

Abstract
Fast mapping of disulfide bonds in proteins containing multiple cysteine residues is often required in order to assess the integrity of tertiary structure of proteins prone to degradation and misfolding or to detect distinct intermediate states generated in the course of oxidative folding. A new method of rapid detection and identification of disulfide-linked peptides in complex proteolytic mixtures utilizes the tendency of collision-activated peptide ions to lose preferentially side chains of select amino acids in the negative ion mode. Cleavages of cysteine side chains result in efficient dissociation of disulfide bonds and produce characteristic signatures in the fragment ion mass spectra. While cleavages of other side chains result in insignificant loss of mass and full retention of the peptide ion charge, dissociation of external disulfide bonds results in physical separation of two peptides and, therefore, significant changes of both mass and charge of fragment ions relative to the precursor ion. This feature allows the fragment ions generated by dissociation of external disulfide bonds to be easily detected and identified even if multiple precursor ions are activated simultaneously. Such broad-band selection of precursor ions for consecutive activation is achieved by lowering the DC/RF amplitude ratio in the first quadrupole filter of a hybrid quadrupole-time-of-flight mass spectrometer. The feasibility of the new method is demonstrated by partial mapping of disulfide bridges within a 37 kDa protein containing sixteen cysteine residues and complete disulfide mapping within lysozyme (14.5 kDa) containing eight cysteine residues. In addition to detecting peptide pairs connected by a single external disulfide, the new method is also shown to be capable of identifying peptides containing both external and internal disulfide bonds. The two major factors determining the efficiency of disulfide mapping using the new methodology are the effectiveness of proteolysis and the ability of the resulting proteolytic fragments to form multiply charged negative ions.
Keywords: mass spectrometry, electrospray ionization, negative ion, peptide ion, disulfide, collision-induced dissociation, side chain loss, precursor ion selection, quadrupole mass filter, isotopic distribution
INTRODUCTION
Mass spectrometry became in recent years an indispensable tool for obtaining information on protein amino acid sequence and post-translational modification patterns,1, 2 as well as characterizing protein higher order structure and conformational heterogeneity.3 Disulfide bonding is one of the most striking examples of a post-translational modification being a major determinant of the tertiary fold topology and conformational stability. Formation of covalent bonds between thiol groups of two cysteine residues that may be distant in the sequence but proximal in the three-dimensional structure provides a significant reinforcement to the native tertiary and quaternary folds by reducing conformational entropy of the unfolded state. Although the number of protein sequences with experimentally determined disulfide bonds has been grown steadily in the past years, the actual number of proteins, where the presence of disulfide bridges is only inferred (based on sequence homology or fold similarity) remains almost an order of magnitude higher. In addition to mapping native disulfides, there are situations when information on non-native disulfide connectivity is also at premium. For example, it is known that oxidative folding of proteins often proceeds through intermediate states with non-native disulfides. Therefore, characterizing of such non-native disulfide bonds provides important information on protein folding pathways.4 Perhaps more important from the practical point of view is the fact that a large fraction of protein pharmaceuticals contain multiple cysteine residues. Aging and degradation of such protein-based products often result in disulfide scrambling, whose extent obviously limits their utility.5 Furthermore, while the efficient expression of such products usually requires utilization of eukaryotic systems, production cost considerations put pressure towards expanding the role of simple prokaryotic systems-based expression strategies.6 The usefulness of bacterial expression is currently limited by difficulties incurred upon production of correctly folded disulfide-bonded proteins, although several examples of successful prokaryotic production of disulfide-bonded protein pharmaceuticals have been reported.7 Clearly, a wider utilization of bacterial expression protocols for production of protein pharmaceuticals will require effective means of mapping disulfide connectivity to ensure correct tertiary fold (for internal disulfides) and/or quaternary topology (for external disulfides) and also to assess conformational heterogeneity.8
Establishing disulfide connectivity patterns in large proteins possessing several cysteine residues is a challenging task. Several mass spectrometry-based strategies have been designed to provide information on disulfide connectivity, most of which rely on a combination of proteolysis in solution and peptide ion fragmentation (such as collision-activated dissociation, CAD) in the gas phase.9 Cleavage of disulfide bonds rarely occurs under collisional activation conditions that are typically used to obtain sequence information of peptide and protein ions. This greatly complicates any efforts (or indeed makes it impossible) to obtain extensive sequence information on large proteins containing multiple disulfide bonds using the so-called “top-down” approach, which bypasses the proteolytic step in solution and instead relies on protein ion dissociation in the gas phase. Straightforward applications of the “bottom-up” approach (e.g., CAD of proteolytic fragments) provide a more reliable way to map disulfides, although it typically increases the time required for analyses several fold and cannot be used for rapid disulfide mapping. More sophisticated methods for assigning disulfide bonds in proteins with multiple cysteine residues have been also developed, for example, those that combine partial reduction of a protein and chemical modification of sulfhydryl groups, followed by partial hydrolysis of a peptide chain, mass tag incorporation and subsequent mass mapping by mass spectrometry.10, 11
Disulfides can readily undergo dissociation during in-source decay when UV-MALDI (337 nm) is used to generate peptide ions, with dissociation efficiency being strongly dependent on the choice of a MALDI matrix.12 This phenomenon can be used to design a protocol for automated screening of complex peptide mixtures for the presence of disulfide bonds.13 Alternatively, electron-capture dissociation (ECD),14 electron transfer ion/ion reactions15 or UV-photodissociation (UVPD)16 can be used to provide disulfide connectivity information. However, data interpretation step in each case can be quite time-consuming owing to the fact that both ECD and UVPD typically generate backbone fragments as well, giving rise to convoluted tandem mass spectra.
Unlike most other protein and peptide ion fragmentation techniques, negative ion CAD under certain conditions leads almost exclusively to dissociation events at few select side chains, leaving the backbone intact.17 Since cysteine is one of the amino acids whose side chain is prone to dissociation under such conditions,18 negative ion CAD is an effective means of disulfide bond cleavage in the gas phase.19 The objective of this work is to design a strategy for screening of complex proteolytic mixtures for rapid and reliable identification of disulfide-linked peptides. This new strategy will have diverse applications ranging from mapping disulfide bonds in proteins whose higher order structure is unknown to monitoring conformational heterogeneity and assessing stability of protein pharmaceuticals to identification of intermediate states in oxidative folding reactions. Furthermore, development of this experimental strategy will benefit the studies of dynamics of disulfide-containing proteins using amide hydrogen/deuterium exchange (HDX), where physical separation of disulfide-linked peptic fragments normally requires use of reducing agents active under acidic conditions (such as phosphines20). This leads to an increase of the analysis time and, as a consequence, the extent of back-exchange. Back-exchange is a major factor that adversely affects the resolution afforded by HDX MS measurements.21 Substitution of chemical reduction of protein in solution with gas phase dissociation of disulfide bonds would result in shorter analysis time due to elimination of a relatively lengthy sample processing step and, therefore, better quality of the hydrogen exchange measurements. Although most of the examples presented in this work use trypsin as a means of protein digestion in solution, proteolysis under slow exchange conditions followed by dissociation of disulfide bonds in the gas phase (by means of CAD of peptide anions) is also feasible.
EXPERIMENTAL
Materials
Recombinant human serum transferrin N-lobe (hTf/2N) was generously provided by Prof. Anne B. Mason (Univ. of Vermont Medical School). Bovine serum albumin (BSA) and chicken egg white lysozyme were purchased from Sigma-Aldrich Chemical Company (Milwaukee, MI). Immobilized TPCK (L-(tosylamido-2-phenyl) ethyl chloromethyl) trypsin was obtained from Pierce Biotechnology Inc. (Rockford, IL) and washed 3 times with 100mM NH4HCO3/NH4OH buffer pH 8.0 prior to digestion. All chemicals were of analytical grade or higher.
Methods
proteolysis
Protein digestion was initiated by dispersing the immobilized trypsin in 50 μM protein (hTf/2N or BSA) solutions in 100mM NH4HCO3, whose pH levels were adjusted with NH4OH to 8.0. The enzyme-to-protein molar ration was kept at ca. 1:1 and digestion was carried out at 37°C for 3 hours. Immobilized TPCK trypsin was removed from the digestion mixtures by centrifugation prior to mass analysis. A combination of tryptic and peptic proteolyses of lysozyme was carried out by placing the mixture of tryptic fragments of this protein (vide supra) to ammonium formate/formic acid buffer whose pH was adjusted to 2.5 and applying this solution to immobilized pepsin for 1 hr at 37°C.
Methods
mass spectrometry
All mass spectra were acquired with a QStar-XL (ABI-Sciex, Framingham, MA) hybrid quadrupole-time-of-flight mass spectrometer. The protein digest solutions were either continuously infused into the standard Turbospray™ source at a flow rate of 5 μL/min (Tf/2N and BSA) or sprayed from a single capillary (Proxeon Biosystems A/S, Odense, Denmark) using a nanospray source (lysozyme). The total protein consumption ranged from ca. 5 nmol (with Turbospray™) to below 0.1 nmol (with nanospray). All mass spectra were acquired in the negative ion mode, unless specified otherwise. Typical operating conditions for acquiring mass spectra were as follow: ionspray voltage, −4500 V; de-clustering potential, −80 V; ion focusing potential, −265 V; de-clustering potential 2, −15 V; source temperature, 80°C. The width of a transmission window of a quadrupole filter (Q) preceding the RF-only collision cell (q) was adjusted using the “offset drop” parameter. Narrow-band selection of peptide ions for fragmentation (collision-activated dissociation, CAD) and MS/MS analysis was carried out using the offset drop value of 0.1. Broadband selection of multiple precursor ions for simultaneous fragmentation of multiple precursor ions was carried out by increasing the offset drop value to 6.5. Typical acquisition time for each broadband MS/MS spectrum was 10 min or less. This allowed the entire m/z region of interest to be fully covered within 2−4 hours, depending on the protein.
Results and Discussion
Although dissociation of both internal and external (i.e., inter-chain) disulfide bonds in peptide cations is rarely achieved when they are subjected to collisional activation, there are several other ion activation methods that allow these bonds to be cleaved in the gas phase. Perhaps most prominent of those is ECD, an ion fragmentation technique that utilizes the ability of low energy (≤ 0.1 eV) electrons to cause dissociation of N-C and disulfide bonds upon attachment to polycationic peptide ions.14 Although the backbone dissociation provides structurally diagnostic c and z. fragment ions, multiplicity of ion fragmentation channels often generates convoluted product ion spectra. Since our main objective is to devise a method that allows fast identification of disulfide-linked peptides in complex mixtures, presence of multiple fragment ions in the spectra (only a fraction of which result from dissociation of disulfide bonds) may obscure the “signature” fragments arising from disulfide dissociation and, therefore, complicate our efforts. This situation is complicated further by our desire to streamline the entire procedure of disulfide mapping using simultaneous activation of several precursor ions, only a fraction of which would possess disulfide bonds. Under these circumstances, interference of a large number of c and z. fragment ions present in mass spectra at high abundance will make confident identification of the signal arising from dissociation of disulfides very challenging.
In contrast to positive ion CAD and ECD, collision-activated fragmentation of peptide anions has not yet become a commonly accepted practical tool to obtain sequence information, as it often promotes side chain cleavages in addition to backbone dissociation. However, under certain conditions it may lead almost exclusively to dissociation at select side chains. Recent theoretical calculations and experimental studies reported by Bilusich et al. demonstrate that backbone cleavage reactions actually follow loss of side chains.22 Such hierarchy of dissociation events allows the backbone fragmentation to be avoided almost completely if the time frame of peptide anion activation is sufficiently short. This situation can be readily achieved in beam-type mass spectrometers, where the second-generation of fragment ions (resulting from the backbone cleavages) is rarely observed.19 Furthermore, fragmentation spectra of disulfide-linked peptide anions acquired with beam-type instruments contain distinct “signatures” arising from the dissociation of disulfide bonds.19 As shown in Scheme 1, cleavages occur at three different locations (Cβ’-S’, S’-S” and S”- Cβ”), giving rise to a group of fragment ions (denoted in Scheme 1 as A, B, B” and C) with characteristic spacing between them.
Scheme 1.

An example of such fragment ion signatures is shown in Figure 1a, where a mass-selected covalent peptide dimer anion (a doubly charged tryptic fragment [8−18]-[43−50] of hTf/2N connected by an external disulfide Cys9-Cys48) was subjected to CAD in a collision cell of a hybrid quadrupole-time-of-flight mass spectrometer. Two distinct groups of peaks are clearly seen in the spectrum (m/z regions 900−980 and 1220−1300), which correspond to peptide ions [8−18]− and [43−50]−, each bearing a single negative charge. Dissociation of the S’-S” bond results in reduction of one cysteine residue and oxidation of another, a process that usually has two different outcomes (B and B” in Scheme 1). As a result, the central peak in each cluster has a peculiar isotopic distribution with contributions from both reduced and oxidized forms of cysteine (mass difference 2 u). Importantly, the only other abundant fragment ions in this mass spectrum correspond to losses of few side chains from the precursor ions and H2O molecules from either the precursor ion or the two separated peptide anions. This leads to a very characteristic appearance of the fragment ions spectrum, which has only three groups of closely clustered peaks, one corresponding to the precursor ion (and its satellites resulting from side chain losses that leave the disulfide bond intact) and the other two corresponding to the separated peptide anions (complementary fragments generated upon dissociation of the disulfide bond).
Figure 1.

Mass spectra of fragment anions generated by CAD of a disulfide-linked peptide dimer [8−18]-[43−50] derived from digestion of hTf/2N with trypsin. The spectrum shown on panel (a) was obtained with a hybrid QqTOF mass spectrometer (CAD carried out in an RF-only quadrupole), and the spectrum shown on panel (b) was acquired with an FT ICR mass spectrometer (SORI CAD). Open squares and circles indicate ions corresponding to intact peptide monomers produced by dissociation of the disulfide bond in the peptide dimer ion without any backbone cleavages (zoomed views are shown in insets).
The appearance of the CAD mass spectrum collected with a hybrid quadrupole-time-of-flight mass spectrometer suggests that the selectivity of the peptide anion dissociation may be exploited to streamline the screening of complex mixtures for peptides containing external disulfide bonds. Indeed, one may think of a strategy that utilizes simultaneous dissociation of a group of precursor peptide ions within a certain m/z range, rather than a single precursor. Only the peptide anions containing external disulfide bonds will give rise to groups of abundant fragment ions that fall outside of the m/z region of the “precursor window.” At the same time, all others would mostly suffer few side chain losses and, as a result, the corresponding fragment ions would have much smaller m/z deviation from their precursors. Even if a certain side chain loss does result in a significant change of the m/z ratio (e.g., a hypothetical process leading to charge reduction upon removal of an acidic side chain), such fragment ions can be easily filtered out, based on the absence of a highly specific spacing of fragment ion peaks in a cluster, characteristic of a disulfide bond dissociation.
Multiplexing of peptide ion dissociation is usually implemented on trapping type mass spectrometers, with Stored Waveform Inverse Fourier Transform (SWIFT)23 being perhaps the most popular technique allowing broadband (or indeed tailor-made band) ion isolation and activation. However, the fragmentation behavior of the disulfide-linked peptide dimer anions in a trapping cell of a Fourier transform mass spectrometer (FTMS) is dramatically different from that observed with a hybrid quadrupole-time-of-flight mass spectrometer (compare Figures 1a and b). SORI CAD24 of a doubly charged tryptic peptide dimer fragment [8−18]-[43−50] of hTf/2N connected by an external disulfide Cys9-Cys48 carried out in a Fourier transform ion cyclotron resonance mass spectrometer gives rise to a convoluted fragmentation pattern, where most fragments result from dissociation of the disulfide bond and losses of side chains followed by backbone cleavages (Figure 1B). Although this spectrum contains ionic contributions from the two separated peptide anions with intact backbones as well, low abundance of these fragments makes them unfit to serve as reliable indicators of the presence of an external disulfide bond in the precursor ion. Therefore, it is unlikely that a streamlined procedure for screening peptide mixtures for the presence of disulfide-linked peptides can be implemented on trapping type mass spectrometers.
Multiplexing of peptide ion fragmentation is rarely practiced on beam-type mass spectrometers, although it is possible to adjust the ion transmission parameters of certain mass analyzers to allow transmission of precursor ions in a broad m/z range. For example, lowering of the DC-to-RF amplitude ratio in a quadrupole mass filter results in broadening of the range of ions passing through the filter.25, 26 Standard operating parameters of a commercial QSTAR XL mass spectrometer allow the DC-to-RF amplitude ratio to be brought down from the apex of the stability region, allowing the ion transmission window to exceed 50 u (Figure 2a). It must be noted, however, that the quality of mass selection deteriorates once the kinetic energy of ions passing through the quadrupole filter is increased. Thus, elevation of ion kinetic energy to a level of 25 V, which is still below the fragmentation threshold, results in appearance of a set of low abundance ions outside the selected transmission window (Figure 2b). All these ions correspond to high-abundance ionic species in MS1 (Figure 2c), a fraction of which apparently passes the quadrupole filter upon increase of their kinetic energy. Such partial transparency of a quadrupole filter becomes particularly noticeable if the intensity of ions within the transmission window itself is very low compared with other ions in the MS1 spectrum. Therefore, implementation of broadband MS/MS experimental scheme on a commercial hybrid quadrupole-time-of-flight mass spectrometer will require acquisition of two data sets, with kinetic energy of ions set below and above the fragmentation threshold, respectively. The former will contain contributions from high-abundance ions outside of the mass-selection window, while the latter will have both these ions and the fragments derived from dissociation of the mass-selected precursors.
Figure 2.

The effect of ion kinetic energy on transparency of the first quadrupole filter (Q) of a hybrid QqTOF mass spectrometer. Top panel shows a broadband selection of a range of low-intensity precursor ions (m/z 1430−1480) derived from digestion of hTf/2N with trypsin. The middle panel shows the broadband MS/MS spectrum acquired below the fragmentation threshold. Origin of all ions outside of the precursor window can be traced to abundant peptide anions in MS1 spectrum (bottom trace).
Simultaneous analysis of both sets of data allows unambiguous identification of fragment ions outside of the precursor ion transmission window to be made. Figure 3 shows a mass spectrum acquired upon activation of peptide ions in m/z window 1430−1480 u (same as in Figure 2a). The precursor ion kinetic energy was set above the dissociation threshold level (65 V), while all other parameters were kept the same as in the case of a spectrum acquired below the fragmentation threshold. Only two new groups of abundant ion peaks appear outside of the precursor ion window, which were not already present in the spectrum acquired below the fragmentation threshold (for clarity, these two spectra are overlaid in Figure 3). A closer look at these two groups of fragment ion peaks (marked with a square and a circle in Figure 3) reveals patterns characteristic of external disulfide bond dissociation (see the inset in Figure 3). Reduced peptide mass for each group of fragment ions can be easily calculated based on the structure of product ions derived from dissociation of disulfide-linked dimers (Scheme 1). Such calculations carried out for the dataset shown in Figure 3 yield 2492.6 Da (peak cluster at m/z 1220−1260) and 1824.0 Da (m/z 1780−1840), which allows these two peptides to be identified as hTf/2N tryptic fragments [233−254] and [218−232]. Each fragment contains a single cysteine residue, which allows a disulfide bond to be easily assigned.
Figure 3.

Detection and identification of a 227−241 disulfide bridge in hTf/2N by broadband CAD of anions (m/z 1430−1480) derived from digestion of this protein with trypsin. The broadband spectra acquired below and above the fragmentation threshold are shown in gray and black, respectively. The inset shows a group of peaks corresponding to peptide [233−254]. Anionic species representing another peptide from the dimer, [218−232], are marked with circles.
The entire population of tryptic peptide ions can be quickly scanned for the presence of other disulfide-linked peptide dmers by dividing the entire m/z range into several “precursor ion windows” and acquiring two broadband MS/MS spectra for each of them (one at precursor ion kinetic energy set at 25 V and another one at 65 V). Out of twenty-two broadband CAD spectra covering the entire range of precursor ions, two more (in addition to one discussed above) contain signatures of disulfide-bonded peptide dimer ion dissociation (Figures 4 and 5). Once again, the identity of disulfide-linked tryptic fragments can be established unequivocally by calculating masses for the reduced forms of peptides corresponding to anionic fragments falling outside of the precursor ion window and not having a match in the broadband spectrum acquired below the fragmentation threshold (gray traces in Figures 4 and 5). These fragments are identified as [8−18] and [43−50] (Figure 4), and [125−143] and [328−337] (Figure 5), each containing a single cysteine residue. Therefore, two more disulfide bonds in hTf/2N can be easily determined. Other broadband CAD spectra yielded no information on disulfide-linked peptides, although occasionally they too contained ions outside of the precursor window, whose origin could not be traced to the abundant peptide ions penetrating the quadrupole filter at elevated kinetic energy. For example, collisional activation of a band of ions in m/z range 810−880 gives rise to an apparent fragment ion at m/z 1340 (Figure 6). However, this spectrum lacks a characteristic disulfide signature, which clearly indicates that disulfide-linked peptide dimer anions are very unlikely to be among the group of precursor ions activated in this experiment.
Figure 4.

Detection and identification of a 9−48 disulfide bridge in hTf/2N by broadband CAD of anions (m/z 1090−1150) derived from digestion of this protein with trypsin. The broadband spectra acquired below and above the fragmentation threshold are shown in gray and black, respectively. The inset shows a group of peaks corresponding to peptide [8−18]. Anionic species representing another peptide from the dimer, [43−50], are marked with squares.
Figure 5.

Detection and identification of a 137−331 disulfide bridge in hTf/2N by broadband CAD of anions (m/z 1550−1620) derived from digestion of this protein with trypsin. The broadband spectra acquired below and above the fragmentation threshold are shown in gray and black, respectively. The inset shows a group of peaks corresponding to peptide [125−143]. Anionic species representing another peptide from the dimer, [328−337], are marked with squares.
Figure 6.

An example of broadband CAD of monomeric peptide anions derived from digestion of hTf/2N with trypsin. The only ion outside of the precursor ion window (m/z 810−860), whose origin cannot be traced to abundant peptide ions in MS1 able to pass the quadrupole filter at elevated kinetic energy, is the one at m/z 1340. The appearance of this ion peak (zoomed view, inset) is inconsistent with a notion of this ion being a product of disulfide dissociation.
In addition to three disulfide bonds identified by broadband negative ion CAD of a mixture of tryptic peptides of hTf/2N, this protein contains five other disulfide bonds (Figure 7). Three of them (158−174, 161−179 and 171−177) are expected to be internal, since they are located within a single tryptic fragment (Ala149-Lys193). Therefore, dissociation of these disulfides will not produce characteristic signatures, which can be seen so clearly in the fragmentation spectra of peptide dimer anions. Obviously, this problem may be rectified if another proteolytic enzyme (or indeed a mixture of several different enzymes) is used to produce cleavages within this cysteine-rich segment of the protein. The other two disulfides that were not detected using the broadband negative ion CAD (19−39 and 118−194) likely escaped detection because the respective tryptic peptides are very small and deficient in acidic residues (there is a single acidic residue in one of them and no acidic residues in the other). As a result, these peptide dimers are very unlikely to form even moderately abundant anionic species, whose intensity would be sufficient to afford production of detectable “signature fragments” in broadband CAD experiments. This is a fundamental problem, which can only be addressed by using orthogonal or complementary methods of disulfide detection. For example, positive ion CAD of intact multiply charged protein ions carried out on FT ICR27 or hybrid quadrupole-time-of-flight28 mass spectrometers produce extensive cleavages of amide bonds. However, the resulting complementary b- and y-fragments would not be physically separated in the gas phase (and, consequently, escape detection) if they are connected to one another by disulfide bonds. Therefore, detection of positive fragment ions of b- and y-type produced upon dissociation of amide bond(s) within a segment separating two cysteine residues indicates that the latter ones are not connected to each another by a disulfide bridge.
Figure 7.
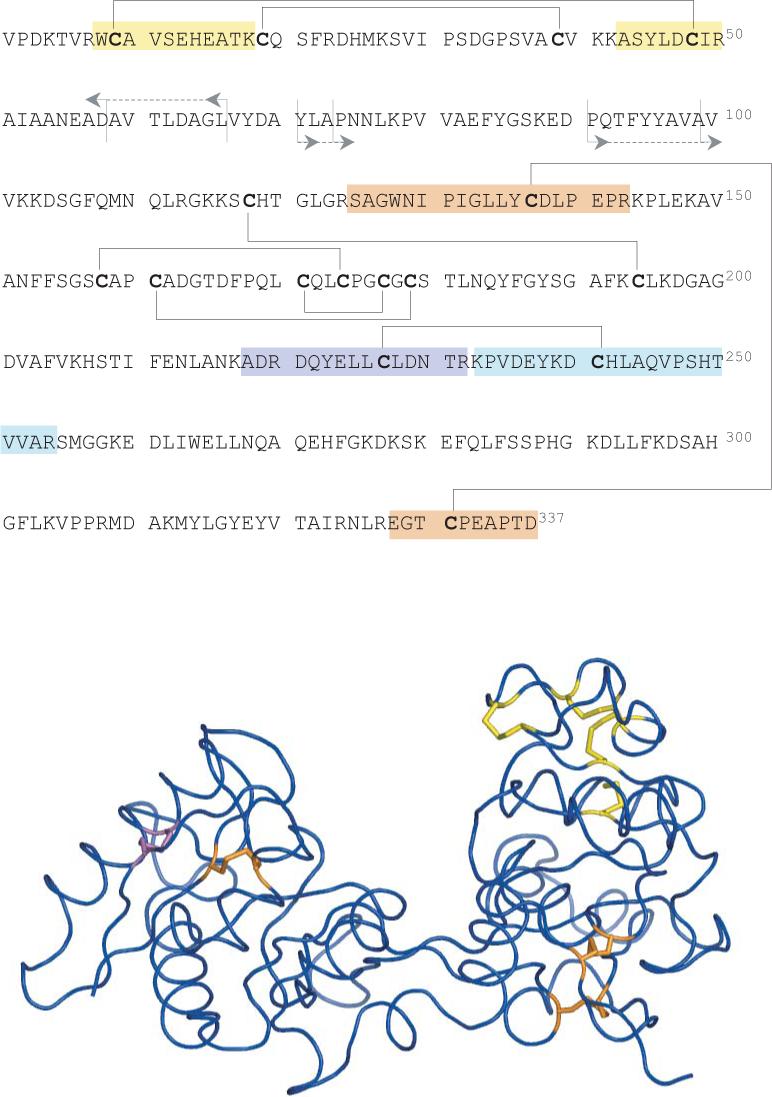
Amino acid sequence (top) and three-dimensional structure of hTf/2N (bottom) showing locations of disulfide bonds. Disulfide bonds whose existence has been determined by broadband CAD in the negative ion mode (tryptic digest) are colored in orange (disulfide-linked tryptic peptide dimers are highlighted in the top diagram). The disulfide bond whose existence is inferred from the results of positive ion CAD of intact protein is colored in purple (positions of amide bond cleavages leading to abundant b- and y-ions are shown on the top diagram).
A CAD spectrum of intact multiply charged hTf/2N ions is shown in Figure 8. In order to increase the signal-to-noise ratio, mass selection of precursor ions was carried out in a broadband regime (charge states +25 and +26). Collisional activation of these two ions produces abundant b- and y-fragments, which are derived from dissociation of amide bonds in a protein segment spanning residues 57 to 98. This allows the disulfide connectivity to be localized in two groups of cysteine residues, one including residues 9, 19, 39 and 48, and the other including the rest of hTf/2N cysteine residues. Since the 9−48 connectivity has been already determined using broadband negative ion CAD of hTf/2N tryptic digest, the existence of a 19−39 disulfide bridge can be unequivocally established. Therefore, the two complementary approaches to mapping disulfide bonds (broadband negative ion CAD of proteolytic fragments and positive ion CAD of intact protein ions) allow four (out of eight) disulfide bonds to be quickly and reliably identified without the need for time-consuming chemical modifications. Since the total number of disulfide bridges that can by formed by random coupling of 2n cysteine residues can be calculated simply as
| (1) |
identification of four disulfides reduces the number of possible combinations from 120 to only 28. Since not all cysteine residues are necessarily oxidized in proteins, the total number of combinations should also include a possibility for some thiol groups to be reduced:
| (2) |
and the reduction of all possible combination is even more significant (from 983,041 for eight pairs of cysteine residues to only 897 for 4 pairs). This number is likely to be decreased further by substituting trypsin with another enzyme (or a mixture of proteases) as a means of enhancing the efficiency of proteolysis.
Figure 8.

Broadband selection (top) and fragmentation (bottom) of intact multiply charged hTf/2N cations carried out with a hybrid QqTOF mass spectrometer. The inset in the top panel shows positive ion ESI mass spectrum of hTf/2N prior to precursor ion selection. The inset in the bottom panel shows a series of y-ions carrying 14 charges produced by dissociation of amide bonds in the [90−99] region of the protein (ladder sequencing).
One other potential complication that needs to be considered when discussing application of broadband negative ion CAD as a means of disulfide mapping, is a possibility that some externally-linked peptide dimer anions may contain more than one disulfide bond. Although such species were not observed among tryptic peptides of hTf/2N, such an unfavorable situation can be encountered when other proteins with high cysteine content are proteolyzed. An example is shown in Figure 9, where a mixture of peptides produced by incomplete digestion of bovine serum albumin (BSA) with trypsin was subjected to broadband (m/z 1650−1740) CAD in the negative ion mode. Four distinct groups of abundant fragment ions are clearly observed in the spectrum acquired above the fragmentation threshold. The appearance of each of these groups of fragment ion peaks suggests that these fragments are produced upon dissociation of disulfide-linked anions. Calculation of peptide masses for these fragments provides close matches with cysteine-containing tryptic peptides of BSA, [529−544] for the group of fragments in m/z range 1810−1890, [569−597] for the group of fragments in m/z range 1520−1590, [89−100] for the group of fragments in m/z range 1310−1400, and [106−138] for the group of fragments in m/z range 1870−1940.
Figure 9.

Detection of four tryptic peptides of BSA involved in formation of external disulfide bridges by broadband CAD in the negative ion mode (m/z 1650−1740). The broadband spectra acquired below and above the fragmentation threshold are shown in gray and black, respectively. The two precursor ions that were identified as disulfide-linked dimers are labeled in the inset.
Identification of four, rather than just two, peptides with external disulfide bonds in one experiment may be a result of dissociation of a single peptide tetramer or, alternatively, presence of two different peptide dimer anions in a precursor window. Narrowing the band of precursor ions selected for collisional activation allows one to establish that there are two distinct peptide dimer anions whose m/z ratios initially fell within the ion selection window (Figure 10a and b). Further complication is due to the fact that one peptide in each pair has three, rather than just one, cysteine residues. Values of the precursor ion masses indicate that all peptides are fully oxidized, i.e. “extra” cysteine residues form an internal disulfide bond. Nevertheless, it is impossible to make a distinction between “internally” and “externally” linked cysteine residues solely based on the data collected in broadband negative ion CAD experiments. This task, however, can be aided in many cases by a complementary fragmentation approach that does not involve any additional sample work-up. For example, after the identity of the disulfide-linked dimers containing internal thiol-thiol bridges is established, corresponding cationic species can be mass-selected and subjected to CAD. Detection of fragment ions derived from amide bond(s) dissociation within a segment separating two cysteine residues will serve as a reliable indicator that this pair does not form an internal disulfide bridge. At the same time, complete absence of fragment ions derived from dissociation of backbone amides within the entire segment separating two cysteine residues is often indicative of the fact that they do form a disulfide bond. This may not always be enough to establish the complete thiol-thiol connectivity patterns, particularly in the case of proteins with closely spaced cysteine residues (e.g., [529−544]-[569−597] dimer discussed above, see Supporting Information for more detail). However, even in these highly unfavorable circumstances it is possible to rule out certain connectivity patterns based on the observed backbone fragmentation pattern. As a result, the range of possible disulfide patterns can be narrowed down considerably when the broadband CAD of peptide anions is complemented with fragmentation of positive ions.
Figure 10.

Narrow-band negative ion CAD of the two dimeric tryptic peptides of BSA whose dissociation reveals the presence of external disulfide bonds. Labeling of fragment ions is the same as in Figure 9. The insets show fragment ions peaks corresponding to peptides with three cysteine residues each, [106−138] (top) and [569−597] (bottom).
A more reliable scheme leading to unequivocal determination of disulfide connectivity patterns within peptide dimers containing both internal and external disulfides utilizes a second proteolytic step prior to peptide anion fragmentation in the gas phase. This will be illustrated using hen egg lysozyme, a 14.5 kDa protein containing four disulfide bridges. Broadband CAD of lysozyme tryptic digest revealed presence of three peptide dimers connected by external disulfide bonds (see Supplementary Material for more detail). One of these dimers (represented by a triply charged anion at m/z 1088) corresponds to a peptide dimer containing four cysteine residues, three of which are located within a single peptide monomer comprising residues 74−96 of lysozyme sequence (Figure 11, top). Although CAD in the positive ion mode fails to establish the cysteine connectivity pattern within this peptide, combination of tryptic and peptic digests (carried out in sequence) generates a peptide trimer, with two monomer containing one cysteine residue each and the two remaining cysteine residues contained within the third peptide monomer. CAD of the corresponding anionic species (a doubly charged ion at m/z 1298) generates a series of characteristic fragments following dissociation of one or more disulfide bonds (Figure 11, bottom).
Figure 11.

Top: detection of a lysozyme tryptic peptide dimer containing two disulfide bonds by broadband CAD in the negative ion mode (the peptide dimer structure is shown in the inset). Bottom: additional processing of the peptide mixture with pepsin generates a peptide trimer connected by two disulfide bonds (amino acid residues cut by pepsin are typed in gray). CAD of the corresponding anionic species gives rise to five groups of fragment ions produced by dissociation of one or both disulfide bonds in the gas phase. Individual peptide monomers are marked with open circles and triangles, as shown on the inset.
Dissociation of both disulfide bonds in the gas phase gives rise to groups of fragment ions at m/z 619 (residues 91−96), 934 (residues 62−68) and 1043 (residues 75−84). Two other groups of fragment ions correspond to peptide dimers generated upon dissociation of single disulfide bonds in the peptide trimer. These dimers containing a single surviving disulfide bond are (75−84)/(91−96) at m/z 1633 and (62−68)/(75−84) at m/z 1979. The only ambiguity that is not completely eliminated at this point is whether Cys63 is connected to Cys76 or Cys80. CAD of a triply charged cationic species corresponding to the peptide trimer at m/z 867 results in physical separation of the complementary fragment ions produced upon amide bond cleavages between Cys76 or Cys80, an observation that would not have been possible if Cys63 were connected to Cys76 (see Supplementary Material for more detail). Thus, combination of proteolysis, broadband CAD in the negative ion mode and narrowband CAD in the positive ion mode allows precise disulfide maps to be obtained even under unfavorable circumstances (close clustering of cysteine residues).
Conclusions
The high specificity of peptide anion dissociation in beam-type mass analyzers allows identification of disulfide-linked peptides in complex mixtures to be streamlined by using broadband ion activation in a hybrid quadrupole-time-of-flight mass spectrometer. Ions derived from dissociation of external disulfide bonds can be easily distinguished from other fragments (mostly side chain losses). The procedure does not require sample work-up beyond ordinary proteolysis, which minimizes time and cost of disulfide mapping. Since internal disulfides cannot be effectively mapped within proteolytic peptides using this approach, inefficient proteolysis generating large peptide fragments containing more than one cysteine residue increases the chance of missing certain disulfide bonds. In addition, external disulfides located on peptides without acidic residues also have an increased chance of escaping the detection. The quality (completeness) of disulfide maps produced by broadband negative ion CAD can be significantly improved by acquiring complementary CAD MS data in the positive ion mode without any additional sample processing. Although the work presented in this article was carried out on a hybrid quadrupole-time-of-flight MS, it seems that broadband CAD can be implemented on other types of mass spectrometers with a quadrupole analyzer (such as triple quadrupole MS). Furthermore, the results of our preliminary work with a linear trap MS (LTQ-MS) suggest that this mass spectrometer can also be used for disulfide mapping using broadband negative ion CAD, although in this case increased trapping time leads to generation of abundant ions produced by backbone cleavage. Presence of such fragment ions in broadband CAD spectra makes selective detection of products of disulfide bond dissociation in the gas phase more difficult (although not impossible). The simplicity of the new method of disulfide mapping within cysteine-rich proteins makes is an attractive option when a large number of protein samples has to be analyzed (e.g., in the studies of oxidative folding, quality control of protein pharmaceuticals, etc.). While all of the examples of disulfide mapping presented in this work were carried out using natively folded proteins with “correct” disulfide patterns, it remains to be seen whether this technique can be applied to characterization of heterogeneous samples (e.g., partially degraded proteins with mixed or scrambled disulfide patterns). A simple yet reliable method of quantitative assessment of disulfide network heterogeneity will be a very valuable tool of evaluating integrity of protein higher order structure.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health R01 GM061666. The authors acknowledge Dr. Anne B. Mason (University of Vermont Medical School) for a generous gift of hTf/2N samples. We also wish to thank Drs. Rinat R. Abzalimov and Wendell P. Griffith (UMass-Amherst) for their technical help and Dr. Stephen J. Eyles (UMass-Amherst) for valuable input to this work and a critical reading of the manuscript. We are grateful to Prof. Roman A. Zubarev for providing access to an LTQ-MS.
References
- 1.Standing KG. Peptide and protein de novo sequencing by mass spectrometry. Curr. Opin. Struct. Biol. 2003;13:595–601. doi: 10.1016/j.sbi.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 2.Coon JJ, Syka JE, Shabanowitz J, Hunt DF. Tandem mass spectrometry for peptide and protein sequence analysis. Biotechniques. 2005;38:519–523. doi: 10.2144/05384TE01. [DOI] [PubMed] [Google Scholar]
- 3.Kaltashov IA, Eyles SJ. Studies of biomolecular conformations and conformational dynamics by mass spectrometry. Mass Spectrom. Rev. 2002;21:37–71. doi: 10.1002/mas.10017. [DOI] [PubMed] [Google Scholar]
- 4.Welker E, Wedemeyer WJ, Narayan M, Scheraga HA. Coupling of conformational folding and disulfide-bond reactions in oxidative folding of proteins. Biochemistry. 2001;40:9059–9064. doi: 10.1021/bi010409g. [DOI] [PubMed] [Google Scholar]
- 5.Yu J. Intentionally degrading protein pharmaceuticals to validate stability-indicating analytical methods. BioPharm. Appl. 2000;13:46–50. [Google Scholar]
- 6.Walsh G. Biopharmaceuticals: recent approvals and likely directions. Trends Biotechnol. 2005;23:553–558. doi: 10.1016/j.tibtech.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Mantile G, Fuchs C, Cordella-Miele E, Peri A, Mukherjee AB, Miele L. Stable, long-term bacterial production of soluble, dimeric, disulfide-bonded protein pharmaceuticals without antibiotic selection. Biotechnol. Progr. 2000;16:17–25. doi: 10.1021/bp990129g. [DOI] [PubMed] [Google Scholar]
- 8.Chang J-Y, Lu B-Y, Li L. Conformational impurity of disulfide proteins: Detection, quantification, and properties. Anal. Biochem. 2005;342:78–85. doi: 10.1016/j.ab.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 9.Gorman JJ, Wallis TP, Pitt JJ. Protein disulfide bond determination by mass spectrometry. Mass Spectrom. Rev. 2002;21:183–216. doi: 10.1002/mas.10025. [DOI] [PubMed] [Google Scholar]
- 10.Qi J, Wu W, Borges CR, Hang D, Rupp M, Torng E, Watson JT. Automated data interpretation based on the concept of “negative signature mass” for mass-mapping disulfide structures of cystinyl proteins. J. Am. Soc. Mass Spectrom. 2003;14:1032–1038. doi: 10.1016/S1044-0305(03)00145-4. [DOI] [PubMed] [Google Scholar]
- 11.Wu W, Huang W, Qi J, Chou Y-T, Torng E, Watson JT. ‘Signature sets’, minimal fragment sets for identifying protein disulfide structures with cyanylation-based mass mapping methodology. J. Proteome Res. 2004;3:770–777. doi: 10.1021/pr049961t. [DOI] [PubMed] [Google Scholar]
- 12.Huwiler KG, Mosher DF, Vestling MM. Optimizing the MALDI-TOF-MS observation of peptides containing disulfide bonds. J. Biomol. Tech. 2003;14:289–297. [PMC free article] [PubMed] [Google Scholar]
- 13.Schnaible V, Wefing S, Resemann A, Suckau D, Bucker A, Wolf-Kummeth S, Hoffmann D. Screening for disulfide bonds in proteins by MALDI in-source decay and LIFT-TOF/TOF-MS. Anal. Chem. 2002;74:4980–4988. doi: 10.1021/ac025807j. [DOI] [PubMed] [Google Scholar]
- 14.Zubarev RA. Reactions of polypeptide ions with electrons in the gas phase. Mass Spectrom. Rev. 2003;22:57–77. doi: 10.1002/mas.10042. [DOI] [PubMed] [Google Scholar]
- 15.Chrisman PA, Pitteri SJ, Hogan JM, McLuckey SA. SO2− electron transfer ion/ion reactions with disulfide linked polypeptide ions. J. Am. Soc. Mass Spectrom. 2005;16:1020–1030. doi: 10.1016/j.jasms.2005.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fung YM, Kjeldsen F, Silivra OA, Chan TW, Zubarev RA. Facile disulfide bond cleavage in gaseous peptide and protein cations by ultraviolet photodissociation at 157 nm. Angew. Chem. Int. Ed. Engl. 2005;44:6399–6403. doi: 10.1002/anie.200501533. [DOI] [PubMed] [Google Scholar]
- 17.Bowie JH, Brinkworth CS, Dua S. Collision-induced fragmentations of the (M-H)− parent anions of underivatized peptides: an aid to structure determination and some unusual negative ion cleavages. Mass Spectrom. Rev. 2002;21:87–107. doi: 10.1002/mas.10022. [DOI] [PubMed] [Google Scholar]
- 18.Bilusich D, Brinkworth CS, McAnoy AM, Bowie JH. The fragmentations of [M-H]− anions derived from underivatised peptides. The side-chain loss of H2S from Cys. A joint experimental and theoretical study. Rapid Commun. Mass Spectrom. 2003;17:2488–2494. doi: 10.1002/rcm.1224. [DOI] [PubMed] [Google Scholar]
- 19.Chrisman PA, McLuckey SA. Dissociations of disulfide-linked gaseous polypeptide/protein anions: ion chemistry with implications for protein identification and characterization. J. Proteome Res. 2002;1:549–557. doi: 10.1021/pr025561z. [DOI] [PubMed] [Google Scholar]
- 20.Burns JA, Butler JC, Moran J, Whitesides GM. Selective reduction of disulfides by Tris(2-carboxyethyl)phosphine. J. Org. Chem. 1991;56:2648–2650. [Google Scholar]
- 21.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 22.Bilusich D, Maselli VM, Brinkworth CS, Samguina T, Lebedev AT, Bowie JH. Direct identification of intramolecular disulfide links in peptides using negative ion electrospray mass spectra of underivatised peptides. A joint experimental and theoretical study. Rapid Commun. Mass Spectrom. 2005;19:3063–3074. doi: 10.1002/rcm.2149. [DOI] [PubMed] [Google Scholar]
- 23.Guan S, Marshall AG. Stored waveform inverse Fourier transform (SWIFT) ion excitation in trapped-ion mass spectometry: Theory and applications. Int. J. Mass Spectrom. Ion Proc. 1996:157–158. 5–37. [Google Scholar]
- 24.Gauthier JW, Trautman TR, Jacobson DB. Sustained off-resonance irradiation for collision-activated dissociation involving Fourier transform mass spectrometry. Collision-activated dissociation technique that emulates infrared multiphoton dissociation. Analyt. Chim. Acta. 1991;246:211–225. [Google Scholar]
- 25.Leary JJ, Schmidt RL. Quadrupole mass spectrometers: An intuitive leak at the math. J. Chem. Educ. 1996;73:1142–1145. [Google Scholar]
- 26.Steel C, Henchman M. Understanding the quadrupole mass filter through computer simulation. J. Chem. Educ. 1998;75:1049–1054. [Google Scholar]
- 27.Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, McLafferty FW. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J. Am. Chem. Soc. 1999;121:806–812. [Google Scholar]
- 28.Thevis M, Loo RR, Loo JA. Mass Spectrom. Mass spectrometric characterization of transferrins and their fragments derived by reduction of disulfide bonds. J. Am. Soc. 2003;14:635–647. doi: 10.1016/S1044-0305(03)00199-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.