

Summary
With the availability of whole genome sequence in many species, linkage analysis, positional cloning, and microarray are gradually becoming powerful tools for investigating the links between phenotype and genotype or genes. However, in these methods, causative genes underlying a quantitative trait locus, or a disease, are usually located within a large genomic region or a large set of genes. Examining the function of every gene is very time-consuming and needs to retrieve and integrate the information from multiple databases or genome resources. PGMapper is a software tool for automatically matching phenotype to genes from a defined genome region or a group of given genes by combining the mapping information from the Ensembl database and gene function information from the OMIM and PubMed databases. PGMapper is currently available for candidate gene search of human, mouse, rat, zebrafish, and 12 other species.
1 INTRODUCTION
Decoding the genetic information from genotype to phenotype is essential in understanding numerous biological processes that contribute to overall cell, tissue, and organism response, particularly in specific disease states. With the completion of genome sequencing in many species, extensive efforts have been made to determine the genetic structure underlying phenotypic traits. One of the most popular methods is linkage analysis, which can identify the genomic region that is closely linked with the causative gene responsible for single-gene diseases or quantitative trait loci (QTL). At present, as the development of new genetic techniques and the availability of inexpensive DNA marker genotyping, QTL or disease loci have become easier to identify (Abiola et al., 2003). However, despite the success in finding QTL or disease loci, identifying genes remains a major challenge (Glazier et al., 2002). Positional cloning is one of the leading methods for identifying the genetic causes of QTL or diseases. However, as a QTL or a disease locus is often positioned within a large genomic region that contains hundreds of genes, this procedure is usually expensive and time-consuming. Manually evaluating the potential connection between every gene within a QTL or a disease locus and the phenotypic trait of interest is time-consuming and tedious and also somewhat incomplete as it needs to retrieve and integrate the information from multiple databases. Several computational tools have been developed to facilitate selecting candidate genes for diseases (Aerts et al., 2006; Adie et al., 2006; Lopez-Bigas et al., 2004); these tools can prioritize candidate genes based on functional annotation and/or sequence features. A website, PhenoGen, can also search candidate genes for a complex trait based on the co-occurrence of differentially expressed genes in microarray experiments and phenotypic QTL (p-QTL) or co-occurrence of p-QTL and expression QTL (e-QTL) (Bhave et al., 2007). However, these existing tools are not intentionally designed to identify the candidate genes for the chromosomal region of a QTL and do not solve two major concerns of experimental researchers working on QTL analysis and positional cloning. Namely, they need to know how many candidate genes exist within the QTL region according to known literature reports and detailed information of those candidates and related reports indicating their candidacy. There is also a need of a computational tool with high flexibility for them to quickly explore or test various possibilities for the associations of genes with phenotype or diseases by their knowledge and experience and then construct further experimental strategies based on these possibilities. PGMapper was developed to fill these gaps. PGMapper automatically matches particular phenotypic traits to genes from QTL or disease loci by integrating the mapping information from the Ensembl database (http://www.ensembl.org/index.html) and gene function information from the OMIM (http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim) and PubMed databases (http://www.ncbi.nlm.nih.gov/sites/entrez) and provides the detailed information about candidates and relevant literature reports. It also allows users to easily construct complex search strategies using different combinations of key words or phrases, field qualifiers, text connectors, and Boolean operators and to test a variety of possible relationships between genes and phenotypic traits. Although PGMapper was developed to identify the candidate genes of a QTL or a disease locus, it can also be used for whole genome analysis of a complex trait or a disease.
Microarray technology is playing an essential role in investigating the link between genotype and phenotype (Sequeira et al., 2006). Considering there may be hundreds or even thousands of up/down-regulated genes in a microarray experiment, and confirming the known association between such a large number of genes and the disease phenotype of interest is a difficult task, we also affix a separate module to search candidate genes for microarray experiments.
2 FUNCTIONALITY
PGMapper is a web application based on Java EE technologies. The interface of the system is user friendly and interactive, and all results can be viewed online or saved in Excel or PDF format for further analysis.
2.1 QTL module and disease locus module
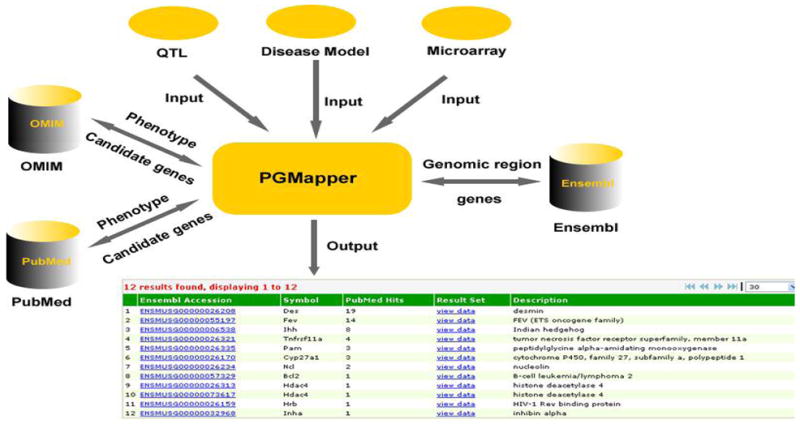
QTL module and disease locus module are developed to search candidate genes for QTL and disease loci, respectively. The inputs to the system are the map location of a QTL or a disease locus and key words that can describe the features of a particular phenotypic trait or disease. First, PGMapper retrieves the information of all genes within a QTL or a disease locus from the Ensembl database and then searches both OMIM and PubMed to obtain information about candidate genes. The search term is the combination of the gene symbol with key words related to phenotype. For each candidate, all references found from OMIM or PubMed are listed on a separate page. Users can follow the link to see more details for each reference. Fig. 1 presents the information flow for candidate gene search in PGMapper.
Figure 1.
2.2 Microarray module
Microarray module is used to search candidate genes for microarray experiments. Users may first choose a set of differentially expressed genes between the experimental group and the control group or those genes of interest as the inputs to the system and then specify the key words possibly associated with specific disease phenotypes. The system will automatically pick out those genes that may have actions on a disease or other traits of interest based on all available reports in both OMIM and PubMed.
3 FUTURE DIRECTIONS
We have developed a web-based tool used to search candidate genes underlying a particular phenotypic trait for a specified genome region. In our method, we do not rank the search result with a complex algorithm. Instead, we simply rank candidate genes by the number of references that could indicate the candidacy of a gene. Moreover, it is possible that our searching method may turn up some candidate genes not functionally related to, but only literally related to, phenotypic traits of interest. Our next step is to develop new algorithms to score and rank candidate genes according to the relevance of the gene with the phenotypic trait by an integrative strategy of combining natural language processing and ontology coupled with other public data sources.
Acknowledgments
Funding comes from NIH, National Institute of Arthritis and Musculoskeletal and Skin Diseases, AR51190 to WKG. This work is also supported by the Center of Excellence for Genomics and Bioinformatics at the University of Tennessee Health Science Center. We thank Dr. David Armbruster, author’s editor at UTHSC, for editing this manuscript.
References
- Abiola O, et al. The nature and identification of quantitative trait loci: a community’s view. Nat Rev Genet. 2003;4:911–916. doi: 10.1038/nrg1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adie EA, et al. SUSPECTS: enabling fast and effective prioritization of positional candidates. Bioinformatics. 2006;22:773–774. doi: 10.1093/bioinformatics/btk031. [DOI] [PubMed] [Google Scholar]
- Aerts S, et al. Gene prioritization through genomic data fusion. Nature biotechnology. 2006;24:537–544. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- Bhave SV, et al. The PhenoGen informatics website: tools for analyses of complex traits. BMC genetics. 2007;8:59. doi: 10.1186/1471-2156-8-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazier A, et al. Finding genes that underlie complex traits. Science. 2002;298:2345–2349. doi: 10.1126/science.1076641. [DOI] [PubMed] [Google Scholar]
- Lopez-Bigas N, Ouzounis CA. Genome-wide identification of genes likely to be involved in human genetic disease. Nucleic acids research. 2004;32:3108–3114. doi: 10.1093/nar/gkh605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira A, Turecki G. Genome wide gene expression studies in mood disorders. Omics. 2006;10:4444–45. doi: 10.1089/omi.2006.10.444. [DOI] [PubMed] [Google Scholar]