

Abstract
In this study benzyl and ethyl groups were appended to pyridine and aniline ancillary ligands in diiron(II) complexes of the type [Fe2(μ-O2CArR)2(O2CArR)2(L)2], where -O2CArR is a sterically hindered terphenyl carboxylate, 2,6-di(p-tolyl)- or 2,6-di(p-fluorophenyl)benzoate (R = Tol or 4-FPh, respectively). These crystallographically characterized compounds were prepared as analogs of the diiron(II) center in the hydroxylase component of soluble methane monooxygenase (MMOH). Use of 2-benzylpyridine (2-Bnpy) afforded doubly-bridged [Fe2(μ-O2CArTol)2(O2CArTol)2(2-Bnpy)2] (1) and [Fe2(μ-O2CAr4-FPh)2(O2CAr4-FPh)2(2-Bnpy)2] (4), whereas tetra-bridged [Fe2(μ-O2CArTol)4(4-Bnpy)2] (3) resulted when 4-benzylpyridine (4-Bnpy) was employed. Similarly, 2-(4-chloro-benzyl)pyridine (2-(4-ClBn)py) and 2-benzylaniline (2-Bnan) were employed as N-donor ligands to prepare [Fe2(μ-O2CArTol)2(O2CArTol)2(2-(4-ClBn)py)2] (2) and [Fe2(μ-O2CArTol)2(O2CArTol)2(2-Bnan)2] (5). Placement of the substituent on the pyridine ring had no effect on the geometry of the diiron(II) compounds isolated when 2-, 3-, or 4-ethylpyridine (2-, 3-, or 4-Etpy) was introduced as the ancillary nitrogen ligand. The isolated [Fe2(μ-O2CArTol)2(O2CArTol)2(2-Etpy)] (6), [Fe2(μ-O2CArTol)2(O2CArTol)2(3-Etpy)] (7), [Fe2(μ-O2CArTol)2(O2CArTol)2(4-Etpy)] (8), and [Fe2(μ-O2CAr4-FPh)2(O2CAr4-FPh)2(2-Etpy)2] (9) complexes all contain doubly-bridged metal centers. The oxygenation of compounds 1 – 9 was studied by GC-MS and NMR analysis of the organic fragments following decomposition of the product complexes. Hydrocarbon fragment oxidation occurred for compounds in which the substrate moiety was in close proximity to the diiron center. The extent of oxidation depended upon the exact makeup of the ligand set.
Introduction
The selective oxidation of hydrocarbons under ambient conditions is important for both industrial and ecological reasons.1,2 Dioxygen can be harnessed by certain iron-containing bacterial enzymes to effect the controlled two-electron oxidation of organic substrates. Bacterial multicomponent monooxygenases that catalyze the selective conversion of hydrocarbons to alcohols are of particular interest because of their wide range of substrates.3 The hydroxylase component of soluble methane monooxygenase (MMOH) performs this function by first activating dioxygen to afford reactive intermediates, which in turn can insert one oxygen atom selectively into the C–H bond.4-7 The benefits to understanding this process on a molecular level are two-fold. Such knowledge would provide insight into the mechanisms of metalloproteins and facilitate the design of catalysts for dioxygen activation and selective hydrocarbon oxidation.
To discern the intricacies of these systems, investigators have turned to small molecule model compounds to sharpen the connection between structure and function in the enzymes. The active site of MMOH houses a diiron center, surrounded by four carboxylate and two histidine residues, which performs the selective hydroxylation.8,9 To represent the core structure of the native enzyme accurately in a synthetic compound requires a carboxylate-rich ligand environment. Isolation of discrete dinuclear iron centers demands a ligand framework that provides sufficient steric bulk around the metal center to avoid oligomer formation10-14, without forming only mononuclear species.15,16
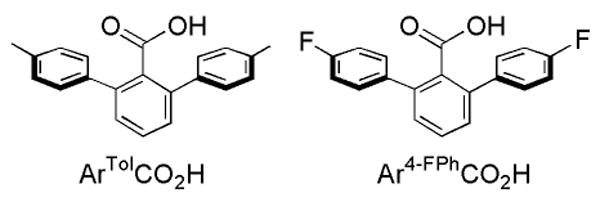
The introduction of m-terphenyl-based carboxylates, depicted in Chart 1, has facilitated the assembly of the desired dinuclear cores.16-20 The protective pocket of aromatic rings prevents bimolecular decomposition reactions but can also block access of substrate to the O2-activated diiron center. To address this concern, substrates have been incorporated into the ancillary N-donor ligands coordinated to the iron atoms, as illustrated by the formation of benzaldehyde from a benzyl moiety following exposure to O2 of the diiron(II) complexes [Fe2(μ-O2CArTol)2(O2CArTol)2(N,N-Bn2en)2],21,22 [Fe2(μ-O2CArTol)4(BAp-OMe)2],23 and [Fe2(μ-O2CArTol)4(NH2(CH2)2SBn)2],24 where ArTolCO2- is 2,6-di(p-tolyl)benzoate, N,N-Bn2en is N,N-dibenzyl-ethylenediamine, and BAp-OMe is p-methoxybenzylamine.
Chart 1.
In the present study we have expanded the family of substrate-tethered carboxylate-bridged diiron(II) compounds having benzyl- or ethyl-derivatized pyridine or aniline donor auxiliary ligands. The oxygenation chemistry of these complexes was evaluated by analysis of the organic fragments produced by introduction of O2 into their methylene chloride solutions. A portion of this work has been previously communicated.25
Experimental Section
General Considerations
All reagents were obtained from commercial suppliers and used as received, unless otherwise noted. Methylene chloride, diethyl ether (Et2O), and pentane were saturated with nitrogen and purified by passing through activated Al2O3 columns under argon.26 Dioxygen (99.994%, BOC Gases) was dried by passing the gas stream through a column of Drierite. The synthesis and characterization of [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] and [Fe2(μ-O2CAr4-FPh)2(O2CAr4-FPh)2(THF)2] were reported previously.27 Purification of 2-benzylpyridine (2-Bnpy), 2-(4-chlorobenzyl)pyridine (2-(4-ClBn)py), 4-benzylpyridine (4-Bnpy), 2-ethylpyridine (2-Etpy), 3-ethylpyridine (3-Etpy), and 4-ethylpyridine (4-Etpy) was accomplished by vacuum distillation prior to use. Authentic samples of α-phenyl-2-pyridylmethanol (2-PhCHOHpy) and α-methyl-2-pyridinemethanol (2-MeCHOHpy) were synthesized in a manner analogous to pyridyl alcohols reported in the literature.28 Air-sensitive manipulations were carried out under nitrogen in an Mbraun drybox. All samples were pulverized and thoroughly dried to remove solvent prior to determining their elemental composition. Solvent molecules included in the elemental composition of 1, 2, 3, and 6 were found in the crystal lattice or detected in the 1H NMR spectra of these compounds. Acceptable analytical data were never obtained for 5 despite the crystallinity of the sample.
[Fe2(μ-O2CArTol)2(O2CArTol)2(2-Bnpy)2] (1)
To a stirred CH2Cl2 (3.2 mL) solution of [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] (232 mg, 0.159 mmol), 2-Bnpy (55.2 mg, 0.326 mmol) in CH2Cl2 (1 mL) was added dropwise and allowed to react for 20 min. Vapor diffusion of diethyl ether into the resulting green solution yielded pale blue-green crystals (229 mg, 87%) of 1 suitable for X-ray crystallography. FT-IR (KBr, cm-1): 3055 (w), 3026 (w), 2917 (w), 2859 (w), 1606 (s), 1570 (s), 1514 (s), 1482 (m), 1453 (s), 1410 (s), 1379 (s), 1242 (w), 1187 (w), 1142 (w), 1109 (w), 1074 (w), 1020 (m), 850 (m), 816 (m), 798 (s), 780 (m), 765 (m), 744 (m), 700 (m), 644 (w), 619 (w), 581 (w), 542 (m), 518 (m), 469 (w). Anal. Calcd. for C108H90N2Fe2O8·CH2Cl2: C, 75.22; H, 5.33; N, 1.61. Found: C, 75.25; H, 5.63; N, 1.55.
[Fe2(μ-O2CArTol)2(O2CArTol)2(2-(4-ClBn)py)2] (2)
Freshly distilled 2-(4-ClBn)py (40.6 mg, 0.199 mmol) in CH2Cl2 (1 mL) was added dropwise to a CH2Cl2 (3 mL) solution of [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] (102 mg, 0.0699 mmol), with stirring for 20 min. Vapor diffusion of diethyl ether into the olive green solution yielded mint green crystals (96.1 mg, 80%) of 2 suitable for X-ray crystallography. FT-IR (KBr, cm-1): 3057 (w), 2971 (w), 2918 (w), 2850 (w), 1607 (s), 1570 (s), 1543 (s), 1514 (s), 1488 (s), 1452 (s), 1406 (s), 1380 (s), 1187 (w), 1100 (w), 1079 (w), 1016 (m), 950 (w), 856 (m), 819 (s), 800 (s), 780 (m), 767 (s), 760 (s), 735 (m), 711 (m), 696 (w), 641 (w), 601 (w), 581 (w), 542 (s), 517 (s). Anal. Calcd. for C108H88N2Fe2O8Cl2·0.25CH2Cl2: C, 74.48; H, 5.11; N, 1.60. Found: C, 74.22; H, 5.49; N, 1.96.
[Fe2(μ-O2CArTol)4(4-Bnpy)2] (3)
Bright green X-ray quality crystals (172 mg) of 3 were isolated in 88% yield from the reaction of 4-benzylpyridine (46.0 mg, 0.272 mmol) with [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] (173 mg, 0.119 mmol) by vapor diffusion of diethyl ether into the CH2Cl2 (4 mL) solution. FT-IR (KBr, cm-1): 3024 (w), 2972 (w), 2916 (w), 2857 (w), 1614 (s), 1584 (m), 1548 (m), 1512 (m), 1429 (s), 1382 (s), 1302 (w), 1183 (w), 1151 (w), 1109 (w), 1069 (w), 1029 (w), 841 (m), 810 (s), 788 (s), 762 (w), 726 (w), 705 (s), 616 (w), 583 (m), 564, 524 (s), 483 (w), 460 (w). Anal. Calcd. for C108H90N2O8Fe2·0.25CH2Cl2: C, 77.54; H, 5.44, N, 1.67. Found: C, 77.55; H, 5.54; N, 1.57.
[Fe2(μ-O2CAr4-FPh)2(O2CAr4-FPh)2(2-Bnpy)2] (4)
2-Benzylpyridine (34.1 mg, 0.202 mmol) was added to [Fe2(μ-O2CAr4-FPh)2(O2CAr4-FPh)2(THF)2] (91.2 mg, 0.0611 mmol) in a CH2Cl2 (4 mL) solution, and immediately the light sea-green solution was subjected to pentane vapor diffusion. Light-green blocks of 4 (97.7 mg, 95%) suitable for X-ray diffraction studies were recovered. FT-IR (KBr, cm-1): 3061 (w), 2959 (w), 2923 (w), 2863 (w), 1604 (s), 1569 (s), 1510 (s), 1454 (s), 1409 (m), 1379 (m), 1224, 1299 (w), 1186 (w), 1159 (s), 1095 (w), 1073 (w), 1013 (w), 855 (m), 837 (s), 806 (s), 793 (m), 702 (m), 622 (w), 581 (w), 559 (m), 526 (m), 469 (w). Anal. Calcd. for C100H66N2Fe2O8F8: C, 71.18; H, 3.94; N, 1.66. Found: C, 70.97; H, 3.91; N, 1.93.
[Fe2(μ-O2CArTol)2(O2CArTol)2(2-Bnan)2] (5)
A pale yellow solution of [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] (87.0 mg, 0.0595 mmol) in 4 mL of CH2Cl2 was allowed to react with 2-benzylaniline (2-Bnan) ( 24.7 mg, 0.135 mmol). After 30 min the solution was reduced almost to dryness under vacuum, at which time 2 mL of ClPh were added. Diffusion of pentane vapor into the solution yielded pale yellow blocks (50.1 mg, 50%) of 5 suitable for X-ray crystallography. FT-IR (KBr, cm-1): 3334, 3281, 3026, 2918, 1598 (s), 1549 (s), 1514 (s), 1494 (m), 1453 (m), 1413 (m), 1377 (s), 1306 (w), 1272 (s), 1233 (w), 1186 (w), 1144 (w), 1109 (w), 1078 (w), 1021 (m), 855 (m), 818 (s), 801 (s), 784 (s), 714 (s), 699 (s), 687 (w), 616 (w), 583 (w), 544 (s), 520 (s), 458 (w), 414 (w).
[Fe2(μ-O2CArTol)2(O2CArTol)2(2-Etpy)2] (6)
A stirred solution of [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] (336 mg, 0.230 mmol) in CH2Cl2 (8 mL) was combined with 2-ethylpyridine (54.5 mg, 0.509 mmol) and allowed to react for 20 min. Vapor diffusion of Et2O into the yellow-green solution resulted in the formation of white needles of 6 (330 mg, 95%). FT-IR (KBr, cm-1): 3053 (w), 2970 (w), 2919 (w), 1604 (s), 1567 (s), 1514 (s), 1481 (m), 1452 (s), 1408 (s), 1367 (s), 1306 (w), 1264 (m), 1213 (w), 1144 (w), 1109 (m), 1062 (w), 1018 (m), 849(s), 799 (s), 780 (s), 767 (s), 736 (s), 709 (s), 644 (w), 609 (w), 558 (w), 542 (m), 522 (m), 444 (w), 416 (w). Anal. Calcd. for C98H86N2O8Fe2·0.5CH2Cl2: C, 75.17; H, 5.57; N, 1.78. Found: C, 75.52; H, 5.57; N, 1.65.
[Fe2(μ-O2CArTol)2(O2CArTol)2(3-Etpy)2] (7)
3-Ethylpyridine (11.3 mg, 0.105 mmol) was added to a CH2Cl2 (4 mL) solution of [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] (73.1 mg, 0.0500 mmol), and the mixture was stirred for 20 min. Diethyl ether vapor diffusion into the resulting pale yellow-green solution led to the formation of colorless block crystals of 7 (63.6 mg, 83%) suitable for X-ray crystallography. FT-IR (KBr, cm-1): 3054 (w), 2962 (w), 2922 (w), 2864 (w), 1604 (s), 1514 (s), 1454 (s), 1413 (m), 1353 (s), 1262 (w), 1188 (w), 1146 (w), 1109 (m), 1018 (m), 845 (m), 819 (s), 799 (s), 781 (s), 765 (s), 735 (m), 700 (m), 653 (w), 601 (w), 582 (w), 543 (m), 517 (s), 492 (w), 451 (w), 413 (w). Anal. Calcd. for C98H86N2O8Fe2: C, 76.86; H, 5.66; N, 1.83. Found: C, 76.63; H, 5.78; N, 2.04.
[Fe2(μ-O2CArTol)2(O2CArTol)2(4-Etpy)2] (8)
Colorless block crystals (66.0 mg, 68%) of 8 were obtained by vapor diffusion of Et2O into a CH2Cl2 (4 mL) reaction mixture of [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] (92.6 mg, 0.0634 mmol) with 4-ethylpyridine (21.6 mg, 0.202 mmol). FT-IR (KBr, cm-1): 3054 (w), 3023 (w), 2966 (w), 2915 (w), 1609 (s),1543 (s), 1514 (s), 1453 (s), 1406 (m), 1380 (s), 1312 (w), 1224 (w), 1186 (w), 1109 (w), 1069 (w), 1025 (w), 968 (w), 910 (w), 856 (m), 816 (s), 799 (s), 785 (s), 732 (s), 712 (s), 609 (w), 584 (m), 545 (m), 519, 489 (w), 448 (w). Anal. Calcd. for C98H86N2O8Fe2: C, 76.86; H, 5.66; N, 1.83. Found: C, 77.04; H, 5.85; N, 1.72.
[Fe2(μ-O2CAr4-FPh)2(O2CAr4-FPh)2(2-Etpy)2] (9)
To a stirred CH2Cl2 (4 mL) solution of [Fe2(μ-O2CAr4-FPh)2(O2CAr4-FPh)2(THF)2] (112 mg, 0.0751 mmol), was added 2-ethylpyridine (34.8 mg, 0.325 mmol), forming a light yellow-green solution that was stirred for 20 min before pentane vapor was diffused into the reaction mixture. Colorless block crystals (107 mg) were isolated in 92% yield. FT-IR (KBr, cm-1): 3062 (w), 2971 (w), 2880 (w), 1606 (s), 1571 (m), 1546 (m), 1510 (s), 1483 (w), 1455 (s), 1413 (m), 1380 (s), 1300 (w), 1222 (s), 1160 (s), 1096 (m), 1021 (w), 856 (s), 846 (s), 836 (s), 806 (s), 787 (m), 775 (m), 737 (w), 713 (m), 644 (w), 582 (w), 555 (s), 528 (m), 472 (w), 455 (w). Anal. Calcd. for C90H62N2Fe2O8F8: C, 69.15; H, 4.00; N, 1.79. Found: C, 69.44; H, 3.98; N, 2.09.
Physical Measurements
FT-IR spectra were recorded on a Thermo Nicolet Avatar 360 spectrometer with OMNIC software. 1H NMR spectra were measured with a Varian 300 spectrometer housed in the Massachusetts Institute of Technology Department of Chemistry Instrument Facility (MIT DCIF). Chemical shifts were referenced to the residual solvent peaks. All spectra were recorded at ambient probe temperature, 293 K.
X-ray Crystallographic Studies
Intensity data were collected on a Bruker (formerly Siemens) APEX CCD diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å), controlled by a Pentium-based PC running the SMART software package.29 Single crystals were mounted on the tips of glass fibers, coated with paratone-N oil, and cooled to –100 °C under a stream of N2 maintained by a KRYO-FLEX low-temperature apparatus. Data collection and reduction protocols are described elsewhere.30 The structures were solved by direct methods and refined on F2 by using the SHELXTL-97 software31 incorporated in the SHELXTL software package.32 Empirical absorption corrections were applied by using the SADABS program,33 and the structures were checked for higher symmetry by using the PLATON software.34 All non-hydrogen atoms were located and their positions refined with anisotropic thermal parameters by least-squares cycles and Fourier syntheses. In general, hydrogen atoms were assigned to idealized positions and given thermal parameters equivalent to either 1.5 (methyl hydrogen atoms) or 1.2 (all other hydrogen atoms) times the thermal parameter of the carbon atom to which they were attached. Compound 1 contains one CH2Cl2 molecule disordered over two positions; the atoms were distributed equally. One Et2O molecule is present in the lattice of compound 3. There is a CH2Cl2 molecule in the lattice of 4. In the asymmetric unit of 6 are contained CH2Cl2 molecules, one of which is half occupied and lies on a center of symmetry and the other is disordered and was refined with the atoms distributed equally over two positions. Data collection and experimental details are summarized in Table S1; those for 1 were reported previously.25 The structures of 1 – 3, and 5 – 9 are shown in Figures 1 and S1 – S8. Selected bond lengths and angles for 1 – 3, and 5 – 9 are provided in Tables 1 and 2.
Figure 1.
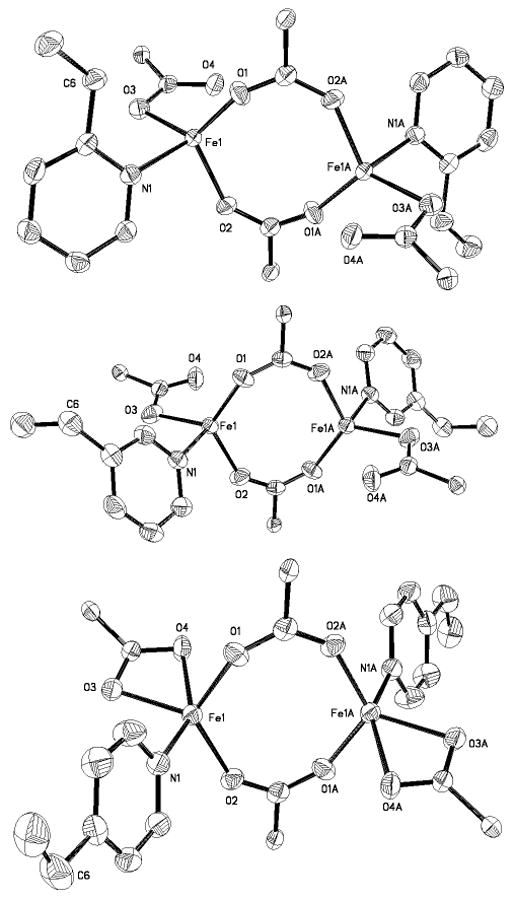
ORTEP drawings of [Fe2(μ-O2CArTol)2(O2CArTol)2(2-Etpy)2] (6) (top), [Fe2(μ-O2CArTol)2(O2CArTol)2(3-Etpy)2] (7) (middle), and [Fe2(μ-O2CArTol)2(O2CArTol)2(4-Etpy)2] (8) (bottom) illustrating 50% probability thermal ellipsoids for all non-hydrogen atoms. The aromatic rings of the -O2CArTol ligands are omitted for clarity.
Table 1.
Selected Bond Lengths (Å) and Angles (deg) for 1, 2, 3, and 5a
 | ||||
|---|---|---|---|---|
| 1 | 2 | 3 | 5 | |
| Fe1…Fe2 | 4.2380(9)b
4.6052(9)c |
4.3908(11) | 2.7715(6) | 4.0646(10) |
| Fe1-N1 | 2.124(2)b
2.150(2)c |
2.146(2) | 2.0830(16)
2.0901(16)e |
2.175(2) |
| Fe1-O1 | 2.0270(19)b
1.977(2)c |
2.001(2) | 2.1349(13)
2.0238(13)e |
1.978(2) |
| Fe1-O2 | 1.956(2)b
2.003(2)c |
1.971(2) | 2.0251(13)
2.1372(14)e |
1.989(2) |
| Fe1-O3 | 2.014(2)b
2.118(2)c |
2.068(2) | 2.1322(13)
2.0176(13)e |
2.0798(19) |
| Fe1-O4 | 2.463(2)b,d
2.281(2)c |
2.308(2) | 2.0319(13)
2.1310(14)e |
2.2150(19) |
| O1-Fe1-N1 | 100.40(9)b
113.05(9)c |
97.59(9) | 95.58(6)
103.97(6)e |
96.36(9) |
| O2-Fe1-N1 | 95.26(9)b
96.61(9)c |
98.87(9) | 101.82(6)
97.19(6)e |
87.29(9) |
| O3-Fe1-N1 | 93.09(9)b
85.70(8)c |
91.46(8) | 98.14(6)
96.69(6)e |
98.26(9) |
| O4-Fe1-N1 | 150.10(8)b
144.90(8)c |
150.39(8) | 99.31(6)
97.05(6)e |
153.99(9) |
| O1-Fe1-O2 | 115.98(9)b
106.16(9)c |
112.41(9) | 87.59(5)
88.65(5)e |
121.42(9) |
| O1-Fe1-O3 | 110.50(8)b
125.43(9)c |
119.39(9) | 166.24(5)
159.29(5)e |
113.22(9) |
| O1-Fe1-O4 | 95.37(8)b
89.56(8)c |
101.99(8) | 91.39(5)
91.06(5)e |
106.18(8) |
| O2-Fe1-O3 | 130.19(9)b
122.66(9)c |
125.10(9) | 90.61(5)
87.26(5)e |
124.06(9) |
| O2-Fe1-O4 | 100.44(8)b
102.55(8)c |
93.97(8) | 158.85(5)
165.38(5)e |
91.75(8) |
| O3-Fe1-O4 | 57.45(8)b
59.20(8)c |
59.56(7) | 85.39(5)
87.86(5)e |
61.18(7) |
Numbers in parentheses are estimated standard deviations of the last significant figures. Atoms are labeled as indicated in Figures S1 – S4.
Molecule 1 in asymmetric unit.
Molecule 2 in the asymmetric unit.
Fe…Onon-coordinating.
Parameter for Fe2 with N2, O5 – O8.
Table 2.
Selected Bond Distances (Å) and Angles (deg) for 6 – 9a
 | ||||
|---|---|---|---|---|
| 6 | 7 | 8 | 9 | |
| Fe1…Fe2 | 4.3366(12) | 4.0861(10) | 4.1542(14) | 4.4830(10)b
4.4434(10)c |
| Fe1-N1 | 2.143(3) | 2.104(2) | 2.130(3) | 2.135(3)b
2.136(3)c |
| Fe1-O1 | 1.959(3) | 1.9569(17) | 2.013(3) | 1.943(2)b
1.952(2)c |
| Fe1-O2 | 2.010(3) | 1.9861(17) | 1.972(2) | 2.013(2)b
2.002(2)c |
| Fe1-O3 | 2.023(3) | 1.9711(18) | 2.061(3) | 2.118(2)b
2.052(2)c |
| Fe1-O4 | 2.517(3)d | 2.715(2)d | 2.323(3) | 2.225(2)b
2.330(2)c |
| O1-Fe1-N1 | 104.61(13) | 97.73(8) | 94.20(11) | 104.54(11)b
98.49(11)c |
| O2-Fe1-N1 | 96.74(12) | 99.99(8) | 95.68(11) | 99.25(10)b
103.44(11)c |
| O3-Fe1-N1 | 92.79(12) | 91.64(8) | 93.37(11) | 91.94(10)b
92.61(9)c |
| O4-Fe1-N1 | 149.18(12) | 144.49(7) | 150.91(10) | 151.20(9)b
151.40(10)c |
| O1-Fe1-O2 | 114.46(13) | 122.82(8) | 120.61(11) | 108.07(11)b
110.24(11)c |
| O1-Fe1-O3 | 125.14(12) | 121.68(8) | 114.31(10) | 133.70(10)b
132.13(11)c |
| O1-Fe1-O4 | 98.37(11) | 96.76(7) | 88.49(10) | 91.44(9)b
96.41(10)c |
| O2-Fe1-O3 | 114.46(13) | 111.64(8) | 123.29(11) | 111.53(10)b
112.06(10)c |
| O2-Fe1-O4 | 93.25(12) | 99.01(7) | 107.80(10) | 98.28(9)b
94.02(9)c |
| O3-Fe1-O4 | 56.43(11) | 53.36(7) | 59.53(9) | 60.29(8)b
59.57(8)c |
Mössbauer Spectroscopy
Mössbauer spectra were recorded on an MSI spectrometer (WEB Research Co.) with a 57Co source in a Rh matrix maintained at room temperature in the MIT DCIF. Solid samples of 1 and 6 were prepared by suspending ∼0.028 mmol of pulverized crystalline material in Apeizon N grease and coating the mixture on the lid of a nylon sample holder. Data were collected at 4.2 K, and the isomer shift (δ) values are reported with respect to natural iron foil that was used for velocity calibration at room temperature. The spectra were fit to Lorentzian lines by using the WMOSS plot and fit program.35
Oxidation Product Analyses
Oxidation reactions were performed by exposing a CH2Cl2 solution of the diiron(II) complex to dioxygen over a period of time. All samples were prepared in an anaerobic glove box prior to bubbling dried dioxygen through them. To work up the oxygenation reactions, Chelex was added to the solutions to remove the iron. The resulting slurry was stirred for 2 h, after which the Chelex was removed by filtration. The resin was washed with CH2Cl2 twice more, and the filtrates were combined. The solvent was removed in vacuo to afford the N-donor ligands for analysis. The products were identified by NMR spectroscopy and/or GC-MS by comparing their NMR spectra and/or their retention times and mass spectral patterns to those of authentic samples. The amount of oxidized ligand obtained for 1, 2, and 4 was quantified by 1H NMR spectroscopy using 1,4-bis(trimethylsilyl)benzene as an internal standard. For compounds 6 and 9 the amount of ligand oxidized was quantitated by GC-MS using 2-bromonaphthalene as an internal standard. Control experiments established that, in the absence of the diiron(II) complexes, neither dioxygen-saturated CH2Cl2 nor the workup process induces significant ligand oxidation.
GC-MS Analyses
Analyses were carried out on a Hewlett-Packard HP-5890 gas chromatograph connected to a HP-5971 mass analyzer. A J&W Scientific HP-5MS capillary column of dimensions (30 m × 0.25 mm × 0.25 μm) was employed for GC-MS studies. The following program was used to effect all separations: initial temperature = 90 °C; initial time = 7 min; temperature ramp = 90 – 210 °C at 20 deg/min; and a final time = 1.5 min. Quantitations were made by comparison of the total ion count of 2-Etpy, 2-MeCHOHpy, and 2-acetylpyridine with that of 2-bromonaphthalene, the internal standard. Calibration plots for the detector response were prepared for authentic samples of 2-Etpy, 2-MeCHOHpy, 2-acetylpyridine, and 2-bromonaphthalene by using stock solutions of known concentrations.
Results
Synthesis and Characterization of Diiron(II) Complexes Containing Benzylpyridine and Benzylaniline Ligands
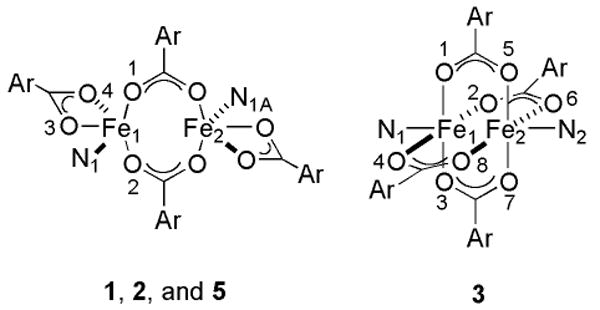
Displacement of THF from [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] by two equiv of 2-Bnpy led to the formation of the diiron(II) complex [Fe2(μ-O2CArTol)2(O2CArTol)2(2-Bnpy)2] (1), Scheme 1. The previously reported structure of 1 is given in Figure S1, and selected bond lengths and angles are listed Table 1.25 Two inequivalent, centrosymmetric diiron(II) complexes are located in the unit cell of 1 with Fe…Fe distances of 4.2385(9) Å and 4.6050(9) Å. In one dimer the geometry of the iron center is distorted trigonal bipyramidal, with two oxygen atoms from bridging carboxylates, two from a bidentate carboxylate, and the N-atom of benzylpyridine ligand forming the coordination sphere. The iron atoms are four-coordinate in the other dimer because the terminal carboxylate is monodentate with a long Fe…O interaction of 2.463(2) Å. Both dimers have windmill structures (Chart 2). The methylene carbon atoms of the benzyl groups are 3.247(3) Å and 3.219(3) Å, respectively, away from the closest iron atoms.
Scheme 1.
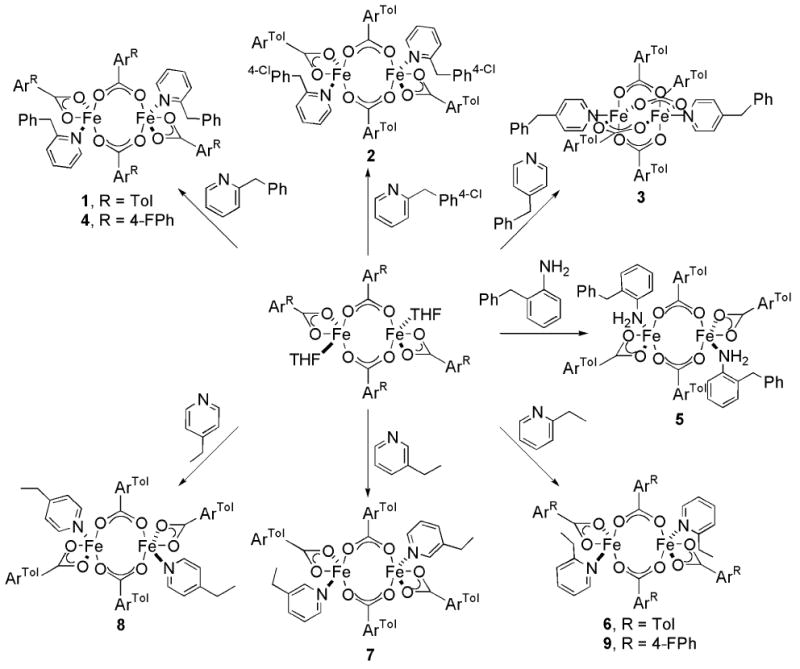
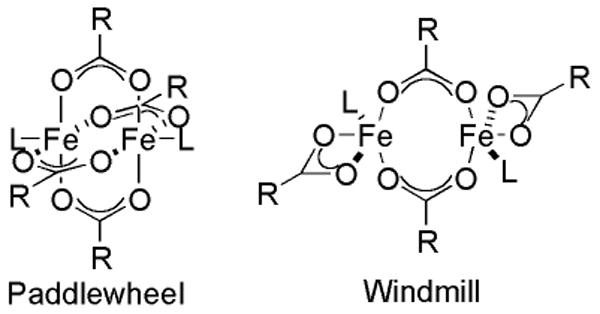
Chart 2.
The 2-(4-ClBn)py analog, [Fe2(μ-O2CArTol)2(O2CArTol)2(2-(4-ClBn)py)2] (2) was also prepared (Scheme 1), the structure of which is presented in Figure S2. Selected bond lengths and angles are listed in Table 1. Scrutiny of these structural parameters reveals close similarities to 1. One metal center comprises the asymmetric unit and is related to the other in the complex by a crystallographically imposed center of symmetry. The separation of the two metal centers in 2 is 4.3907(11) Å and the distance between Fe1 and the benzyl carbon atom (C6) is 3.254(3) Å.
Bright green crystals of 3 were isolated from the reaction of [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] with 4-Bnpy (Scheme 1). The X-ray structure confirms the formation of the diiron(II) complex, (Figure S3), and Table S2 lists selected bond lengths and angles. The diiron core is bridged by four carboxylate ligands, the O-atoms of which form the base of a square pyramid that is capped by 4-Bnpy ligands. The distance between the two metal centers in this paddlewheel structure (Chart 2) is 2.7715(6) Å, almost 2 Å less than the 4.6050(9) Å separation in 1; the iron-to-benzyl carbon distance in 3 is 6.392(2) Å, ∼3 Å longer than that observed in 1.
Treatment of the [Fe2(μ-O2CAr4-FPh)2(O2CAr4-FPh)2(THF)2] complex with 2-Bnpy results in the formation of 4, Scheme 1. The structure of 4 could not be completely solved due to severe solvent disorder, however the connectivity of the metal fragment could be ascertained. The windmill geometry is adopted, and the bond lengths and angles are comparable to those in 1. Distorted trigonal bipyramidal iron centers, related by an inversion center, are coordinated by two bridging carboxylates, one bidentate carboxylate, and one benzylpyridine.
A doubly carboxylate-bridged diiron(II) complex [Fe2(μ-O2CArTol)2(O2CArTol)2(2-Bnan)2] (5) is isolated from the reaction of 2-Bnan with [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] (Scheme 1). A structural determination revealed two iron centers related by an inversion center. Figure S4 shows the structure of 5 and pertinent bond lengths and angles are listed in Table 1. These atoms are bridged by two carboxylates, which coordinate in an unsymmetrical syn,syn-bidentate mode between the two metal centers.36 The Fe…Fe separation is 4.0644(10) Å. In addition, each metal center is ligated to one bidentate carboxylate ligand and a benzylaniline moiety. The methylene carbon atom of the benzyl group is 4.522(3) Å away from the closest iron atom.
Synthesis and Characterization of Diiron(II) Complexes Containing Ethylpyridine Ligands
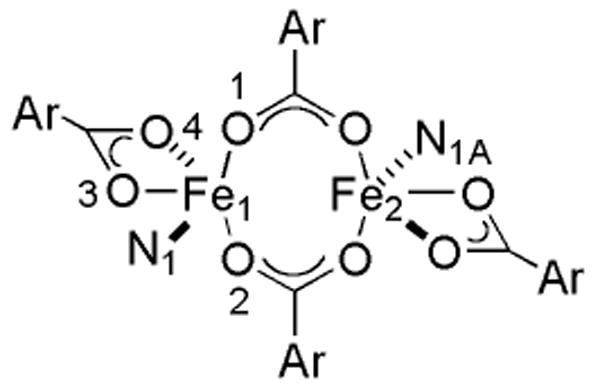
Compounds 6, 7, and 8 were prepared by displacement of the THF molecules in [Fe2(μ-O2CArTol)2(O2CArTol)2(THF)2] with 2 equiv of 2-, 3-, or 4-ethylpyridine, respectively (Scheme 1). Their X-ray structures are depicted in Figures 1 and S5 – S7, and selected bond lengths and angles are available in Table 2. Each compound adopts a windmill geometry in which the iron centers, related by crystallographically imposed centers of symmetry, are coordinated by two bridging carboxylate O-atoms, one from a terminal carboxylate, and one N-atom from ethylpyridine. A fourth O-atom from the terminal carboxylate binding in a bidentate fashion completes the coordination sphere of 8. Among the three compounds, the Fe…Fe distance in 7 is shortest at 4.0872(10) Å, followed by 8 with a distance of 4.1546(13) Å. A significantly larger separation between metal centers of 4.3361(12) Å occurs in 6, probably because its pyridine ortho substituent offers the most steric bulk near the diiron core. The nearest iron-to-substrate (Fe1…C6) distance is 3.237(4) Å in the 2-Etpy compound, 6, being 5.446(3) Å in 7 and 6.439(4) Å in 8.
Treatment of [Fe2(μ-O2CAr4-FPh)2(O2CAr4-FPh)2(THF)2] with 2-Etpy results in the formation of 9 (Scheme 1). Figures 1 and S8 display the structure, and Table 2 lists selected bond lengths and angles. The structure of 9 has two centrosymmetric molecules in the unit cell that exhibit the windmill geometry with Fe…Fe separations of 4.4826(9) Å for Fe1…Fe1A and 4.4433(9) Å for Fe2…Fe2A. Distorted trigonal bipyramidal iron centers are coordinated by two bridging carboxylate O-atoms, two from a bidentate carboxylate, and one N-atom from ethylpyridine. The iron-to-methylene carbon distances are 3.197(3) Å for Fe1…C6 and 3.214(3) Å for Fe2…C51.
Mössbauer Spectroscopy
The zero-field Mössbauer spectra, measured at 4.2 K, of powder samples of 1 and 6 are portrayed in Figures 2 and S9. The following parameters refer to 1 and 6, respectively. Both spectra exhibit a single sharp (Γ = 0.23 – 0.30 and 0.25 – 0.27 mm s-1), slightly asymmetric quadrupole doublet. The quadrupole splitting (ΔEQ = 3.05(2) and 2.96(2) mm s-1) and isomer shift (δ = 1.22(2) and 1.20(2) mm s-1) values are consistent with those of high spin diiron(II) complexes having oxygen-rich coordination environments, indicating that 1 and 6 have high spin S = 2 ground states.20,37,38
Figure 2.
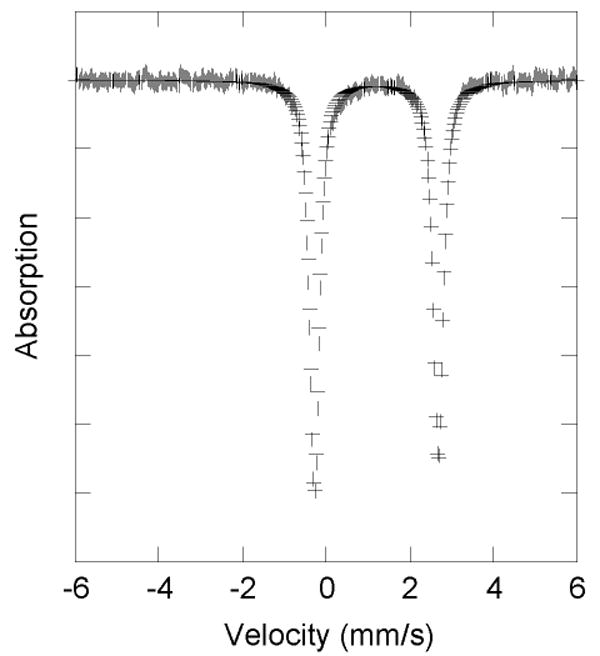
Mössbauer spectrum (experimental data (|), calculated (–)) recorded at 4.2 K for solid sample of [Fe2(μ-O2CArTol)2(O2CArTol)2(2-Etpy)2] (6).
Dioxygen Reactivity of Diiron(II) Complexes Containing Benzyl Substrates
Table 3 summarizes the conditions and amount of oxidation product isolated for the reactions of compounds 1 – 5 with dioxygen in CH2Cl2. The product formed upon exposure of a CH2Cl2 solution of 1 to dioxygen at ambient temperature, identified by GC-MS and quantitated by 1H NMR spectroscopy, is α-phenyl-2-pyridylmethanol, in an 82% yield based on 1. The remaining recovered material is unmodified benzylpyridine ligand. Similarly, upon exposure of 2 to dioxygen, the 2-(4-ClBn)py was oxidized at the benzyl position. The alcohol is produced in an average yield of 40% based on 2, as established by 1H NMR spectroscopy. Reaction of 4 with dioxygen resulted in the formation of α-phenyl-2-pyridylmethanol, with a yield of 66% based on 4. Product analysis following the oxygenation of 3 or 5 revealed only unmodified 4-benzylpyridine or 2-benzylaniline. No evidence of the alcohol was obtained.
Table 3.
Summary of the Conditions and Amount of Oxidation Product Isolated Following the Reaction of Compounds 1 – 5 with Dioxygen in CH2Cl2 at Room Temperature.
| [Fe2] (mM) | Reaction Time (min) | % Oxidized Ligand Recovereda | |
|---|---|---|---|
| 1 | 4.0 | 40 | 82 |
| 2 | 1.8 | 30 | 40 |
| 3 | 2.4 | 30 | 0 |
| 4 | 4.3 | 30 | 66 |
| 5 | 1.8 | 30 | 0 |
Based on [Fe2]
Dioxygen Reactivity of Diiron(II) Complexes Containing Ethyl Substrates
A summary of the conditions and amount of oxidation products isolated for the reaction of compounds 6 – 9 with dioxygen in CH2Cl2 is included in Table 4. The reactivity of the diiron(II) complexes 6 – 9 was probed by decomposing the oxygenated CH2Cl2 solution and analyzing the contents by GC-MS. Despite the different carboxylate ligands, analysis of the reaction solutions of 6 and 9 with dioxygen by GC-MS displayed three peaks with M+ = 107, 123, and 121. By comparing their retention times with those of authentic samples, we identified them as 2-ethylpyridine, α-methyl-2-pyridinemethanol, and 2-acetylpyridine, respectively. 1H NMR spectroscopy confirmed that α-methyl-2-pyridinemethanol is indeed the product and not 2-pyridylethanol, the other possible isomer with a molecular weight of 123. Slightly more oxidized ligand is isolated for 6 than 9, 46% vs 35% based on starting dimer, but in both systems four times more 2-acetylpyridine was recovered than the pyridylalcohol product. Analysis of the analogous dioxygen reactions of 7 and 8 revealed a species with M+ = 107, corresponding to unmodified 3- or 4-ethylpyridine.
Table 4.
Summary of the Conditions and Amount of Oxidation Product Isolated Following the Reaction of Compounds 6 – 9 with Dioxygen in CH2Cl2 at Room Temperature.
| [Fe2] (mM) | Reaction Time (min) | % Oxidized Ligand Recovereda | ||
|---|---|---|---|---|
| 2-acetylpyridine | 2-MeCHOHpy | |||
| 6 | 7.0 | 60 | 40 | 5 |
| 7 | 3.3 | 60 | 0 | 0 |
| 8 | 3.3 | 60 | 0 | 0 |
| 9 | 6.6 | 60 | 29 | 6 |
Based on [Fe2]
Discussion
Intramolecular oxidation of benzylic C–H bonds of coordinated aliphatic amine ligands in carboxylate-rich diiron(II) complexes upon exposure to dioxygen has been achieved.21-24 There are many examples of mononuclear and dinuclear iron complexes with pyridine or pyridine-modified ligands in both nitrogen- and oxygen-rich ligand environments.20,39 Aniline is not commonly employed as a ligand in iron complexes, but aniline derivatives, including bidentate ligands such as 1,2-phenylenediamine and o-aminothiophenol,40-42 a macrocycle bearing pendant anilino donors on the 1,4,7-triazacyclononane scaffold,43 and other aliphatic amines have successfully coordinated ferrous metal centers.15,21-23 In preparing carboxylate-bridged diiron(II) complexes described herein, pyridine and aniline were chosen as alternative ancillary nitrogen donors. The choice of benzylpyridines and benzylaniline was made to preserve the same dangling substrate moiety.
To provide access to the oxidizing intermediates formed upon reaction of the diiron(II) center with O2, substrates were placed in the ortho position of the ancillary ligands. In all cases resulting steric demands of the ligands afforded windmill structures, which were isolated and crystallographically characterized for 1, 2, 4, 5, 6, and 9. The position of the substrate on the pyridine ring can affect both the stereochemistry and dioxygen reactivity of the resultant diiron(II) complex.25 To investigate further this relationship for C–H activation, two series of ligands were employed, namely, 2-, 3-, or 4-benzylpyridine and 2-, 3-, or 4-ethylpyridine. Using 3-Bnpy as the N-donor for carboxylate-bridged diiron(II) complexes resulted only in the formation of insoluble powders,44 but isolation of the 4-Bnpy complex, 3, revealed a tetrabridged-carboxylate diiron(II) complex. Movement of the ethyl-substituent from the ortho to meta and para positions with 3- and 4-Etpy respectively did not alter the geometry of the resultant diiron(II) centers. Both 7 and 8 contain doubly carboxylate-bridged dimetallic units.
The use of terphenyl carboxylate ligands has enabled the synthesis of coordinatively unsaturated carboxylate-bridged diiron(II) complexes that are stoichiometrically analogous to those of reduced RNR-R2,45 MMOH,8,46 and Δ9D47 enzyme active sites. The windmill structures of diiron(II) benzylpyridine compounds 1, 2, and 4 are very similar. Each metal atom is coordinated by one N-atom from benzylpyridine and three O-atoms from one terminal and two bridging carboxylate ligands. The iron atoms are four- or five-coordinate depending on the length of the fourth Fe–O interaction from the terminal carboxylate. The exchange of -O2CArTol for -O2CAr4-FPh, or 2-benzyl- for 2-(4-chloro)benzylpyridine, causes little difference in the resultant diiron(II) complexes 4 and 2 compared to 1. There are two inequivalent centrosymmetric molecules in the crystal lattice of 1. A four-coordinate iron (Fe1) has three short Fe–O bonds, the average of which, denoted as Fe–Oav, being 1.999(2) Å, and one long bond (Fe–OL), 2.463(3) Å. A second molecule of 1, which includes Fe2, has Fe–Oav = 2.033(2) Å and an Fe–OL of 2.281(2) Å, thus rendering the Fe2 center five-coordinate. As a result, the Fe…Fe separation is larger for five-coordinate Fe2 compared to four-coordinate Fe1, the respective distances being 4.6052(9) Å and 4.2380(9) Å. The five-coordinate iron center in 2 has Fe-Oav = 2.013(2) with the Fe–OL bond equal to 2.308(2). Its metal-metal separation is 4.3908(11) Å. A smaller difference between Fe–OL and Fe–Oav correlates with a larger separation between the two metal centers. Typically, when the Fe–Oav and Fe–OL distances are disparate, the coordination number of the iron center reduces from 5 to 4 and the metal-metal distance is shortened. A summary of this information is available in Table S2 (Supporting Information).
The windmill structure of the diiron(II) benzylaniline derivative 5 is closely related to those of the benzylpyridine compounds, the differences being a slightly longer Fe–N distance (∼0.05 Å) and shorter Fe–O bonds. The Fe–Oav is 2.016(3) Å, with the Fe–OL being 2.214(2) Å. Despite the five-coordinate metal centers, the Fe…Fe separation of 4.0656(10) Å is the shortest yet observed. This result reflects the decreased steric crowding at the metal center for benzylaniline compared to benzylpyridine, because the aromatic ring of the N-donor is an additional atom removed from the metal center.
When the benzyl group is shifted from the ortho to the para position of the pyridine ring, the structure converts to the paddlewheel isomer. The tetracarboxylate-bridged diiron center of 3 differs greatly from the other benzylpyridine complexes. Each metal atom has two long and two short Fe–O bonds, with the average distances for each of 2.1338(14) Å and 2.0246(13) Å. The Fe–N bond is ∼0.05 Å shorter than those of the windmill isomers, and the metal-metal distance is 2.7715(6) Å, 1.3 Å shorter than the smallest separation observed in a windmill structure.
The structural parameters of diiron(II) ethylpyridine complexes 6 – 9 are comparable, irrespective of the geometry of the ethylpyridine moiety or the substituents on the terphenyl carboxylate ligands. The Fe–N bond lengths of 6 and 9 are within 0.01 Å of one another. The four-coordinate metal center has Fe–Oav = 1.998(3) Å and Fe–OL = 2.517(3) Å. There are two five-coordinate centrosymmetric molecules in 9. The Fe–Oav is 2.025(2) Å for Fe1 and 2.002(2) Å for Fe2, and the Fe–OL bond lengths are 2.225(2) Å and 2.330(2) Å for Fe1 and Fe2, respectively, ∼0.2-0.3 Å shorter than in 6. As a result the respective Fe…Fe separations are longer for 9 than for 6, 4.4830(10) and 4.4434(10) Å vs 4.3361(12) Å. Both 7 and 8 have much smaller Fe…Fe distances, 4.0861(10) Å and 4.1542(14) Å, respectively. The removal of the ethyl substituent from the ortho position of the pyridine ring relieves steric strain. In addition four-coordinate iron atoms occur in 7 resulting in the shortest Fe–Oav of 1.9714(17) Å and longest Fe–OL interaction of 2.715(2) Å. The metal-metal distance of 8, short for five-coordinate iron, is balanced by a longer Fe–Oav of 2.068(3) Å. This information is included in Table S2.
The single sharp quadrupole doublet exhibited in the Mössbauer spectra of 1 and 6 reflects the fact that, for both compounds, the iron centers are in equivalent coordination environments that are related by a virtual C2 axis. The quadrupole splitting (ΔEQ = 3.05(2) and 2.96(2) mm s-1) and isomer shift (δ = 1.22(2) and 1.20(2) mm s-1) values of 1 and 6, respectively, are consistent with those of high spin diiron(II) complexes in nitrogen and oxygen coordination environments.20,37 Even though both 1 and 6 have iron(II) sites with coordination number 4, their isomer shifts are slightly higher than those usually observed (1.0 – 1.1 mm s-1) for this geometry.38 The quadrupole splitting and isomer shift parameters obtained for 1 and 6 are analogous to those of MMOHred, which are 3.01 and 1.3 mm s-1, respectively.48 This similarity reflects their oxygen-rich ligand environment, mimicking that of the enzyme active site.
The reactivity of the benzylpyridine complexes is greater than that of the ethylpyridine analogs, as expected from the slightly weaker ∼2 kcal/mol49-51 C–H bond energy of the benzyl moiety. In both systems, C–H activation only occurs when the substrate is located at the ortho position of the coordinated pyridine ligand. No oxidation occurs when pyridine is replaced by aniline as coordinating ligand to which the substrate is appended. Although 1 and 5 have similar solid state geometries, the benzyl moiety is 1.5 Å farther away from the closest iron atom in the unreactive 2-benzylaniline, compared to the 2-benzylpyridine, complex.
When the benzyl group is moved from the ortho to the para position of the pyridine ring, the substrate is more distant from the diiron core than in 5, and the reactivity of the resultant diiron(II) complex is affected. Upon addition of dioxygen to a CH2Cl2 solution of 3, no ancillary ligand oxidation occurs. The benzyl carbon of the 4-Bnpy is more than 3 Å farther away from the closest iron atom than it is in 1 and is directed outward by the geometry of the ligand. Only by ligand dissociation or intermolecular reactions would it be possible to access any oxidizing species formed at the diiron core.
The core structures of the four ethylpyridine complexes 6 – 9 are virtually identical. Despite differences in the carboxylate ligands between 6 and 9 and in the pyridine ring substitution position for 6, 7, and 8, the compounds all adopt windmill structures with two ethyl-substituted pyridines as the N-donor ligands. Only where the ethyl group is in the ortho position, however, are 2-pyridylmethanol and 2-acetylpyridine formed upon addition of dioxygen. For 7 and 8, ethylpyridine is quantitatively recovered. This result suggests that an O2-activated diiron species is responsible for hydroxylation of the ethyl group. The presence of the ketone product, however, which was not observed in the benzylpyridine system, raises the possibility that radical chemistry may be involved. This and other mechanistic issues are targeted in our ongoing investigations of the present and related systems.
The carboxylate ligands with p-tolyl substituents form more reactive diiron oxidants than their p-fluorophenyl substituent analogs for both the benzyl- and ethylpyridine systems. Since the steric requirements of these two terphenyl carboxylate groups are similar, we attribute the decreased yield of alcohol product for the -O2CArTol vs the -O2CAr4-FPh systems to electronic factors. The pKa values of HO2CAr4-FPh and HO2CArTol are 6.14 and 6.50, respectively,52 but there are four such ligands, which may be sufficient to alter the reactivity of O2-generated species required to oxidize the C–H bonds and alter the yield.
Electronic factors are similarly likely to influence the relative reactivities of 1 and 2. The chloro substituent of the benzylpyridine in 2-(4-ClBn)py is far enough removed from the diiron center so as not to affect steric properties at the dimetallic center, but will withdraw electron density from the benzylic position. The pKa values of pyridinium and 4-chloropyridinium are 5.25 and 3.84, respectively.53 This difference in donating ability of H vs Cl may be responsible for the diminished amount of substrate that is oxidized in 2 compared to 1.
Conclusions
A series of diiron(II) complexes were prepared with benzyl- and ethylpyridine ligands to serve as substrates for C–H activation following introduction of dioxygen. Oxidation of organic moieties tethered in this manner to the diiron core was accomplished without the adjacent heteroatom employed in previous work.21-24 The current range of substrates was also extended from benzyl C–H to include less reactive ethyl C–H bonds. For the latter case the presence of the ketone product may indicate a reaction pathway involving radical chemistry. Substrate oxidation occurs only when the group is in close proximity to the diiron core by positioning it ortho to the coordinated N-atom of pyridine ring. No oxidation was observed for substituents placed in the meta or para positions of the pyridine. When steric factors remain constant, more electron-donating carboxylate and pyridine ligands increase the amount of oxidized product compared to their electron more deficient counterparts, suggesting the need to stabilize an electrophilic intermediate to carry out these transformations.
Supplementary Material
Supporting Information Available: Table S1 summarizing X-ray crystallographic information for 1 – 3, 5 – 9; Table S2 comparing the Fe…Fe and Fe–O distances in 1, 2, and 5 – 9 with their coordination number; Figures S1 – S8 showing ORTEP diagrams of 1 – 3, 5 – 9; Figure S9 illustrating Mössbauer data and fits for 1; and corresponding X-ray crystallographic files in CIF format for 2, 3, and 5 – 9. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by grant GM-32134 from the National Institute of General Medical Sciences. We thank Dr. Jane Kuzelka and Dr. Sungho Yoon for acquiring the Mössbauer spectra, Mr. Leslie J. Murray help with the GC-MS measurements, and Dr. Peter Müller for assistance with X-ray crystallography.
References
- 1.Erwin DP, Erickson IK, Delwiche ME, Colwell FS, Strap JL, Crawford RL. Appl Environ Microbiol. 2005;71:2016–2025. doi: 10.1128/AEM.71.4.2016-2025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vardar G, Wood TK. Appl Environ Microbiol. 2004;70:3253–3262. doi: 10.1128/AEM.70.6.3253-3262.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colby J, Stirling DI, Dalton H. Biochem J. 1977;165:395–402. doi: 10.1042/bj1650395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feig AL, Lippard SJ. Chem Rev. 1994;94:759–805. [Google Scholar]
- 5.Wallar BJ, Lipscomb JD. Chem Rev. 1996;96:2625–2657. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 6.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Angew Chem Int Ed Engl. 2001;40:2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 7.Baik MH, Newcomb M, Friesner RA, Lippard SJ. Chem Rev. 2003;103:2385–2419. doi: 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]
- 8.Whittington DA, Lippard SJ. J Am Chem Soc. 2001;123:827–838. doi: 10.1021/ja003240n. [DOI] [PubMed] [Google Scholar]
- 9.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 10.Lee D, Sorace L, Caneschi A, Lippard SJ. Inorg Chem. 2001;40:6774–6781. doi: 10.1021/ic010726b. [DOI] [PubMed] [Google Scholar]
- 11.Mandal SK, Young VG, Jr, Que L., Jr Inorg Chem. 2000;39:1831–1833. doi: 10.1021/ic9912676. [DOI] [PubMed] [Google Scholar]
- 12.Herold S, Lippard SJ. J Am Chem Soc. 1997;119:145–156. [Google Scholar]
- 13.Goldberg DP, Telser J, Bastos CM, Lippard SJ. Inorg Chem. 1995;34:3011–3024. [Google Scholar]
- 14.Rardin RL, Poganiuch P, Bino A, Goldberg DP, Tolman WB, Liu S, Lippard SJ. J Am Chem Soc. 1992;114:5240–5249. [Google Scholar]
- 15.Lee D, Lippard SJ. Inorg Chim Acta. 2002;341:1–11. [Google Scholar]
- 16.Hagadorn JR, Que L, Jr, Tolman WB. J Am Chem Soc. 1998;120:13531–13532. doi: 10.1021/ja983333t. [DOI] [PubMed] [Google Scholar]
- 17.Lee D, Lippard SJ. J Am Chem Soc. 1998;120:12153–12154. [Google Scholar]
- 18.Tolman WB, Que L., Jr J Chem Soc Dalton Trans. 2002:653–660. [Google Scholar]
- 19.Lee D, Lippard SJ. In: Comprehensive Coordination Chemistry II Bio-Coordination Chemistry. Que L Jr, Tolman WB, editors. Vol. 8. Elsevier Inc.; San Diego, CA: 2004. pp. 309–342. [Google Scholar]
- 20.Tshuva EY, Lippard SJ. Chem Rev. 2004;104:987–1012. doi: 10.1021/cr020622y. [DOI] [PubMed] [Google Scholar]
- 21.Lee D, Lippard SJ. J Am Chem Soc. 2001;123:4611–4612. doi: 10.1021/ja0100930. [DOI] [PubMed] [Google Scholar]
- 22.Lee D, Lippard SJ. Inorg Chem. 2002;41:827–837. doi: 10.1021/ic011008s. [DOI] [PubMed] [Google Scholar]
- 23.Yoon S, Lippard SJ. Inorg Chem. 2003;42:8606–8608. doi: 10.1021/ic0350133. [DOI] [PubMed] [Google Scholar]
- 24.Carson EC, Lippard SJ. Manuscript in preparation 2005 [Google Scholar]
- 25.Carson EC, Lippard SJ. J Am Chem Soc. 2004;126:3412–3413. doi: 10.1021/ja031806c. [DOI] [PubMed] [Google Scholar]
- 26.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 27.Lee D, Lippard SJ. Inorg Chem. 2002;41:2704–2719. doi: 10.1021/ic020186y. [DOI] [PubMed] [Google Scholar]
- 28.Bellezza F, Cipiciani A, Cruciani G, Fringuelli F. J Chem Soc Perkin Trans. 2000;1:4439–4444. [Google Scholar]
- 29.SMART v5.626: Software for the CCD Detector System. Bruker AXS; Madison, WI: 2000. [Google Scholar]
- 30.Kuzelka J, Mukhopadhyay S, Spingler B, Lippard SJ. Inorg Chem. 2003;42:6447–6457. doi: 10.1021/ic0345976. [DOI] [PubMed] [Google Scholar]
- 31.Sheldrick GM. SHELXTL97-2: Program for Refinement of Crystal Structures. University of Göttingen; Germany: 1997. [Google Scholar]
- 32.SHELXTL v5.10: Program Library for Structure Solution and Molecular Graphics. Bruker AXS; Madison, WI: 1998. [Google Scholar]
- 33.Sheldrick GM. SADABS: Area-Detector Absorption Correction. University of Göttingen; Germany: 1996. [Google Scholar]
- 34.Spek AL. PLATON, A Multipurpose Crystallographic Tool. Utrecht University; Utrecht, The Netherlands: 1998. [Google Scholar]
- 35.Kent TA. WMOSS v2.5: Mössbauer Spectral Analysis Software. WEB Research Co.; Minneapolis: 1998. [Google Scholar]
- 36.Rardin RL, Tolman WB, Lippard SJ. New J Chem. 1991;15:417–430. [Google Scholar]
- 37.Münck E. In: Physical Methods in Bioinorganic Chemistry: Spectroscopy and Magnetism. Que L Jr, editor. University Science Books; Sausalito, CA: 2000. pp. 287–319. [Google Scholar]
- 38.Yoon S, Lippard SJ. J Am Chem Soc. 2005;127:8386–8397. doi: 10.1021/ja0512531. [DOI] [PubMed] [Google Scholar]
- 39.Costas M, Mehn MP, Jensen MP, Que L., Jr Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 40.Takács J, Soós E, Nagy-Magos Z, Markó L, Gervasio G, Hoffmann T. Inorg Chim Acta. 1989;166:39–46. [Google Scholar]
- 41.Dickman MH. Acta Cryst. 2000;C56:58–60. doi: 10.1107/s0108270199013487. [DOI] [PubMed] [Google Scholar]
- 42.Ghosh P, Begum A, Bill E, Weyhermüller T, Wieghardt K. Inorg Chem. 2003;42:3208–3215. doi: 10.1021/ic020617c. [DOI] [PubMed] [Google Scholar]
- 43.Fallis IA, Farley RD, Malik KMA, Murphy DM, Smith HJ. J Chem Soc Dalton Trans. 2000:3632–3639. [Google Scholar]
- 44.Carson, E. C.; Lippard, S. J. Unpublished results.
- 45.Logan DT, Su XD, Åberg A, Regnström K, Hajdu J, Eklund H, Nordlund P. Structure. 1996;4:1053–1064. doi: 10.1016/s0969-2126(96)00112-8. [DOI] [PubMed] [Google Scholar]
- 46.Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ. Chem Biol. 1995;2:409–418. [PubMed] [Google Scholar]
- 47.Lindqvist Y, Huang W, Schneider G, Shanklin J. EMBO J. 1996;15:4081–4092. [PMC free article] [PubMed] [Google Scholar]
- 48.DeWitt JG, Bentsen JG, Rosenzweig AC, Hedman B, Green J, Pilkington S, Papaefthymiou GC, Dalton H, Hodgson KO, Lippard SJ. J Am Chem Soc. 1991;113:9219–9235. [Google Scholar]
- 49.McMillen DF, Golden DM. Ann Rev Phys Chem. 1982;33:493–532. [Google Scholar]
- 50.Barton BD, Stein SE. J Chem Soc Faraday Trans 1. 1981;77:1755–1762. [Google Scholar]
- 51.Rossi MJ, McMillen DF, Golden DM. J Phys Chem. 1984;88:5031–5039. [Google Scholar]
- 52.Chen CT, Siegel JS. J Am Chem Soc. 1994;116:5959–5960. [Google Scholar]
- 53.Asperger S, Cetina-Cizmek B. Inorg Chem. 1996;35:5232–5236. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Table S1 summarizing X-ray crystallographic information for 1 – 3, 5 – 9; Table S2 comparing the Fe…Fe and Fe–O distances in 1, 2, and 5 – 9 with their coordination number; Figures S1 – S8 showing ORTEP diagrams of 1 – 3, 5 – 9; Figure S9 illustrating Mössbauer data and fits for 1; and corresponding X-ray crystallographic files in CIF format for 2, 3, and 5 – 9. This material is available free of charge via the Internet at http://pubs.acs.org.