, 3H), 2.02-1.57 (m, 11H), 1.54-0.89 (m, 6H). 31P NMR (CDCl3): δ -18.9, -19.0 (1:1 mixture of diastereomers). HRMS (ESI) C34H44ClN4O9P calculated 719.2613 (M+H)+, found 719.2600. Anal. (C34H44ClN4O9 P. 2H2O) C, H, N. H: calcd, 6.41, found, 5.89.
N-Methyl-N-(4-chlorobutyl) O-(5-nitrofuryl-2-methyl) O-(4-((S)-2-acetylamino-2-(1-(3-carbamoyl-4-cyclohexylmethoxyphenyl)ethylcarbamoyl)ethyl))phenyl phosphoramidate (10) was obtained from 5c (0.15 g, 0.27 mmol) as described for the preparation of 8. The crude product was purified using column chromatography (10% MeOH in EtOAc) to yield 10 (0.13 g, 58%) as a brown foam. Rf = 0.34 (10% MeOH in EtOAc). 1H NMR (CDCl3): δ 8.26-8.11 (m, 1H), 8.09-7.99 (m, 1H), 7.95-7.76 (m, 1H), 7.24-7.09 (m, 2H), 7.08-6.99 (m, 2H), 6.96-6.82 (m, 1H), 6.72-6.55 (m, 1H), 6.50-6.20 (m, 1H), 6.18-6.54 (m, 1H), 5.16-4.82 (m, 3H), 4.79-4.46 (m, 1H), 4.03-3.78 (m, 2H), 3.62-3.43 (m, 2H), 3.15-2.81 (m, 5H), 2.80-2.51 (m, 3H), 2.04-1.89 (m, 3H), 1.87-1.56 (m, 10H), 1.49-0.90 (m, 8H). 31P NMR (CDCl3): δ -18.9. HRMS (ESI) C37H49ClN5O10P calculated 790.2984 (M+H)+, found 790.2981. Anal. (C37H49ClN5O10P.H2O) C, H, N.
N-(4-methylphenyl)phosphoramidic dichloride (12a). A solution of diisopropylethylamine (0.81 mL, 4.7 mmol) in CH2CL2 (3 mL) was added slowly to a precooled solution of p-toluidine (0.50 g, 4.7 mmol) and POCl3 (0.43 mL, 4.7 mmol) in CH2CL2 (30 mL) at - 10° C. The cooling bath was removed, and the reaction was stirred overnight at room temperature. Saturated ammonium chloride was added, the layers were separated, and the aqueous layer was extracted with CH2CL2. The combined organic layers were washed with brine, dried over MgSO4, filtered and evaporated. The crude product was purified by column chromatography (10:1 CHCl3:EtOAc) to yield 12a (0.72 g, 69%) as an oil. NMR (CDCl3): δ 7.22 (d, 2H), 7.16 (d, 2H), 2.30 (s, 3H). 31P NMR (CDCl3): δ -16.6.
Kinetics
A solution of 12a (20 mg) in acetonitrile (300 μL) was added to a solution of Na2CO3 (20 mg) in water (300 μL). The solution was mixed for 5 min, passed through a short plug of celite, and diluted in cacodylate buffer (400 μl, 0.4 M). The pH was adjusted to 7.4 and the resulting solution was monitored by 31P NMR at 37 °C.
N-(4-methylphenyl)phosphoramidic acid (12b). A solution of 12a (0.50 g, 2.2 mmol) in acetonitrile (5 mL) was added slowly to a solution of NaOH (0.18 g, 4.5 mmol) in H2O (10 mL) at 0 °C. The pH was monitored near the end of the addition to assure that the pH remained > 11. The solution was then stirred for 30 min, warmed to room temperature, and filtered through Celite. This solution of 12b as its disodium salt was suitable for all further experiments. 31P NMR (D2O): δ -24.8.
Allyl 4-aminobenzoate (13a). Allyl bromide (0.99 mL, 11 mmol) was added to a suspension of potassium p-aminobenzoate (2.0 g, 11 mmol) in DMF (40 mL). The reaction mixture was stirred overnight, diluted with water and extracted with EtOAc (3 × 15 mL). The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to yield an oil. The crude product was purified by column chromatography (10:1 CHCl3/EtOAc) to yield 13a (2.0 g, 70%) as a foam. 1H NMR (CDCl3): δ 7.99 (d, 2H), 7.08 (d, 2H), 6.05-5.97 (m, 1H), 5.51-5.30 (m, 2H), 4.82-4.78 (m, 2H).
Allyl 4-Aminophenylacetate (13b) A mixture of 4-aminophenylacetic acid (0.50 g, 3.3 mmol), allyl alcohol (20 mL) and concentrated H2SO4 (1 mL) was refluxed for 2 h and then allowed to cool to room temperature. The reaction mixture was poured over ice, basified (pH ∼ 10) with Na2CO3 and extracted with EtOAc (3 × 10 mL). The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to dryness to yield an oil. The crude product was purified using column chromatography (10:1 CHCl3/EtOAc) to yield 13b (0.63 g, 51%) as an oil. Rf = 0.34 (10:1 CHCl3/EtOAc). 1H NMR (CDCl3): δ 7.06 (d, 2H), 6.61 (d, 2H), 6.00-5.80 (m, 1H), 5.32-5.11 (m, 2H), 4.57 (d, 2H), 3.50 (s, 2H)
N-Acetyl-4-aminophenylalanine allyl ester (13c). Ac2O was added slowly to a solution of 4-nitrophenylalanine (3.0 g, 13 mmol) and NaOH (1.1 g, 26 mmol) in a 3:1 mixture of H2O/acetone (120 mL). After stirring for 1 h, acetone was removed under reduced pressure. The remaining aqueous solution was basified (pH ∼10) with saturated sodium bicarbonate and extracted with EtOAc (3 × 10 mL). The organic extracts were discarded. The aqueous layer was acidified (pH ∼ 3) with dilute HCl and extracted with EtOAc. The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to give N-acetyl-4-nitrophenylalanine (3.4 g, 74%) as a white solid. 1H NMR (DMSO-d6): δ 8.16 (d, 1H), 8.11 (d, 2H), 7.48 (d, 2H), 4.45 (m, 1H), 3.20-2.87 (m, 2H), 1.71 (s, 3H). A portion of this product (2.5 g, 9.6 mmol) was dissolved in ethanol (100 mL), 10% Pd/C (0.5 g) was added, and the mixture was agitated in a hydrogen atmosphere (60 psi) for 1 h. The reaction mixture was filtered and evaporated to give N-acetyl-4-aminophenylalanine (2.13 g, 100%) as a white solid. 1H NMR (DMSO-d6): δ 8.01 (d, 1H), 6.81 (d, 2H), 6.40 (d, 2H), 4.23 (m, 1H), 2.90-2.40 (m, 2H), 1.72 (s, 3H). This compound (5.19 g, 23.4 mmol) was converted to the allyl ester 13c as described for the preparation of 13b. The crude product was purified using column chromatography (5% MeOH in CH2CL2) to yield 13c (3.6 g, 60%) as an oil. 1H NMR (CDCl3): δ 6.86 (d, 2H), 6.60 (d, 2H), 6.00-5.79 (m, 1H), 5.40-5.21 (m, 2H), 4.82 (m, 1H), 4.60 (d, 2H), 3.02 (m, 2H), 1.98 (s, 3H).
N-Methyl-N-(4-chlorobutyl) N′-(4-carboallyloxy)phenyl phosphorodiamidic - chloride (14a). Diisopropylethylamine (0.51 mL, 2.9 mmol) was added to a mixture of phosphoramidic dichloride 3 (0.70 g, 2.9 mmol) and allyl ester 13a (0.52 g, 2.9 mmol) in dry dichloroethane (10 mL). The reaction mixture was refluxed for 10 days, allowed to cool room temperature, diluted with CH2CL2 (10 mL) and quenched with saturated ammonium chloride. The layers were separated, the organic layer was washed with brine, dried over MgSO4, filtered and evaporated to yield an oil. The crude product was purified by column chromatography (10:1 CHCl3/EtOAc) to yield 14a (0.22 g, 20%) as an oil. 1H NMR (CDCl3): δ 7.99 (d, 2H), 7.08 (d, 2H), 6.05-5.97 (m, 1H), 5.57 (d, 1H), 5.51-5.30 (m, 2H), 4.82-4.78 (m, 2H), 3.54 (t, 2H), 3.36-3.13 (m, 2H), 2.74 (d, 3H), 1.87-1.64 (m, 4H). 31P NMR (CDCl3): δ -11.3. MS (ESI) m/z 379/381 (M+H)+.
N-Methyl-N-(4-chlorobutyl) N-(4-carboallyloxymethyl)phenyl phosphorodiamidic chloride (14b). Diisopropylethylamine (0.27 mL, 1.6 mmol) was added to a pre-cooled solution of 13b (0.3 g, 1.6 mmol) and POCl3 (0.2 mL, 1.6 mmol) in dry CH2CL2 (3 mL) at −5 °C. The reaction mixture was stirred for 10 min. N-methyl-N-(4-chlorobutyl)amine hydrochloride (0.37 g, 2.4 mmol) was dissolved in dry CH2CL2 (2 mL) and added to the reaction mixture at −5 °C. Diisopropylethylamine (0.82 mL, 4.7 mmol) in dry CH2CL2 (1 mL) was then added slowly to the mixture. The reaction mixture was stirred and warmed to room temperature over 3 h, saturated ammonium chloride was added, and the aqueous layer was extracted with CH2CL2 (3 × 10 mL). The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to yield an oil. The crude product was purified by column chromatography (10:1 CHCl3/EtOAc) to yield 14b (0.62 g, 34%) as a yellow oil. 1H NMR (CDCl3): δ 7.19 (d, 2H), 7.01 (d, 2H), 6.00-5.79 (m, 1H), 5.41-5.17 (m, 3H), 4.59 (d, 2H), 3.60 (s, 2H), 3.49 (bs, 2H), 3.27-3.09 (m, 2H), 2.71 (d, 3H), 1.84-1.59 (m, 4H). 31P NMR (CDCl3): δ -10.6. MS (ESI) m/z 393/395 (M+H)+.
N-Methyl-N-(4-chlorobutyl) N-(4-(2-((S)-acetylamino-2-carboallyloxy)ethyl))phenyl phosphorodiamidic chloride (14c). Phosphorodiamidic chloride 14c was obtained from 13c (0.50 g, 1.9 mmol) as described for the preparation of 14b. The crude product was purified by column chromatography to yield 14c (0.30 g, 33%) as a white foam. 1H NMR (CDCl3): δ 7.19-6.91 (m, 4H), 6.00-5.79 (m, 2H), 5.41-5.22 (m, 2H), 4.97-4.78 (m, 1H), 4.60 (d, 2H), 3.59-3.46 (m, 2H), 3.28-2.97 (m, 4H), 2.71 (d, 3H), 1.99 (s, 3H), 1.81-1.57 (m, 4H). 31P NMR (CDCl3): δ -10.5. MS (ESI) m/z 464/466 (M+H)+.
N-Methyl-N-(4-chlorobutyl) O-(5-nitrofuryl-2-methyl) O-(4-carboxy)phenyl phosphorodiamidate (15a). LiHMDS (1.0 M solution in THF, 0.65 mL, 0.65 mmol) was added slowly to a pre-cooled solution of nitrofurfuryl alcohol (0.08 g, 0.59 mmol) in dry THF (1 mL) at -78°C. The mixture was then cannulated to a pre-cooled solution of 14a (0.22 g, 0.59 mmol) in dry THF (1 mL) at −20 °C. The reaction was stirred for 4 h at −20 °C and then quenched with saturated ammonium chloride. The reaction mixture was extracted with ethyl acetate (3 × 5 mL). The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to yield an oil. The crude product was purified by column chromatography (10% MeOH in EtOAc) to give the allyl ester (0.11 g, 40%) as a brown foam. 1H NMR (CDCl3): δ 7.96 9 (d, 2H), 7.26 (d, 1H), 6.99 (d, 2H), 6.65 (d, 1H), 6.16-5.84 (m, 1H), 5.51-5.19 (m, 3H), 5.09 (d, 2H), 4.92-4.73 (m, 2H), 3.52 (t, 2H), 3.22-2.99 (m, 2H), 2.69 (d, 3H), 1.85-1.61 (m, 4H). 31P NMR (CDCl3): δ -15.6. MS (ESI) m/z 486/488 (M+H)+.
Sodium p-toluenesulfinate (45 mg, 0.25 mmol) was dissolved in water (0.6 mL) and added to a solution of the allyl ester (0.11 g, 0.23 mmol) and Pd(PPh3)4 (13 mg, 11 μmol) in THF (1.2 mL). The reaction mixture was stirred for 45 min at room temperature, and then diluted with diethyl ether and washed with water (3 × 2 mL). The aqueous extracts were combined, washed with ether (1 × 4 mL), acidified to pH 3 with 2% HCl and extracted with EtOAc (3 × 3 mL). The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to yield 15a (80 mg, 76%) as a yellow foam. 1H NMR (CDCl3, TMS) δ: 7.96 (d, 2H), 7.26 (d, 1H), 6.99 (d, 2H), 6.65 (d, 1H), 6.45-6.25 (m, 1H), 5.09 (d, 2H), 3.52 (t, 2H), 3.22-2.99 (m, 2H), 2.69 (d, 3H), 1.85-1.61 (m, 4H). 31P NMR (CDCl3) δ: -15.4. HPLC (Gradient 30% to 100% CH3CN/H2O [0.1% TFA] over 35 min): 9.37 min; 100%. MS (ESI) m/z 444/446 (M-H)-.
N-Methyl-N-(4-chlorobutyl) O-(5-nitrofuryl-2-methyl) O-(4-carboxymethyl)phenyl phosphorodiamidate (15b). Phosphorodiamidate 15b was obtained as a yellow foam (0.10 g, 40%) from 14b (0.21 g, 0.54 mmol) by the two step sequence described for the preparation of 15a. 1H NMR (CDCl3): δ 7.19-7.06 (m, 3H), 7.88 (d, 2H), 6.51 (d, 1H), 5.82 (d, 1H), 5.01 (d, 2H), 3.51 (s, 2H), 3.48 (t, 2H), 3.11-2.97 (m, 2H), 2.61 (d, 3H), 1.79-1.51 (m, 4H). 31P NMR (CDCl3): δ -14.4. HPLC (Gradient 30% to 100% CH3CN/H2O [0.1% TFA] over 35 min): 11.48 min; 100%. MS (ESI) m/z 456/458 (M-H)-.
N-Methyl-N-(4-chlorobutyl) O-(5-nitrofuryl-2-methyl) O-(4-(2-((S)-acetylamino-2-carboxy)ethyl))phenyl phosphorodiamidate (15c). Phosphorodiamidate 15c was obtained as a foam (0.12 g, 38%) from 14c (0.29 g, 0.63 mmol) by the two step sequence described for the preparation of 15a. 1H NMR (CDCl3): δ 7.00-6.81 (m, 3H), 6.75-6.62 (d, 3H), 6.28-6.15 9(m, 2H), 5.21-5.00 (m, 2H), 4.92-4.61 (bs, 1H), 3.51 (bs, 2H), 3.19-2.92 (m, 4H), 2.62 (m, 3H), 1.99 (bs, 3H), 1.69-1.52 (m, 4H). 31P NMR (CDCl3): δ -15.1. HPLC (Gradient 30% to 100% CH3CN/H2O [0.1% TFA] over 35 min): 10.12 min; 100%. MS (ESI) m/z 531/533 (M+H)+.
N-Methyl-N-(4-chlorobutyl) O-(5-nitrofuryl-2-methyl) O-(4-(1-(3-carbamoyl-4-cyclohexylmethoxyphenyl)ethylcarbamoyl))phenyl phosphorodiamidate (16). Diisopropylethylamine (0.07 mL, 0.38 mmol) was added to a pre-cooled solution of phosphoramidate 15a (80 mg, 0.17 mmol), amine 7 (50 mg, 0.19 mmol) and PyBOP (90 mg, 0.31 mmol) in dry CH2CL2 (2 mL) at 0 °C. The reaction was stirred for 5 min at 0 °C and 30 min at room temperature. The reaction mixture was then quenched with saturated ammonium chloride and extracted with CH2CL2 (3 × 5 mL). The organic extracts were combined, washed with brine, dried over MgSO4, filtered and evaporated to yield an oil. The crude product was purified by column chromatography (10% MeOH in EtOAc) to yield 16 (70 mg, 60%) as a foam. 1H NMR (CDCl3): δ 8.22 (d, 1H), 7.96-7.81 (m, 1H), 7.61 (d, 2H), 7.56-7.40 (m, 1H), 7.25-7.13 (m, 1H), 6.92 (d, 2H), 6.82-6.66 (m, 1H), 6.66-6.53 (m, 1H), 6.27-6.00 (m, 2H), 5.35-5.10 (m, 1H), 5.03 (d, 2H), 3.90 (d, 2H), 3.48 (t, 2H), 3.18-2.92 (m, 2H), 2.64 (d, 3H), 1.94-1.47 (m, 10H), 1.38-0.98 (m, 7H). 31P NMR (CDCl3): δ -14.9. HPLC (Gradient 30% to 100% CH3CN/H2O [0.1% TFA] over 35 min): 17.28 min; 100%. HRMS (ESI) m/z C33H43ClN5O8P calculated 704.2616 (M+H)+, found 704.2633. Anal. (C33H43ClN5O8P .3H2O) C, H, N. H: calcd, 6.51; found, 5.90.
N-Methyl-N-(4-chlorobutyl) O-(5-nitrofuryl-2-methyl) O-(4-(1-(3-carbamoyl-4-cyclohexylmethoxyphenyl)ethylcarbamoyl)methyl)phenyl phosphorodiamidate (17) Phosphorodiamidate 17 was obtained from 15b (90 mg, 0.19 mmol) as described for the synthesis of 16. The crude product was purified by column chromatography (10% MeOH in EtOAc) to yield 15b (30 mg, 70%) as a yellow foam. 1H NMR (CDCl3): δ 8.08 (d, 1H), 7.87 (bs, 1H), 7.34 (dd, 1H), 7.20 (d, 1H), 7.11 (d, 2H), 6.99-6.81 (m, 4H), 6.60 (d, 1H), 6.13 (bs, 1H), 6.01 (d, 1H), 5.83 (d, 1H), 5.08 (d, 3H), 3.91 (d, 2H), 3.51-3.41 (m, 4H), 3.12-3.01 (m, 2H), 2.61 (d, 3H), 1.82-1.53 (m, 10H), 1.46-0.93 (m, 7H). 31P NMR (CDCl3): δ -15.0. HPLC (Gradient 30% to 100% CH3CN/H2O [0.1% TFA] over 35 min): 19.27 min; 100%. HRMS (ESI) m/z C34H45ClN5O8P calculated 718.2773 (M+H)+, found 718.2766. Anal. (C34H45ClN5O8P.H2O) C, H, N.
N-Methyl-N-(4-chlorobutyl) O-(5-nitrofuryl-2-methyl) O-(4-(2-((S)-acetylamino-2-(1-(3-carbamoyl-4-cyclohexylmethoxyphenyl)ethylcarbamoyl)ethyl)))phenyl phosphorodiamidate (18). Phosphorodiamidate 18 was obtained from 15c (0.12 g, 0.23 mmol) as described for the synthesis of 16. The crude product was purified by column chromatography (10% MeOH in EtOAc) to yield 18 (0.14 g, 68%) as a foam. 1H NMR (CDCl3): δ 8.11 (bs, 1H), 8.02 (bs, 1H), 7.89-7.81 (m, 1H), 7.39-7.18 (m, 2H), 7.12-7.01 (m, 2H), 7.00-6.78 (m, 4H), 6.67-6.52 (m, 1H), 6.42 (bs, 1H), 6.31 (bs, 1H), 5.97 (d, 1H), 5.08-4.88 (m, 3H), 4.79-4.52 (m, 1H), 3.99-3.81 (m, 2H), 3.52-3.39 (m, 2H), 3.13-2.80 (m, 4H), 2.70-2.51 (t, 3H), 1.98-1.51 (m, 13H), 1.39-0.91 (m,, 7H). 31P NMR (CDCl3): δ -14.2, -14.3 (1:1 mixture of diastereomers). HPLC (Gradient 30% to 100% CH3CN/H2O [0.1% TFA] over 35 min): 18.25 min, 50%; 18.42 min, 50%. HRMS (ESI) m/z C37H50ClN6O9P calculated 789.3147 (M+H)+, found 789.3144. Anal. (C37H50ClN6O9P.3H2O) C, H, N. H: calcd, 6.69; found, 6.13. N: calcd, 9.97; found, 9.36.
Supplementary Material
Supporting Information Available: Elemental analyses of 8 – 10 and 16 – 18. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
Financial Support from the National Cancer Institute (Grant R01 CA34619) is gratefully acknowledged. Support from the Purdue Cancer Center Support Grant P30 CA23168 for services provided by the NMR and Mass Spectrometry Shared Resources is appreciated.
References
- 1.Koch CA, Anderson D, Moran MF, Ellis C, Pawson T. SH2 and SH3 Domains: Elements that Control Interactions of Cytoplasmic Signaling Proteins. Science. 1991;252:668–74. doi: 10.1126/science.1708916. [DOI] [PubMed] [Google Scholar]
- 2.Beattie J. SH2 Domain Protein Interaction and Possibilities for Pharmacological Intervention. Cell Signal. 1996;8:75–86. doi: 10.1016/0898-6568(95)02033-0. [DOI] [PubMed] [Google Scholar]
- 3.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, Nell BG, Birge RB, Fajardo JE, Chou MM, Hanafusa H, Schafhausen B, Cantley LC. SH2 Domains Recognize Specific Phosphopeptide Seqeunces. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 4.Songyang Z, Shoelson SE, McGlade J, Oliver P, Pawson T, Bustelo XR, Barbacid M, Sabe H, Hanafusa H, Yi T, Ren R, Baltimore D, Ratnosky S, Feldman RA, Cantley LC. Specific Motifs Recognized by the SH2 Domains of Csk, 3BP2, Fps/Fes, GRB-2, HCP, SHC, Syk and Vav. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luttrell DK, Lee A, Lansing TJ, Crosby RM, Jung KD, Willard D, Luther M, Rodriguez M, Berman J, Gilmer TM. Involvement of pp60c-src with Two Major Signaling Pathways in Human Breast Cancer. Proc Natl Acad Sci USA. 1994;91:83–7. doi: 10.1073/pnas.91.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cartwright CA, Meisler AI, Eckhart W. Activation of the pp60c-src Protein Kinase is an Early Event in Colonic Carcinogenesis. Proc Natl Acad Sci USA. 1990;87:558–62. doi: 10.1073/pnas.87.2.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ottenhoff-Kalff AE, Rijsken G, van Beurden EACM, Hennipman A, Michles AA, Staal GEJ. Characterization of protein tyrosine kinases from human breast cancer: involvement of the c-src oncogene product. Cancer Res. 1992;52:4773–8. [PubMed] [Google Scholar]
- 8.Lewis LA, Chung CD, Chen JPJR, Moran M, Patel VP, Miceli MC. The Lck SH2 Phosphotyrosine Binding Site Is Critical for Efficient TCR-Induced Procesive Tyrosine Phosphorylation of the ζ-Chain and IL-2 Production. J Immunol. 1997;159:2292–300. [PubMed] [Google Scholar]
- 9.Sawyer TK. Src homology-2 domains: Structure, mechanisms, and drug discovery. Biopolymers. 1998;47:243–61. doi: 10.1002/(SICI)1097-0282(1998)47:3<243::AID-BIP4>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 10.Lee T, Lawrence DS. Nonpeptidic ligands that target the SH2 domain of the lck tyrosine kinase. J Med Chem. 2000;43:1173–9. doi: 10.1021/jm990462r. [DOI] [PubMed] [Google Scholar]
- 11.Beaulieu PL, Cameron DR, Ferland JM, Gauthier J, Ghiro E, Gillard J, Gorys V, Poirier M, Rancourt J, Wernic D, Llinas-Brunet M, Betageri R, Cardozo M, Hickey ER, Ingraham R, Jakes S, Kabcenell A, Kirrane T, Lukas S, Patel U, Proudfoot J, Sharma R, Tong L, Moss N. Ligands for the tyrosine kinase p56lck SH2 domain: Discovery of potent dipeptide derivatives with monocharged, nonhydrolyzable phosphate replacements. J Med Chem. 1999;42:1757–66. doi: 10.1021/jm980676t. [DOI] [PubMed] [Google Scholar]
- 12.Proudfoot JR, Betageri R, Cardozo M, Gilmore TA, Glynn S, Hickey ER, Jakes S, Kabcenell A, Kirrane TM, Tibolla AK, Lukas S, Patel UR, Sharma R, Yazdanian M, Moss N. Nonpeptidic, Monocharged, Cell Permeable Ligands for the p56Lck SH2 Domain. J Med Chem. 2001;44:2421–31. doi: 10.1021/jm000446q. [DOI] [PubMed] [Google Scholar]
- 13.Bohacek R, Dalgarno DC, Hatada M, Jacobsen VA, Lynch Ba, Macek KJ, Merry T, Metcalf CAI, Narula SS, Sawyer TK, Shakespeare WC, Violette SM, Weigele M. X-ray Structure of Citrate Bound to Src SH2 Leads to a High-Affinity, Bone-Targeted Src SH2 Inhibitor. J Med Chem. 2001:660–3. doi: 10.1021/jm0002681. [DOI] [PubMed] [Google Scholar]
- 14.Fu JM, Castelhano AL. Design and Synthesis of a Pyridone-Based Phosphotyrosine Mimetic. Bioorg Med Chem Lett. 1998;8:2813–6. doi: 10.1016/s0960-894x(98)00503-4. [DOI] [PubMed] [Google Scholar]
- 15.Mathe C, Periguad C, Gosselin G, Imbach JL. Phosphopeptide Prodrug Bearing an S-Acyl-2-thioethyl Enzyme-Labile Phosphate Protection. J Org Chem. 1998;63:8547–50. [Google Scholar]
- 16.Meyers CLF, Hong L, Joswig C, Borch RF. Synthesis and biological activity of novel 5-fluoro-2′-deoxyuridine phosphoramidate prodrugs. J Med Chem. 2000;43:4313–8. doi: 10.1021/jm000301j. [DOI] [PubMed] [Google Scholar]
- 17.Tobias SC, Borch RF. Synthesis and biological studies of novel nucleoside phosphoramidate prodrugs. J Med Chem. 2001;44:4475–80. doi: 10.1021/jm010337r. [DOI] [PubMed] [Google Scholar]
- 18.Tobias SC, Borch RF. Synthesis and biological evaluation of a cytarabine phosphoramidate prodrug. Mol Pharmaceutics. 2004;1:112–6. doi: 10.1021/mp034019v. [DOI] [PubMed] [Google Scholar]
- 19.Fries KM, Joswig C, Borch RF. Synthesis and biological evaluation of 5-fluoro-2′-deoxyuridine phosphoramidate analogs. J Med Chem. 1995;38:2672–80. doi: 10.1021/jm00014a019. [DOI] [PubMed] [Google Scholar]
- 20.Freel Meyers CL, Borch RF. Activation mechanisms of nucleoside phosphoramidate prodrugs. J Med Chem. 2000;43:4319–27. doi: 10.1021/jm000302b. [DOI] [PubMed] [Google Scholar]
- 21.Lunney EA, Para KS, Rubin JR, Humblet C, Fergus JH, Marks JS, Sawyer TK. Structure-based design of a novel series of nonpeptide ligands that bind to the pp60src SH2 domain. J Am Chem Soc. 1997;119:12471–6. [Google Scholar]
- 22.Steinberg G, Borch RF. Synthesis and evaluation of pteroic acid-conjugated nitroheterocyclic phosphoramidates as folate receptor-targeted alkylating agents. J Med Chem. 2001;44:69–73. doi: 10.1021/jm000306g. [DOI] [PubMed] [Google Scholar]
- 23.Alsina J, Rabanal F, Chiva C, Giralt E, Albericio F. Active Carbonate Resins: Application to the Solid-Phase Synthesis of Alcohol, Carbamate and Cyclic Peptides. Tetrahedron. 1998;54:10125–52. [Google Scholar]
- 24.Smyth MS, Ford H, Burke TR. A General Method for the Preparation of Benzylic α,α-Difluorophosphonic Acids; Non-Hydrolyzable Mimetics of Phosphotyrosine. Tet Lett. 1992;33:4137–40. [Google Scholar]
- 25.Domchek SM, Auger KR, Chatterjee S, Burke TR, Jr, Shoelson SE. Inhibition of SH2 domain/phosphoprotein association by a nonhydrolyzable phosphonopeptide. Biochemistry. 1992;31:9865–70. doi: 10.1021/bi00156a002. [DOI] [PubMed] [Google Scholar]
- 26.Gamcsik MP, Ludeman SM, Shulman-Roskes EM, McLennan IJ, Colvin ME, Colvin OM. Protonation of phosphoramide mustard and other phosphoramides. J Med Chem. 1993;36:3636–45. doi: 10.1021/jm00075a019. [DOI] [PubMed] [Google Scholar]
- 27.Millis KK, Colvin ME, Shulman-Roskes EM, Ludeman SM, Colvin OM, Gamcsik MP. Comparison of the Protonation of Isophosphoramide Mustard and Phosphoramide Mustard. J Med Chem. 1995;38:2166–75. doi: 10.1021/jm00012a017. [DOI] [PubMed] [Google Scholar]
- 28.Hiroi K, Abe J, Suya K, Sato S, Koyama T. The Palladium-Catalyzed Asymmetric α–Allylation of Carbonyl Compounds with Chiral Allyl Esters via Enamines and Imines. J Org Chem. 1994;59:203–13. [Google Scholar]
- 29.Straus DB, Chan AC, Patai B, Weiss A. SH2 domain function is essential for the role of the Lck tyrosine kinase in T cell receptor signal transduction. J Biol Chem. 1996;271:9976–81. doi: 10.1074/jbc.271.17.9976. [DOI] [PubMed] [Google Scholar]
- 30.Won J, Hur YG, Hur EM, Park SH, Kang MA, Choi Y, Park C, Lee KH, Yun Y. Rosmarinic acid inhibits TCR-induced T cell activation and proliferation in an Lck-dependent manner. Eur J Immunol. 2003;33:870–9. doi: 10.1002/eji.200323010. [DOI] [PubMed] [Google Scholar]
- 31.Hanke JH, Gardner JP, Dow RL, Changelain PS, Brissette WH, Weringer EJ, Pollok BA, Connely PA. Discovery of a Novel, Potent, and Src Family-selective Tyrosine Kinase Inhibitor. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Elemental analyses of 8 – 10 and 16 – 18. This material is available free of charge via the Internet at http://pubs.acs.org.