

Abstract
Prokaryotic DNA repair nucleases are useful reagents for detecting DNA lesions. UvrABC endonuclease, encoded by the UvrA, UvrB and UvrC genes can incise DNA containing bulky nucleotide adducts and intra-strand cross-links. UvrA, UvrB and UvrC were cloned from Bacillus caldotenax (Bca) and UvrC from Thermatoga maritima (Tma) and recombinant proteins were overexpressed in and purified from E. coli. Incision activities of UvrABC composed of all Bca derived subunits (UvrABCBca) and an interspecies combination UvrABC composed of Bca derived UvrA and UvrB and Tma derived UvrC (UvrABCTma) were compared on benoz[a]pyrene-7,8-dihyrodiol-9,10-epoxide (BPDE) adducted substrates. Both UvrABCBca and UvrABCTma specifically incised both BPDE adducted plasmid DNAs and site-specifically modified 50 bp oligonucleotides containing a single (+)-trans- or (+)-cis-BPDE-adduct. Incision activity was maximal at 55 - 60 °C. However UvrABCTma was more robust than UvrABCBca with 4-fold greater incision activity on BPDE adducted oligonucleotides and 1.5-fold greater on [3H]BPDE-adducted plasmid DNAs. Remarkably, UvrABCBca incised only at the 8th phosphodiester bond 5′ to the BPDE modified guanosine. In contrast, UvrABCTma performed dual incision, cutting at both the 5th phosphodiester bond 3′ and 8th phosphodiester bond 5′ from BPDE modified guanosine. BPDE adduct stereochemistry influenced incision activity and cis-adducts on oligonucleotide substrates were incised more efficiently than trans-adducts by both UvrABCBca and UvrABCTma. UvrAB-DNA complex formation was similar with (+)-trans- and (+)-cis-BPDE-adducted substrates suggesting that UvrAB binds both adducts equally and that adduct configuration modifies UvrC recognition of UvrAB-DNA complex. Dual incision capability and higher incision activity of UvrABCTma makes it a robust tool for DNA adduct studies.
Keywords: UvrABC endonuclease, Benzo[a]pyrene-diol-epoxide, DNA damage, DNA repair, preferential incision
1. Introduction
UvrABC endonuclease is a DNA repair nuclease consisting of three subunits encoded by the UvrA, UvrB and UvrC genes. This damage-specific endonuclease incises DNA containing a wide variety of structurally unrelated lesions including bulky chemical DNA-adducts and intra-strand cross-links (1-4). This system has been widely used for specifically detecting and mapping DNA adducts in human genes. Until recently, the only available UvrABC endonuclease has been from E. coli. A major disadvantage of E. coli UvrABC endonuclease is its thermal instability, especially the UvrA and UvrC subunits (5). This limited stability leads to difficulty in estimating incision efficiency and quantitative adduct detection.
In order to develop a more stable reagent with greater utility for DNA adduct detection, UvrABC endonuclease subunits were cloned from the thermophilic eubacteria Bacillus caldotenax (Bca) and Thermatoga maritima (Tma), organisms that have the remarkable property of growing at a temperature of 70 °C or above (6;7). Individual recombinant protein subunits were overexpressed in and purified from E. coli. DNA adducted by the highly reactive and mutagenic metabolite of benzo[a]pyrene, [(+)-7R, 8S-dihydroxy-9S,10R-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene (BPDE)] (8;9) was used as substrate to characterize the specific incision activity of recombinant Bca UvrABC (UvrABCBca) and interspecies combination of Bca UvrAB with Tma UvrC (UvrABCTma). We also used BPDE adduct stereochemistry as a tool to investigate the dependence on helix distortion in repair of DNA-adducts by both thermoresistant UvrABC endonucleases by digesting oligonucleotide substrates site specifically modified with (+)-cis- and (+)-trans-BPDE adducts. Efficient detection of BPDE-DNA adducts induced in human cells is very important for environmental carcinogen and cancer prevention studies. The results show that thermoresistant UvrABCBca and UvrABCTma are stable endonucleases with great utility for detecting BPDE-DNA adducts, both UvrABCBca and UvrABCTma preferentially incised cis-BPDE-adducted DNA. The interspecies combination of UvrABCTma not only makes a more robust endonuclease but confers dual incision capability not observed with UvrABCBca.
2. Materials and methods
2.1. Cloning, overexpression and purification of Bacillus caldotenax (Bca) UvrA, UvrB and UvrC, and Thermatoga maritima (Tma) UvrC and mutant Bca UvrC
B. caldotenax (Bca) UvrA, UvrB and UvrC proteins were overexpressed in E. coli cells and purified individually following the T7 IMPACT™ system manual (NEB) with some modifications as described in detail (10). T. maritima (Tma) UvrC was cloned, overexpressed and purified using the same procedures used for Bca UvrC. The QuickChange Site-Directed mutagenesis kit (Stratagene) was used to generate the plasmids encoding the Bca UvrC mutants. The following primer sets were used in conjunction with the parent vector, pTXB1-uvrCBca to prepare the indicated mutants: GGAGCAGCCGGGCGTGTATTTGATGAAAGAC and GTCTTTCATCAAATACACGCCCGGCTGCTCC for C18V; AAGTCGCTGAAAAATCGT-GTCCGCTCGTAT and ATACGAGCGGACAC-GATTTTTCAGCGACTTC for E41R; CGAAGTCGCTGAAAAAACGTGTCCGCTCGTATT and AATACGAGCGGACACGTTTTTTCAGCGACTTCG for E41K,. Correct clones were selected by sequencing to confirm that the mutation had been introduced. The mutant forms of pTXB1-uvrCBca were transformed into BL21(DE3) RIL cells (Stratagene). Mutants of Bca UvrC were overexpressed and purified using the same procedures used for wild type Bca UvrC. The proteins were maintained at -20 °C in storage buffer (50 mM Tris-HCl, pH 8.5, 500 mM KCl, 0.1 mM EDTA and 50% glycerol).
2.2. Preparation of BPDE damaged plasmid DNA
[3H](+)-7R,8S-dihydroxy-9S,10R-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene ([3H]BPDE, specific activity 1120 mCi/mmol) was obtained from the NCI Chemical Carcinogen Repository (Chemsyn, Kansas City, MO) and handled according to manufacturer's safety instructions. [3H]BPDE-adducted Form I plasmid DNAs (pTHQB04, containing a 1.5 kbp fragment of the human β-globin gene; pTHQ008, containing a 1.8 kbp fragment of the human p53 gene) were prepared as described previously (10). Briefly, DNAs were incubated with [3H]BPDE or tetrahydrofuran (THF, [3H]BPDE stock solvent) in TE buffer [10 mM Tris-HCl pH 7.5, 1 mM Na2- EDTA] 16 hrs at 37 °C. Unbound [3H]BPDE and its hydrolysis products were removed by ethyl acetate extraction (11). The extent of DNA adducts formed by [3H]BPDE in plasmid DNAs were determined by scintillation spectrometry.
2.3. Plasmid relaxation assay
Cleavage of [3H]BPDE-DNA substrates were monitored by following the conversion of plasmids from form I to form II in a plasmid relaxation assay as described (10). Briefly, [3H]BPDE treated substrates (20 fmol, equivalent to 71 ng pTHQB04 and 75 ng pTHQ008) were incubated with UvrA, UvrB and UvrC in 20 μl of UvrABC buffer (50 mM Tris-HCI, pH 7.5, 50 mM KCI, 10 mM MgCI2, 5 mM DTT, 1 mM ATP). Forms I and II were resolved by 1% agarose gel electrophoresis, visualized by staining with SYBR-Gold (Molecular Probes, Eugene Oregon, USA). Fluorescence emitted by SYBR-Gold of resolved bands was quantified using fluorescence detection mode (Blue excitation, 537 nm) of a Molecular Dynamics Storm 860 PhosphorImager (Amersham Pharmacia Biotech Inc. Piscataway, NJ). The average numbers of incisions and incision efficiencies were calculated using Poisson distribution (10).
2.4 Preparation of oligonucleotide substrates
2.4.1. DNA substrates containing stereoisomeric BPDE-adducts
Adducted oligonucleotides (11-mers site specifically modified with (+)-trans- and (+)-cis-anti-BPDE adducts) were the kind gifts of Dr. Nicholas Geacintov, NYU. The (+)-trans-, (+)-cis-, and no BPDE control 11-mers were ligated with equal moles of a 19-mer and a 20-mer in the presence of equal moles of the complementary 50-mer strands as described (12;13). The (+)-anti-BPDE-N2-dG-adducted strand was labeled with [32P] internally 21 bp from 5′-end or 19 bp from 3′ end, at the 3′- end or at the 5′- end as indicated in Figure 4. To construct the substrate labeled 21 bp from the 5′ end, the 11-mers [ (+)-trans, (+)-cis, or no BPDE] were 5′-phosphorylated using γ-[ 32P]-ATP and T4 polynucleotide kinase, 19-mer was 5′-phosphorylated using unlabeled ATP and T4 polynucleotide kinase prior to assembly. To construct the substrate labeled 19 bp from 3′- end, the 19-mer was 5′-phosphorylated using γ-[ 32P]ATP and T4 polynucleotide kinase, and 11-mers [(+)-trans, (+)-cis, or no BPDE control] were 5′-phosphorylated using unlabeled ATP and T4 polynucleotide kinase prior to assembly. To construct 5′end-labeled substrate, the 20-mer was replaced by a 19-mer (ACTACGTACTGTTACGGCT; 20-mer with 5′ G deleted) to produce an overhanging end to prevent self-ligation of 5′- end labeled double stranded logionucleotides. This 19-mer (20 mer with 5′ G deleted) was 5′-phosphorylated using γ-[ 32P]-ATP and T4 polynucleotide kinase, 11-mers [(+)-trans, (+)-cis, or no BPDE control] and right hand 19 mer were 5′-phosphorylated using unlabeled ATP and T4 polynucleotide kinase. To construct 3′ end-labeled substrate, the 19-mer was replaced by an 18-mer (GCAATCAGGCCAGATCTG; 19-mer with 3′ C deleted). The 18-mer and 11-mer [(+)-trans, (+)-cis, or no BPDE control] were 5′-phosphorylated using unlabeled ATP. Phosphorylated trans, (+)-cis, or control 11-mers were ligated with equal moles of phosphorylated 18-mer and unphosphorylated 20-mer in the presence of equal moles of the complementary 50-mer strand. The ligated oligonucleotides were modified with α-[32P]dCTP at 3′- end in the presence of Klenow DNA polymerase (14). [32P]-labelled 49- or 50-mers (5′-end labelled substrate is a 49 mer oligo) were purified by electrophoresis in a 12% polyacrylamide gel containing 7.5 M urea and were reannealed with fresh 50-mer complementary strand (bottom strand, Figure 4). The constructed 50-bp substrates were purified by electrophoresis in a non-denaturing 10% polyacrylamide gel containing 1 × TBE.
Figure 4. Incision maps of UvrABCBca and UvrABCTma on BPDE adducted oligonucleotide substrates. A. Substrates labeled internally 19 bp from 3′ end. B. Substrate labeled internally 21 bp from 5′ end. C. Substrate labeled at 3′ end.
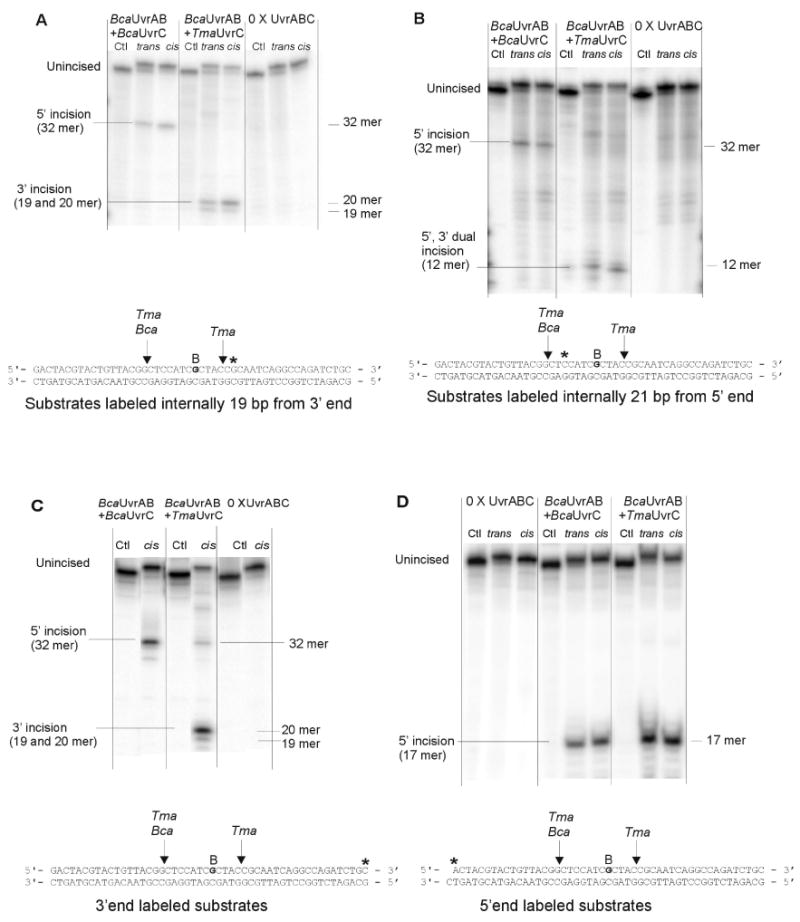
BPDE modified 50 bp double stranded oligonucleotides (1 nM) labeled at different sites were pre-incubated with UvrA (10 nM) and UvrB (250 nM) for 30 min at 60 °C, Tma UvrC or Bca UvrC (100 nM) was added and incubation continued at 60 °C for 60 min. Reaction products were denatured and resolved by electrophoresis in 15% polyacrylamide gels containing 7.5 M urea.
2. 4.2. Oligonucleotide substrates containing fluorescein (FldT) adduct
DNA substrates were synthesized by Sigma-Genosys (Woodlands, TX). The DNA sequence of the 50mer double-stranded substrate containing a single internal fluorescein (FldT) adduct was: F26, (GACTACGTACTGTTACGGCTCCATC[FldT]CTACCGCAATCAGGCCAGATCT GC) while the non-damaged complementary bottom strand was NDB, (GCAGATCTGGCCTGATTGCGGTAGCGATGGAGCCGTAACAGTACGTAGTC). The F26, 50-mer strand was either 5′ end-labeled or 3′ end-labeled. The 5′ end-labeling used Optikinase (USB) and 32P γ-ATP (3000 Ci/mmol, Amersham Biosciences) according to standard procedures. While the 3′ end-labeling was achieved by using Terminal transferase (Roche) and 32P α-dideoxyATP. The reactions were terminated by the addition of EDTA (20 mM) and the enzymes heat denatured by incubation for 10 min at 65 °C. Unincorporated radioactive nucleotides were removed by gel filtration chromatography (Biospin-6, BioRad). The labeled oligonucleotide was annealed with the complementary oligonucleotide using equimolar amounts. The double-stranded character of the oligonucleotide duplex was analyzed on a native 10% polyacrylamide gel.
2.5. UvrABC incision of oligonucleotide substrates
2.5.1. UvrABC incision of oligonucleotide substrates containing (+)-anti-BPDE stereoisomeric adducts
Fifty bp substrates (20 fmol) were preincubated with B. caldotenax UvrA (10 nM) and UvrB (250 nM) at 60 °C for 30 min. Bca UvrC or Tma UvrC was added to 100 nM and the reactions were incubated for an additional 60 min or as indicated in figures at 60 °C in 20 μl of UvrABC buffer. To investigate the temperature dependence of incision, experiments were performed under the following conditions: preincubation with 10 nM UvrA plus 250 nM UvrB at selected temperatures from 37 to 60 °C for 30 minutes, then UvrC was added to 100 nM and reactions continued at same temperatures for 30 min as indicated in Figure 2. Time courses of specific incision by UvrABCBca and UvrABCTma on trans- and cis-BPDE damaged 50 bp oligonucleotide substrates were examined in a kinetic assay at 37 °C and 60 °C (Figure 3). For time course of incision, UvrABC reactions were performed with 20 fmol of substrates and reactions conducted at 37 and 60 °C. Substrates were preincubated with 10 nM UvrA and 250 nM UvrB for 30 min, then UvrC was added to 100 nM and incubation continued for selected times from 0 to 60 min as indicated in Figure 3. Reactions were terminated by adding 2 μl of stop buffer (1% SDS, 200 mM EDTA-Na2). DNAs were precipitated by addition of 1/10 volume 3 M Na-acetate (pH 5.7) and 2.5 volumes 95%ethanol, and precipitates were collected by centrifugation. The pellets were redissolved in 96% formamide loading buffer (96% formamide, 0.05% xylene cyanole, 0.05% bromphenol blue, 10 mM Na2EDTA). DNAs were denatured at 90 °C for 5 min and resolved by electrophoresis in denaturing 15% polyacrylamide gel containing 7.5 M urea in 1× TBE buffer (89 mM Tris, 89 mM Boric acid, 10 mM EDTA, pH 8.4). Resolved band intensities were quantitated with a Molecular Dynamics Storm 860 PhosphorImager and Imagequant software.
Figure 2. Temperature dependent incision of trans- and cis-BPDE adducted oligonucleotide substrates. A. UvrABCTma B. UvrABCBca.
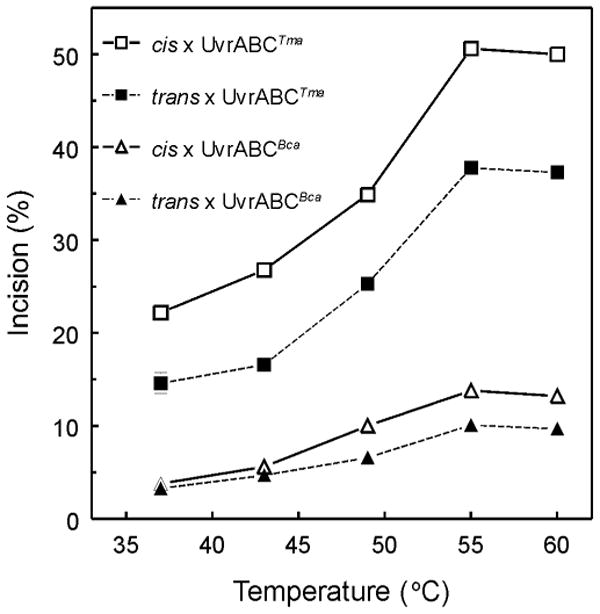
Trans- and cis-BPDE adducted 50 bp double stranded oligonucleotides labeled internally 19 bp from 3′-end (1 nM) were pre-incubated with UvrA (10 nM) and UvrB (250 nM) for 30 min at 37, 43, 49, 55 or 60 °C as indicated. UvrCTma (A.) or UvrCBca (B.) was added to 100 nM and the reaction was incubated for an additional 30 min at the indicated temperatures. Reaction products were denatured and resolved by electrophoresis in 12% polyacrylamide gels containing 7.5 M urea. The intensities of the resolved bands were quantitated and the average incision ratio was calculated. Means ± SD from triplicates reaction are plotted.
Figure 3. Time course of UvrABCTma and UvrABCBca incisions of trans- and cis-BPDE adducted oligonucelotide substrates. A. 60 °C B. 37 °C.
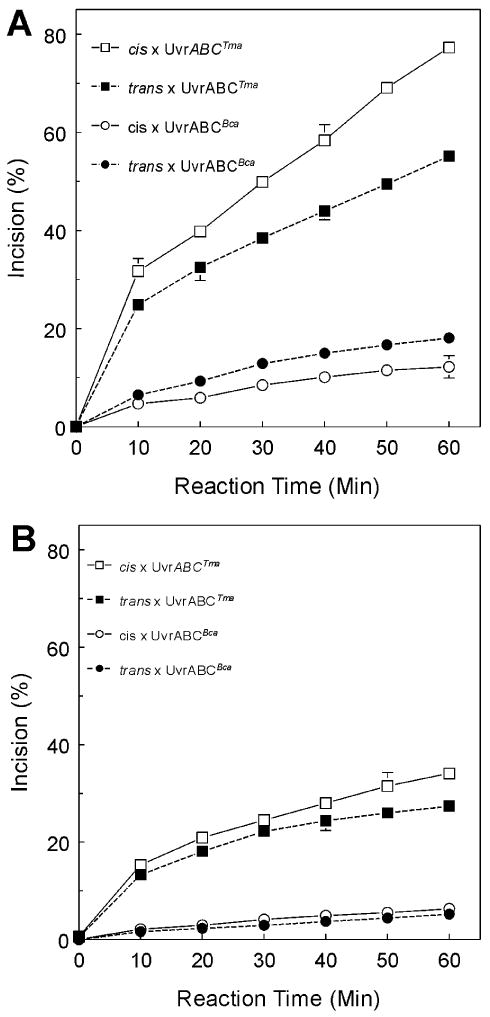
Oligonucleotides containing either a cis-, a trans- or no BPDE-adduct were site specifically labeled with [32P] internally 19 bp from 3′-end to monitor the 5′ incision (producing a 32-mer fragment) for UvrABCBca and 3′ incision (producing a 20-mer fragment) for UvrABCTma, labeled 50-bp oligonucleotides were used as substrates in UvrABC reactions. UvrABC reactions were performed with twenty femto moles of substrates. Preincubation of substrates with UvrA 10 nM and UvrB 250 nM for 30 minutes at 60 °C, then add UvrC (100 nM) with incubation at varied time as indicated in the figure. Reaction products were denatured and resolved on 12% polyacrylamide sequencing gel. The intensities of the resolved bands were quantitated and the average incision ratio was calculated. Means ± SD from triplicate reactions are plotted.
2.5.2. UvrABC incision of DNA substrates containing fluorescein (FldT) adduct
Prior to initiation of the incision assay, the UvrABC proteins were heated to 65 °C for 10 min. The 5′ or 3′ end-labeled duplex DNA (2 nM, F2650/NDB) was treated with wild type UvrABC or UvrAB with mutant Bca UvrC (20 nM Bca UvrA, 100 nM Bca UvrB and 50 nM Bca UvrC) in 20 μl of UvrABC buffer (50 mM Tris-HCl pH 7.5, 50 mM KCl, 10 mM MgCl2, 1 mM ATP and 5 mM DTT) at 55 °C for the 30 min. The reactions were terminated by addition of EDTA (20 mM). The DNA from reaction was denatured by addition of formamide and heating to 85 °C for 5 min. Incision products were resolved on a 10% denaturing polyacrylamide gel and electrophoresis was performed at 325 V in 1 × TBE buffer for 40 min. Gels were dried and exposed to a phosphorImager screen (Molecular Dynamics) overnight. The percent of the DNA incised was calculated using the band intensities within the lane and the Molecular Dynamics software, ImageQuant.
2.6. Binding affinity assay of UvrA or UvrAB to (+)-anti-BPDE stereoisomeric adducts
To investigate the role of BPDE-adduct stereochemistry on repair of DNA-adducts, binding affinity of UvrA or UvrA /UvrB with (+)trans- and (+)cis-BPDE adducted substrates was examined. [32P]-labeled DNA substrates (1 nM) containing stereospecific adducts were incubated with B. caldotenax UvrA alone or UvrA plus UvrB at 60 °C for 30 min in a 10 μl reaction containing UvrABC buffer. Following incubation, 2 μl of 80% glycerol was added and the mixture was immediately loaded onto a 4% native polyacrylamide gel containing 1× TBE buffer plus 1 mM ATP and 10 mM MgCl2 and run at 4 °C. The binding ratios of UvrA or UvrAB with (+)trans- or (+)cis-BPDE substrates were quantitated with a Molecular Dynamics Storm 860 PhosphorImager and Imagequant software.
3. Results
3.1. UvrA, UvrB and UvrC expression and purification
Purification of recombinant Bacillus caldotenax (Bca) UvrA, UvrB and UvrC subunits was reported elsewhere (10). Thermatoga maritima (Tma) UvrC was cloned, overexpressed in E. coli and purified using a similar approach. The activities of UvrC from Bca and Tma were compared when used in conjunction with Bca UvrA and UvrB. The resulting endonuclease systems are referred to as UvrABCBca and UvrABCTma.
3.2. Incision capability of UvrABCBca and UvrABCTma on BPDE damaged plasmid DNAs
A plasmid relaxation assay was used to compare incision capacities of UvrABCBca and UvrABCTma endonucleases. When adducts formed on supercoiled DNA are incised by UvrABC nucleases, the DNA helix is relaxed and Form I DNA (supercoiled) is changed to Form II (nicked/open circular DNA). The two forms are readily resolved by agarose gel electrophoresis. Two plasmids (pTHQB04 and pTHQ008) were used as substrates for UvrABC incision assays. Plasmid DNAs untreated (THF control) and treated with [3H]BPDE to form 2 adducts per plasmid molecule (10) were used as substrates for the cleavage reactions. The substrates were incubated with UvrABCBca or UvrABCTma with UvrC at 0, 12.5 or 25 nM as indicated in Figure 1. Maximal activity for each was achieved with UvrC at 25 nM. Specific incision on [3H]BPDE damaged pTHQ008 was 48% with UvrABCBca. Subsitution of UvrABCTma for UvrABCBca increased incision to 70%. Incision of UvrABCBca on [3H]BPDE damaged pTHQB04 was 41%, while reactions with UvrABCTma exhibited incision of 58%. Thus, UvrABCTma appears to be more active than UvrABCBca on BPDE-adducted plasmid DNAs.
Figure 1. Incision of [3H]BPDE treated plasmids by UvrABCTma and UvrABCBca A. [3H]BPDE treated pTHQ008. B. [3H]BPDE treated pTHQB04.
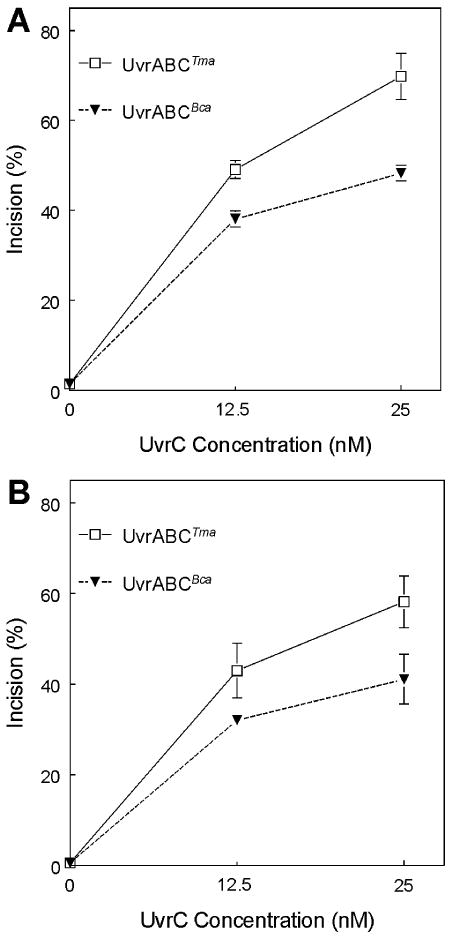
Plasmids pTHQ008 and pTHQB04 treated with (+)-[3H]BPDE-anti (2 lesions/plasmid) were used as substrates for UvrA, UvrB and UvrC in a plasmid relaxation assay. Twenty fmol of DNA substrate were incubated with UvrABC in 20 μl of UvrABC buffer. Bca UvrA (2.5 nM) and Bca UvrB (62.5 nM) were held constant. Tma UvrC and Bca UvrC concentrations were varied as indicated. Form I and Form II plasmids were resolved by agaraose gel electrophoresis, quantitated and the average incision was calculated by applying the Poisson distribution and expressed as % of lesions incised. Means ± SD from quadruplicates reactions are plotted.
3.3. Temperature dependent incision of UvrABCBca and UvrABCTma on BPDE adducted DNAs
To demonstrate the thermophilic character UvrABCBca and UvrABCTma incision activity, temperature dependence of incision was determined. Oligonucleotides (50-mers) containing either a (+)cis-, a (+)trans- or no BPDE-adduct in one strand were site-specifically labeled (internally 19 bp from 3′-end of 50-bp substrates) with [32P] and used as substrates for UvrABC endonuclease. Incision by UvrABCBca produced a 32 nucleotide fragment (5′ incision only), whereas incision by UvrABCTma produced a 20 nucleotide fragment (3′ incision monitored). Efficient incision required increased UvrABC concentration up to 4-fold as compared to reaction conditions with BPDE-damaged plasmid DNAs. Incision activity was temperature-dependent and reached a plateau at 55 - 60 °C (Figure 2). At 37 °C, both UvrABCBca and UvrABCTma exhibited low incision activity. At all temperatures monitored, both UvrABCBca and UvrABCTma incised cis-BPDE-adducted DNA substrates more than trans-BPDE-adducted DNA substrates. Incision by UvrABCTma was 3- to 4-fold greater than incision by UvrABCBca at all temperatures and on both substrates.
3.4. Time course of specific incision of UvrABCBca and UvrABCTma on trans- and cis-BPDE damaged DNA
The selectivity of UvrABCBca and UvrABCTma for damaged DNAs was examined further by testing the ability of UvrABC specifically to recognize and to incise (+)trans- and (+)cis-BPDE-damaged 50 bp oligonucleotide substrates in a kinetic assay (Figure 3). The time course of lesion specific incision was determined at 37 °C and 60 °C. Oligonucleotides containing either a cis-, a trans- or no BPDE-adduct were site specifically labeled with [32P] 19 bp from the 3′ end to monitor incision (5′ incision for UvrABCBca, producing a 32-mer fragment; 3′ incision for UvrABCTma, producing a 20-mer fragment). Incision activities of both UvrABCBca and UvrABCTma were time dependent on both (+)trans- and (+)cis-BPDE DNA substrates. Incision continued to increase for at least 60 min at 60 °C. Incision was less robust at 37 °C for both UvrABCBca and UvrABCTma. Incision was not observed on control substrates that did not contain a BPDE adduct. Incision activity of UvrABCTma was about 3.5-fold greater than UvrABCBca. Again, both UvrABCBca and UvrABCTma showed greater incision activity on (+)cis-BPDE-adducted substrates than on (+)trans-BPDE-adducted substrates.
3.5. UvrABCTma exhibits dual incision capability not present in UvrABCBca
As noted above, UvrABCBca incised only 5′ of the lesion, whereas UvrABCTma incised 3′ of the lesion. In order to map the specific incision sites and to demonstrate that UvrABCTma performed dual incision, (+)-trans, (+)-cis, and no BPDE 50-bp substrates were constructed and labeled on the (+)-anti-BPDE-N2-dG-adducted strand with [32P] either internally 21 bp from 5′ end, internally 19 bp from 3′ end, at the 3′- end or at the 5′ end (49/50-mer, see methods 2.4). The results of incision of substrates labeled internally 21 bp from 5′ end, internally 19 bp from 3′ end, and at the 3′- end showed that UvrABCBca only produces a 32-mer fragment from substrates labeled at all 3 sites (Figure 4, panels A-C). The results of incision of substrates labeled at 5′ end showed that UvrABCBca only produces a 17-mer fragment (Figure 4, panel D).These results indicate that Bca UvrC only incises at the 8th phosphodiester bond 5′ to the BPDE modified guanosine. In contrast, UvrABCTma generates different patterns with different sites of labelling (Figure 4, panels A-D). UvrABCTma incision of oligonucleotides labelled 19 bp from the 3′ end or at the 3′ end generates a doublet corresponding to 19 and 20-mers (Figure 4, panels A and C). A 12-mer is released by incision of oligonucleotides labeled 21 bp from the 5′ end (Figure 4B). A 17-mer fragment is produced by Tma UvrC incision of substrates labeled at the 5′ end (Figure 4, panel D). These results indicate that UvrABCTma performs dual incision, cutting both the 5th phosphodiester bond 3′ and 8th phosphodiester bond 5′ from BPDE-modified guanosine.
3.6. The interrelation of UvrABCTma and UvrABCBca in UvrABC incision reaction
To investigate potential interaction between Tma UvrC and Bca UvrC in the UvrABC incision reaction, Tma UvrC and Bca UvrC were added individually, sequentially or concurrently into the reactions after preincubation of substrate oligos with Bca UvrA and UvrB. Incision on cis-BPDE-adducted oligonucelotides was monitored with substrates [32P]-labeled 19 bp from 3′ end. Incision 5′ to the adducted nucleotide without 3′ incision yields a labeled 32-mer. Incision 3′ of the adducted nucleotide (with or without 5′ incision) yields a labeled 20-mer. Bca UvrC alone yielded the 32-mer as seen previously. Likewise, Tma UvrC alone yielded only the 20-mer product and in greater yield than the Bca UvrC generated 32 mer product. Sequential incubation with Tma UvrC followed by Bca UvrC yielded 20-mer in amounts equal to Tma UvrC alone and a small amount of 32-mer. This result suggests that Bca UvrC incised a small number of substrates not incised by Tma UvrC. Sequential incubation first with Bca UvrC then Tma UvrC also yielded both 32-mer and 20-mer bands. However, both were less intense than observed with either Bca UvrC or Tma UvrC alone. Reduction in 32-mer generated by Bca UvrC after addition of Tma UvrC suggests that Tma UvrC is making a 3′ incision on substrates already incised 5′ of the lesion by Bca UvrC. Concurrent addition of Bca UvrC and Tma UvrC yielded both the 32-mer and the 20-mer. Howerver, although the 20-mer was more intense than the 32-mer, the intensities of both bands were less than with either UvrC alone. This result suggests that the Tma UvrC clearly is more active than the Bca UvrC, the two UvrC's do not cooperate and may interfere with each other.
3.7. UvrA and UvrAB recognize and bind both (+)-trans- and (+)-cis-BPDE modified substrates with equal affinity
Incision of cis-BPDE adducted substrates was greater than incision of trans-BPDE-adducted substrates by both UvrABCBca and UvrABCTma. To investigate whether BPDE stereochemistry affected repair by altering recognition of the lesion by UvrA or in loading UvrB by UvrAB, the binding affinity of UvrA and UvrA plus UvrB with trans- and cis-BPDE adducted substrates was examined by electrophoretic mobility shift assay (EMSA).
UvrA-DNA complex formation was the same for both (+)cis- and (+)trans-BPDE adducted substrates (Figure 6A). Likewise, UvrAB complex formation also was the same for the two substrates (Figure 6B). These results indicate that UvrAB complex recognizes and binds both adducts with equal affinity and that UvrA and UvrAB do not distinguish between cis- and trans-BPDE-adducts. Thus, the preferential incision of (+)-cis-BPDE-adducts suggests that adduct configuration modifies recognition of UvrB-DNA lesion complex by both Bca and Tma UvrC.
Figure 6. Protein-DNA complex formation with (+)-trans- and (+)-cis-BPDE adducted oligonucleotides. A. UvrA-DNA complex formation. B. UvrAB-DNA complex formation.

Trans and cis-BPDE modified 50 bp double stranded oligonucleotides (1 nM) were incubated with different concentrations of Bca UvrA or UvrA + UvrB as indicated in the figure. UvrA and UvrB relative units: 1 = UvrA 2.5 nM, UvrB 62.5 nM. The reaction was performed at 60 °C for 30 min in a 10 μl reaction containing 50 mM Tris-CI, pH7.5, 50 mM KCI, 10 mM MgCI2, 5 mM DTT and 1 mM ATP. The reaction samples were resolved on 4% native polyacylamide gel run with TBE buffer containing 1 mM ATP and 10 mM MgCI2 at 4 °C. The intensities of the resolved bands were quantitated and the average binding ratios of UvrA or UvrA B with trans- or cis-BPDE substrates were calculated. Means ± SD of four independent experiments are plotted.
3.8. The Bca UvrC mutants exhibit similar 5′ incision property as wild type Bca UvrC
3.8.1. Analysis of the Bca UvrC amino acid sequence
Comparison of the Bca UvrC sequence to 22 UvrC proteins including Tma and E. coli UvrC, revealed several differences in the N-terminal nuclease domain, specifically at Cys18 and Glu39 of the Bca UvrC sequence. An alignment of Bca UvrC, Tma UvrC and E. coli UvrC is depicted in Figure 7. Within the Bca UvrC sequence, Cys18 lies within the first conserved element of the GIY-YIG endonuclease domain, however, among UvrCs, Val is the most common amino acid between the absolutely conserved Gly and Tyr. Unlike almost all other UvrC sequences, Bca UvrC contains an acidic amino acid, Glu39, adjacent to the catalytic Arg, Arg40. We hypothesized that these amino acids may interfere with the 3′ incision event and therefore via site-directed mutatagenesis these amino acids were converted to the most common amino acids present at these position. The amino acid changes were: Cys18Val, Glu39Asn and Glu39Lys. The recombinant proteins were purified as described for the Wt proteins and an image of the proteins is shown in Figure 8, panel A.
Figure 7. Alignment of UvrC sequences of E. coli, B. caldotenax and T. maritima. A. 3′ nuclease domain. B. Uvr interaction domain.

Amino acids shared by all 3 UvrC proteins are highlighted in light blue; by E. coli UvrC (EcoC) and B. caldotenax UvrC (BcaC), in yellow; by EcoC and T. maritima UvrC (TmaC), in magenta; by BcaC and TmaC, in green. Alignment performed using Clustal W. In panel A, the amino acids (C18 and E39) that were mutated in Bca UvrC are highlighted in red and marked with a † above the sequence.
Figure 8. Mutation of C18 or E39 does not restore 3′ endonuclease activity.
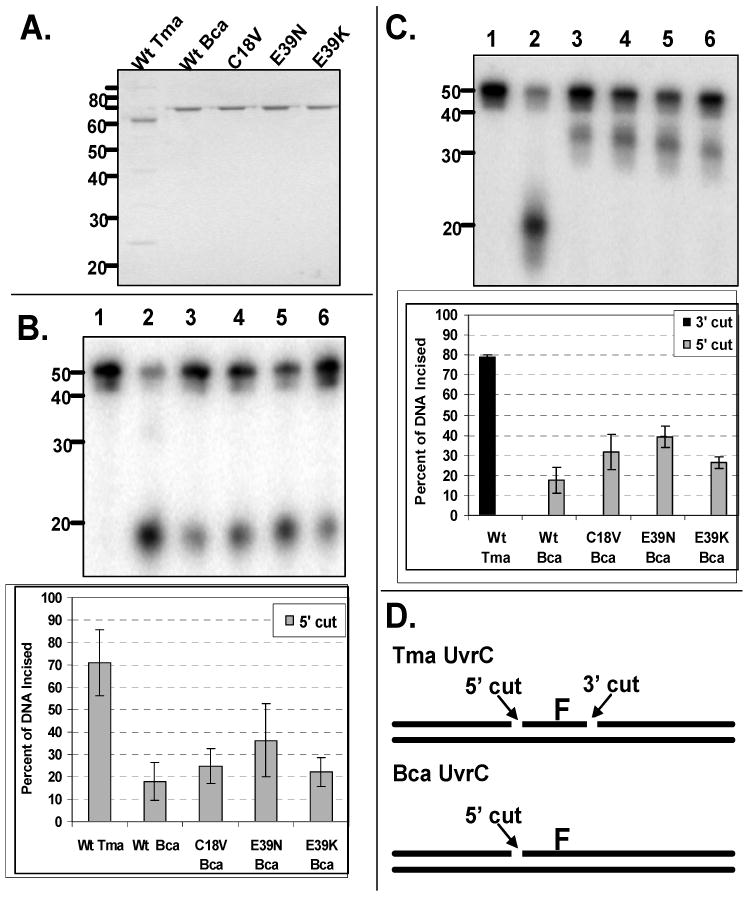
A. Protein gel of proteins used in study. Fifteen microliters of each protein (500 nM) was applied to a 10% Tris-Bis NuPAGE gel. Electrophoresis was carried out for 55 min at 200 V. The protein gel was stained with SimplyBlue and destained with water.
B. Oligonucleotide incision products and graphic representation of the data obtained from the 5′–[32P]-end-labeled duplex. The fluorescein adducted 50 bp duplex (2 nM F2650/NDB) was incubated with UvrA (20 nM), UvrB (100 nM), and the indicated UvrC (50 nM) at 55 °C for 30 min in reaction buffer. The reactions were terminated with stop buffer, and the incision products were analyzed on a 10% denaturing polyacrylamide gel. Graphic representation of the data is reported as the mean±the standard deviation (n=3).
C. Oligonucleotide incision products and graphic representation of the data obtained from the 3′-[32P]-end-labeled duplex. The fluorescein adducted 50 bp duplex (2 nM F2650/NDB) was incubated with UvrA (20 nM), UvrB (100 nM), and the indicated UvrC (50 nM) at 55 °C for 30 min in reaction buffer. The reactions were terminated with stop buffer, and the incision products were analyzed on a 10% denaturing polyacrylamide gel. Graphic representation of the data is reported as the mean±the standard deviation (n=3).
D. Graphic summarizing the observed endonuclease sites produced by the different UvrC proteins. Tma UvrC creates the dual incision while Bca UvrC only makes the cut on the 5′ side of the DNA lesion.
3.8.2. Oligonucleotides incision assays of wild type and mutated Bca UvrC
Oligonucleotide incision assays were performed with wild type (Wt) Tma, Wt Bca and mutant Bca UvrC proteins to compare the incision patterns and relative activity (Fig. 8). The DNA substrate was a fluorescein-adducted thymine centrally located within a 50 bp duplex. As shown in Fig. 8 panels B and C, the Tma and the Bca UvrC proteins created the same incision pattern when the DNA was 5′ end-labeled with [32P], a 18 nucleotide product. Curiously, Wt Bca UvrC possessed about 25% of the incision activity as Wt Tma, while the mutants ranged between 31 - 51%. When the DNA was labeled at the 3′ end of the duplex with [32P], the Bca UvrC proteins failed to make the expected 3′ cut, because a 20 bp product was expected while a 32 bp product is observed. This is consistent with the observation that Bca UvrC only makes the incision on the 5′ side of the DNA lesion while Tma UvrC makes the dual incision (Fig. 8, panel D). Therefore, the Bca UvrC amino acids Cys18 and Glu39 play no role in determining whether the 3′ cut will occur.
Discussion
Nucleotide excision repair (NER) is the major pathway that removes UV photoproducts and “bulky” chemical adducts from DNA. In prokaryotes, lesion recognition and excision is performed by the UvrABC endonuclease. UvrABC endonuclease detects and removes DNA damage induced by a wide range of chemical carcinogens. UvrABC consists of three subunits (UvrA, UvrB, UvrC) that act sequentially. Studies with E. coli UvrABC indicate that damage recognition begins with the dimerization of UvrA. The UvrA2 dimer loads UvrB onto the lesion, resulting in a stable UvrB-DNA pre-incision complex at the lesion, and UvrA2 is released. This model is corroborated by recent studies by Della Vecchia et al (15) using Bca UvrA and UvrB. The UvrB-DNA preincision complex is recognized by UvrC and leads to incision of the damaged strand at the fourth or fifth phosphodiester bond 3′ to the lesion. Generally, this 3′ incision is immediately followed by hydrolysis of the eighth phosphodiester bond 5′ to the lesion to complete excision (16-20). Two notable exceptions to this paradigm have been reported. CHO, which incises only 3′ of a lesion, and UvrCII, a high salt induced E. coli UvrC tetramer that incisies only 5′ of certain lesions. Our studies of Bca UvrC, which incises only 5′ of both (+)trans- and (+)cis-BPDE-DNA adducts, indicate a third exception to the paradigm.
Moolenaar et al (21;22) reported an E. coli UvrC homologue named Cho that incises only at the 9th phosphodiester bond 3′ to the modified nucleotide, four nucleotides beyond the site of the normal UvrC incision. Cho can incise some DNA lesions more efficiently than Eco UvrC. Eco UvrC can make the 5′ incision on a substrate already incised 3′ by Cho indicating potential for interaction between UvrC and Cho. In the present study, we observed that Tma UvrC could incise 3′ of the lesion on substrates already incised 5′ by Bca UvrC. Thus, Tma UvrC can complement Bca UvrC to make a 3′ incision which is not observed in the absence of Tma UvrC. Also, sequential incubation of substrates with UvrABCTma followed by addition of Bca UvrC, we found 5′ incision fragment produced by UvrABCBca indicating incision on substrates not incised by UvrABCTma. These results raise the possibility that Bca UvrC might be a B. caldotenax homologue of UvrC with only 5′ incision activity and that another as yet unidentified UvrC homologue providing 3′ incision activity in B. caldotenax has not yet been identified. Thus analogous to UvrC/Cho interaction in E. coli, UvrC homologues may work together for efficient dual incision on particular lesions.
Tang et al (23) separated two forms of E. coli UvrC eluting at different ionic strengths from DNA-cellulose: UvrCI (eluted with 0.4 M KCI) and UvrCII (eluted with 0.6 M KCI). The molecular weight of UvrCII is four times that of UvrCI suggesting that UvrCII is a tetramer. The specific incision activity of UvrCII is only one-fourth that of UvrCI correlating with much lower binding affinity of UvrCII than UvrCI to UvrAB-DNA complex. The sites of incision at 5′ and 3′ of a UV-induced pyrimidine dimer are same for both UvrCI and UvrCII (23). However, UvrCI and UvrCII have different nuclease incision activity on a CC-1605-N3-adenine adduct. UvrCI exhibits dual incision activity, cutting 5′ and 3′ of the adduct, whereas UvrCII makes only the 5′ incision (24). Our UvrC preparations are eluted from the chitin binding column in 2 M NaCl and dialyzed to 500 mM NaCl (10). It is possible that the high salt conditions are converting Bca UvrC but not Tma UvrC into a tetramer and the tetrameric form exhibits 5′-only incision on certain DNA lesions. Tetrameric UvrCII was able to make incisions at lesions in the absence of UvrB. We have not been able to detect tetramerization of Bca UvrC. Furthermore, specific incision at lesions is observed only in the presence of UvrA/UvrB (10).
Results of the present study show that UvrABCBca and UvrABCTma both exhibit better incision of (+)cis-BPDE adducts than (+)trans-BPDE adducts, similar to the preference exhibited by E. coli UvrABC (13;14). This differential in incision is not as great as in human nucleotide excision repair extracts (25). Our results combined with these earlier reports by others clearly indicate that UvrA binding affinity is not the only determinant in UvrABC recognition and excision of lesions.
(+)BPDE-anti forms two stereoisomeric adducts: (+)trans- and (+)cis-BPDE adducts. The predominant conformation of the (+)trans-adduct has the pyrene ring system lying in DNA minor groove (causing a minor helix distortion). The predominant conformation of the (+)cis-adduct has the pyrene ring system “intercalated” in DNA causing a major helix distortion (26-29). A widely held hypothesis is that the configurational and conformational differences play a major role in recognition of BPDE-induced DNA lesions by NER. This conformational difference is proposed to contribute to the preferential recognition of the (+)cis-BPDE-adduct by the NER systems of both humans and prokaryotes. To answer the question of whether poor incision of (+)trans-BPDE-adducts by UvrABC is caused by poor formation of UvrB-DNA pre-incision complex to trans-BPDE-adducts, we tested the binding affinity of UvrA and UvrA/UvrB with trans- and cis-BPDE adducted substrates. Neither UvrA nor UvrA/UvrB displayed differential binding affinity for (+)-trans or (+)-cis-BPDE modified substrates (Figure 6). This result indicates that UvrA/UvrB recognizes and binds both adducts with equal affinity. Zou et al (14) reported that E. coli UvrA and UvrB form complexes better with (+)cis-BPDE-adducted DNA than with (+)trans-BPDE-adducted DNA. Thus, the efficiency of incision by E. coli UvrABC correlated with UvrAB binding affinity for the (+) isomers. However, this correlation did not hold when comparing binding and incision of (+) and (-) isomers, suggesting that factors other than UvrAB affinity were important. In our studies, preferential incision of (+)-cis-BPDE-adducts in the face of equal binding by Bca UvrAB of trans- and cis-BPDE adducts supports the suggestion that UvrC plays a final role in DNA lesion recognition. Since UvrC incision of UvrB-DNA complex is adduct configuration dependent for both Bca and Tma UvrC, the structure of the DNA adduct appears to affect the interactions between UvrC and the UvrB/DNA complex. UvrB has ATPase and DNA helicase activity. When it binds DNA-adducts, UvrB uses its helicase fold to bend the DNA and push the damaged DNA strand into the nuclease cleft in UvrC (19;30-32). UvrC is capable of recognizing some secondary structure, such as UvrB opened DNA complex (18;31;33;34). It is very possible that UvrC will not cut unless UvrB has bound and altered the conformation of damaged DNA, or if the DNA is not bent sufficiently to enter into the UvrC nuclease site. We propose that the (+)cis-BPDE adduct may more easily adopt the preferential structure upon binding by Bca UvrB at the elevated temperatures of the assay, thus facilitating UvrC binding and incision. Clearly, the mechanism of UvrC discrimination between similar lesions is not well understood and further study is needed.
The exciting discovery in this study is that interspecies use of UvrABCTma not only makes a more robust endonuclease but also confers dual incision capability (cutting at both the 4th or 5th phosphodiester bond 3′ and the 8th phosphodiester bond 5′ to BPDE modified guanosine) not present in UvrABCBca. Bca UvrC incises only at the 8th phosphodiester bond 5′ to the BPDE modified guanosine. The results were confirmed on substrates modified with both (+)trans- and (+)cis-BPDE [32P]-labeled in four different positions. This result raises an intriguing question: why does Bca UvrC incise only at the 8th phosphodiester bond 5′ to the BPDE lesion but not on the 3′ side? The mechanism of UvrC incision defined with E. Coli UvrABC is a sequential process. When UvrC recognizes UvrB-DNA pre-incision complex, it first incises the damaged strand at the 4th or 5th phosphodiester bond 3′ to the lesion and then immediately follows this 3′ incision by hydrolysis of the 8th phosphodiester bond 5′ to the lesion. Our results clearly show that 5′ incision is not always dependent on prior 3′ incision.
UvrC has two functional nuclease domains (34;35;35-37). The N-terminal domain is responsible for cutting the damaged strand most commonly at the 5th phosphodiester bond 3′ to the modified nucleotide, while the C-terminal domain is responsible for the second incision at the 8th phosphodiester bond 5′ to the damaged nucleotide. The N-terminal nuclease domain contains a region that interacts with a homologous domain of the UvrB (C-terminal) in the UvrBC-DNA incision complex (35;36). In E. coli, the C-terminal nuclease domain contains all the elements for 5′ incision (34;35). Tma UvrC incises with high efficiency and produces 5′ and 3′ dual incision.
Sequence divergence between Bca UvrC and Tma UvrC may highlight residues important for 3′ nuclease activity. The region of UvrC most likely to impact 3′ incision activity is the 3′ nuclease domain. Comparison of this region in of Tma, Bca and Eco UvrC (Figure 7A) revealed that Bca UvrC is more similar to Eco UvrC than Tma UvrC. Bca UvrC shares all but 4 amino acids with Eco UvrC, whereas Tma UvrC has 8 amino acid differences, 3 of which are at divergent positions in Bca UvrC. Because Tma UvrC and not Bca UvrC retains dual incision activity, one might infer that the 8 divergent amino acids in the Tma sequence must not be essential for 3′ nuclease activity. The arginine residue at position 42 in Eco UvrC (marked with # in Figure 7A) is critical to 3′ nuclease activity and is flanked by divergent amino acids in Bca UvrC. However, the amino acid preceding is a significant change in charge (K > E) in Bca UvrC but changed to neutral charge (K>N) in Tma UvrC, whereas the following amino acid is a conservative change in Bca UvrC (L>V) and unchanged in Tma UvrC. We tested whether two of the divergent amino acids in Bca UvrC were responsible for the loss of 3′ incision activity. Changing neither Cys18 to Val, nor Glu39 to Asn or Lys, was able to confer 3′ incision activity on Bca UvrC. There are numerous other amino acid differences between Bca UvrC and other UvrC proteins that have dual incision activity. Extensive site directed mutation studies are required to elucidate which of these amino acid changes are responsible for loss of the 3′ nuclease activity in Bca UvrC.
It is possible that the interactions of Bca UvrC and Tma UvrC with UvrB via the Uvr domain are different causing the respective UvrC's to align differently thus abrogating 3′ nuclease activity in Bca UvrC. As shown in Figure 7B, the Uvr regions of Bca, Tma and Eco UvrC share some sequence identity but diverge considerably. Again, although Bca is slightly less divergent from Eco UvrC overall than is Tma UvrC, there are amino acids conserved in Tma UvrC that are not conserved in Bca UvrC. The fact that we have examined functionality with Bca UvrA, UvrB and UvrC and under these conditions Bca UvrC has only 5′ endonuclease activity, suggests that B. caldotenax may have a second UvrC homologue analogous to E. coli CHO that performs the 3′ incision.
In conclusion, recombinant thermoresistant UvrABC endonucleases composed of subunits cloned from B. caldotenax and T.maritima were used as an analytical tools to detect BPDE-DNA adducts. The results show that both UvrABCBca and interspecies UvrABCTma specifically incise (+)-trans- and (+)-cis-BPDE-DNA adducts with greater activity on (+)-cis-BPDE- adducts. Interestingly, the interspecies UvrABCTma not only performs dual incision but also is more active than UvrABCBca. UvrA/UvrAB complex recognizes and binds both trans- and cis-adducts with equal affinity, but UvrC from both species preferentially incise (+)-cis-BPDE-adducts suggesting that UvrC recognition of UvrB-DNA lesion complex plays a final role in DNA lesion recognition. Comparison of these two UvrC proteins offers the opportunity to learn more about the specific functions of UvrC in the recognition and removal of DNA lesions.
Figure 5. The interrelation of UvrABCTma and UvrABCBca in UvrABC incision reaction.
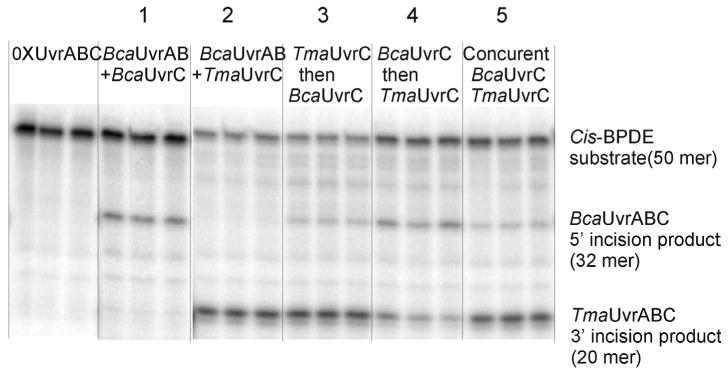
UvrABC incision reaction were performed in UvrABC buffer with subunit UvrA (10 nM), UvrB (250 nM) and UvrC (100 nM) in the presence of 32P-labeled cis-BPDE DNA substrates. Substrates were preincubated with Bca UvrAB at 60 °C for 30 min, then Bca UvrC or Tma UvrC were individually added in reaction 1 and 2; or Tma UvrC and Bca UvrC were concurrently added in reaction5, with incubation 60 min at 60 °C. After preincubation of UvrAB with substrates, at reaction 3, Tma UvrC was added and incubated for 60 min then Bca UvrC was added sequentially for additional 60 min incubation; At reaction 4, Bca UvrC was added and incubated for 60 min then Tma UvrC was added sequentially for additional 60 min incubation at 60 °C. Reaction products were denatured and resolved by electrophoresis in 15% polyacrylamide gels containing 7.5 M urea.
Acknowledgments
The authors thank Dr. Mark Melton for technical assistance in purifying the Bca UvrC mutants and Ms. Heather Miller for technical assistance in analyzing sequences.
Footnotes
This work was supported in part by USPHS Grants R01ES06460 and R01ES011314 and by the Intramural Research Program of the NIEHS, NIH.
References
- 1.Denissenko MF, Pao A, Pfeifer GP, Tang M. Oncogene. 1998;16:1241–1247. doi: 10.1038/sj.onc.1201647. [DOI] [PubMed] [Google Scholar]
- 2.Hoare S, Zou Y, Purohit V, Krishnasamy R, Skorvaga M, Van Houten B, Geacintov NE, Basu AK. Biochemistry. 2000;39:12252–12261. doi: 10.1021/bi0013187. [DOI] [PubMed] [Google Scholar]
- 3.Kowalczyk A, Carmical JR, Zou Y, Van Houten B, Lloyd RS, Harris CM, Harris TM. Biochemistry. 2002;41:3109–3118. doi: 10.1021/bi010450j. [DOI] [PubMed] [Google Scholar]
- 4.Mekhovich O, Tang M, Romano LJ. Biochemistry. 1998;37:571–579. doi: 10.1021/bi971544p. [DOI] [PubMed] [Google Scholar]
- 5.Zou Y, Crowley DJ, Van Houten B. J Biol Chem. 1998;273:12887–12892. doi: 10.1074/jbc.273.21.12887. [DOI] [PubMed] [Google Scholar]
- 6.Huang YP, Ito J. Nucleic Acids Res. 1998;26:5300–5309. doi: 10.1093/nar/26.23.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, McDonald L, Utterback TR, Malek JA, Linher KD, Garrett MM, Stewart AM, Cotton MD, Pratt MS, Phillips CA, Richardson D, Heidelberg J, Sutton GG, Fleischmann RD, Eisen JA, White O, Salzberg SL, Smith HO, Venter JC, Fraser CM. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- 8.Guengerich FP. Pharmacol Ther. 1992;54:17–61. doi: 10.1016/0163-7258(92)90050-a. [DOI] [PubMed] [Google Scholar]
- 9.Quan T, Reiners JJ, Jr, Culp SJ, Richter P, States JC. Mol Carcinog. 1995;12:91–102. doi: 10.1002/mc.2940120206. [DOI] [PubMed] [Google Scholar]
- 10.Jiang G, Skorvaga M, Van Houten B, States JC. Protein Expr Purif. 2003;31:88–98. doi: 10.1016/s1046-5928(03)00137-2. [DOI] [PubMed] [Google Scholar]
- 11.Straub KM, Meehan T, Burlingame AL, Calvin M. Proc Natl Acad Sci U S A. 1977;74:5285–5289. doi: 10.1073/pnas.74.12.5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang G, Jankowiak R, Grubor N, Banasiewicz M, Small GJ, Skorvaga M, Van Houten B, States JC. Chem Res Toxicol. 2004;17:330–339. doi: 10.1021/tx034184h. [DOI] [PubMed] [Google Scholar]
- 13.Zou Y, Luo C, Geacintov NE. Biochemistry. 2001;40:2923–2931. doi: 10.1021/bi001504c. [DOI] [PubMed] [Google Scholar]
- 14.Zou Y, Liu TM, Geacintov NE, Van Houten B. Biochemistry. 1995;34:13582–13593. doi: 10.1021/bi00041a038. [DOI] [PubMed] [Google Scholar]
- 15.DellaVecchia MJ, Croteau DL, Skorvaga M, Dezhurov SV, Lavrik OI, Van Houten B. J Biol Chem. 2004 doi: 10.1074/jbc.M408659200. [DOI] [PubMed] [Google Scholar]
- 16.Moolenaar GF, Visse R, Ortiz-Buysse M, Goosen N, van de PP. J Mol Biol. 1994;240:294–307. doi: 10.1006/jmbi.1994.1447. [DOI] [PubMed] [Google Scholar]
- 17.Moolenaar GF, Bazuine M, van Knippenberg IC, Visse R, Goosen N. J Biol Chem. 1998;273:34896–34903. doi: 10.1074/jbc.273.52.34896. [DOI] [PubMed] [Google Scholar]
- 18.Moolenaar GF, Monaco V, van der Marel GA, van Boom JH, Visse R, Goosen N. J Biol Chem. 2000;275:8038–8043. doi: 10.1074/jbc.275.11.8038. [DOI] [PubMed] [Google Scholar]
- 19.Verhoeven EE, Wyman C, Moolenaar GF, Goosen N. EMBO J. 2002;21:4196–4205. doi: 10.1093/emboj/cdf396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou Y, Van Houten B. EMBO J. 1999;18:4889–4901. doi: 10.1093/emboj/18.17.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moolenaar GF, Rossum-Fikkert S, van Kesteren M, Goosen N. Proc Natl Acad Sci U S A. 2002;99:1467–1472. doi: 10.1073/pnas.032584099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Houten B, Eisen JA, Hanawalt PC. Proc Natl Acad Sci U S A. 2002;99:2581–2583. doi: 10.1073/pnas.062062599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang M, Nazimiec M, Ye X, Iyer GH, Eveleigh J, Zheng Y, Zhou W, Tang YY. J Biol Chem. 2001;276:3904–3910. doi: 10.1074/jbc.M008538200. [DOI] [PubMed] [Google Scholar]
- 24.Nazimiec M, Lee CS, Tang YL, Ye X, Case R, Tang M. Biochemistry. 2001;40:11073–11081. doi: 10.1021/bi010953p. [DOI] [PubMed] [Google Scholar]
- 25.Hess MT, Gunz D, Luneva N, Geacintov NE, Naegeli H. Mol Cell Biol. 1997;17:7069–7076. doi: 10.1128/mcb.17.12.7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jankowiak R, Lin CH, Zamzow D, Roberts KP, Li KM, Small GJ. Chem Res Toxicol. 1999;12:768–777. doi: 10.1021/tx980233s. [DOI] [PubMed] [Google Scholar]
- 27.Lu PQ, Jeong H, Jankowiak R, Small GJ, Kim SK, Cosman M, Gracintov NE. Chem Res Toxicol. 1991;4:58–69. doi: 10.1021/tx00019a008. [DOI] [PubMed] [Google Scholar]
- 28.Huang W, Amin S, Geacintov NE. Chem Res Toxicol. 2002;15:118–126. doi: 10.1021/tx010135y. [DOI] [PubMed] [Google Scholar]
- 29.Xie XM, Geacintov NE, Broyde S. Chem Res Toxicol. 1999;12:597–609. doi: 10.1021/tx990021a. [DOI] [PubMed] [Google Scholar]
- 30.Delagoutte E, Bertrand-Burggraf E, Dunand J, Fuchs RP. J Mol Biol. 1997;266:703–710. doi: 10.1006/jmbi.1996.0830. [DOI] [PubMed] [Google Scholar]
- 31.Delagoutte E, Fuchs RP, Bertrand-Burggraf E. J Mol Biol. 2002;320:73–84. doi: 10.1016/S0022-2836(02)00401-1. [DOI] [PubMed] [Google Scholar]
- 32.Moolenaar GF, Herron MF, Monaco V, van der Marel GA, van Boom JH, Visse R, Goosen N. J Biol Chem. 2000;275:8044–8050. doi: 10.1074/jbc.275.11.8044. [DOI] [PubMed] [Google Scholar]
- 33.Skorvaga M, Theis K, Mandavilli BS, Kisker C, Van Houten B. J Biol Chem. 2002;277:1553–1559. doi: 10.1074/jbc.M108847200. [DOI] [PubMed] [Google Scholar]
- 34.Verhoeven EE, van Kesteren M, Turner JJ, van der Marel GA, van Boom JH, Moolenaar GF, Goosen N. Nucleic Acids Res. 2002;30:2492–2500. doi: 10.1093/nar/30.11.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moolenaar GF, Franken KL, van de PP, Goosen N. Mutat Res. 1997;385:195–203. doi: 10.1016/s0921-8777(97)00042-6. [DOI] [PubMed] [Google Scholar]
- 36.Moolenaar GF, Franken KL, Dijkstra DM, Thomas-Oates JE, Visse R, van de PP, Goosen N. J Biol Chem. 1995;270:30508–30515. doi: 10.1074/jbc.270.51.30508. [DOI] [PubMed] [Google Scholar]
- 37.Verhoeven EE, van Kesteren M, Moolenaar GF, Visse R, Goosen N. J Biol Chem. 2000;275:5120–5123. doi: 10.1074/jbc.275.7.5120. [DOI] [PubMed] [Google Scholar]