

Abstract
Purpose
These studies were designed to determine whether the synthetic steroid mifepristone inhibits ovarian cancer growth in vitro and in vivo and the molecular mechanisms involved.
Experimental Design
The effect of mifepristone on ovarian cancer cell growth in vitro was studied in ovarian cancer cell lines of different genetic backgrounds (SK-OV-3, Caov-3, OV2008, and IGROV-1). In addition, the growth inhibition capacity of mifepristone on ovarian carcinoma xenografts was tested in nude mice.
Results
Mifepristone inhibited ovarian cancer cell proliferation in a dose- and time-dependent manner. The cytostatic effect of mifepristone was confirmed in a clonogenic survival assay and was not linked to loss of viability. Mifepristone blocked DNA synthesis, arrested the cell cycle at the G1-to-S transition, up-regulated cyclin-dependent kinase (cdk) inhibitors p21cip1 and p27kip1, down-regulated transcription factor E2F1, decreased expression of the E2F1 regulated genes, cdk1 (cdc2) and cyclin A, and modestly decreased cdk2 and cyclin E levels. The abrupt arrest in cell growth induced by mifepristone correlated with reduced cdk2 activity, increased association of cdk2 with p21cip1 and p27kip1, increased nuclear localization of the cdk inhibitors, and reduced nuclear abundance of cdk2 and cyclin E. In vivo, mifepristone significantly delayed the growth of ovarian carcinoma xenografts in a dose-dependent manner and without apparent toxic effects for the animals.
Conclusions
These preclinical studies demonstrate that mifepristone is effective as a single agent in vitro and in vivo, inhibiting the growth of human epithelial ovarian cancer cells. Mifepristone markedly reduces cdk2 activity likely due to increased association of cdk2 with the cdk inhibitors p21cip1 and p27kip1 and reduced nuclear cdk2/cyclin E complex availability. Acting as a cytostatic agent mifepristone promises to be of translational significance in ovarian cancer therapeutics.
Keywords: mifepristone, ovarian cancer, cdk2, p21cip1, p27kip1
Introduction
Mifepristone, commonly known as RU486, was first synthesized in the early 1980’s and described as a progesterone receptor antagonist (1). The potent antagonism of mifepristone on uterine progesterone receptors led to its clinical application for termination of pregnancy, emergency contraception, luteal phase contraception, and menstrual regulation (2). However, when targeting cells other than uterine cells, the progesterone antagonistic activity of mifepristone is less clear. For instance, in fibroblasts and T47D breast cancer cells treatment with activators of protein kinase A abrogated the antagonistic activity of mifepristone that instead elicited partial agonistic effects (3, 4). In HeLa cells, mifepristone significantly induced progesterone-regulated reporter genes and this agonistic effect was synergistically enhanced by elevating cAMP or by overexpressing the catalytic subunit of protein kinase A (5). The molecular basis for the mixed agonist/antagonist transcriptional regulation of mifepristone in a cell-type specific manner appears to depend on the ratio of coactivators and corepressors of the steroid receptor-mediated transactivation (6).
Apart from its usage as a contraceptive agent, mifepristone has also been effective at diminishing the pain associated with pelvic endometriosis (7, 8), reducing the size of uterine fibroids (9, 10), and inhibiting the growth of meningioma cells in vitro, in experimental animal models, and in patients with inoperable meningiomas (11-13). Another non-contraceptive action of mifepristone is the interference with cancer cell growth. Mifepristone, when given with tamoxifen, inhibited the proliferation of MCF-7 and MDA-231 breast cancer cells in an additive manner (14, 15). In MCF-7 cells, mifepristone displayed synergistic cytotoxic activity with the protein kinase C inhibitor, 7-hydroxystaurosporine (UCN-01) (16) and arrested the growth of MCF-7 sublines made resistant to 4-hydroxytamoxifen (17). Preliminary clinical trials on metastatic breast cancers have shown a promising response to mifepristone in terms of growth inhibition (18, 19). Furthermore, in a recent study using p53/BRCA1 deficient mice, mifepristone prevented the formation of breast tumors (20), indicating its efficacy not only impairing growth of established mammary tumors but also inhibiting mammary tumorigenesis. Apart from breast cancer cells, mifepristone was reported to inhibit proliferation of normal and malignant endometrial cells (21-24), prostate cancer cells (25), and gastric adenocarcinoma cell lines (26).
The action of mifepristone on ovarian cancer has received limited attention. In 1996 Rose and Barnea (27) showed that mifepristone arrested OVCAR-3 and A2780 ovarian cancer cells at the G1 phase of the cell cycle. Years later, it was reported outside the mainstream literature that mifepristone enhanced the toxic effect of cisplatin on COC1 ovarian cancer cells in vitro and in xenografted immunosuppressed mice (28, 29). In the only small clinical trial conducted with 44 patients having recurrent epithelial ovarian cancer whose tumors had become resistant to standard chemotherapy, mifepristone administration showed promising effects against some of the tumors (30). These initial studies indicate an anti-ovarian cancer activity of mifepristone, yet the molecular targets involved in mediating such an effect have not been defined. The aim of the present study was to investigate the molecular mechanisms of cell growth arrest induced by mifepristone in ovarian cancer cells and to further define its efficacy in an in vivo preclinical setting. Exposure of ovarian cancer cells to concentrations of mifepristone likely to be achieved in vivo inhibited their growth by inducing G1 cell cycle arrest without triggering cell death. This growth arrest was accompanied by a decline in cdk2 protein level and activity, which correlated with increased association of cdk2 with the cdk inhibitors p21cip1 and p27kip1, and with reduced levels of transcription factor E2F1 needed for S phase progression. Finally, dose-dependent tumor growth inhibition by mifepristone monotherapy was shown in vivo in nude mice carrying subcutaneous tumors derived from human ovarian cancer cells.
Materials and Methods
Drugs
Mifepristone (RU486), D (-)-norgestrel (levonorgestrel), medroxyprogesterone acetate (Depo-Provera) and progesterone were from Sigma Chemical Company (St. Louis, MO).
Cell lines
SK-OV-3 and Caov-3 cells were obtained in 2003 from the American Tissue Culture Collection (Manassas, VA) at passages 23 and 33, respectively. Cells were routinely maintained in RPMI 1640 (Mediatech, Herndon, VA) supplemented with 5% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA), 10 mmol/L HEPES (Mediatech), 4 mmol/L L-glutamine (Mediatech), 0.45% D(+) glucose (Sigma), 1 mmol/L sodium pyruvate (Mediatech), 1 × nonessential amino acids (Mediatech), 100 IU penicillin (Mediatech), 100 μg/ml streptomycin (Mediatech), and 0.01 mg/ml human insulin (Sigma). The human ovarian carcinoma cell lines OV2008 and IGROV-1 were obtained from Dr. Stephen Howell (University of California, San Diego) and were maintained in RPMI 1640 (Mediatech) supplemented with 5% (OV2008) or 10% (IGROV-1) heat inactivated fetal bovine serum (Atlanta Biologicals) and 10 mmol/L HEPES (Mediatech), 4 mmol/L L-glutamine (Mediatech), 1 mmol/L sodium pyruvate (Mediatech), 1 × nonessential amino acids (Mediatech), 100 IU penicillin (Mediatech) and 100 μg/ml streptomycin (Mediatech). All cell lines were cultured at 37 °C in a humidified atmosphere in the presence of 5% CO2.
Cell proliferation
Cell growth was evaluated in the various ovarian cancer cell lines that were subjected to dose-response or time course treatments. Media containing each of the doses of fresh steroids was replaced every 24 h. Control groups of cells were treated with vehicle ethanol at a final concentration of less than 0.05%. Number of viable cells was evaluated by trypsinization and counting in a hemocytometer chamber using trypan blue dye exclusion (Sigma). Experiments were conducted in media without phenol red and supplemented with charcoal extracted fetal bovine serum, or media containing unextracted serum and having phenol red. Similar results were obtained with both media preparations; therefore, after performing the growth curves, all subsequent experiments were conducted using media with unextracted serum and in the presence of phenol red. When indicated, the proliferation IC50s were calculated using software designed to study drug interaction (Calcusyn, Biosoft, Cambridge, UK).
Clonogenic assay
OV2008 or SK-OV-3 cells were seeded into 6-well plates at 250 cells per well in a final volume of 2 ml of medium containing either vehicle or appropriate drug concentrations. Triplicate cultures were used for each drug concentration and time tested. At the end of the drug exposures, the drug-containing media was replaced with fresh media. All cultures were incubated for an additional 7 days until colonies were large enough to be clearly discerned. At this point, the medium was aspirated, the dishes washed once with PBS, fixed with 100% methanol for 30 minutes, and stained with a filtered solution of 0.5% w/v crystal violet (Sigma) for 10 minutes. The wells were then washed with tap water and dried at room temperature. The colonies, defined as groups of ≥ 50 cells, were scored manually with the aid of an Olympus IMT-2 inverted microscope. Clonogenic survival was expressed as the percentage of colonies formed in mifepristone-treated cells with respect to vehicle-treated cells.
Viability assay
Cells were cultured in sterile 8-well chamber slides at a concentration of 10,000 cells per well and subjected to treatment with vehicle or mifepristone. At the end of the incubation, cells were analyzed by a Live/Dead viability/cytotoxicity assay following the instructions of the manufacturer (Molecular Probes, Eugene, OR). Briefly, cells were washed with PBS, and incubated for 45 min at room temperature in the presence of appropriate concentrations of cell-permeant calcein AM, a substrate for ubiquitous intracellular esterases, and ethidium homodimer (EthD-1), which enters the cells having damaged plasma membranes and stains their DNA. When permeabilization of the plasma membrane was needed, cells were exposed to 0.1% saponin (Acros Organics, Morris Plains, NJ) for 5 min. At the end of the incubation, excess solutions were removed, the slides were mounted, and the labeled cells were observed with a Leica DM/LB microscope (Leica Microsystems, Plymouth, MN) and photographed with a Leica DC 300F digital camera (Leica Microsystems). Calcein formed from calcein AM is well retained within live cells, producing an intense uniform green fluorescence. EthD-1 undergoes enhancement of fluorescence upon binding to DNA, producing a bright red fluorescence in dead cells, but it is excluded by the intact plasma membrane of live cells.
Bromodeoxyuridine (BrdU) labeling and immunocytochemical analysis
Cells were cultured on sterile multi-well chamber slides at a concentration of 10,000 cells per well and treated with vehicle or mifepristone for 24 h. Cells were exposed to 10 μmol/L BrdU (Molecular Probes) for 12 h. At the end of the incubation, cells were fixed with 4% paraformaldehyde, and BrdU incorporated into the DNA was visualized by immunocytochemistry. The slides were incubated overnight at 4 °C with an anti-BrdU monoclonal antibody conjugated to biotin (A-21303; Molecular Probes), washed in PBS, incubated with streptavidin-peroxidase (JacksonImmunoResearch Laboratories, West Grove, PA), and stained with diaminobenzidine (DAB; Sigma) solution for developing peroxidase activity. After washing with tap water the sections were counterstained with hematoxylin, dehydrated in serial alcohols to xylene, and mounted. In the negative control slides the primary antibody was replaced with PBS. Random fields were viewed with a Leica DM/LB microscope (Leica Microsystems), and the number of labeled and unlabeled cells was determined. At least 500 cells were counted for each sample.
Cell cycle analysis
After drug treatment, cells were trypsinized, pelleted by centrifugation at 500 g for 5 min, washed with PBS, and resuspended in cell cycle buffer [3.8 mmol/L sodium citrate (Sigma), 7 U/ml RNase A (Sigma), 0.1 % v/v Triton X-100 (Sigma), and 0.05 mg/mL propidium iodide (Sigma)] at a concentration of 1 × 106 cells/ml and stored in the dark at 4 °C until analysis (24 h). Cells were analyzed using a Becton-Dickinson FACScan flow cytometer and Verity Winlist software (Verity Software, Topsham, ME).
SDS-PAGE and Western blotting
Cells were washed twice with PBS, scrapped, pelleted and lysed by the addition of radioimmunoprecipitation assay (RIPA) buffer containing 50 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 1% NP-40 (Sigma), 0.25% sodium deoxycholate (Sigma), 1 mmol/L EDTA, 1 mmol/L PMSF (Sigma), 1 μg/mL pepstatin (Sigma), 1 mmol/L orthovanadate (Sigma) and 1 mmol/L sodium fluoride (Sigma). Cells were disrupted by passage through a 21 gauge needle, and gently rocked on ice for 30 min. Lysates were centrifuged at 16,000 × g at 4 °C for 15 min, and the supernatant was considered the whole cell extract. This was assayed for protein content using the bicinchoninic acid method (BCA; Pierce, Rockford, IL). The whole cell extracts were appropriately diluted in 6 × concentrated electrophoresis sample buffer, boiled for 10 min, and stored at -80 °C until electrophoresed. Equivalent amounts of protein (50 μg) per point were loaded in 10 or 12 % (w/v) acrylamide gels, subjected to SDS-PAGE and transferred to PVDF membranes. The blots were blocked in 5% (w/v) nonfat milk in Tris-buffered saline (TBS) containing 0.1% (v/v) Tween 20 (T). Blots were then probed overnight with the appropriate dilution of each of the primary antibodies. The membranes were washed 3 × 5 min in TBS-T and incubated with 1:5,000 to 10,000 dilution of peroxidase-conjugate secondary antibody for 1 h at room temperature. The blots were again washed and developed by chemiluminescence and exposed to radiographic film. Blots were stripped and reprobed with an antibody directed against the ubiquitous proteins glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or beta actin to control for protein loading.
Antibodies for Western blot analysis
Primary antibodies for the following proteins were used at the indicated dilutions. Cyclin E (clone HE12; 0.5 μg/mL), cyclin A (clone BF683; 1 μg/mL), E2F1 (clone KH95/E2F; 0.5 μg/mL), p21cip1 (clone 6B6; 2 μg/mL) were from BD Pharmigen (San Diego, CA); p27kip1 (clone 57; 1: 2,000), and Cdk1/cdc2 (clone 1; 1: 2,500) were from BD Transduction Laboratories (San Diego, CA); cyclin B1 (Clone V152; 1/2,000) and poly (ADP-ribose) polymerase (PARP; #9592; 1:1,000) were from Cell Signaling Technology (Danvers, MA); cdk2 (M2; 1:1,000) was from Santa Cruz Biotechnology (Santa Cruz, CA); GAPDH (ab9485; 1:10,000) was from Abcam Inc. (Cambridge, MA); beta actin (clone AC-15; 1:20,000) was from Sigma. Peroxidase-conjugated secondary antibodies were obtained from Jackson ImmunoResearch Laboratories.
Cdk2 immunoprecipitation and histone H1 kinase assay
An aliquot (600 μg of protein) from each cell lysate [NP-40 lysis buffer; 50 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 0.5% NP-40, 1 mmol/L DTT, 2 μg/ml aprotinin, 2 μg/ml leupeptin, 2 μg/ml pepstatin, 1 mmol/L PMSF, 50 mmol/L sodium fluoride, 1 mm/L activated sodium orthovanadate] was incubated with 2 μg polyclonal rabbit antibody to cdk2 (M2; Santa Cruz Biotechnology) overnight at 4 °C. Immunocomplexes associated with cdk2 were collected after incubating for 2 h with protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology). The immune-complexes were washed twice with lysis buffer, mixed with 2 × electrophoresis sample buffer, boiled, and then resolved by SDS-PAGE and analyzed for their components by immunoblotting using the corresponding antibodies for cdk2, cyclin E, p27kip1, or p21cip1 as described above.
For cdk2-dependent kinase assays, cell lysates (100 μg protein) were incubated overnight at 4 °C with constant rotation in 0.5 ml of NP-40 lysis buffer containing 1 μg of anti-cdk2 antibody (M2; Santa Cruz Biotechnology). The mixture was then incubated for 2 h at 4 °C with 25 μl of protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology). Immunocomplexes were washed three times with lysis buffer and twice with kinase buffer [50 mmol/L HEPES (pH 7.2), 10 mmol/L MgCl2 1 mm/L DTT, 1 mmol/L sodium fluoride, and 10 mmol/L β-glycerophosphate]. Subsequently, the beads were resuspended in 30 μl of kinase buffer containing 2 μg of histone H1 (Upstate Cell Signaling Solutions, Lake Placid, NY), 5 μmol/L ATP (Upstate), 5 μCi of [γ32P] ATP (MP Biomedicals, Irvine, CA). The reaction mixtures were incubated at 30 °C for 30 min, the reaction terminated with 30 μl of 2 × electrophoresis sample buffer, boiled, and separated on 12% SDS/polyacrylamide gels. Gels were stained with Coomassie Blue (Sigma) to visualize the histone H1 bands, dried, and autoradiographed.
Subcellular fractionation
Upon treatment with vehicle or mifepristone, cells were washed in PBS, scrapped, pelleted, and resuspended in low salt lysis buffer [10 mmol/L HEPES (pH 7.9), 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L DTT, 1 mmol/L PMSF, 1 mmol/L sodium orthovanadate, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 2 μg/ml pepstatin], incubated on ice for 15 min and homogenized with a hand pestle. The lysates where then centrifuged for 10 min at 800 × g at 4 °C, and the supernatant was collected (cytosolic fraction). The pellet was resuspended in NP-40 lysis buffer (described above) and rocked for 1 h at 4 °C. The suspension was centrifuged at 16,000 × g for 20 min to remove debris, and the supernatant was considered the nuclear fraction. The protein concentrations of both factions were determined as described above. 50 μg of each of the protein fractions were subjected to SDS-PAGE, transferred to PVDF membranes, and immunoblotted with antibodies against p27 kip1, p21 cip1, cdk2, cyclin E, PARP (for nuclear fraction purity) and actin (for cytoplasmic fraction purity and loading control).
Nude mouse studies of SK-OV-3 tumor xenografts
In vivo studies were done following approval by the University of South Dakota Institutional Animal Care and Use Committee (study protocol 50-01-05-08B). Immunodeficient (athymic nude-Foxn1nu) female mice (Harlan, Indianapolis, IN) at 6-8 weeks of age (approx. 23 g in weight) were inoculated s.c. with 1 × 106 log-phase SK-OV-3 cells per site at two sites (right and left flanks). When tumors had developed and the average tumor volume was approximately 50 mm3, animals were randomized into 3 groups of 10 animals each and were implanted s.c. with constant release pellets (Innovative Research of America, Sarasota, FL) containing mifepristone (0.5 or 1 mg/day) or placebo, using a 10-gauge precision trochar. Tumor size and animal weight were measured every 5 days beginning on the day of pellet implantation (day 0). After forty days the animals were sacrificed. Tumors were measured, excised, fixed in 4% paraformaldehyde and embedded in paraffin for histological studies. Tumor volume (in mm3) was calculated according to the following formula: [(W)2 × L]/2 where W and L are the width (perpendicular smaller diameter) and length (perpendicular larger diameter) of the tumor, respectively. The W and L of the tumors were measured using an electronic digital caliper with a resolution of 0.01 mm. Because the range of volumes of experimental tumors was normally large (typically a 5-fold range between the largest and the smallest tumor used in an experiment) we compared relative tumor volumes for individual tumors rather than absolute values. For an individual tumor, the relative tumor volume (Vt/V0) was calculated from its volume at a particular time (Vt) divided by its volume at the start of treatment (V0) on day 0. The mean relative tumor volume for the group was plotted against time for each group to compare effects. The tumor mass doubling times (DT) were estimated by calculating the tumor volume variations over forty days using the following formula (31): DT = 40 × log2/log(V40/V0), where V40 is the volume of the tumor before sacrifice and V0 is the volume of the tumor at the beginning of treatment (day 0). Body weight was expressed in grams and was used to monitor relative systemic toxicity of mifepristone.
Statistics
All data are reported as means ± standard error and statistical significance was defined at P < 0.05. To compare the efficacy of steroid compounds on cell growth inhibition, clonogenic survival, and cell cycle distribution, one-way analysis of variance followed by the Dunnett’s multiple comparison test or two-way analysis of variance followed by the Bonferroni’s multiple comparison test were used as appropriate. To determine significant differences in relative tumor volume (Vt/V0) between groups, repeated-measures two-way (time × treatment) analysis of variance followed by the Bonferroni method as post test was used. The variance of tumor volume increased as the mean volume increased with time in accordance with exponential growth. The variation of tumor volume was much larger on any given day in the control group than in the treated groups, suggesting greater differences in tumor growth among the animals in the control group. A transformation of the data [Y=SQRT (Y)] was done to reduce this variance difference and to normalize the data before analysis.
Results
Mifepristone inhibits ovarian cancer cell growth displaying progesterone-like effect
The effect of mifepristone was first studied together with a panel of steroids composed of progesterone and synthetic progestins, on the growth of SK-OV-3 ovarian cancer cells. The cells were subjected for 4 days to treatment with different doses of progesterone, the synthetic progestins levonorgestrel and medroxyprogesterone acetate, and the progesterone receptor modulator mifepristone. In all cases the number of attached cells was counted for each experimental group before the initiation of the treatment. Media containing each of the doses of fresh steroids was replaced every 24 h for 4 days. At the end of the experiment, the number of viable cells was evaluated by trypsinization and counting in a hemacytometer chamber using trypan blue dye exclusion. Micromolar concentrations of progesterone, levonorgestrel, medroxyprogesterone acetate and mifepristone significantly inhibited the growth of SK-OV-3 cells when compared to the growth achieved by cells treated only with vehicle (Fig. 1). The results also show that mifepristone was a more potent suppressor of SK-OV-3 cell growth than the progestins tested. Overall the results depicted in Fig. 1 clearly show the efficacy of progestins and mifepristone as growth inhibitors in SK-OV-3 cells, and demonstrate that, in terms of growth suppression, mifepristone has progesterone-like effects.
Fig. 1.
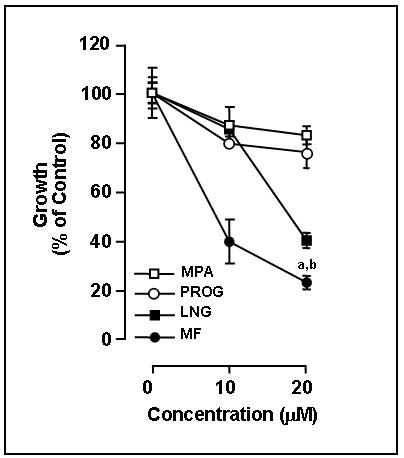
Inhibition of SK-OV-3 cell growth by progestins and mifepristone. SK-OV-3 cells were cultured in the presence of the indicated concentrations of progesterone (PROG), levonorgestrel (LNG), medroxyprogesterone acetate (MPA), or mifepristone (MF). The number of viable cells was recorded at the beginning of the experiment and after 4 days of treatment. The difference between number of cells in vehicle-treated controls at 0 h and after 4 days of culture was considered to be 100%. The growth of the treated groups is expressed as percentage of control. Results are the average of triplicate counts ± SEM. a, P < 0.05 vs. LNG; b, P < 0.001 vs. PROG and MPA.
Mifepristone inhibits growth of ovarian cancer cells of different genetic backgrounds
To define whether mifepristone inhibits growth of ovarian cancer cells of different genetic backgrounds, we subjected various ovarian cancer cell lines (SK-OV-3, Caov-3, IGROV-1, and OV2008) to a fixed, 20 μM concentration of mifepristone for 3 days, with the compound being replaced every 24 h. In all cell lines exposure to 20 μM mifepristone for 24 h was sufficient to arrest cell growth. The arrest persisted during the 3 days studied (Fig. 2A-D). Furthermore, a dose-response experiment was done with SK-OV-3 and OV2008 cells, which were exposed to various concentrations of mifepristone for 3 days. Results shown in Fig. 2E indicate that mifepristone inhibits ovarian cancer cell growth in a dose-dependent manner with a 50%-inhibition concentration achieved with approx. 6-7 μM (proliferation IC50s, 6.25 μM for SK-OV-3 and 6.91 μM for OV2008).
Fig. 2.
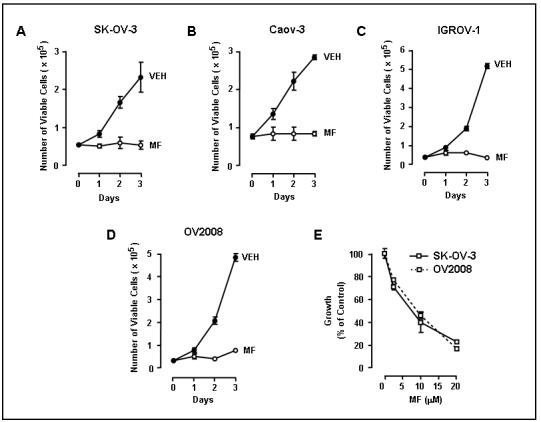
Inhibition of cell growth by mifepristone in various ovarian cancer cell lines. SK-OV-3 (A), Caov-3 (B), IGROV-1 (C) or OV2008 (D) cells were cultured in the presence of 20 μM mifepristone (MF) for the indicated times and the number of viable cells was recorded. In E, a dose response experiment is depicted for SK-OV-3 and OV2008 cells. Because SK-OV-3 and OV2008 have a different pattern of growth, the results of this experiment, in order to be combined within one diagram, are displayed as percent of vehicle-treated control, whose growth was considered to be 100%.
Mifepristone decreases cloning efficiency of ovarian cancer cells inducing cytostasis
To further confirm the cytostatic effect of mifepristone, we performed a clonogenic survival assay using SK-OV-3 and OV2008 cells. Cells were plated at a low density and treated with vehicle or various doses of mifepristone for different times. Treatment was then removed and cells were cultured in mifepristone-free media for 7 days until colonies having more than 50 cells became visible in vehicle treated controls. Increasing the concentration of mifepristone or extending the exposure time of the cells to mifepristone reduced the number of colonies, confirming the growth inhibitory effect of the steroid (Fig. 3A and 3B). To determine whether this growth inhibitory effect was reversible, a characteristic of cytostatic agents (32), cells were treated with various concentrations of mifepristone for 3 days, trypsinized and re-plated at low density together with cells that had been pretreated with vehicle. Results in Fig. 3C show that the number of colonies did not significantly vary between controls and mifepristone-pretreated groups, suggesting that the inhibition of growth by mifepristone is elicited only when the drug is present, and it does not involve cell toxicity. Data shown in Figs. 1-3 suggest that the action of mifepristone as a cell growth inhibitor in ovarian cancer cells is limited to cytostasis at doses up to 20 μM. To confirm the lack of cytotoxicity upon exposure to 20 μM mifepristone, cells that were cultured in the presence of this dose of mifepristone for 3 days, were exposed to the combination of fluorochromes calcein AM and EthD-1. Calcein AM is a cell permeant compound that is converted by cytoplasmic esterases into a green fluorescent product. EthD-1 binds DNA with red fluorescence, but only when the plasma membrane of the cells is compromised (33). The viability of the cells after mifepristone exposure was clearly indicated by the catalysis of calcein AM into a green fluorescence product. This was further confirmed by the lack of red fluorescence in the nuclei, indicative of a lack of binding of EthD-1 to DNA, confirming the integrity of the plasma membrane of mifepristone-treated cells (Fig. 3D). As a control, mifepristone treated cells were exposed to calcein AM and EthD-1 in the presence of saponin to permeabilize the plasma membrane (34). Fig. 3E shows that the presence of saponin was sufficient to allow EthD-1 to enter the cells and to bind DNA as indicated by the red nuclear fluorescence. In conclusion, our results demonstrate that, at doses up to 20 μM, mifepristone is cytostatic but not cytotoxic to ovarian cancer cells.
Fig. 3.
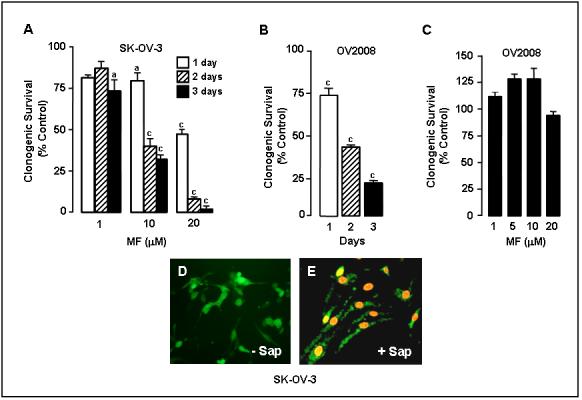
Effect of mifepristone on ovarian cancer cell clonogenic survival. In A, 250 SK-OV-3 cells were seeded in 6-well plates and exposed to 0, 1, 10 or 20 μM mifepristone (MF) for 1, 2, or 3 days. Following treatment the cells were cultured in regular growth medium for 7 days. At the end of the experiment the cells were fixed with 100% methanol and stained with 0.1% crystal violet. Colonies containing ≥ 50 cells were counted. The results are shown as number of colonies counted for the different experimental groups expressed as percentage of the control, which was considered to be 100%; a, P < 0.01 and c, P < 0.001 compared with control. In B, an experiment similar to that in A was done with OV2008 cells. The cells were exposed to 20 μM MF for 1, 2, or 3 days (c, P < 0.001 vs. control). In C, OV2008 cells were incubated with the indicated concentration of MF for 3 days, after which the cells were trypsinized, plated in a clonogenic survival assay, and their growth compared tocells that had not been treated with MF and whose growth was considered to be 100%. D and E show the results of a viability/cytotoxicity assay of SK-OV-3 cells upon exposure to MF. Cells were treated with 20 μM MF for 72 h and exposed to calcein AM plus EthD-1 in the absence (-Sap) (D) or presence (+ Sap) (E) of 0.1% saponin. Magnification × 200.
Mifepristone inhibits DNA synthesis and induces G1 phase arrest in ovarian cancer cells
To examine the effect of mifepristone on the cell cycle in SK-OV-3 and OV2008 cells, the number of cells cycling was assessed by analyzing DNA synthesis with BrdU labeling. Cells were exposed to vehicle or mifepristone for 24 h. Twelve hours before the end of the experiment the cells were pulsed with 10 μM BrdU. Incorporation of BrdU into newly synthesized DNA was evaluated by immunocytochemistry. The bar graphs in Fig. 4A show that the percent of BrdU labeled nuclei abruptly decreased in mifepristone-treated cells compared to vehicle-treated controls. The decrease was ∼4 fold in SK-OV-3 cells and ∼10 fold in OV2008 cells. To further evaluate the effect of mifepristone on cell cycle traverse, SK-OV-3 and OV2008 cells were cultured in the presence of vehicle or 20 μM mifepristone for 1, 2, or 3 days. At the end of the experiment, the cells were stained with propidium iodide and the distribution of cells within the cell cycle was analyzed by flow cytometry. Results, shown in Fig. 4B and 4C indicate that exposure of cells to mifepristone for 1 day significantly reduced the percentage of cells in S phase while inducing a reciprocal increase in the percentage of cells in G0/G1. Percentage of cells in G2/M was significantly decreased in OV2008, yet was unchanged in SK-OV-3. Exposure of the cells to mifepristone longer than 1 day induced a further decrease in the S-phase fraction while inducing additional accumulation of cells in G0/G1. No cells had DNA content within the sub-G0/G1 region of the histogram, indicating the absence of cells with apoptotic characteristics in both control and treated groups.
Fig. 4.
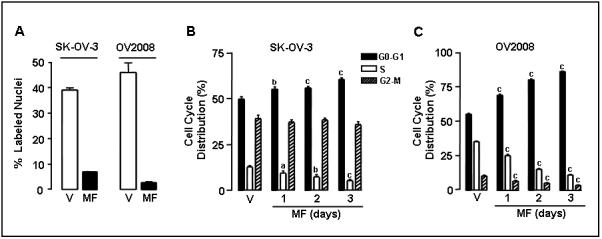
Effect of mifepristone on BrdU incorporation into DNA and on cell cycle kinetics in ovarian cancer cells. A, SK-OV-3 and OV2008 cells were cultured on multi-well slides and treated with either vehicle (V) or 20 μM mifepristone (MF) for 24 h. Cells were exposed to 10 μM BrdU for 12 h. At the end of the experiment, the cells were fixed with 4% paraformaldehyde, and BrdU incorporated into the DNA was visualized by immunocytochemistry using an anti-BrdU monoclonal antibody. BrdU positive nuclei were quantified using a microscope with a 100 × magnification and at least 500 cells were counted for each sample. The experiment was done in triplicate and the results are expressed as percent of BrdU positive cells. B, Logarithmically growing SK-OV-3 and OV2008 ovarian cancer cells were exposed to vehicle (V) or 20 μM MF for 1, 2, or 3 days. The percentage of cells in the G0/G1-, S-, and G2/M-phases of the cell cycle was determined by cytometric analysis of propidium iodide-stained cells. Values represent the means of two separate experiments performed in triplicate ± SEM. a, P < 0.05; b, P < 0.01; and c, P < 0.001 relative to the control group.
Mifepristone changes the expression of several cell cycle regulatory proteins
To study the molecular mechanisms involved in the G1 cell cycle arrest induced by mifepristone in ovarian cancer cells, we explored the G1-to-S transition, which is ultimately controlled by the cyclin E in association with cdk2. This complex phosphorylates the retinoblastoma tumor suppressor which leads to the release of the E2F transcription factor. E2F controls progression through the S phase (35). Mifepristone provoked slight decreases in the expression of cdk2 and cyclin E after 2 days of treatment (Fig. 5A). In addition, expression of p21cip1 and p27kip1 was abruptly up-regulated 1 day after exposure to mifepristone (Fig. 5A). Mifepristone also induced downregulation of the E2F1 transcription factor (Fig. 5B). This was associated with the downregulation of genes whose expression is dependent on the progression of S phase of the cell cycle, such as cyclin A, cyclin B1, and cdk1 (also known as cdc2) (Fig. 5B). Reduction in expression of the S phase cyclin (cyclin A) and the M phase cyclin (cyclin B1) is most likely the cause of a reduced number of cycling cells in mifepristone-treated cells. Together DNA synthesis studies, cytometry data, and cell cycle regulatory protein expression suggest that mifepristone arrests ovarian cancer cells at the late G1 phase of the cell cycle.
Fig. 5.
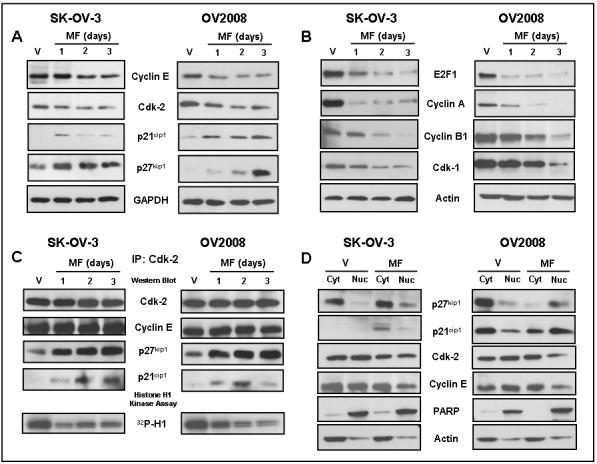
Effect of mifepristone on the expression of cell cycle regulatory proteins, cdk2 activity, association of cdk2 with p21cip1 and p27kip1, and subcellular localization of p27kip1, p21cip1, cdk2 and cyclin E. SK-OV3 or OV2008 cells were exposed to vehicle or 20 μM mifepristone (MF) for the indicated times or for 24 h when not indicated. Whole protein extracts were obtained and separated by electrophoresis, and immunoblots were probed with the indicated antibodies. The housekeeping genes GAPDH (panel A) and beta actin (panel B) were used as loading controls. C, whole protein extracts were immunoprecipitated with anti-cdk2 antibody. The immunoprecipitates were Western blotted for cdk2, cyclin E or the cdk inhibitors p21cip1 and p27kip1. The immunoprecipitates were also assayed for their capacity to phosphorylate histone H1 in vitro in the presence of 32P ATP. Products of the kinase reaction were run on polyacrylamide gels and the phosphorylated histone H1 (32P-H1) was analyzed by autoradiography. In a third experiment shown in panel D, cells were treated with vehicle or 20 μM mifepristone for 24 h. The cells were then fractionated into nuclear and cytosolic fractions and proteins from each respective fraction were subjected to immunoblot analysis with antibodies to p27kip1, p21cip1, cdk2, or cyclin E. PARP and actin were used to assess the purity of the subcellular fractions. Actin was further utilized to control for protein loading. The results were repeated twice with similar outcome.
Mifepristone increases association of p21cip1 and p27kip1 with cdk2, inhibits cdk2 activity, promotes nuclear localization of p21cip1 and p27kip1, and reduces nuclear abundances of cdk2 and cyclin E
Because we observed a decrease in cdk2 abundance together with a large increase in the abundance of cdk inhibitors, we asked whether the activity of cdk2 was also affected by mifepristone. We immunoprecipitated cdk2 from control cells or from cells treated with mifepristone and performed an in vitro kinase assay using histone H1 as substrate. The results, shown in Fig. 5C, demonstrate that within 1 day of exposure to mifepristone, the activity of cdk2 was inhibited in the absence of any change in the abundance of the enzyme that was immunoprecipitated. In addition, there is a remarkable increase in the amounts of p21cip1 and p27kip1 that co-immunoprecipitates with cdk2, suggesting that the inhibition is most likely due to an increase in the binding of the inhibitors p21cip1 and p27kip1. To evaluate whether mifepristone affects the nucleocytoplasmic trafficking of the cdk inhibitors and cdk2, vehicle- and mifepristone-treated cells were subjected to subcellular fractionation and Western blot analysis. Results shown in Fig. 5D reveal that 24 h incubation with mifepristone induced an increase in nuclear localization of p21cip1 and p27kip1, what correlated with decreased cdk2 and cyclin E nuclear levels. These results suggest that mifepristone-mediated inhibition of cdk2 activity also involves decreased cdk2 and cyclin E nuclear abundances, together with increased nuclear abundances of the cdk inhibitors p21cip1 and p27kip1.
Mifepristone inhibits growth of ovarian cancer xenografts in nude mice
In order to study whether mifepristone is capable of inhibiting ovarian cancer tumor growth in vivo, we generated s.c. xenografts of SK-OV-3 cells in immunosuppressed mice. Female mice of approx. 7 weeks of age were inoculated s.c. with 1 × 106 SK-OV-3 cells per site at two sites (right and left flanks). When tumors developed and the average tumor volume was approx. 50 mm3, animals were randomized into three groups that were implanted s.c. with pellets customized to release either placebo, or mifepristone at rates of either 0.5 mg/day or 1 mg/day. The volume of the tumors was measured every 5 days. For an individual tumor, the relative tumor volumes (Vt/V0) (n = 12-19 per group) was calculated from its volume at a particular time (Vt) divided by its volume at the start of treatment (V0) on day 0. The results show that the growth rate of tumors was significantly inhibited by mifepristone at either dose of mifepristone beginning 20 days after the beginning of the treatment (Fig. 6A). The dose-dependent growth inhibition was also confirmed by the fact that the rate of tumor growth achieved after 40 days of treatment with 1 mg/day mifepristone was significantly lower than that of animals treated with 0.5 mg/day of the drug. The calculated doubling times (DT) over the 40 days of the study for the placebo-treated tumors was 9.5 ± 0.6 days (n=12), 14.1 ± 1.6 days for the 0.5 mg/day mifepristone-treated tumors (n=16), and 17.2 ± 1.6 days for the 1 mg/day mifepristone-treated tumors (n=12). This further highlight the capacity of mifepristone to impair the growth of tumors generated upon subcutaneous inoculation of SK-OV-3 ovarian cancer cells into immunosuppressed mice. The animals appeared to tolerate the chronic exposure to mifepristone well as evidenced by stable animal weights that were indistinguishable between the arms of the study (Fig. 6B).
Fig. 6.
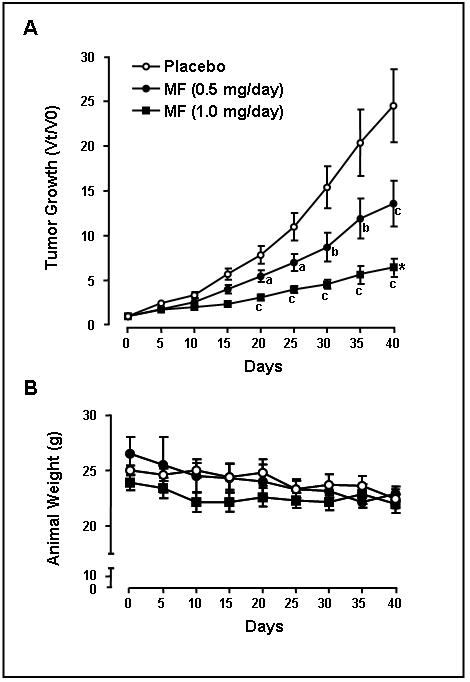
Effect of mifepristone on tumor growth in vivo. A, growth curve for SK-OV-3 tumors in nude mice that were treated with placebo (open circles), 0.5 mg/day mifepristone (closed circles), or 1 mg/day mifepristone (closed squares). Tumor sizes are expressed as relative volume at a particular time (Vt) divided by its volume at the beginning of the treatment (V0) on day 0. a, P < 0.05; b, P < 0.01; and c, P < 0.001 compared to its time-matched placebos, and * P < 0.05 compared with mifepristone-treated at 0.5 mg/day. B, body weights were stable during treatment and similar for the placebo-treated (open circles), mifepristone-treated at 0.5 mg/day (closed circles) or mifepristone-treated at 1 mg/day (closed squares) animals.
Discussion
Major clinical problems in the treatment of ovarian cancer include the resistance to current therapy, the elevated toxicity of standard drugs, and the repopulation of cells that escape chemotherapy. Therefore, the development of novel strategies for ovarian cancer treatment is of significant importance. In this report, we demonstrate that the contraceptive steroid mifepristone is a potent cytostatic agent to human ovarian cancer cells of different genetic backgrounds, inducing cell cycle arrest and blocking the G1-to-S phase transition. Mifepristone-induced G1 arrest involves inhibition of cdk2 activity likely due to a combination of decreased nuclear cdk2 abundance and increased recruitment of cdk inhibitors p21cip1 and p27kip1 to the nucleus. The decline in cdk2 activity is associated with decreased abundance of the transcription factor E2F1 that drives S phase progression. Moreover, we provide evidence that mifepristone, at doses that are likely to be achieved clinically, has an antiproliferative action not associated with cell death.
Blockage of ovarian cancer cells at the G1-to-S transition was shown by the interference of mifepristone with the incorporation of BrdU into newly synthesized DNA molecules. The remarkable inhibition of DNA synthesis induced by mifepristone was consistent with the abrupt interruption in cell growth observed within 24 h of exposure to the drug, the reduction in the number of cells in S phase, and the accumulation of cells in G1 phase. Blockage of the cell cycle in G1 after exposure to mifepristone has also been reported in human gastric adenocarcinoma cells (26), T-47D breast cancer cells (36), and in OVCAR-3 and A2780 ovarian cancer cells (27). In T-47D breast cancer cells, the inhibition of cell cycle progression by mifepristone was accompanied by induction of the cyclin-dependent kinase inhibitor p21cip1 (36). Similar to mifepristone, the synthetic progestin Org2058 also induced T-47D cell cycle arrest mediated through the cooperation of the cell cycle inhibitors p27kip1 and p18ink4 (37). The antiproliferative effect of mifepristone in ovarian SK-OV-3 and OV2008 cells was associated with an increase in the levels of p21cip1 and p27kip1 and with their relocalization to the nucleus; these proteins are likely responsible for the inhibition of cdk2 activity since both co-immunoprecipitated with cdk2. Further inhibition of cdk2 activity may be a result of the decrease of cdk2 protein levels in the nucleus upon sustained exposure to mifepristone. This is because nuclear localization of cdk2 is required for its full activation, leading to the progression of the cell cycle (38, 39). Hence, in ovarian cancer cells cdk2, upon which the cells depend to pass through the G1-S interface (40), had a dual regulation by mifepristone, which reduces its activity and its nuclear abundance. Because cdk2 is frequently up-regulated in ovarian tumors (41), the potent inhibitory effect on cdk2 displayed by mifepristone in ovarian cancer cells may be of central importance in the development of new therapeutic approaches using the steroid. Moreover, the increased abundance of p21cip1 and p27kip1 and their localization to the nucleus in response to mifepristone is of significance because it is generally accepted that nuclear but not cytoplasmic cdk inhibitors are the catalytic inhibitors of cdk2 (42), and also because cytoplasmic localization of p27kip1 in ovarian cancer patients is associated with poor prognosis (43). Thus, mifepristone may be particularly useful in rescuing the tight inhibitory control of cdk inhibitors on the activity of cdk2, which is frequently lost in ovarian cancer. In addition, nuclear p21cip1 and p27kip1, apart from blocking cdk2 activity in response to mifepristone, might be involved as transcriptional cofactors leading to the inhibition of transcription of cell cycle promoting genes (42).
E2F1 is a key transcription factor required for the transactivation of target genes involved in the G1-to-S transition and for correct progression through the cell cycle (44). E2F1 expression was greatly inhibited by mifepristone. This was in agreement with the lack of cell cycle progression observed in ovarian cancer cells after exposure to the drug. E2F1 downregulation may have two components. On one hand the abrupt decline in cdk2 activity may reduce the degree of phosphorylation of the retinoblastoma protein making E2F1 less available for gene regulation. Further, a downregulation of E2F1 by mifepristone is also evident. Because commitment of the cell to S phase progression requires cdk2 activity and E2F1 availability, the downregulation of cdk2 (activity and abundance), together with the downregulation of E2F1, explains the cell cycle arrest of ovarian cancer cells in G1. As expected, mifepristone led to the downregulation of genes needed to progress through S phase, such as cyclin A, cyclin B1, and cdk1 (40).
The cytostatic effect of mifepristone displayed similar potency among the ovarian cancer cell lines studied. This effect occurred irrespective of the p53 background of the cell lines, as SK-OV-3 is p53 null (45), Caov-3 cells express mutant p53 (46), whereas IGROV-1 and OV2008 cells express wild-type p53 (47, 48). This observation is of particular importance because mutations of the p53 tumor suppressor occur at extremely high frequencies in ovarian cancer (49). Thus, mifepristone would be equally effective in tumors with either wild-type p53 or mutant p53 expression. Furthermore, the similar potency of growth inhibition achieved by mifepristone in SK-OV-3 (IC50, 6.25 μM), which are intrinsically resistant to cisplatin (50), and OV2008 (IC50, 6.91 μM) that are sensitive to cisplatin therapy (51), suggest that mifepristone could be useful in abrogating cell growth in ovarian cancer irrespective of the resistance to cisplatin. This is of particular significance when considering that acquisition of resistance to cisplatin occurs with high frequency in patients with recurrent ovarian cancer disease.
At the doses used in this study, the effect of mifepristone was limited to cytostasis demonstrated by the reversibility of the growth inhibition observed when the drug was removed from the culture media, in association with the lack of measurable cell death. It has been reported previously that mifepristone, at concentrations higher than 20 μM, can trigger cell death in OVCAR-3 cells (27). In the cell lines used in our study, concentrations of mifepristone 30 μM or higher display cytotoxic activity (results not shown), suggesting that 20 μM appears to be the threshold for cell cycle inhibition in these ovarian cancer cell lines. Several pharmacokinetic studies in humans report that, when administered orally in doses ranging from 10 to 800 milligrams, mifepristone reaches concentrations in circulation ranging from 2.5 to 20 μM, but not higher (52-55). Although most studies using mifepristone in humans were conducted using short-term treatment schedules, a recent study in patients with non-resectable meningioma who received long-term (months to years) therapy showed that the drug can be safely administered for prolonged periods of time with manageable side effects (56). In our study the pellets of mifepristone used were designed to release either 0.5 or 1 mg/day. Although the actual steady-state concentrations of mifepristone attained in circulation in these mice were not measured, the fact that tumor growth rate was reduced in a dose-dependent manner and without apparent discomfort to the animals, confirms the effectiveness of the compound in vivo. The dose of mifepristone used in our study is within the range used in another investigation in which growth inhibition of prostate cancer xenografts was observed (25); as in our study, no significant changes in the overall weight of the animals were observed upon chronic exposure to mifepristone.
Mifepristone behaved similar to synthetic progestins inducing cell growth arrest, although with greater potency. It is possible that the cytostatic effect of mifepristone is mediated by an agonistic action on progesterone receptors. Support for this idea is that in breast cancer cells, mifepristone interfered with cell proliferation displaying progesterone-like effects (57). In MDA-MB-231 breast cancer cells transfected with the progesterone receptor, mifepristone, akin to progesterone, inhibited cell growth by arresting the cells in the G0/G1 phase of the cell cycle (58). In another study the antiproliferative effect of mifepristone was additive to tamoxifen in MCF-7 cells expressing both estrogen and progesterone receptors (14). However, such antiproliferative action of mifepristone was also observed in estrogen receptor and progesterone receptor negative MDA-231 cells (15), suggesting that the presence of progesterone receptors may not be required for mifepristone to elicit its growth inhibitory effect. Furthermore, the reported level of expression of progesterone receptors in ovarian cancer cell lines is not without controversy. For example Caov-3 cells were reported to express progesterone receptor mRNA in one study (59) but not in another report (60). Similarly, studies in SK-OV-3 cells showing some and no expression of progesterone receptor mRNA have been published (60-62). We have detected progesterone receptor immunoreactive proteins in OV2008 cells but failed to show any reactive proteins in SK-OV-3 cells (data not shown), leading to more doubt about the involvement of progesterone receptors in mifepristone-induced growth inhibition.
Another possibility is that mifepristone may drive its anticancer action through glucocorticoid receptors. This is because it can bind to glucocorticoid receptors with an affinity similar to that for progesterone receptors (63), and ovarian cancer cells have been reported to express glucocorticoid receptors (64). In this regard, we have detected abundant levels of glucocorticoid receptor immunoreactive proteins in both SK-OV-3 and OV2008 cell lines (results not shown). However, when SK-OV-3 and OV2008 cells were cultured in the presence of the glucocorticoid agonist dexamethasone at concentrations equimolar to cytostatic mifepristone, no modifications in cell growth were observed (data not shown), suggesting that even if mifepristone binds glucocorticoid receptors in the ovarian cancer cells it may not trigger receptor transactivation.
A final possibility is that mifepristone may have an effect that does not involve specific hormone receptors. In this regard, mifepristone was shown to have a potent antioxidant effect manifested at micromolar concentrations and attributed to the presence of the dimethylamino phenyl side chain of the molecule (65). Further, the growth inhibitory effect of mifepristone in endometrial cells and macrophages has been attributed, at least in part, to the antioxidant property of the compound (23, 66). A putative antioxidant effect of mifepristone on ovarian cancer cells could be interesting in the context of G1 arrest associated with p21cip1 upregulation, because p21cip1 can be induced in response to some antioxidants in a p53 independent fashion (67, 68). We have shown that mifepristone-induced cell growth arrest is associated with p21cip1 increase in both wild-type p53 expressing OV2008 cells and in p53 null SK-OV3, opening the possibility that mifepristone acts as an antioxidant to drive G1 arrest through a p53-independent p21cip1 upregulation.
In summary, we have shown that mifepristone is a potent blocker of ovarian cancer growth in vitro and in vivo. The feasibility of using mifepristone to enhance the efficacy of conventional chemotherapy for ovarian cancer is encouraging and requires further investigation.
Acknowledgments
We thank to Drs. Barbara Goodman, Ronald Lindahl and Robin Miskimins for the critical reading of the article.
Grant support: This research was supported by NIH Grant Number 2 P20 RR016479 from the INBRE Program of the National Center for Research Resources and funds from the University of South Dakota
References
- 1.Philibert D, Moguilewsky M, Mary I, et al. Pharmacological profile of RU486 in animals. In: Baulieu EE, Segal SJ, editors. The Antiprogesterone Steroid RU486 and Human Fertility Control. Plenum Press; NY: 1985. [Google Scholar]
- 2.Ho PC, Yu Ng EH, Tang OS. Mifepristone: contraceptive and non-contraceptive uses. Curr Opin Obstet Gynecol. 2002;14:325–30. doi: 10.1097/00001703-200206000-00013. [DOI] [PubMed] [Google Scholar]
- 3.Nordeen SK, Bona BJ, Moyer ML. Latent agonist activity of the steroid antagonist, RU486, is unmasked in cells treated with activators of protein kinase A. Mol Endocrinol. 1993;7:731–42. doi: 10.1210/mend.7.6.8395651. [DOI] [PubMed] [Google Scholar]
- 4.Sartorius CA, Tung L, Takimoto GS, Horwitz KB. Antagonist-occupied human progesterone receptors bound to DNA are functionally switched to transcriptional agonists by cAMP. J Biol Chem. 1993;268:9262–6. [PubMed] [Google Scholar]
- 5.Kahmann S, Vassen L, Klein-Hitpass L. Synergistic enhancement of PRB-mediated RU486 and R5020 agonist activities through cyclic adenosine 3′,5′-monophosphate represents a delayed primary response. Mol Endocrinol. 1998;12:278–89. doi: 10.1210/mend.12.2.0067. [DOI] [PubMed] [Google Scholar]
- 6.Liu Z, Auboeuf D, Wong J, et al. Coactivator/corepressor ratios modulate PR-mediated transcription by the selective receptor modulator RU486. Proc Natl Acad Sci U S A. 2002;99:7940–4. doi: 10.1073/pnas.122225699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kettel LM, Murphy AA, Morales AJ, Yen SS. Clinical efficacy of the antiprogesterone RU486 in the treatment of endometriosis and uterine fibroids. Hum Reprod. 1994;9(Suppl 1):116–20. doi: 10.1093/humrep/9.suppl_1.116. [DOI] [PubMed] [Google Scholar]
- 8.Fedele L, Berlanda N. Emerging drugs for endometriosis. Expert Opin Emerg Drugs. 2004;9:167–77. doi: 10.1517/eoed.9.1.167.32945. [DOI] [PubMed] [Google Scholar]
- 9.Murphy AA, Morales AJ, Kettel LM, Yen SS. Regression of uterine leiomyomata to the antiprogesterone RU486: dose-response effect. Fertil Steril. 1995;64:187–90. [PubMed] [Google Scholar]
- 10.Steinauer J, Pritts EA, Jackson R, Jacoby AF. Systematic review of mifepristone for the treatment of uterine leiomyomata. Obstet Gynecol. 2004;103:1331–6. doi: 10.1097/01.AOG.0000127622.63269.8b. [DOI] [PubMed] [Google Scholar]
- 11.Matsuda Y, Kawamoto K, Kiya K, Kurisu K, Sugiyama K, Uozumi T. Antitumor effects of antiprogesterones on human meningioma cells in vitro and in vivo. J Neurosurg. 1994;80:527–34. doi: 10.3171/jns.1994.80.3.0527. [DOI] [PubMed] [Google Scholar]
- 12.Grunberg SM, Weiss MH, Spitz IM, et al. Treatment of unresectable meningiomas with the antiprogesterone agent mifepristone. J Neurosurg. 1991;74:861–6. doi: 10.3171/jns.1991.74.6.0861. [DOI] [PubMed] [Google Scholar]
- 13.Grunberg SM. Role of antiprogestational therapy for meningiomas. Hum Reprod. 1994;9(Suppl 1):202–7. doi: 10.1093/humrep/9.suppl_1.202. [DOI] [PubMed] [Google Scholar]
- 14.El Etreby MF, Liang Y, Wrenn RW, Schoenlein PV. Additive effect of mifepristone and tamoxifen on apoptotic pathways in MCF-7 human breast cancer cells. Breast Cancer Res Treat. 1998;51:149–68. doi: 10.1023/a:1006078032287. [DOI] [PubMed] [Google Scholar]
- 15.Liang Y, Hou M, Kallab AM, Barrett JT, El Etreby F, Schoenlein PV. Induction of antiproliferation and apoptosis in estrogen receptor negative MDA-231 human breast cancer cells by mifepristone and 4-hydroxytamoxifen combination therapy: a role for TGFbeta1. Int J Oncol. 2003;23:369–80. [PubMed] [Google Scholar]
- 16.Yokoyama Y, Shinohara A, Takahashi Y, et al. Synergistic effects of danazol and mifepristone on the cytotoxicity of UCN-01 in hormone-responsive breast cancer cells. Anticancer Res. 2000;20:3131–5. [PubMed] [Google Scholar]
- 17.Gaddy VT, Barrett JT, Delk JN, Kallab AM, Porter AG, Schoenlein PV. Mifepristone induces growth arrest, caspase activation, and apoptosis of estrogen receptor-expressing, antiestrogen-resistant breast cancer cells. Clin Cancer Res. 2004;10:5215–25. doi: 10.1158/1078-0432.CCR-03-0637. [DOI] [PubMed] [Google Scholar]
- 18.Klijn JG, de Jong FH, Bakker GH, Lamberts SW, Rodenburg CJ, Alexieva-Figusch J. Antiprogestins, a new form of endocrine therapy for human breast cancer. Cancer Res. 1989;49:2851–6. [PubMed] [Google Scholar]
- 19.Romieu G, Maudelonde T, Ulmann A, et al. The antiprogestin RU486 in advanced breast cancer: preliminary clinical trial. Bull Cancer. 1987;74:455–61. [PubMed] [Google Scholar]
- 20.Poole AJ, Li Y, Kim Y, Lin SC, Lee WH, Lee EY. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006;314:1467–70. doi: 10.1126/science.1130471. [DOI] [PubMed] [Google Scholar]
- 21.Narvekar N, Cameron S, Critchley HO, Lin S, Cheng L, Baird DT. Low-dose mifepristone inhibits endometrial proliferation and up-regulates androgen receptor. J Clin Endocrinol Metab. 2004;89:2491–7. doi: 10.1210/jc.2003-031945. [DOI] [PubMed] [Google Scholar]
- 22.Schneider CC, Gibb RK, Taylor DD, Wan T, Gercel-Taylor C. Inhibition of endometrial cancer cell lines by mifepristone (RU 486) J Soc Gynecol Investig. 1998;5:334–8. doi: 10.1016/s1071-5576(98)00037-9. [DOI] [PubMed] [Google Scholar]
- 23.Murphy AA, Zhou MH, Malkapuram S, Santanam N, Parthasarathy S, Sidell N. RU486-induced growth inhibition of human endometrial cells. Fertil Steril. 2000;74:1014–9. doi: 10.1016/s0015-0282(00)01606-x. [DOI] [PubMed] [Google Scholar]
- 24.Han S, Sidell N. RU486-induced growth inhibition of human endometrial cells involves the nuclear factor-kappa B signaling pathway. J Clin Endocrinol Metab. 2003;88:713–9. doi: 10.1210/jc.2002-020876. [DOI] [PubMed] [Google Scholar]
- 25.El Etreby MF, Liang Y, Johnson MH, Lewis RW. Antitumor activity of mifepristone in the human LNCaP, LNCaP-C4, and LNCaP-C4-2 prostate cancer models in nude mice. Prostate. 2000;42:99–106. doi: 10.1002/(sici)1097-0045(20000201)42:2<99::aid-pros3>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 26.Li DQ, Wang ZB, Bai J, et al. Effects of mifepristone on proliferation of human gastric adenocarcinoma cell line SGC-7901 in vitro. World J Gastroenterol. 2004;10:2628–31. doi: 10.3748/wjg.v10.i18.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rose FV, Barnea ER. Response of human ovarian carcinoma cell lines to antiprogestin mifepristone. Oncogene. 1996;12:999–1003. [PubMed] [Google Scholar]
- 28.Qin TN, Wang LL. Enhanced sensitivity of ovarian cell line to cisplatin induced by mifepristone and its mechanism. Di Yi Jun Yi Da Xue Xue Bao. 2002;22:344–6. [PubMed] [Google Scholar]
- 29.Liu Y, Wang LL, Deng Y. Enhancement of antitumor effect of cisplatin against human ovarian carcinoma cells by mifepristone in vivo. Di Yi Jun Yi Da Xue Xue Bao. 2003;23:242–4. [PubMed] [Google Scholar]
- 30.Rocereto TF, Saul HM, Aikins JA, Jr., Paulson J. Phase II study of mifepristone (RU486) in refractory ovarian cancer. Gynecol Oncol. 2000;77:429–32. doi: 10.1006/gyno.2000.5789. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz M. A biomathematical approach to clinical tumor growth. Cancer. 1961;14:1272–94. doi: 10.1002/1097-0142(196111/12)14:6<1272::aid-cncr2820140618>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 32.Freshney I. Application of cell cultures to toxicology. Cell Biol Toxicol. 2001;17:213–30. doi: 10.1023/a:1012572930721. [DOI] [PubMed] [Google Scholar]
- 33.Decherchi P, Cochard P, Gauthier P. Dual staining assessment of Schwann cell viability within whole peripheral nerves using calcein-AM and ethidium homodimer. J Neurosci Methods. 1997;71:205–13. doi: 10.1016/s0165-0270(96)00146-x. [DOI] [PubMed] [Google Scholar]
- 34.Ishida H, Hirota Y, Nakazawa H. Effect of sub-skinning concentrations of saponin on intracellular Ca2+ and plasma membrane fluidity in cultured cardiac cells. Biochim Biophys Acta. 1993;1145:58–62. doi: 10.1016/0005-2736(93)90381-9. [DOI] [PubMed] [Google Scholar]
- 35.Seville LL, Shah N, Westwell AD, Chan WC. Modulation of pRB/E2F functions in the regulation of cell cycle and in cancer. Curr Cancer Drug Targets. 2005;5:159–70. doi: 10.2174/1568009053765816. [DOI] [PubMed] [Google Scholar]
- 36.Musgrove EA, Lee CS, Cornish AL, Swarbrick A, Sutherland RL. Antiprogestin inhibition of cell cycle progression in T-47D breast cancer cells is accompanied by induction of the cyclin-dependent kinase inhibitor p21. Mol Endocrinol. 1997;11:54–66. doi: 10.1210/mend.11.1.9869. [DOI] [PubMed] [Google Scholar]
- 37.Swarbrick A, Lee CS, Sutherland RL, Musgrove EA. Cooperation of p27(Kip1) and p18(INK4c) in progestin-mediated cell cycle arrest in T-47D breast cancer cells. Mol Cell Biol. 2000;20:2581–91. doi: 10.1128/mcb.20.7.2581-2591.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang ES, Burnstein KL. Vitamin D inhibits G1 to S progression in LNCaP prostate cancer cells through p27Kip1 stabilization and Cdk2 mislocalization to the cytoplasm. J Biol Chem. 2003;278:46862–8. doi: 10.1074/jbc.M306340200. [DOI] [PubMed] [Google Scholar]
- 39.Brown KA, Roberts RL, Arteaga CL, Law BK. Transforming growth factor-beta induces Cdk2 relocalization to the cytoplasm coincident with dephosphorylation of retinoblastoma tumor suppressor protein. Breast Cancer Res. 2004;6:R130–9. doi: 10.1186/bcr762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–49. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sui L, Dong Y, Ohno M, et al. Implication of malignancy and prognosis of p27(kip1), Cyclin E, and Cdk2 expression in epithelial ovarian tumors. Gynecol Oncol. 2001;83:56–63. doi: 10.1006/gyno.2001.6308. [DOI] [PubMed] [Google Scholar]
- 42.Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003;13:65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 43.Rosen DG, Yang G, Cai KQ, et al. Subcellular localization of p27kip1 expression predicts poor prognosis in human ovarian cancer. Clin Cancer Res. 2005;11:632–7. [PubMed] [Google Scholar]
- 44.Attwooll C, Denchi EL, Helin K. The E2F family: specific functions and overlapping interests. Embo J. 2004;23:4709–16. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Graniela Sire EA, Vikhanskaya F, Broggini M. Sensitivity and cellular response to different anticancer agents of a human ovarian cancer cell line expressing wild-type, mutated or no p53. Ann Oncol. 1995;6:589–93. doi: 10.1093/oxfordjournals.annonc.a059249. [DOI] [PubMed] [Google Scholar]
- 46.Reid T, Jin X, Song H, Tang HJ, Reynolds RK, Lin J. Modulation of Janus kinase 2 by p53 in ovarian cancer cells. Biochem Biophys Res Commun. 2004;321:441–7. doi: 10.1016/j.bbrc.2004.06.169. [DOI] [PubMed] [Google Scholar]
- 47.Fraser M, Leung BM, Yan X, Dan HC, Cheng JQ, Tsang BK. p53 is a determinant of X-linked inhibitor of apoptosis protein/Akt-mediated chemoresistance in human ovarian cancer cells. Cancer Res. 2003;63:7081–8. [PubMed] [Google Scholar]
- 48.Casalini P, Botta L, Menard S. Role of p53 in HER2-induced proliferation or apoptosis. J Biol Chem. 2001;276:12449–53. doi: 10.1074/jbc.M009732200. [DOI] [PubMed] [Google Scholar]
- 49.Havrilesky L, Darcy M, Hamdan H, et al. Prognostic significance of p53 mutation and p53 overexpression in advanced epithelial ovarian cancer: a Gynecologic Oncology Group Study. J Clin Oncol. 2003;21:3814–25. doi: 10.1200/JCO.2003.11.052. [DOI] [PubMed] [Google Scholar]
- 50.Ormerod MG, O’Neill C, Robertson D, Kelland LR, Harrap KR. cis-Diamminedichloroplatinum(II)-induced cell death through apoptosis in sensitive and resistant human ovarian carcinoma cell lines. Cancer Chemother Pharmacol. 1996;37:463–71. doi: 10.1007/s002800050413. [DOI] [PubMed] [Google Scholar]
- 51.Katano K, Kondo A, Safaei R, et al. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002;62:6559–65. [PubMed] [Google Scholar]
- 52.Heikinheimo O. Pharmacokinetics of the antiprogesterone RU 486 in women during multiple dose administration. J Steroid Biochem. 1989;32:21–5. doi: 10.1016/0022-4731(89)90008-3. [DOI] [PubMed] [Google Scholar]
- 53.Heikinheimo O, Lahteenmaki PL, Koivunen E, et al. Metabolism and serum binding of RU 486 in women after various single doses. Hum Reprod. 1987;2:379–85. doi: 10.1093/oxfordjournals.humrep.a136554. [DOI] [PubMed] [Google Scholar]
- 54.Shoupe D, Mishell DR, Jr., Lahteenmaki P, et al. Effects of the antiprogesterone RU 486 in normal women. I. Single-dose administration in the midluteal phase. Am J Obstet Gynecol. 1987;157:1415–20. doi: 10.1016/s0002-9378(87)80235-1. [DOI] [PubMed] [Google Scholar]
- 55.Kawai S, Nieman LK, Brandon DD, Udelsman R, Loriaux DL, Chrousos GP. Pharmacokinetic properties of the antiglucocorticoid and antiprogesterone steroid RU 486 in man. J Pharmacol Exp Ther. 1987;241:401–6. [PubMed] [Google Scholar]
- 56.Spitz IM, Grunberg SM, Chabbert-Buffet N, Lindenberg T, Gelber H, Sitruk-Ware R. Management of patients receiving long-term treatment with mifepristone. Fertil Steril. 2005;84:1719–26. doi: 10.1016/j.fertnstert.2005.05.056. [DOI] [PubMed] [Google Scholar]
- 57.Horwitz KB. The antiprogestin RU38 486: receptor-mediated progestin versus antiprogestin actions screened in estrogen-insensitive T47Dco human breast cancer cells. Endocrinology. 1985;116:2236–45. doi: 10.1210/endo-116-6-2236. [DOI] [PubMed] [Google Scholar]
- 58.Lin VC, Aw SE, Ng EH, Tan MG. Demonstration of mixed properties of RU486 in progesterone receptor (PR)-transfected MDA-MB-231 cells: a model for studying the functions of progesterone analogues. Br J Cancer. 2001;85:1978–86. doi: 10.1054/bjoc.2001.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akahira J, Suzuki T, Ito K, et al. Differential expression of progesterone receptor isoforms A and B in the normal ovary, and in benign, borderline, and malignant ovarian tumors. Jpn J Cancer Res. 2002;93:807–15. doi: 10.1111/j.1349-7006.2002.tb01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hamilton TC, Behrens BC, Louie KG, Ozols RF. Induction of progesterone receptor with 17 beta-estradiol in human ovarian cancer. J Clin Endocrinol Metab. 1984;59:561–3. doi: 10.1210/jcem-59-3-561. [DOI] [PubMed] [Google Scholar]
- 61.Keith Bechtel M, Bonavida B. Inhibitory effects of 17beta-estradiol and progesterone on ovarian carcinoma cell proliferation: a potential role for inducible nitric oxide synthase. Gynecol Oncol. 2001;82:127–38. doi: 10.1006/gyno.2001.6221. [DOI] [PubMed] [Google Scholar]
- 62.McDonnel AC, Murdoch WJ. High-dose progesterone inhibition of urokinase secretion and invasive activity by SKOV-3 ovarian carcinoma cells: evidence for a receptor-independent nongenomic effect on the plasma membrane. J Steroid Biochem Mol Biol. 2001;78:185–91. doi: 10.1016/s0960-0760(01)00081-4. [DOI] [PubMed] [Google Scholar]
- 63.Mao J, Regelson W, Kalimi M. Molecular mechanism of RU 486 action: a review. Mol Cell Biochem. 1992;109:1–8. doi: 10.1007/BF00230867. [DOI] [PubMed] [Google Scholar]
- 64.Xu M, Song L, Wang Z. Effects of Dexamethasone on glucocorticoid receptor expression in a human ovarian carcinoma cell line 3AO. Chin Med J (Engl) 2003;116:392–5. [PubMed] [Google Scholar]
- 65.Parthasarathy S, Morales AJ, Murphy AA. Antioxidant: a new role for RU-486 and related compounds. J Clin Invest. 1994;94:1990–5. doi: 10.1172/JCI117551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roberts CP, Parthasarathy S, Gulati R, Horowitz I, Murphy AA. Effect of RU-486 and related compounds on the proliferation of cultured macrophages. Am J Reprod Immunol. 1995;34:248–56. doi: 10.1111/j.1600-0897.1995.tb00949.x. [DOI] [PubMed] [Google Scholar]
- 67.Liberto M, Cobrinik D. Growth factor-dependent induction of p21(CIP1) by the green tea polyphenol, epigallocatechin gallate. Cancer letters. 2000;154:151–61. doi: 10.1016/s0304-3835(00)00378-5. [DOI] [PubMed] [Google Scholar]
- 68.Liu M, Wikonkal NM, Brash DE. Induction of cyclin-dependent kinase inhibitors and G(1) prolongation by the chemopreventive agent N-acetylcysteine. Carcinogenesis. 1999;20:1869–72. doi: 10.1093/carcin/20.9.1869. [DOI] [PubMed] [Google Scholar]