

1. Introduction
Cellular DNA is susceptible to chemical modifications when exposed to various environmental agents, chemotherapeutic agents, or endogenous agents produced by cellular processes. The lesions produced by these chemical reactions can lead to mutations and/or cell death. Many of these lesions, in the form of base or phosphate modifications, occur on one or the other strand of DNA. Certain agents, however, including bifunctional alkylating agents, platinum compounds, and psoralen can produce covalent adducts with DNA bases on both strands of DNA, leading ultimately to the formation of interstrand cross-links (Figure 1). Interstrand cross-links prevent DNA strand separation and can thus act as absolute blocks to DNA replication and/or DNA transcription. If left intact, interstrand cross-links can lead to cell death. It has been estimated that as few as 20 interstrand cross-links in the bacterial or mammalian genome can be lethal to cells that lack the ability to remove the cross-link.1,2 It is this cytotoxic effect that forms the mechanistic basis of action of many of the anticancer drugs currently in use today.1,3-7
Figure 1.

Interstrand cross-linked DNA.
Modified or otherwise damaged DNA bases can be mutagenic and are potentially cytotoxic. Cells have repair systems to remove these harmful lesions and thus preserve the integrity of their genomes.8 The majority of these repair pathways preserve the genetic information without introducing errors either by directly reversing the damage or by using an undamaged template strand to accomplish error-free repair (Figure 2). The most straightforward method of repairing DNA damage is the direct reversal of the lesion. For example, DNA photolyases [e.g., cyclobutane pyrimidine dimer photolyase and (6–4) photolyase] can directly reverse the two major forms of UV-induced lesions.9 O6-alkylguanine alkyltransferases, found in both bacterial and mammalian cells, remove methyl and other alkyl groups from O6-alkylguanine and to a lesser extent O4-alkylthymine, restoring the base to its original form.10 More recently, the Escherichia coli AlkB protein has been shown to repair 1-methyladenine and 3-methylcytosine lesions in DNA and RNA by direct reversal involving the removal of the methyl group via oxidative demethylation.11-13
Figure 2.
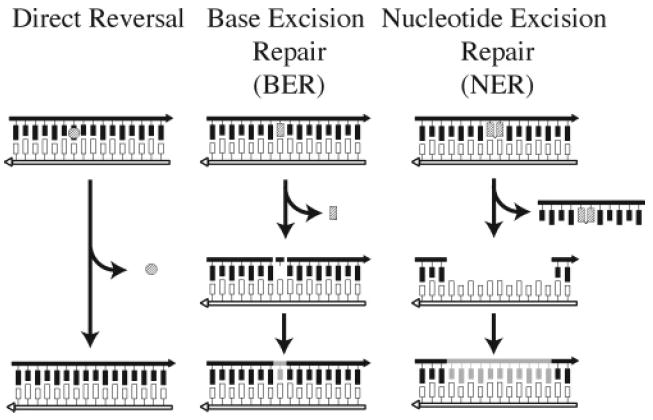
Basic DNA repair pathways.
The importance of an undamaged template strand is evident in the base excision repair (BER) and nucleotide excision repair (NER) pathways (Figure 2). In the BER pathway, the N-glycosyl bond of the damaged base is cleaved by a specific DNA glycosylase, releasing the damaged base. The abasic site that remains on the damaged strand is nicked on the 5′ side by AP endonuclease and on the 3′ side by deoxyribophosphodiesterase (dRpase); the resulting gap is filled in by DNA polymerase, which uses the nondamaged strand as a template, and the nick is sealed by DNA ligase. In NER, incisions are made by endonucleases at some distance 3′ and 5′ to the site of the damaged base. The resulting oligonucleotide containing the damaged base is removed, and the gap is filled in by DNA polymerase, again using the undamaged strand as a template. The remaining nick in the newly synthesized strand is then sealed by DNA ligase.
The physical constraints that interstrand cross-linking agents impart onto the DNA helix in conjunction with the requirement that repair necessarily requires repair of damage on both strands of the DNA makes cross-linking agents particularly deleterious compounds. In a comparative analysis of 234 chemicals where compounds were ranked by in vivo and in vitro genotoxicity, interstrand cross-linking agents consistently ranked at the top.14,15 Indeed, of the 20 most potent genotoxic agents, 12 were bifunctional alkylating agents capable of forming interstrand DNA cross-links.16 The potent genotoxic nature of interstrand cross-linking agents is mirrored in their ability to cause mutagenic events capable of leading to tumor formation. Thus, the rodent TD50 (the total lifetime dose of carcinogen required to increase the probability of tumor formation to 50%) is 10–1000-fold less for cross-linking agents than for monofunctional alkylating agents.17
A direct comparison of bifunctional compounds that are capable of forming interstrand cross-links and their chemically related monofunctional analogues has revealed striking differences in the mutational spectra elicited by each compound.18,19 Monofunctional alkylating agents largely produce point mutations that are readily explained by the type of alkylation product formed. In contrast, the bifunctional cross-linking agents cause chromosomal alterations consistent with DNA strand breakage and hyper-recombination. Thus, when the monofunctional half-mustard analogue of chlorambucil (1) was compared to the cross-link forming chlorambucil (1a) in Chinese hamster cells, the majority of the mutations caused by the monofunctional alkylating agent were point mutations (Figure 3).18 In contrast, the majority of the mutations caused by the bifunctional alkylating agent, chlorambucil, were major deletions. Similar results were obtained when the monofunctional alkylating agent 2-chlor-ethylamine (2) was compared to the bifunctional alkylating agent mechlorethamine (2a) in Drosophila melanogaster.19
Figure 3.
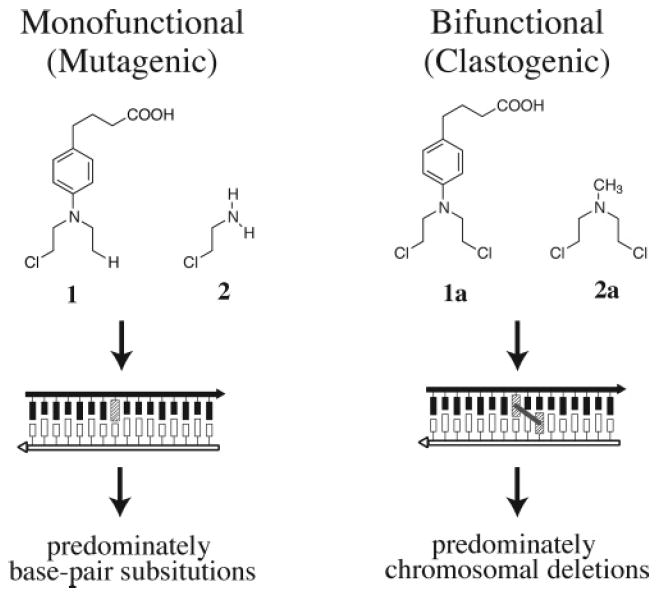
Clastogenic effects of interstrand cross-linking agents.
Cross-linking agents are typically clastogenic rather than mutagenic. Clastogens are agents that cause damage at the chromosomal level rather than at the DNA sequence level; i.e., a clastogen produces the gain, loss, or rearrangement of chromosomal segments and/or causes sister chromatid exchanges. Indeed, cross-species comparisons revealed that compounds capable of forming DNA interstrand cross-links show good correlation between their carcinogenicity and their propensity to cause gross chromosomal alterations.17 This ability of DNA cross-linking agents to cause gross-chromosomal aberrations including chromosomal loss, intra-or multilocus deletions, and breaks is likely to be the source of their genotoxicity. Cross-linking agents are 100–5000-fold more likely to produce chromosomal losses than the half-mustard chlorethylamine, and the monofunctional aziridines are 102–107-fold less active clastogens then their bi-, tri-, and tetrafunctional analogues.16 Furthermore, cross-linking agents are significantly more effective at inducing sister chromatid exchanges (splice recombination between sister chromatids) than their monofunctional analogues and are among the most efficient inducers of mitotic recombination.20-23
Drugs that form interstrand cross-links include bifunctional alkylating agents such as the nitrogen mustards (3, e.g., mechlorethamine R = CH3; chlorambucil, see 1a) and mitomycin C (4) and platinum compounds such as cis-Diamminedichloroplatinum(II) (cis-DDP) (5) and di- and trinuclear cis-DDP analogues (6a and 6b) (Figure 4). These compounds react with the N7 of guanine to form interstrand cross-links.24-27 Psoralen (7) and its derivatives form inter-strand cross-links with DNA when activated by irradiation with long-wavelength UV light.28,29 Psoralens are used therapeutically in the treatment of psoriasis, vitilago, and cutaneous T-cell lymphoma.30-35 It has been proposed that interstrand cross-links can also be produced endogenously in cells by the reaction of cellular DNA with unsaturated aldehydes such as acrolein or crotonaldehyde or dialdehydes such as malondialdehyde.36,37 These compounds, which act essentially as bifunctional alkylating agents, are products of lipid peroxidation. Although there have been a number of investigations on the structure and repair of interstrand cross-links of this type,38-41 the extent to which they form in cellular DNA and their possible biological effects remain unclear.
Figure 4.
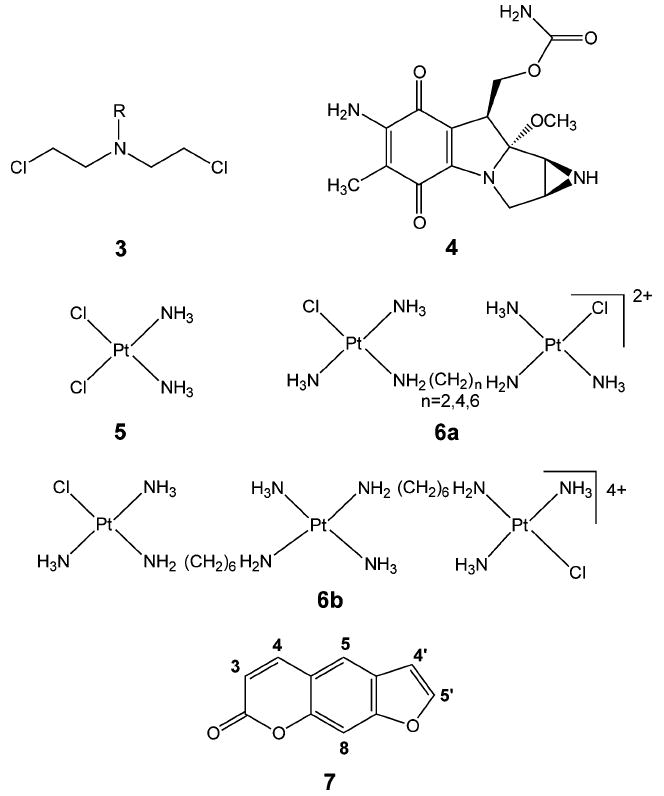
Compounds that can form DNA interstrand cross-links.
Understanding how interstrand cross-links are repaired has been and continues to be a challenging problem. A better understanding of the mechanism(s) involved is not only of academic interest but may be relevant to cancer chemotherapy as well. Tumors often develop resistance to bifunctional anticancer drugs, and one the mechanisms by which resistance occurs is the ability of the tumor cell to repair interstrand cross-links.7,42-47 Thus, a better understanding of the mechanism(s) of DNA interstrand cross-link repair could lead to more effective strategies for the treatment of cancer and/or the design of drugs that produce interstrand cross-links that can evade repair.
This review will focus primarily on DNA interstrand cross-links for which there is a substantial body of information about their repair. These include interstrand cross-links produced by nitrogen mustards, mitomycin C, platinum compounds and psoralen. The formation and structures of these cross-links will first be briefly reviewed. The mechanism of repair of these cross-links as derived from both genetic studies and experiments in cell-free systems will then be described as they occur in both prokaryotic and eukaryotic organisms. It will become apparent from the following discussion that our understanding of interstrand cross-link repair is at best incomplete both at the genetic and certainly at the molecular levels. Nevertheless, an examination of the literature from the past decade suggests that a consensus is emerging on the broader if not the detailed mechanistic aspects of the process.
2. Formation of Interstrand Cross-Links
The reactions of various antitumor drugs with DNA to form interstrand cross-links have been described in recent reviews by Rajski and Williams27 and by Noll et al.48 Consequently, in the following discussion, we will only briefly summarize these reactions. The interested reader is referred to these reviews for more detailed descriptions of the chemical and biochemical reactions involved.
2.1. Nitrogen Mustards
Nitrogen mustards (3, Figure 4) are bifunctional alkylating agents that contain a reactive N,N-bis-(2-chloroethyl)amine functional group. Clinically important examples include chlorambucil (1), mechlorethamine (2) (Figure 3), and phosphamide mustard (8), which is derived from the metabolism of the drug cyclophosphamide (9) (Figure 5). These compounds react with guanine residues in DNA via the aziridinium intermediate (10) to form an N7-alkylated guanine derivative (11) as shown in Figure 6. This guanine monoadduct can then form another reactive aziridinium intermediate (12), which can react either with water to form a 2-hydroxyethyl monoadduct (13) or with a second guanine residue to form the interstrand cross-link (14).
Figure 5.
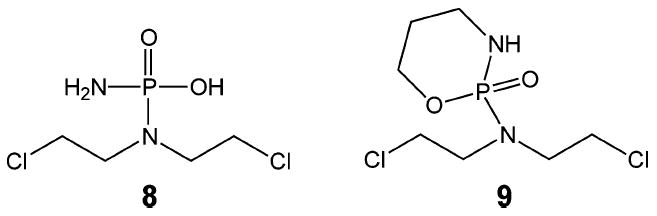
Phosphamide mustards.
Figure 6.
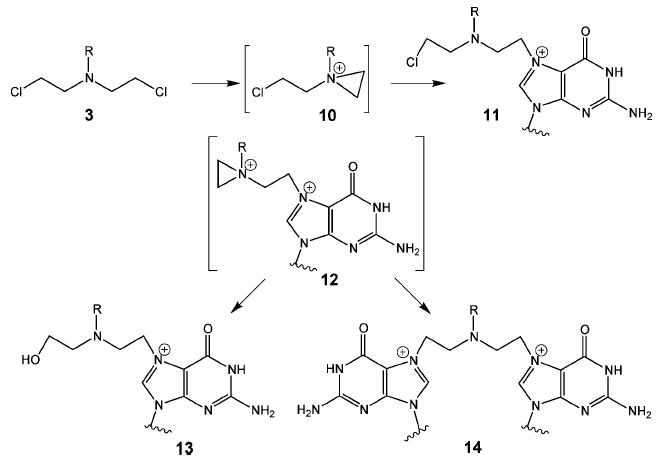
Nitrogen mustard interstrand cross-link.
Interstrand cross-link formation occurs almost exclusively with guanine residues in 5′-GNC-3′ sequences in DNA.49-53 The minimal distance between the guanines in this sequence is approximately 6.8 Å in B-form DNA, which is more than the 5.1 Å distance that can be spanned by the five atoms that comprise the cross-link.27 This constraint would be expected to produce a distortion of the helix in the region of the cross-link. Although detailed structural studies have not as yet been carried out on DNA containing these cross-links, studies by Rink and Hopkins have shown that the mechlorethamine cross-link produces a static bend of approximately 14° in DNA.54
N7-alkylated guanines (15) are to some extent unstable and can undergo a further reaction resulting in cleavage of the N-glycosyl bond (Figure 7). This creates an abasic site in the DNA (16) and in effect cleaves the interstrand cross-link. Alternatively, the imidazole ring of the alkylated guanine can undergo hydrolysis to produce a formamido-pyrimdine (FAPY) derivative (17), which is relatively resistant to further chemical reaction.49 In this case, the interstrand cross-link would remain intact, albeit in a somewhat different structural form. It is apparent then that reaction of nitrogen mustards with DNA can lead to a variety of products. In addition to forming both mono- and cross-linked adducts, the adducts themselves are subject to further chemical transformations. It is these additional chemical reactions that can potentially complicate studies on nitrogen mustard interstrand cross-link repair.
Figure 7.
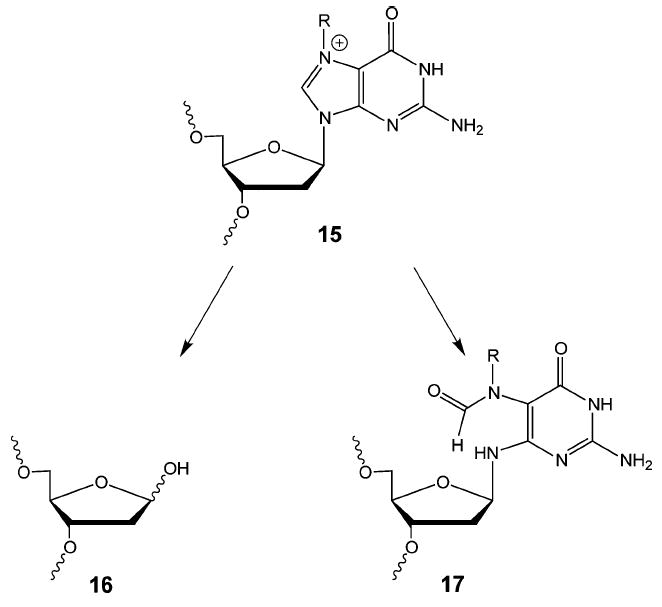
Reactions of N7-alkylated guanine.
2.2. Mitomycin C
Mitomycin C (4, Figure 4), a product of the mold Streptomyces caespitosis, is a bifunctional alkylating agent used to treat a variety of tumors. Unlike the nitrogen mustards, which form cross-links with 5′-GNC-3′ sequences through the major groove of DNA, mitomycin C reacts with guanine residues of 5′-CG-3′ sequences through the minor groove of DNA. Mitomycin C itself is relatively inert and requires chemical or enzymatic reduction of its quinone ring to initiate alkylation. Extensive studies have been carried out on the biosynthesis of mitomycin C, and its reactions with DNA have been reviewed recently.27
When interstrand cross-links are formed with DNA, mitomycin C undergoes a rather complicated series of reactions and rearrangements whose salient features are summarized in Figure 8. Two-electron reduction of the quinone ring of 4 facilitates the loss of the methoxy group, leading to the formation of the hydroquinone intermediate (18). Tautomerization followed by the reaction with the N2-amino group of guanine produces monoadduct 19. Elimination of the carbamoyl group produces the highly reactive vinylogous hydroquinone methide intermediate (20), which alkylates the guanine on the opposite strand of DNA to produce, after oxidation, an interstrand cross-link (21).
Figure 8.
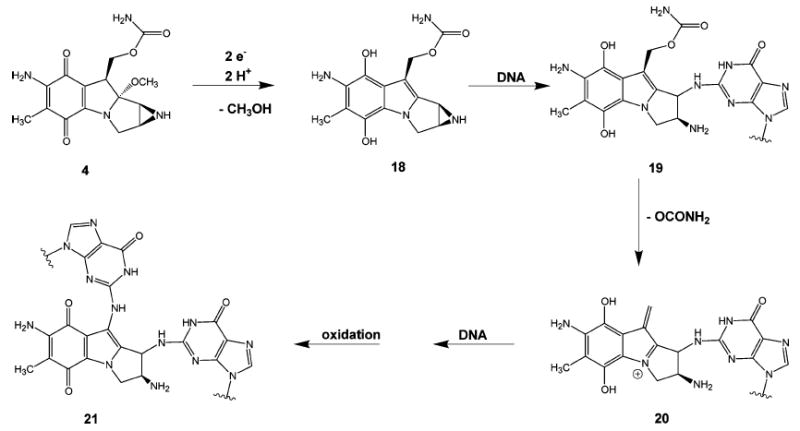
Formation of mitomycin C interstrand cross-link.
Again, in contrast to the nitrogen mustard cross-link, molecular modeling studies suggest that the mitosene moiety of the cross-link is readily accommodated in the minor groove and minimally perturbs the structure of the DNA at the site of the cross-link.27 Electrophoretic mobility shift analysis (EMSA) of ligated duplexes that contain mitomycin interstrand cross-links show that the cross-link does not induce bending of the DNA.55 NMR studies on a short DNA duplex containing a mitomycin C interstrand cross-link revealed, however, that the cross-link does cause some widening of the minor groove.56 When these results are taken together, they suggest that the presence of the mitomycin C interstrand cross-link causes minimal perturbation to DNA structure, a factor that could be of importance when considering how interstrand cross-links are detected in cellular DNA.
2.3. Platinum Compounds
cis-Diamminedichloroplatinum (5) or cis-DDP is a bi-functional antitumor agent that is used primarily in the treatment of testicular cancer. It forms adducts with the N7-nitrogen of purines, reacting mainly with guanine. The active form of the compound is believed to be the aquated species, cis-[Pt(NH3)2(H2O)2]2+, formed by the displacement of the chlorides by water.57 The main products produced by the reaction of cis-DDP and DNA are intrastrand cross-links between guanine residues in 5′-GG-3′, 5′-GNG-3′, and between adenine and guanine in 5′-AG-3′ sequences. These intrastrand cross-links account for greater than 90% of the adducts formed. cis-DDP interstrand cross-links (22) (Figure 9) are formed between guanines in 5′-GC- sequences to the extent of 2–5%.58 Unlike an N7-alkylated deoxyguanosine adduct or an N7-protonated deoxyguanosine, both of which are prone to depurination, N7-platinum dG adducts are resistant to such a reaction. Recent theoretical studies suggest that the N-glycosyl bonds of dG and platinated dG are very similar in nature, in contrast to the N-glycosyl bond of N7-protonated dG.59 Thus, the kinetic barrier for N-glycosyl bond scission is less for the protonated dG, whereas it is essentially the same for both dG and platinated dG.
Figure 9.
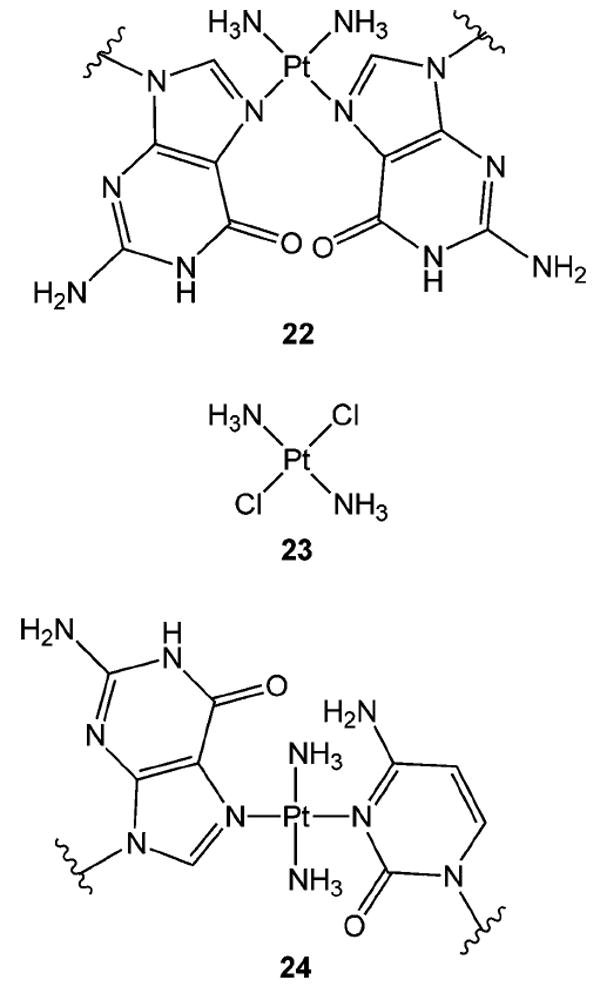
cis-Diamminedichloro- and trans-diamminedichloroplatinum interstrand cross-links.
The presence of the cis-DDP interstrand cross-link results in a significant deformation of the DNA helix at the site of the cross-link. Gel EMSA of multimers produced by ligating short DNA duplexes that contain a single interstrand cross-link show that the helix is unwound by 79° and bent by 45°.60 Solution61,62 and crystal63 structures, derived respectively by high-resolution NMR and X-ray crystallographic analyses of decamers that contain a single interstrand cross-link, are in essential agreement with mobility shift results. These studies also show that the cytosines that were originally paired with now platinated guanines assume an extrahelical conformation. The cis-Diammineplatinum moiety lies in the minor groove, and bending is toward the minor groove. Thus, the helix conformation at the site of the cross-link is similar to that of Z-form DNA.
trans-DDP (23) (Figure 9) cannot form an intrastrand 5′-GG-3′ cross-link because of steric constraints. Rather this compound forms N7-guanine monoadducts64 and interstrand cross-links (24) between the N7-nitrogen of guanine and the N3-nitrogen of its base-paired cytosine.65 Intrastrand cross-links between two nonadjacent guanines or a guanine and cytosine that are separated by at least one base residue are also observed.66,67 Leng and co-workers have observed that 5′-GNG-3′ intrastrand cross-links in single-stranded DNA rapidly isomerize to form the interstrand cross-link when hybridized with their complementary DNA strand.68
The trans-DDP interstrand cross-link causes less distortion of the DNA helix than does the cis-DDP interstrand cross-link. Thus, the cross-linked DNA is unwound by 12° and is bent by 26°.65 NMR studies show that the guanine base of the cross-link is in the syn conformation and forms a Hoogsteen-type base pair with the platinated cytosine.69
In contrast to cis-DDP, trans-DDP is not an effective therapeutic agent. This is presumably due to the slow rate at which the interstrand cross-link is formed65 and the likelihood that the monoadducts are inactivated or removed by reactions with cellular nucleophiles such as glutathione.64,67,70,71
2.4. Psoralens
Psoralen (7) and its derivatives, a class of furocoumarins found in leafy vegetables and other plants, have found use as medicinal agents since the times of the ancient Egyptians, who used plant extracts containing these compounds to treat skin diseases such as vitiligo.72 These planar, tricyclic, semiaromatic compounds interact with DNA via intercalation (Figure 10). The binding affinities vary from milli- to micromolar concentrations, depending upon the substituents on the psoralen ring. Thus, 8-methoxypsoralen (25) has a dissociation constant, Kd, of 2.5 × 10−3 M with calf thymus DNA, whereas 4,5′,8-trimethylpsoralen (26) and 4′-amino-methyl-4,5′,8-trimethylpsoralen (27) have Kd values of 5.6 × 10−5 and 6.6 × 10−6 M, respectively, as measured by equilibrium dialysis.73
Figure 10.
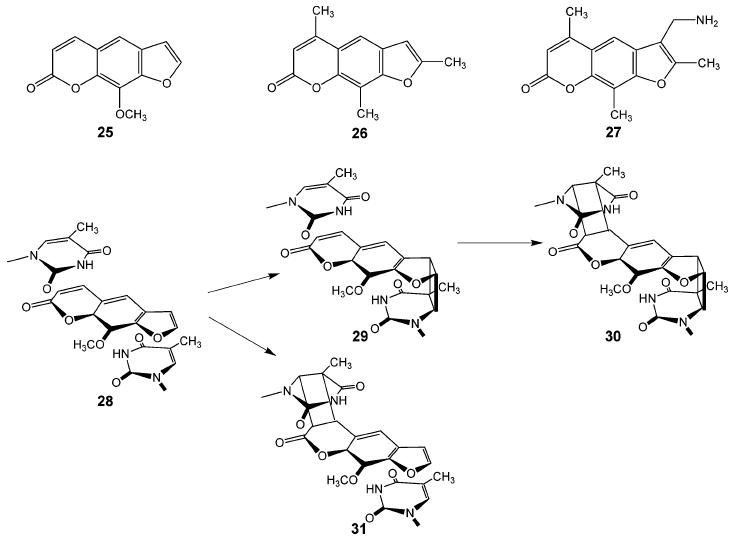
Psoralens and their DNA photoadducts.
Psoralens can be induced to form covalent adducts with DNA by irradiation with long wavelength ultraviolet light.28,29 Although psoralens show no particular sequence specificity when binding to DNA, they do preferentially form covalent adducts with thymines in 5′-TA-3′ and 5′-AT-3′ sequences in DNA. When intercalated with these sequences in B-form DNA, the 3,4-pyrone ring and 4′,5′-furan ring double bonds are oriented directly above or below the 5,6-double bonds of the thymines (28) (Figure 10). Absorption of light by the drug triggers a 2+2 cycloaddition reaction between the furan or pyrone ring and the adjacent thymine, leading to the formation of a monoadduct containing a cyclobutane bridge. The furan-side monoadduct (29) can absorb a second quantum of light, triggering a second cycloaddition between the pyrone double bond and the thymine on the opposite DNA strand. This reaction results in the formation of an interstrand cross-link (30). In contrast, the pyrone-side monoadduct (31) lacks the ability to absorb a second quantum of light and thus remains as the monoadduct.
The steric constraints imposed upon psoralen intercalation by the DNA helix result in psoralen–thymine adducts that have a cis-syn configuration. NMR studies have been carried out on eight base pair DNA duplexes that contain a 4′-hydroxymethy-4,5′,8-trimethylpsoralen74,75 or a 4′-amino-methyl-4,5′,8-trimethylpsoralen76 interstrand cross-link in the center of the duplex. These studies show that, although there is considerable local distortion at the site of the cross-link, the duplex retains its B-form conformation three base pairs from the cross-link site. The cross-link causes the duplex to unwind approximately 25° as would be expected for an intercalating agent but does not induce any significant bending of the helix.77
Unlike nitrogen mustard and mitomycin C interstrand cross-links, psoralen interstrand cross-linked DNA is relatively easy to synthesize, and the cross-link itself is chemically very stable. Consequently, as will become evident in the following sections, the majority of studies on the repair of interstrand cross-links in defined DNA substrates have been carried out on the psoralen interstrand cross-link.
2.5. Synthesis of Interstrand Cross-Linked DNA Duplexes
One of the challenges to studying the repair of interstrand cross-links at the molecular level is the preparation of DNA duplex substrates that have a single cross-link at a defined site in the duplex. Treatment of preformed DNA duplexes with bifunctional alkylating agents often results in the production of a broad spectrum of products, only a small percentage of which contain the cross-linked duplex of interest. In the case of the nitrogen mustards, the initial products are often unstable and can undergo further chemical transformations. Although it is possible to direct cross-linking agents such as platinum and psoralen to specific sites in the duplex, the yields of cross-linked material are often quite low.
Recent advances in oligonucleotide syntheses have led to the development of methodologies that facilitate the preparation of duplexes with defined interstrand cross-links. For example, in some cases, it is possible to prepare a single-stranded oligonucleotide that contains the monoadducted version of the cross-link positioned at a specific site in the oligonucleotide. Hybridization with a complementary strand and further chemical or photochemical reaction produces the interstrand cross-link. This strategy has been used to synthesize short duplexes that contain mitomycin C,78 psoralen,79,80 or trimethylene81,82 interstrand cross-links. Methods have also been developed that allow the complete de novo synthesis of short interstrand cross-linked duplexes on an automated DNA synthesizer.83-87 This approach can potentially produce relatively large amounts of material for both physical and biochemical studies. The interested reader is directed to a recent review that describes these various synthetic strategies in greater detail.48 Suffice to say that the preparation of DNA duplexes with interstrand cross-links of defined structure remains an interesting challenge for the synthetic chemist. Progress in this area should provide substrates for studies that will lead to a more detailed understanding of interstrand cross-link repair at the molecular level.
3. Repair of Interstrand Cross-Links
The repair of DNA interstrand cross-links is less thoroughly characterized than many of the other DNA repair pathways. Three hurdles to the study of DNA interstrand cross-link repair have presented themselves. The first, briefly mentioned above, is the technical challenge associated with producing substrates suitable for studying the mechanisms of interstrand cross-link repair. The second difficulty is the growing realization that different cross-links and in some cases different orientations of the same type of cross-link result in differences in the way cells repair the cross-link, thus making generalizations difficult. The third and perhaps most challenging obstacle to studying the repair of interstrand cross-links has been the involvement not of a single pathway or process but rather the involvement of multiple overlapping and redundant repair processes. Keeping these limitations in mind, we have endeavored in the following sections to summarize what is known about the repair of DNA inter-strand cross-links in E. coli, Saccharomyces cerevisiae, and mammalian cells.
In the broadest terms, there appear to be two basic processes by which interstrand cross-link lesions are repaired. The first is an error-free process dependent upon homologous recombination, and the second is an error-prone process dependent upon translesion synthesis. Both the error-free and error-prone processes draw heavily upon proteins with known roles in the NER, homologous recombination, and translesion DNA synthesis pathways. Before attempting to describe the complicated and as yet poorly understood interplay of these pathways, we will provide a brief overview of these repair pathways focusing exclusively on subpathways known to be involved in cross-link repair.
Nucleotide Excision Repair (NER)
The NER pathway was first discovered in E. coli as a mechanism involved in the repair of UV-radiation-induced DNA damage. Subsequently, it was shown to be involved in the repair of bulky DNA adducts induced by UV irradiation and chemical modification in all organisms. Extensive reviews of NER in bacteria, yeast, and mammalian cells are available.8,88-90 In its simplest form, NER involves four steps (Figure 11): (i) damage recognition and formation of a preincision complex, (ii) asymmetric incisions on the 5′ and 3′ sides of the DNA damage to produce a short DNA fragment that contains the adduct, (iii) DNA repair synthesis using the undamaged template strand, and (iv) ligation to complete the repair of the DNA.
Figure 11.
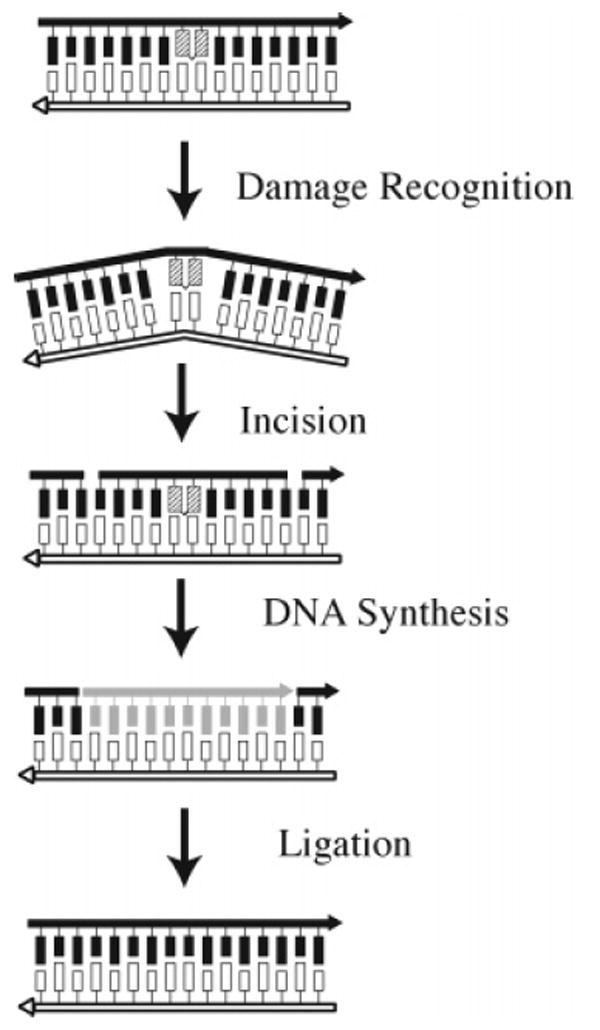
Nucleotide excision repair in E. coli.
Proteins involved in the repair of interstrand cross-links and that belong in the NER pathway are listed in Table 1. In E. coli, NER-mediated DNA damage recognition is an ATP-dependent process mediated by the (UvrA)2(UvrB)1 complex that recognizes damaged DNA and results in the loading of UvrB onto the DNA. After dissociation of UvrA, the remaining UvrB recruits UvrC to the damage site, which is in turn followed by DNA incisions. Dissociation of the incision complex is followed by DNA synthesis in which Pol I uses the complementary, undamaged strand as a template. The process is completed by the action of DNA ligase. In mammalian cells, NER is initiated when the XPC/ hHR23B complex (see Table 1 for S. cerevisiae names) binds to DNA damage and recruits TFIIH. TFIIH and its component helicases form a preincison bubble at the site of damage. The subsequent recruitment of XPA–RPA, XPG, and XPF/ ERCC1 leads to the formation of a catalytically competent incision complex. DNA synthesis or gap-filling is carried out by either DNA Pol δ or Pol ∈ followed by ligation by human DNA ligase I.
Table 1.
| bacterial | yeast, S. cerevisiae | mammalian | Function/DNA |
|---|---|---|---|
| NER Proteins | |||
| RAD1 | XPF | nuclease, 5′ incision; in complex with ERCC1 | |
| RAD2 | XPG | nuclease, 3′ incision | |
| RAD3 | XPD | helicase | |
| RAD4 | XPC | damage recognition factor | |
| RAD10 | ERCC1 | Part of the 5′ incision complex with XPF | |
| RAD14 | XPA | damage verification factor | |
| RAD23B | hHR23B | cofactor of XPC | |
| RAD25/SSL2 | XPB | helicase | |
| RPA 1, 2, 3 | RPA p70, p34, p14 | binds in the preincision complex | |
| REV7 | MAD2B | mitotic spindle assembly checkpoint protein | |
| PCNA | PCNA | sliding clamp loader for polymerases | |
| UvrA | damage recognition; ATPase, DNA binding | ||
| UvrB | ATPase, helicase, recognition with UvrA | ||
| UvrC | nuclease | ||
| UvrD | helicase II; initiates unwinding from a nick | ||
| DNA Pol I | resynthesizes excised DNA fragment | ||
| POL δ | Pol δ | normal DNA replication polymerase; responsible for elongation of Okazaki fragments (A-family pol.) | |
| POL2 | Pol ∈ | normal DNA leading strand replication polymerase | |
| DNA ligase | DNA ligase | DNA ligase I | seals resynthesized DNA fragment |
| Homologous Recombination Proteins | |||
| RecA | RAD51 | RAD51 | mediates pairing between homologous DNA molecules |
| RecB, C, D | RAD50/MRE11/XRS2 complex | Rad50/Mre11/NBS1 complex | exonuclease/helicase; binds to ends of linear ds ends, precede RecA/Rad51 |
| RAD52 | RAD52 | RAD51 paralogue, mediates HR | |
| XRCC2 | break and cross-link repair; interacts with RAD51 | ||
| XRCC3 | break and cross-link repair; interacts with RAD51 | ||
| RAD55/RAD57 | RAD51 paralogue, mediates HR | ||
| RAD54 | RAD54 | induction of supercoiling | |
| mus81/mms4 | heterodimer branch-specific endonuclease | ||
| RecJ | 5′ → 3′ single-strand specific DNA exonuclease | ||
| RuvA, B | helicase, specifically recognizes Holliday junctions and promotes branch migration | ||
| RuvC | endonuclease, resolves Holliday junctions during HR | ||
| γ-H2AX | phosphorylated histone H2AX; accumulates at DSB; function in repair unknown | ||
| EXO1 | EXO1 | 3′ exonuclease and flap-endonuclease involved in recombination and double-strand break repair | |
| PSO2 | Artemis | single-strand DNA-specific 5′ → 3′ exonuclease; functions in mitotic stress checkpoint | |
| Translesion Repair Proteins | |||
| REV1/[REV3/REV7] | REV1/Pol ζ | DNA polymerase ζ extends misrepair following Pol ι (B-family pol.) | |
| POLη/RAD30 | Pol η | replicates past photodimers (Y-family pol.) | |
| dinB; PolIV | Pol κ; DINB1 | replicates past abasic sites and oxidative lesions in an error-prone manner; =mus308 in Drosophila melanogaster (Y-family pol.) | |
| Pol ι | paralogue of Pol η; inserts deoxynucleotides across from DNA lesions (Y-family pol.) | ||
| Pol2; PolB; dinA | induced by SOS response; results in frameshifts | ||
| RAD5 | putative ubiquitin ligase; binds ssDNA; interacts with RAD18 | ||
| RAD6 | RAD6 | ubiquitin-conjugating enzyme | |
| RAD18 | RAD18 | E3 ubiquitin ligase; post-replication repair ssDNA binding | |
Homologous Recombination
The primary function of homologous recombination is to repair double-strand breaks or single-strand gaps in DNA that are formed as a result of the collapse of replication forks and/or exposure to DNA-damaging agents. A number of distinct recombination processes generally fall under the rubric of homologous recombination, including homology-mediated double-strand break repair, break-induced replication, as well as single-strand annealing. Extensive reviews of homologous recombination in bacteria, yeast, and mammalian cells are available.91-97 In its most widely recognized form, homologous recombination involves four steps (Figure 12): (i) double-strand break recognition followed by nucleolytic processing to produce single-stranded 3′ ends of DNA, (ii) protein-mediated strand invasion of the single-stranded DNA with the homologous chromosome, (iii) DNA synthesis, which regenerates the degraded DNA using the undamaged homologous chromosome as a template, and (iv) resolution of the Holliday-junction intermediates to generate the double-stranded DNA with crossover and noncrossover products possible.
Figure 12.
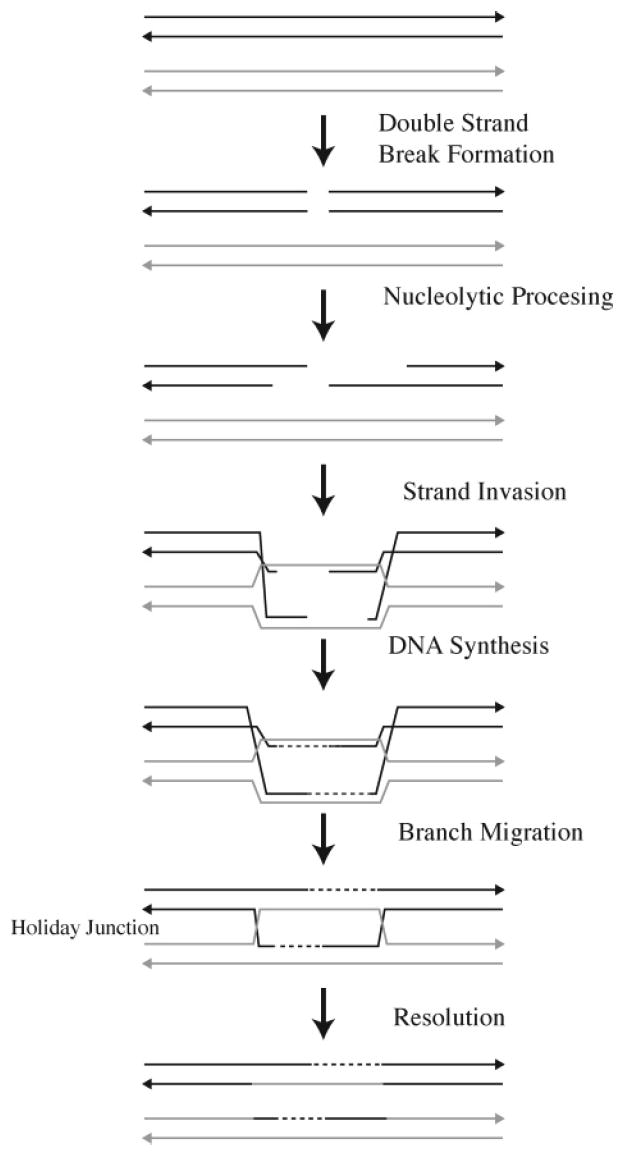
Homologous recombination.
Proteins that are involved in the repair of interstrand cross-links and that belong in the homologous recombination pathway are listed in Table 1. In E. coli, the RecBCD complex initiates recombination by acting on the break to create a single-stranded DNA substrate suitable for RecA protein activity. The RecBCD complex is a combination nuclease/ helicase that binds to the end of linear double-stranded DNA. After unwinding and degradation, RecA is loaded onto the resected double-stranded DNA with a 3′-terminal single-stranded DNA tail. RecA, in combination with the single-strand-binding (SSB) protein, forms a continuous presynaptic filament on the DNA. This nucleoprotein complex acts as the functional unit to perform a rapid and efficient search for homology within the double-stranded DNA recipient, resulting in the formation of a joint molecule. After joint molecule formation, DNA polymerase I regenerates the DNA sequence. The resultant Holliday junction is resolved by the RuvC protein. RuvC acts in concert with the RuvAB proteins to coordinate the steps of branch migration and Holliday-junction resolution.
In yeast, the resection of the DNA (see Table 1 for mammalian names) is carried out by an unidentified nuclease. Rad51, the yeast homologue to RecA, in conjunction Rad52 displaces the single-stranded DNA that is presumably covered by RPA. The resultant nucleofilament along with Rad54 and Rad55/57 promotes DNA unwinding and strand annealing between donor DNA and the incoming Rad51 nucleofilament. The resolution of the resultant recombination intermediates is carried out in yeast by a diverse set of mechanisms including the mus81-mms4 nuclease and/or a suspected RuvABC-like activity called Resolvase A.
Translesion Synthesis
The primary function of translesion DNA synthesis is to facilitate replication past DNA lesions that block replication. Despite the evolution of DNA repair processes such as direct reversal, BER, and NER (Figures 2 and 11) to repair DNA damage, these processes are not completely efficient. To escape the lethal effect of blocked replication, translesion DNA synthesis allows the replication of DNA containing persistent DNA damage. Extensive reviews of translesion DNA synthesis in bacteria, yeast, and mammalian cells are available.98-106 In its simplest form, translesion synthesis involves three steps (Figure 13): (i) normal bidirectional DNA replication is interrupted by the presence of a DNA lesion, and the replicative polymerase is released; (ii) a specialized translesion DNA polymerase is loaded onto the site and replicates a short distance past the lesion; and (iii) the translesion polymerase is replaced by the replicative DNA polymerase, which continues replication. Thus, translesion synthesis past the damage regenerates the double-stranded DNA in what can be an error-free or error-prone manner depending upon the type of lesion.
Figure 13.
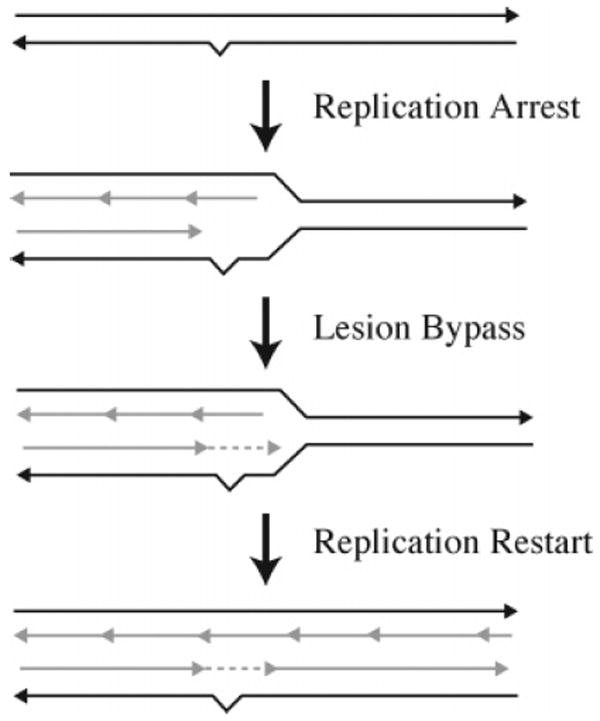
Translesion synthesis.
Proteins that are involved in the repair of interstrand cross-links and that belong in the translesion DNA synthesis pathway are listed in Table 1. Recent work has dramatically increased our understanding of the role of translesion DNA synthesis in lesion bypass mechanisms that allow blocked replication forks to progress. The Y family of DNA polymerases in mammalian cells are the most thoroughly characterized of the polymerases involved in translesion DNA synthesis; however, these DNA polymerases have not been implicated in the repair of interstrand cross-links. In E. coli, DNA polymerase II has been implicated. This poorly understood DNA polymerase is not a member of the Y family but is one of three damage-inducible DNA polymerases in E. coli. In yeast, Rev3 and Rev7 form a heterodimer that constitutes DNA polymerase ζ, a member of the B family of DNA of polymerases. Rev3 and Rev7 have been implicated in the translesion DNA replication of intermediates generated by the repair of interstrand cross-links.
As described in the following sections, the repair of interstrand cross-links in both prokaryotic and eukaryotic cells has been shown to involve components of the NER, homologous recombination, and error-prone translesion replication pathways. In addition to these known components, there are still others whose involvement has been identified genetically but whose mechanistic role is not understood, as well as yet to be identified components whose action can be inferred. Other recent reviews of DNA interstrand cross-link repair are available.1,7,45,107-109 As described below and in these other reviews, although a good deal is known regarding the repair of interstrand cross-links, much remains to be understood.
3.1. Repair in Prokaryotes
Very early studies showed that wild-type E. coli were able to survive treatment with bifunctional alkylating agents that produced up to 70 DNA interstrand cross-links in the bacterial genome. Strains of bacteria that lacked the functioning recombination protein, RecA, or were deficient in NER activity were also able to survive such treatment, although they could tolerate fewer (5–20) interstrand cross-links.110,111 Double mutants that lacked both recombination as well as NER activity were, however, unable to survive more than one cross-link per genome.111 These and other observations led Cole, who studied repair of psoralen interstrand cross-links in E. coli, to propose a repair model that involved a combination of NER and homologous recombination.112 In this model, the cross-link is first partially excised by components of the NER pathway. This is followed by RecA-mediated strand exchange between the damaged DNA and additional copies of homologous DNA. Branch migration and repair synthesis in effect replaces one of the damaged strands with undamaged DNA from the homologous DNA, leaving the other strand with the remnant of the cross-link. A second round of NER then removes this lesion.
Biochemical experiments employing purified E. coli UvrABC, RecA, and replication proteins have been carried out in vitro to study the repair of psoralen interstrand cross-links. As described above, UvrABC, which is composed of UvrA, UvrB, and UvrC proteins, is an ATP-dependent enzyme that is responsible for the first step of NER; that is, it creates incisions on either side of the DNA lesion. Experiments by Sancar and co-workers using DNA that contained randomly placed furan- and pyrone-side psoralen monoadducts showed that incisions are made at the eighth phosphodiester bond 5′ to the monoadduct and at the fifth phosphodiester bond 3′ to the monoadduct.113
A somewhat different picture emerged for incisions made around the psoralen interstrand cross-link. van Houten et al. developed methodology that allowed preparation of a 40-base-pair duplex that contained a single 4′-hydroxymethyl-4,5′,8-trimethylpsoralen interstrand cross-link at a 5′-TA-3′ sequence located in the middle of the duplex.114 Incubation of this duplex with UvrABC and analysis of the products by polyacrylamide gel electrophoresis showed that incisions were made at the ninth phosphodiester bond 5′ to the psoralen cross-link and at the third phosphodiester bond 3′ to the psoralen cross-link.114,115 The products of this reaction appeared to be generated in equal amounts, suggesting that the incisions occurred in a coupled manner. Surprisingly, incisions were observed only on the strand bearing the furan adduct. No cutting was observed on either side of the pyrone adduct. This behavior is in stark contrast to that of the monoadducts, where incisions are made on either side of the furan or pyrone monoadducts. Although the reason for this selectivity is unclear, it is likely dependent upon the cross-link-induced distortion of the helix.115
A model for the repair of the psoralen interstrand cross-link was proposed on the basis of these in vitro experiments (Figure 14).115 In the first step, UvrABC makes two incisions, one on the 5′ and one on the 3′ side of the furan adduct, creating an 11-nucleotide fragment linked via the cross-link to the complementary strand. This oligonucleotide fragment is then displaced by a homologous strand of a sister DNA duplex, which is then incorporated into the DNA duplex. This strand exchange reaction, which is mediated by RecA, creates a three-stranded structure, one strand of which is the excised and displaced cross-link containing 11-mer. UvrABC then cuts on either side of the pyrone adduct of the three-stranded structure. The product of this incision reaction, which contains the two original strands of the DNA joined by the psoralen cross-link, is then displaced by DNA polymerase I in combination with helicase II. This reaction uses the newly acquired homologous strand as a template to fill in the gap created by the excised psoralen cross-linked duplex. DNA ligase then seals the nick left by the displacement/polymerization reaction.
Figure 14.
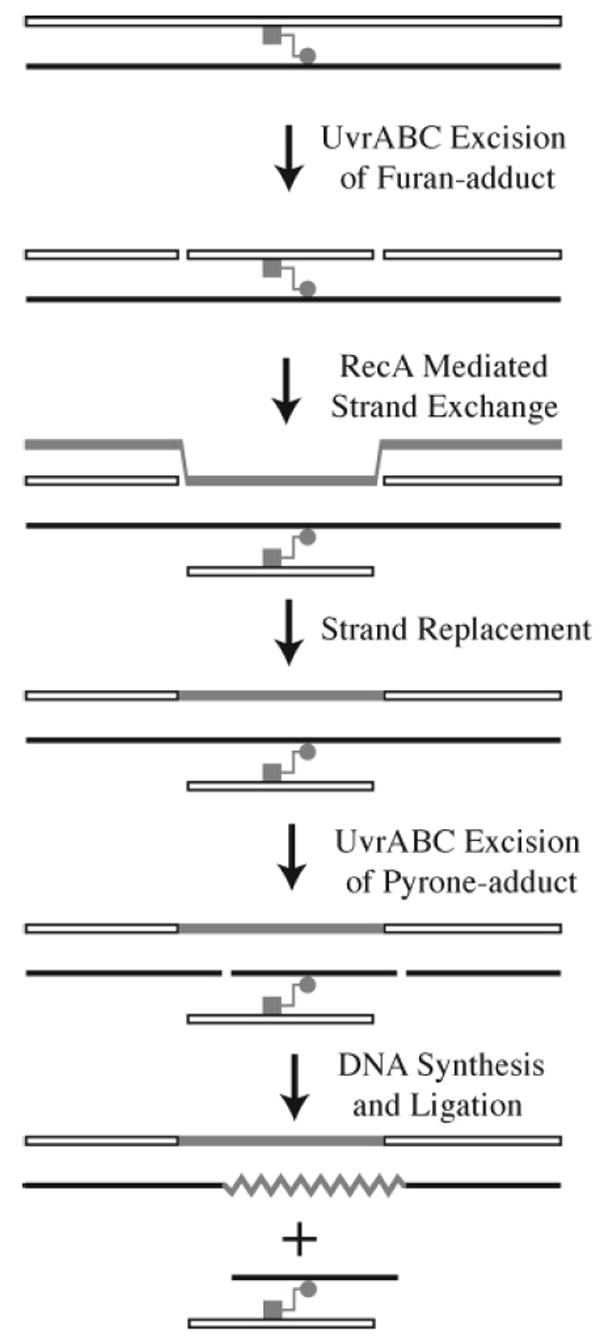
Model for interstrand cross-link repair in E. coli. The furan adduct is indicated by the square, and the pyrone adduct is indicated by the circle.
This model was tested by preparing three-stranded complexes that contained a 4′-hydroxymethyl-4,5′,8-trimethylpsoralen interstrand cross-link.116,117 The complex was prepared by incubating an oligonucleotide that contained a specifically placed furan-side monoadduct with plasmid DNA in the presence of RecA. Because the sequence of the psoralen-derivatized oligonucleotide was homologous to that of the plasmid DNA, strand invasion occurred. Irradiation of the complex then resulted in the formation of an interstrand cross-link, where the pyrone adduct formed with the plasmid strand that is complementary to the oligonucleotide. This three-stranded complex thus resembles the three-stranded complex postulated to form after the first incision step and RecA-mediated strand invasion. When this three-stranded plasmid complex was incubated with UvrABC, only incision on the plasmid (pyrone adduct) strand was observed. This is consistent with the proposed model. Furthermore, the addition of DNA polymerase I, helicase II, and DNA ligase resulted in incorporation of nucleoside triphosphates, an indication that repair of the plasmid DNA had occurred.
Experiments by Sladek et al.118 shed additional light on the strand exchange process. A single-stranded plasmid DNA was constructed that contained a 4′-hydroxymethyl-4,5′,8-trimethylpsoralen cross-linked oligonucleotide (19-mer) linked to the plasmid DNA via a pyrone adduct (Figure 15a). This cross-linked substrate was shown to undergo strand exchange with a homologous 7250-bp linear DNA duplex in the presence of RecA, producing a nicked circular double-stranded molecule. This reaction demonstrates that RecA-mediated strand transfer can occur past the cross-link. The nicked circular DNA was then ligated to form a covalently closed circular DNA that contained the cross-linked oligonucleotide as a third strand. When this plasmid DNA was incubated with UvrABC, a cross-linked duplex was excised whose size was consistent with the creation of incisions on either side of the pyrone adduct in the plasmid DNA strand.
Figure 15.
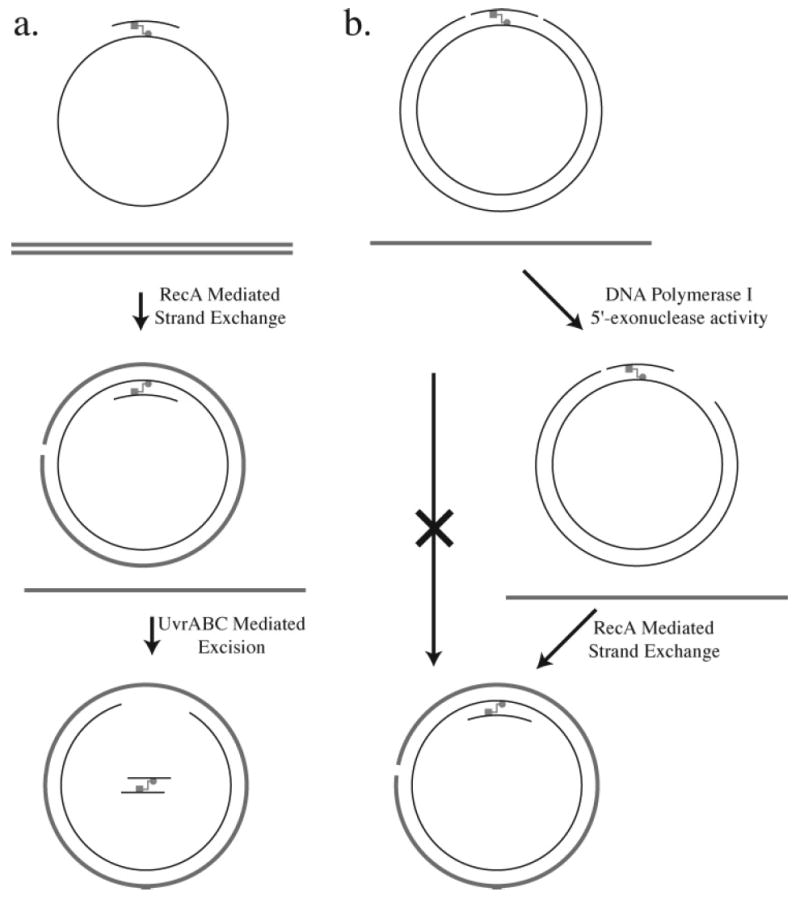
In vitro strand exchange reactions with psoralen cross-linked DNA. The furan adduct is indicated by the square, and the pyrone adduct is indicated by the circle.
In a separate set of experiments, a 4′-hydroxymethyl-4,5′,8-trimethylpsoralen cross-linked circular double-stranded DNA plasmid was prepared that contained nicks at the eighth and fourth phosphodiester positions on the 5′ and 3′ sides, respectively, of the furan adduct (Figure 15b). This plasmid DNA thus mimics psoralen cross-linked DNA that has undergone the first UvrABC-mediated incision step. No strand exchange was observed when this plasmid DNA was incubated with homologous single-stranded DNA in the presence of RecA. This result shows that the nicks are not sufficient to allow strand exchange to occur and is consistent with the observation that RecA-mediated strand exchange requires the presence of single-stranded gaps. The result also suggests that an additional step must occur between the initial UvrABC incisions and the strand exchange reaction. To test this hypothesis, the nicked plasmid was incubated with DNA polymerase I, whose 5′-exonuclease activity would create a single-stranded gap on the 3′ side of the cross-link. When this gapped plasmid DNA was incubated with homologous linear DNA in the presence of RecA, strand exchange was observed. Interestingly, DNA polymerase I was unable to extend the 3′ end adjacent to the psoralen cross-link.
These observations require modification of the model originally proposed by van Houten et al.115 After the initial incisions made by UvrABC on the furan-adduct strand, the 5′-exonuclease activity of DNA Pol I creates a gap on the 3′ side of the adduct.118 This gap serves as a site for the initiation of the RecA-mediated strand exchange reaction. The remaining steps then follow those of the originally proposed model.
The experiments described above were carried out using DNA cross-linked with 4′-hydroxymethyl-4,5′,8-trimethylp-soralen. In experiments examining incisions by UvrABC on DNA containing 4,5′,8-trimethylpsoralen interstrand cross-links, Jones and Yeung found that initial incisions could be made on either the furan-adducted or pyrone-adducted strand of the cross-linked DNA.119,120 It appeared that a preference for one strand or the other correlated with the sequences surrounding the cross-link. Thus, incisions were made on the furan-adducted strand when there were G/C-rich sequences with 6–12 bases on the 5′ side of this lesion. When there was a non-G/C-rich sequence or when the G/C-rich sequences were located only on the 3′ side of the furan adduct, incisions were observed on either strand. In contrast to this behavior, Cheng et al. found incisions only on the furan-adduct strand of a 4′-hydroxymethyl-4,5′,8-trimethylpsoralen cross-linked DNA, even when there were G/C-rich sequences with 6–12 nucleotides 5′ to this lesion.117 These results led to the suggestion that the observed strand specificity of incision is a function of the psoralen 4′-hydroxymethyl group, which enhances the asymmetry between the furan and pyrone sides of the 4′-hydroxymethyl-4,5′,8-trimethylpsoralen cross-link.117
Results from studies by Munn and Rupp suggested that psoralen cross-linked DNA may undergo a conformational change before the initial incision by UvrABC can occur.121 These experiments showed that a complex of UvrA and UvrB bound equally well to linear plasmid DNA that contained either a 4′-hydroxymethyl-4,5′,8-trimethylpsoralen furan-side monoadduct or an interstrand cross-link. However, the efficiency of incision when UvrC was added was 30% after 3.3 min for the monoadduct but only 4% for the cross-link. When closed circular plasmid DNA carrying a single furan-side monoadduct was tested, the efficiency of the incision reaction was found to be essentially independent of the superhelical density. In contrast, incision efficiency on closed circular plasmid DNA carrying a single interstrand cross-link increased in direct proportion to the superhelical density. These results suggested that the UvrA/UvrB preincision complex may partially unwind the cross-linked DNA before incision occurs.
Similar results were obtained with a covalently closed circular plasmid that contained a single N-methylmitomycin A (32, Figure 16) interstrand cross-link.122 Similar to mitomycin C, N-methylmitomycin A forms an interstrand cross-link by alkylating the N2-exocyclic amino groups of guanine in a 5′-CG-3′ sequence. Unlike the psoralen inter-strand cross-link, which resides in the major groove and creates local distortions of the DNA helix, the mitomycin interstrand cross-link resides in the minor groove and produces little or no distortion of the helix. The rate of UvrABC incision of the supercoiled form of the mitomycin cross-linked plasmid DNA was found to be approximately 200 times greater than that of the relaxed form of the cross-linked plasmid. In contrast, the rate of incision on supercoiled plasmid DNA that contained N-methylmitomycin A monoadducts was only twice that of the relaxed form of the plasmid.
Figure 16.
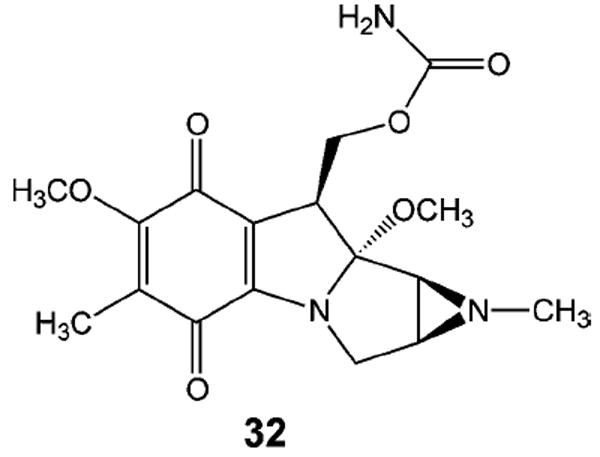
N-Methylmitomycin A.
These observations and those showing that UvrABC recognizes and cuts a wide variety of chemically and structurally diverse adducts123 led Pu et al.122 to propose that the UvrAB complex, which is believed to be responsible for damage recognition, may sense lesion-imposed conformational restrictions on DNA. In this model, the UvrAB complex would induce conformational changes in the DNA upon binding. If no lesion were present, normal binding interactions would take place and the complex would translocate or dissociate along or from the DNA. If, on the other hand, the lesion restricted the UvrAB complex-induced conformational change, this would in effect trap the complex and serve as a signal for further processing of the lesion. Thus, even if the lesion does not significantly perturb the structure of the DNA, as is the case with the mitomycin interstrand cross-link, it can still be recognized if it restricts the “flexibility” of the DNA. Thus, structural distortions per se are not recognized, but rather impediments to dynamic motions in the DNA signal the presence of the damage.122-124 Consistent with this proposed recognition model is the observation that UvrA and the UvrAB complex both exhibit DNA winding and helicase activities.125,126
More recent experiments have suggested the existence of an additional pathway for the repair of psoralen and mitomycin interstrand cross-links.127 In these studies, psoralen or mitomycin interstrand cross-links were shown to be lethal in E. coli mutants that lack UvrB but not in UvrA UvrC double mutants. This observation led the authors to suggest that UvrB plays a role in recognizing the interstrand cross-link. However, the other proteins required for processing or repair of the damage have not been identified.
Psoralen interstrand cross-links in E. coli are repaired primarily if not exclusively by a mechanism that involves NER and homologous recombination. It appears that at least two distinct pathways can be used to repair nitrogen mustard interstrand cross-links. Loechler and co-workers studied the repair of mechlorethamine interstrand cross-links in E. coli. The nitrogen mustard cross-link was inserted into a 5′-GAC-3′/5′-GTC-3′ sequence in a short DNA duplex by reacting the duplex with mechlorethamine.49 The cross-link was stabilized by treating the cross-linked duplex with base, which converted the N7-alkylated guanines to their formamidopyrimdine derivatives. The cross-linked duplex was then ligated into a SV40-based human shuttle vector, which contained an ampicillin resistance gene, to give plasmid DNA that contained a single interstrand cross-link.128 The cross-linked plasmid DNA was transfected into E. coli, and the replication efficiency, defined as the ratio of the number of colonies formed by cross-linked plasmid versus the number of colonies formed by noncross-linked plasmid, was determined.129,130
The results showed that the cross-linked plasmid was able to replicate and was therefore repaired with essentially the same efficiency in wild-type cells and in cells that contained a recA deletion. This result was not unexpected because (1) the plasmid DNA was not homologous to E. coli DNA and (2) the cells were transfected only with cross-linked plasmid. Thus, under these conditions, RecA-mediated homologous recombination is not possible. When E. coli cells were treated with mechlorethamine, cell survival was significantly reduced in the recA deletion mutant versus wild-type cells.130 These experiments point to the participation of homologous recombination in cross-link repair when homologous DNA is available.
The replication efficiency of the nitrogen mustard cross-linked plasmid was significantly diminished in cells that lacked functioning UvrA and UvrB, components of the NER pathway.129 Other possible repair pathways did not seem to be involved, however. For example, replication efficiency in mutants that lacked BER activities, including formamidopyrimdine DNA glycosylase, 3-methyladenine glycosylase, or DNA deoxyribophosphodiesterase, was found to be very similar to that in wild-type cells. Replication efficiency was also not reduced in cells that lacked the 5′ → 3′ exonuclease activities of DNA polymerase I, exonuclease VII, or RecJ.
These results suggested that repair of the nitrogen mustard cross-linked plasmid involved components of the NER pathway coupled with translesion synthesis. Experiments carried out in a deletion mutant, ΔpolB, that lacks DNA polymerase II activity showed a significant decrease in replication efficiency.130 This observation suggests that DNA polymerase II, a DNA polymerase implicated previously to play a role in DNA repair, serves as the translesion polymerase in this pathway. Its role in this pathway appears to be physiologically relevant because cell survival is diminished in a ΔpolB E. coli strain treated with mechor-ethamine.130
The picture that emerges then is one in which nitrogen mustard interstrand cross-links can be repaired by one of two parallel but nonredundant pathways. Both pathways require components of the NER pathway to make incisions on either side of the interstrand cross-link. In the homologous recombination-dependent pathway, homologous recombination in effect supplies an undamaged oligonucleotide to fill the gap created by incision reactions and thus replaces one of the damaged strands. In the other pathway (Figure 17), the gap is filled by DNA polymerase II-mediated translesion synthesis using the other damaged strand as a template. In both pathways, the remnant cross-link lesion can be removed by a second round of NER.
Figure 17.
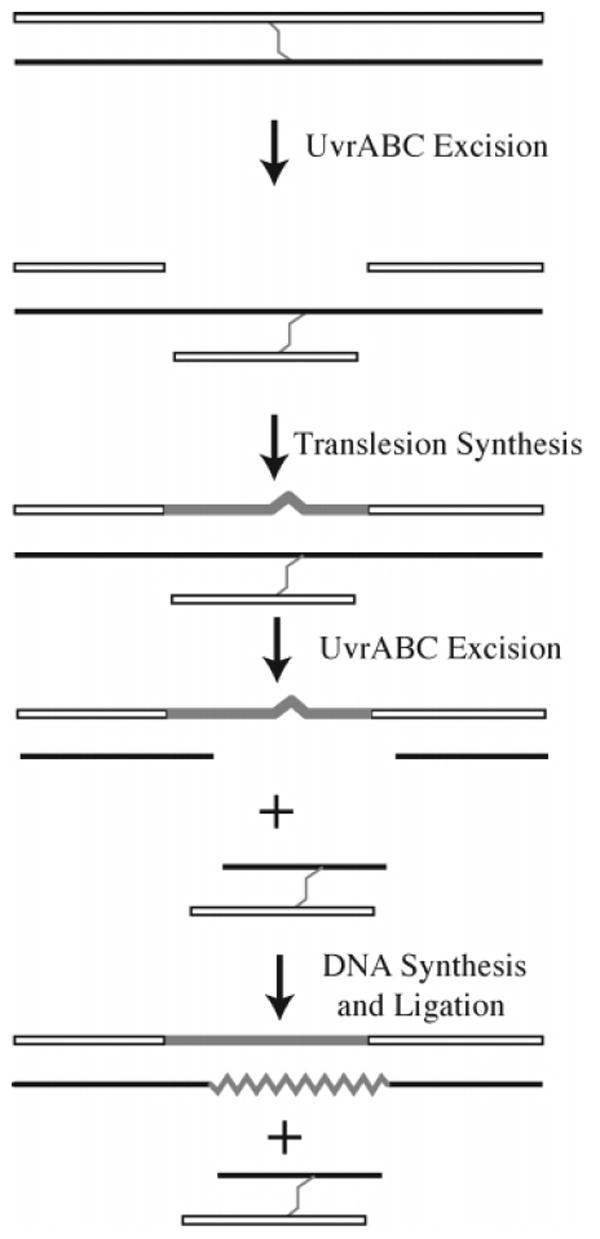
Nitrogen mustard interstrand cross-link repair in E. coli.
The NER/Pol II pathway does not appear to be involved with the repair of psoralen interstrand cross-links.129,131 This led Loechler and co-workers to suggest that the structure of the interstrand cross-link may dictate the pathway that is used to repair the cross-link.129 They point out that the psoralen interstrand cross-link is much more rigid than the relatively flexible nitrogen mustard cross-link. Recent studies on the repair of a single N4C-ethyl-N4C interstrand cross-link (33, Figure 18) in plasmid DNA would seem to support this conjecture.132 In these studies, the cross-link was placed in a 5′-CG-3′ or 5′-GC-3′ sequence. Assays in which plasmids carrying a single copy of the N4C-ethyl-N4C interstrand cross-link were transfected into E. coli showed that as in the case of the nitrogen mustard cross-link, replication efficiency was similar in wild-type and RecA-deficient cells. Replication efficiency was significantly reduced but not eliminated in a UvrA defective mutant, suggesting that an additional pathway not involving homologous recombination or NER might be involved. Interestingly, the relative replication efficiency of the plasmid with the 5′-GC-3′ cross-link was approximately twice that of the 5′-CG-3′ cross-link. Physical studies showed that the 5′-CG-3′ cross-link induces little or no distortion in the DNA helix, whereas the 5′-GC-3′ cross-link induces a significant distortion and most likely increases the flexibility of the helix at the site of the cross-link. This increased flexibility may account for the greater efficiency of repair as suggested in the nitrogen mustard cross-link repair studies.
Figure 18.
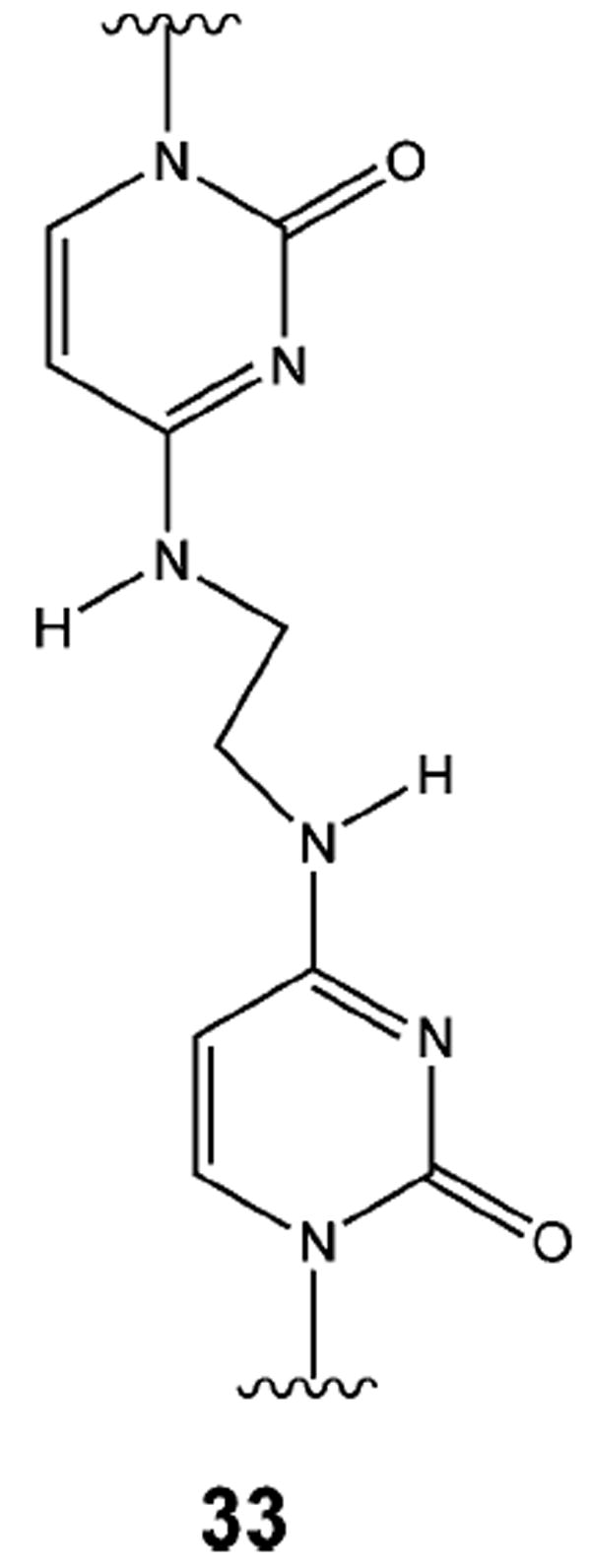
N4C-Ethyl-N4C interstrand cross-link.
3.2. Repair in Eukaryotes
The repair of interstrand DNA cross-links in eukaryotic cells is considerably more complicated than the repair in prokaryotic cells, and as such, significant aspects of cross-link repair remain in question. As described below, despite the complexities and outstanding questions regarding cross-link repair in higher organisms, it appears that the repair roughly fits into the two broad categories, one involving homologous recombination in an error-free process and the other involving an error-prone process dependent upon lesion bypass.
3.2.1. S. cerevisiae
Genetic and biochemical studies in S. cerevisiae have contributed considerably to our understanding of the repair of eukaryotic DNA repair. Early genetic studies in yeast identified numerous mutants that were sensitive to various types of DNA damage (e.g., UV-induced damage and ionizing radiation). These mutants were grouped together into broad categories (called epistasis groups) based upon the ability of one gene to suppress the effect of a second gene. Three epistasis groups were identified and have been subsequently shown to play a role in the repair of DNA interstrand cross-links in yeast cells. These epistasis groups, not surprisingly, correspond with NER (RAD3 epistasis group),133 homologous recombination (RAD52 epistasis group),134 and the translesion DNA synthesis subpathway of postreplication repair (RAD6 epistasis group).133,135
Recently, it was demonstrated that the simultaneous disruption of one gene from each of these epistasis groups was sufficient to render yeast cells incapable of surviving the presence of a single interstrand cross-link (Figure 19). Wild-type or repair-competent S. cerevisiae are able to withstand the cytotoxic effects of having approximately 120 psoralen interstrand cross-links or 200 cis-DDP DNA inter-strand cross-links;136-140 however, this repair capacity can be lowered and ultimately eliminated by disrupting the REV3, SNM1, and RAD51 genes. The pairwise disruption of these genes produced strains that were more sensitive than any of the single mutants to the presence of DNA interstrand cross-links, with the triple mutant displaying an even greater sensitivity in which a single interstrand cross-link is lethal.140 Given the role of NER, homologous recombination, and postreplication repair in bacterial repair of DNA interstrand cross-links, it is not surprising to learn that the SNM1, REV3, and RAD51 are epistatic with these repair processes in S. cerevisiae. Thus, repair processes in which REV3, SNM1, and RAD51 function appear to represent the entire interstrand cross-link repair capacity of S. cerevisiae (Figure 19).140
Figure 19.
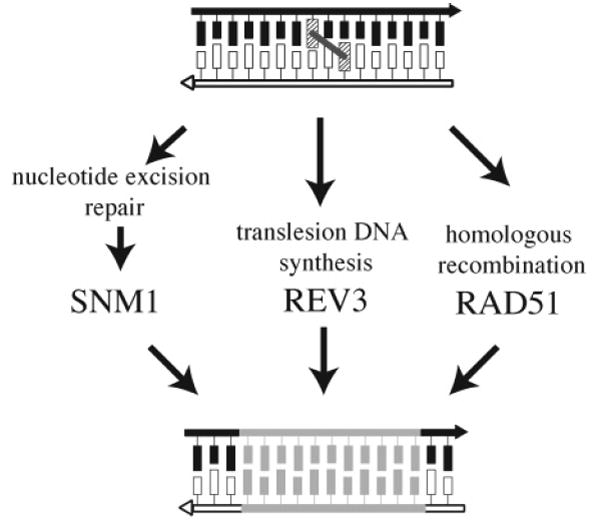
Interstrand cross-link repair pathways in S. cerevisiae.
Each of these three genes had been identified earlier as playing a role in the repair of interstrand cross-links, and two of the three had been identified previously in early yeast genetic studies as sensitive to UV and/or ionizing radiation. REV3, the catalytic subunit of polymerase ζ, a nonessential DNA polymerase that in complex with the Rev7p can efficiently replicate past a cis-syn cyclobutane dimer in an error-prone process,141,142 is a member of the RAD3 epistasis group. RAD51 is a member of the RAD52 epistasis group and encodes the yeast homologue of the bacterial RecA protein. SNM1, unlike REV3 and RAD51, does not appear to play a role in noninterstrand cross-link repair, rather SNM1 (also known as PSO2) was identified in a screen for mutants that were specifically sensitive to interstrand cross-linking by psoralen.143,144 While SNM1 does not participate in the NER of monoadducts, it has been shown to function downstream of the NER proteins in interstrand cross-link repair processes,133 thus making it epistatic with members of the RAD3 epistasis group in regard to interstrand cross-link repair. It is interesting to note that REV3 (or PSO1) was also identified as a mutant specifically sensitive to interstrand cross-linking by psoralen.143,144 As is the case in much of the literature regarding the repair of interstrand DNA cross-links, it is important to note that other groups have reported different epistatic relationships between SNM1 and NER, homologous recombination, and REV3.133,145 Indeed, recent work suggests that the epistatic relationship between SNM1, RAD52, and RAD4 may even be cell-cycle-dependent.146
The ability to manipulate the genetics of S. cerevisiae has facilitated a growing understanding of the plasticity of interstrand cross-link repair and, in particular, the capacity of these repair processes to compensate for one another. Studies that have examined the contribution of individual repair processes to the aggregate repair capacity of yeast cells can be divided into two categories: (1) studies in which cell survival in the presence of DNA interstrand cross-linking agents is measured and (2) studies in which the fate of plasmids containing single defined interstrand cross-links (i.e., psoralen cross-links) is followed.
In a recent study, the cytotoxicities of four interstrand cross-linking agents, cis-DDP, mechlorethamine, mitomycin C, and bischloroethylnitrosurea (BCNU), were determined in S. cerevisiae strains deficient in NER (RAD1Δ), homologous recombination (RAD52Δ), translesion synthesis (REV3Δ) and pairwise combinations of these deletions.147 When the cytotoxicity of these different agents was directly compared, it was observed that different interstrand cross-links rely upon different combinations of pathways. For instance, the RAD52Δ REV3Δ double mutant was the most sensitive to cis-DDP, mitomycin C, and BCNU; however, nitrogen mustard cytotoxicity was greatest in the REV3Δ RAD1Δ strain. Studies such as these can provide useful information regarding the overall repair capacity of cells; however, analysis of this type can be complicated by the fact that many of these cross-linking agents form monoadducts whose presence might interfere with the analysis.
These limitations can be avoided by studying the repair of plasmid substrates containing site-specific interstrand cross-links. These studies have been principally carried out using single psoralen cross-links located in a region of the plasmid that is homologous to a sequence in the yeast chromosome. These plasmid substrates have proven useful tools for examining the contribution and overlap of the NER, homologous recombination, and postreplication repair processes.148,149 In these studies, yeast cells are transformed with cross-linked plasmid DNA and the repair of the cross-link is determined by comparing factors such as survival and/or chromosomal integration to those in either undamaged plasmid DNA or plasmid DNA carrying a related type of damage, oftentimes a double-strand break or a psoralen monoadduct. When yeast strains are transformed in which various genes suspected of playing a role in the repair of interstrand cross-links have been deleted or disrupted, it is possible to characterize the contribution of these genes to the repair of interstrand cross-links. For example, the deletion of the genes belonging to the translesion DNA synthesis pathway (e.g., RAD5, RAD6, and RAD18) eliminated mutations associated with the error-prone repair of interstrand cross-linked DNA via translesion DNA replication. However, disruption of this pathway did result in an increased level of recombination.148 Preventing yeast cells from processing interstrand cross-links using the translesion apparatus reduced the mutational frequency and thus led to an increase in recombination, presumably by forcing the cross-link to be repaired by recombination.148 Similarly, deletion of genes in the NER pathway (e.g., RAD1, RAD3, RAD4, and RAD10) or homologous repair pathway (e.g., RAD51, RAD52, RAD55, and RAD57) resulted in increased mutation frequencies, presumably by channeling more of the cross-link repair into error-prone postreplication repair processes. The deletion of NER genes also produced lower levels of cross-link-induced recombination, suggesting that some of the cross-links processed by the NER apparatus are subsequently channeled into the homologous recombination repair pathway.
Efforts to characterize the biochemical steps in interstrand cross-link repair in S. cerevisiae are not nearly as comprehensive as those for E. coli. The earliest studies that followed the physical consequences, as opposed to the genetic consequences, of interstrand cross-link damage primarily examined the effect that psoralen interstrand cross-linking had on chromosomal DNA. These early studies relied upon equilibrium density centrifugation to monitor the fate of psoralen interstrand cross-links. DNA that has interstrand cross-links undergoes rapid renaturation when compared to noncross-linked DNA; thus, the kinetics of converting cross-linked DNA to noncross-linked DNA can be followed by monitoring the conversion of double-stranded DNA to single-stranded DNA.139,150-152 Wild-type cells treated in the exponential phase were able to completely convert rapidly, renaturing cross-linked DNA to noncross-linked DNA in approximately 2 h. This step has often been referred to as “unhooking” and at a minimum requires that the interstrand DNA cross-link be excised from one of the damaged strands. Unhooking was accompanied by the appearance of single-and double-strand breaks.139,152-154
From work done in yeast as well as in mammalian cells, there is an emerging consensus that in rapidly growing eukaryotic cells the formation of cross-linked induced DNA strand breaks and the unhooking of the cross-linked DNA strands occurs at or near the site of a stalled or collapsed replication fork (Figure 20). These one-ended DNA strand breaks are often referred to as a double-strand break but differ from classical double-strand breaks that arise in noninter-strand cross-linked DNA. The cross-link-induced DNA strand break (CID) depicted in Figure 20 can be formed independently of the unhooking reaction that results in the formation of the slowly renaturing single-stranded DNA, observed in early biochemical studies in yeast.
Figure 20.
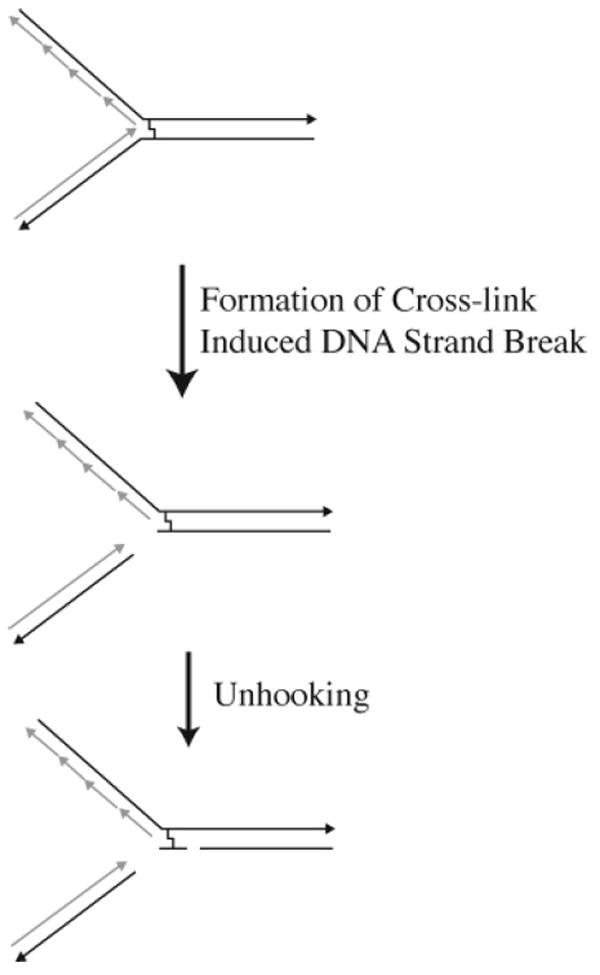
Cross-link-induced DNA strand breaks.
The relationship between the formation of CIDs and the unhooking of the DNA cross-link continues to be unclear. Evidence shows that the formation of CIDs when psoralen is the cross-linking agent is dependent upon the function of NER proteins;139,152-154 however, when nitrogen mustard is used as the cross-linking agent, the formation of CIDs occurs in the absence of NER proteins.146,155,156 Early observations that the formation of CIDs and unhooking of psoralen interstrand cross-links requires the activity of members of the NER pathway139,150,151 has led to considerable confusion within the field of interstrand cross-link repair. As described above, the use of equilibrium centrifugation clearly demonstrated the absolute dependence of NER proteins on both the unhooking of psoralen interstrand cross-links and the formation of CIDs.139,150-152 This led to the general notion that the unhooking of cross-linked DNA and the formation of DNA strand breaks resulted as a function of NER components. However, S. cerevisiae cells treated with nitrogen mustard give rise to CIDs that occur in the absence of functional NER proteins (i.e., RAD1, RAD2, and RAD4).146,155,156 The formation of CIDs in the absence of NER proteins has also been observed with nitrogen mustard, mitomycin C, and cis-DDP in mammalian cells.157,158 These observations suggest that the NER-dependent formation of CIDs may not be a general feature of interstrand cross-link repair but rather is specific to psoralen interstrand cross-links.
It seems likely that the formation of cross-link-induced DNA strand breaks is one of the early steps in the repair of interstrand DNA cross-links in rapidly growing cells as opposed to stationary phase cells. When the size of DNA fragments are monitored using equilibrium centrifugation, the reassembly of these DNA fragments was shown to be complete within approximately 2 h.139 The repair of these DNA strand breaks was found to be dependent upon not only homologous recombination (RAD51)152 but also upon the activity of SNM1.139,159,160 A role for RAD51 in the repair of DNA strand breaks is not surprising, because that is precisely the role that it plays in the repair of ionizing radiation induced DNA double-strand breaks.134 The mechanism by which SNM1 functions in rejoining DNA interstrand cross-link-induced strand breaks is still unknown. SNM1 was first identified in screens for mutants that were sensitive to the formation of psoralen and nitrogen mustard interstrand DNA cross-links.143,161,162 As described above, SNM1 is epistatic with genes of the NER pathway (specifically RAD1, RAD3, RAD4, and RAD10) in regard to the repair of DNA interstrand cross-links,143,146 but it demonstrates wild-type resistance to monofunctional alkylating agents and ionizing radiation.161 While the mechanistic role of SNM1 in the NER-dependent repair of DNA interstrand cross-links is unclear, it is known to function following the unhooking reaction.
The Snm1 protein is a member of the β-CASP metallo-β-lactamase superfamily of enzymes that includes the human paralogue, Artemis.163 Artemis is the gene that is mutated in radiation-sensitive severe combined immune deficiency (RS-SCID), a disease that results from defects in V(D)J recombination.164 The Artemis protein is a single-strand DNA-specific 5′ → 3′ exonuclease, and it has been suggested that the yeast paralogue, Snm1p, may have a nucleolytic role in the processing of DNA interstrand cross-links.165 Recent work by McHugh and colleagues showed that the function of Snm1p overlaps that of Exo1p, a 5′ → 3′ exonuclease and flap endonuclease involved in recombination, double-strand break repair, and DNA mismatch repair, and Msh2p, which forms heterodimers with Msh3p and Msh6p, which bind to DNA mismatches to initiate the mismatch repair process.146,166,167
The role of translesion DNA synthesis in the repair or tolerance of DNA interstrand cross-links is the least well-characterized of the interstrand cross-link repair processes. The RAD6 postreplicative repair pathway is largely involved in the repair/tolerance of UV-induced DNA damage and other types of DNA damage that block replication. Postreplication repair involves a number of different proteins and represents a complicated set of repair pathways, including both error-prone and error-free processes. The principle components known to be involved in the postreplication repair of DNA interstrand cross-links include RAD5, RAD6, RAD18, and REV3 and comprise one of the error-prone pathways of postreplication repair.
The frequency of induced mutation at the site of a defined psoralen interstrand cross-link is quite high in repair-proficient yeast cells (~5%).148 The disruption of the homologous recombination pathway increased the observed mutation frequency by approximately 4-fold, while the disruption of RAD5, RAD6, or RAD18 resulted in a decrease in mutations.148,168 RAD6 encodes a ubiquitin-conjugating enzyme that plays a role in signaling for proteosomal degradation via polyubiquitination169,170 and monoubiquitination of histones.171 The DNA repair function of Rad6p is facilitated by a strong interaction with Rad18.172,173 Rad18p, unlike Rad6p, interacts with single-stranded DNA and appears to be responsible for targeting the Rad6p/Rad18p complex to the site of DNA damage.174 Rad5p also shows affinity for single-stranded DNA and is known to interact with Rad18p in vitro.175 The exact role that these protein complexes play in translesion DNA synthesis is unclear, although it has been suggested that it allows recruitment of polymerase ζ, which is encoded by REV3 and REV7 (the catalytic and regulatory subunits, respectively), to the site of a stalled replication fork.172
REV3-dependent error-prone repair of DNA interstrand cross-links is linked to the cell cycle.143,155 The disruption of REV3 renders stationary-phase cells much more sensitive to damage by nitrogen mustards than cells undergoing exponential growth.155 These studies suggest that when cells are held in G1 phase, where there is no sister chromatid to allow for homologous recombinational repair pathways, the error-prone REV3-dependent lesion bypass is important. This repair process would be predicted to be less important in cells that have passed through S and G2 phases and thus have a homologous sequence present within the cell. As with many aspects of the repair of interstrand cross-links in eukaryotes, the role of translesion DNA synthesis in yeast is more complicated than simply playing a role in only the error-prone repair. While REV3 disruptants were more sensitive in stationary phase than in exponentially growing cells, disruption of RAD6 and RAD18 showed sensitivity in both phases.155 This suggests that some of the other error-free postreplication repair subpathways that do not rely upon REV3 also play a role in the repair of interstrand DNA cross-links.
As described in the following section on DNA interstrand cross-link repair in mammalian cells, there are many conserved elements in the repair of DNA interstrand cross-links between S. cerevisiae and higher eukaryotes (e.g., mammalian cells); however, there are also some significant differences. It is important to bear in mind that these differences may be less dramatic than is typically thought. The majority of the studies described above have utilized haploid strains of yeast that contain a single set of chromosomes (i.e., half of the full set of genetic material). The response of yeast to DNA damage is oftentimes different in haploid cells compared to diploid yeast.176 For instance, the loss of proofreading activities of DNA polymerases δ and ∈ is lethal in haploid cells but not in diploid cells;177 the loss of DNA polymerase proofreading coupled with mismatch repair deficiency is lethal in haploid cells but tolerated in diploids;178 and diploids are significantly more resistant to γ radiation than are haploid cells.179 Indeed, increasing numbers of chromosomal copies (the number of copies of chromosomes can be manipulated in yeast to produce haploid, diploid, and higher ploidy numbers) result in increased resistance to treatment with the cross-linking agent psoralen180 and with nitrous acid,181 a compound capable of producing interstrand DNA cross-links.182-185 Thus, some of the differences observed between haploid yeast strains and diploid mammalian cells may not necessarily reflect fundamental differences in the repair processes.
3.2.2. Mammalian Cells: Genetic Studies
The majority of studies on the repair of interstrand cross-links in mammalian cells have been carried out using different assays, different substrates, and different cell lines. Consequently, it is difficult to compare directly the results from different experiments, and indeed, at times, the results from these different studies appear to be contradictory. In the following sections, we have attempted to summarize the observations derived from these studies. However, we feel that it is still premature to attempt to derive definitive mechanisms for the repair of interstrand cross-links in mammalian cells based on these observations.
In studying the repair of interstrand cross-links in mammalian cells, researchers have taken cues from studies carried out in yeast. Generally, in cell-based assays, the involvement of repair pathways or specific proteins is deduced through the systematic elimination of protein players. This is achieved by using human tumor cell lines that are know to be deficient in a particular protein or Chinese hamster ovary (CHO) cell lines that have been identified as deficient in NER and/or homologous recombination proteins. There are three main types of cellular assays: (1) assays that monitor sensitivity/ cross resistance, (2) assays that look at changes in protein expression or increases in protein density at nuclear foci following the insult with an interstrand cross-linking agent, and (3) assays that analyze post-treatment evidence of repair, including determination of chromosomal breakage/interstrand cross-linking, repair synthesis, and/or host-cell reactivation of a reporter gene. While these studies are important and suggestive, they cannot unequivocally determine if the measured response is exclusively due to interstrand cross-links. Thus, although lesions formed by compounds such as psoralen and mitomycin C are primarily interstrand cross-links, other agents commonly studied, such as nitrogen mustards and cis-DDP, form a small percentage of interstrand cross-links relative to the total number of lesions produced. This makes studies with specifically prepared interstrand cross-linked substrates indispensable for interpreting data acquired through genetic studies.
Cytotoxicity assays such as those performed by Bramson et al.43 were helpful in elucidating the causes of innate or acquired resistance to chemotherapeutics used in treating cancer. In these studies, B-cell chronic lymphocytic leukemia (B-CLL), which is known to develop resistance to nitrogen mustards, was chosen as a model to study resistance to this class of alkylating agents. Analysis showed no cross-resistance to UV-damaged DNA, a substrate for NER, or methyl methanesulfonate-damaged DNA, a substrate for BER, a result that indicated a lack of exclusive involvement of these pathways in the repair of nitrogen mustard inter-strand adducts. Through this and other studies, which were designed to monitor changes in sensitivity to cross-linking agents following overexpression of DNA repair proteins, it was determined that the repair of nitrogen mustards most likely involves an incision–recombination–incision set of repair events.43
Other variations on this type of survival study used Western blotting to examine up- or downregulation of repair proteins following treatment with a cross-linking agent. For example, Aloyz et al.186 noted that several DNA repair genes, including those involved in NER were implicated in the development of cis-DDP resistance. This study, which used the National Cancer Institute’s tumor cell line panel, showed that overexpression of XPD protein imparted a 2–4-fold increase in cis-DDP resistance and was accompanied by an increased Rad51 nuclear foci density and by sister chromatid exchanges, thus indirectly implicating homologous recombination in the repair process.186
Western blotting is used to monitor changes in cellular protein levels, while changes in protein density at sites of DNA damage can be determined by staining cultured cells with fluorescently labeled antibodies that recognize the proteins under investigation. For example, Wang and coworkers187 used these techniques to examine the levels and location of repair proteins in 14 human cell lines after treatment with melphalan, a nitrogen mustard. They showed that the level of XRCC3 was elevated in these cells, whereas the levels of RAD51, RAD52, KU70, KU86, and DNA-PK were unaffected. The unchanged levels of KU70, KU86, and DNA-PK led the authors to conclude that nonhomologous end joining (NHEJ) is not a potential repair pathway. Immunostain examination of RAD51 foci formation in eight cell lines with variable resistance to melphalan showed an increased density of RAD51 foci in both sensitive and resistant cells. Because the levels of RAD51 remained unchanged in these cells, the results suggested a redistribution of cellular RAD51 protein in response to the melphalan-mediated DNA damage. Similar experiments by Nan Lui also showed a defect in the formation of RAD51 foci in XRCC2 null CHO cells following treatment by mitomycin C.188 Again, the defect in the formation of nuclear foci did not correspond to reduced levels of the RAD51 protein but rather a redistribution of cellular stores after damage. These and other experiments have consistently shown that RAD51 is localized at sites of damage induced by psoralen, mitomycin C, and melphalan,47,189,190 results that strongly implicate the involvement of homologous recombination in interstrand cross-link resistance and repair.
Neidernhofer et al. examined nuclear foci formation to determine if the processing of interstrand cross-links results in the formation of CIDs and whether the XPF/ERCC1 endonuclease complex is required.158 This was done by immunostaining for the phosphoepitope of γ-H2AX protein at foci in mitomycin C-treated ERCC1 null cells. γ-H2AX is the phosphorylated form of histone H2A and binds to DNA that contains double-strand breaks. It is one of the earliest proteins detected at sites of DNA damage. Although mitomycin C did mediate an increase in the number of γ-H2AX foci, the appearance of CIDs was not caused by the endonuclease activity of XPF/ERCC1. However, it was subsequently determined that ERCC1 was necessary for the resolution of the breaks, a step that is distinct from CIDs in this proposed model.
The presence of additional factors in nuclear foci was shown by Pichierri and co-workers in experiments with Fanconi’s anemia (FA) cells.191 These cells are hypersensitive to treatment with interstrand cross-linking agents. Wild-type cells were found to respond to psoralen or mitomycin C treatment by forming foci that include a RAD50/MRE11/ NBS1 complex, which has recently been shown to function as a double-strand break sensor.192 Because a yeast mre11 strain showed sensitivity to interstrand cross-linking agents, Mre11p, which has double-stranded DNA exonuclease and single-stranded DNA endonuclease activities, has been suggested to play a role in interstrand cross-link repair in yeast and mammalian cells. FA cells were unable to activate Rad50p/Mre11p/Xrs2p focus formation when treated with mitomycin C or psoralen. This deficiency was not due to an inability to respond to double-strand breaks because normal foci were seen when double-strand breaks were induced by ionizing radiation. It was also determined that the defect was not dependent upon cross-link unhooking or incision by the XPF/ERCC1 complex. These observations lead to the hypothesis that formation of the Rad50p/Mre11p/Xrs2p complex at the site of damage is an important contributor to interstrand cross-link repair and that a factor absent in FA cells contributes to the assembly of the RMN/DNA complex. Although there is no evidence showing that FA proteins remove interstrand cross-links, they appear to be central components in the cellular response to DNA cross-linking agents.193-195
Another repair pathway, translesion synthesis, or lesion bypass has been examined by looking for nuclear foci involving DNA polymerases. It had already been established that when stalled replication forks occur, because of the presence of UV damage or bulky adducts, a group of special DNA polymerases can act on the replication complex to bypass the lesion. These polymerases, including Pol ζ, Pol η, Pol ι, and a more recently discovered Pol κ, participate in the replication of damage that might not otherwise be tolerated by the cell.196,197 A study by Bergoglio and coworkers looked at the formation of nuclear foci containing Pol κ in MRC5 (human lung fibroblasts) and XP12ROSV40 (XP-A) cells transfected with Pol κ in response to treatment with cis-DDP.198 The Pol κ was not immunostained as outlined in previous studies but was tagged with green fluorescent protein. These Pol κ foci were seen to colocalize with PCNA, which was detected by immunostaining. Previous studies had shown the Pol κ was unable to perform translesion synthesis opposite a cis-DDP intrastrand adduct in vitro.199 However, the authors point out that, in this study, the PCNA served as a target for the localization of Pol κ, as has been shown for Pol δ and Pol ∈ and as had been suggested for Pol ζ and Pol η. The 2-fold overexpression of Pol κ was also responsible for a 7–9-fold increase in spontaneous mutagenesis, indicating an involvement in an error-prone pathway of repair.198
A considerable number of genetic studies of the repair of interstrand cross-links involve some form of postprocessing of the cellular DNA to ascertain the fate of the cross-link. Vos and Hanawalt developed an assay that can be used to detect interstrand cross-links in cellular DNA.200 Using this assay, they determined the repair efficiency of psoralen interstrand cross-links and psoralen monoadducts in an actively transcribed dihydrofolate reductase (DHFR) gene. Recovered cellular DNA was denatured and electrophoresed on a neutral agarose gel. Under these conditions, psoralen cross-linked DNA will renature and migrate as double-stranded DNA, whereas DNA with psoralen monoadducts remains denatured and migrates as single-stranded DNA. The DHFR gene was then identified by Southern blotting. After 24 h, 80% of the introduced interstrand psoralen cross-links were removed, whereas only 45% of the monoadducts were removed.
Larminat and Bohr used this assay to study the effect of cis-DDP treatment on the actively transcribed DHFR gene in wild-type and ERCC1 mutant CHO cells.201 Survival of the ERCC1-deficient cells was greatly reduced after minimal dosing with cis-DDP, and these cells were able to remove only a small percentage of the interstrand cross-links. Furthermore, the replication activity of cellular DNA in ERCC1 mutant cells was less than half that measured in wild-type cells. When the ERCC1-deficient cells were complemented by stably transfecting with ERCC1, the amount of replication was partially restored. This shows a clear involvement of ERCC1 and presumably its XPF counterpart in the repair of cis-DDP cross-links.
In an attempt to circumvent resistance to cis-DDP, Farrell and co-workers have developed several multinuclear platinum compounds (6a and 6b) that demonstrate improved potency even in cis-DDP-resistant cell lines.202 Paradoxically, Colella found that overexpression of ERCC1, the protein that when in complex with XPF is responsible for the incision of damage in NER, actually sensitizes the cells to lesions of 6b. This occurs presumably because “… a greater number of DNA lesions are induced than other enzymes in the pathway can repair”.203
The Comet assay204 and pulsed field gel electrophoresis (PFGE)157,205 are two methods commonly used to monitor the processing of interstrand cross-links. In the Comet assay, cells that have been exposed to the cross-linking agent are further irradiated with X-rays, a process that induces random strand breaks in the DNA. The irradiated cells are then lysed in situ in an agarose gel plug and electrophoresed under alkaline conditions. This produces a comet-shaped streak of DNA. The length of the comet tail is an indicator of the extent of DNA damage because DNA that contains inter-strand cross-links migrates more slowly than denatured DNA that contains no cross-links. The extent of cross-link unhooking can then be determined by monitoring the length of the tail as a function of the time after treatment with the cross-linking agent. The Comet assay does not monitor the extent of CIDs. On the other hand, PFGE provides a much more rigorous measure of double-strand breaks.
Chinese hamster-derived cell lines deficient in NER proteins, XPB, XPD, XPF, ERCC1, or XPG, homologous recombination proteins, XRCC2 or XRCC3, or the nonhomologous end-joining protein, XRCC5, were exposed to increasing concentrations of the nitrogen mustard, mechlorethamine, and cell survival was determined relative to CHO wild-type cells.157 The XPF and ERCC1 mutants were 15 times more sensitive than wild-type cells, whereas XPB, XPD, and XPG mutants were less than 2 times as sensitive. As measured by the modified Comet assay, the XPF and ERCC1 mutants were both incapable of unhooking the interstrand cross-link, a preliminary step in cross-link processing. As expected, the wild type and XPB, XPD, and XPG mutants were all able to unhook the cross-link. These results suggested that XPF and ERCC1 were necessary for cell survival and cross-link unhooking, whereas the other NER proteins were dispensable. XRCC2 and XRCC3 mutants were 12 and 26 times more sensitive, respectively, than wild-type cells in the cell survival assay, and these mutants were able to unhook the interstrand cross-link, suggesting a role for homologous recombination in the repair of mustard-induced damage. The observation that XRCC5 mutants were not sensitive to mechlorethamine suggested that the NHEJ was not involved in the repair of this interstrand cross-link.
Exposure of wild-type CHO cells to mechlorethamine also produced CIDs as determined by PFGE.157 When monitored as a function of time, the cross-link-induced breaks disappeared, which indicated that repair of the cross-linked DNA had occurred. A similar repair was observed in the XPF and XRCC5 mutants but not in the XRCC2 and XRCC3 mutants. Together, these results suggested a DNA repair model157 in which a CID is produced when a replication fork stalls in the vicinity of the cross-link (Figure 20). XPF/ERCC1 is then involved in unhooking the cross-link, and homologous recombination facilitates further repair.
Similar studies were also carried out in CHO cells exposed to cis-DDP.205 Cell survival studies showed that, of the NER mutant cell lines tested, only XPF and ERCC1 cells showed increased sensitivity (40- and 37-fold, respectively) to the drug, while those deficient in XPB, XPD, and XPG showed a slightly (1.3–3.1-fold) increased sensitivity. Cell lines deficient in XRCC2 and XRCC3 demonstrated 50- and 38-fold increased sensitivity to cis-DDP treatment, while the NHEJ mutant, XRCC5, showed no increase in sensitivity. These results, which were similar to those with the nitrogen mustard, suggested that cell survival after cis-DDP treatment depended upon XPF/ERCC1 and proteins involved in homologous recombination.
As was the case with the nitrogen mustard, both XPF and ERCC1 mutants were unable to unhook the cis-DDP cross-link as determined by the modified Comet assay. Unexpectedly, both the XPG and XPD mutants, which were insensitive to drug treatment, were also unable to unhook the cross-link. Even more surprising was the observation that the XRCC3 mutant, which was hypersensitive to cis-DDP, was unable to unhook the cross-link. Thus, there was no clear correlation between cell survival and the ability to remove the cis-DDP interstrand cross-link. Furthermore, it would appear that unhooking and perhaps removal of the cross-link was dependent upon the full complement of NER proteins as well as those involved in homologous recombination.205
Interestingly, a separate study by Simpson and Sale showed that ΔRev1-DT40 chicken B-lymphocyte cells are 6.8-fold more sensitive to cis-DDP treatment that wild-type DT40 cells.206 This implicates another repair pathway, error-pone replication bypass in the processing of these types of adducts and supports the yeast studies that show a decreased tolerance for the effects of cis-DDP when the REV3 subunit is eliminated.140 Although the requirement for REV3 in translesion synthesis has been established in yeast, in vertebrates, the involvement of this repair pathway has only been inferred by mutational analysis after the repair of an interstrand cross-link. Therefore, this study supports the involvement of translesion repair in the tolerance of inter-strand cross-links of cis-DDP, and the authors suggested that it may be involved in conjunction with other pathways.206
It is not universally agreed upon whether a homologous recombination event precedes an incision event in interstrand cross-link repair or if a cross-linked-induced break is necessary to initiate repair events. Some researchers have looked for evidence of homologous recombination by examining genomic DNA after the treatment with a cross-linking agent. Sister chromatid exchanges are mediated by homologous recombination.207 Sasaki et al. recently looked for the disparity in the amounts of sister chromatid exchanges and other chromosomal aberrations such as single or isoch-romatid breaks and chromatid exchanges between wild type and various mutant DT-40 chicken B-lymphocyte cells that were treated with mitomycin C.208 In wild-type cells, treatment with mitomycin C resulted in a 2- and 100-fold increase in sister chromatid exchanges and chromosomal aberrations, respectively. The authors proposed that the frequencies of these events reflected a life-or-death decision pathway in which either CIDs or incision intermediates generated by XPF/ERCC1 are resolved through homologous recombination. In cases where homologous recombination is deficient, the repair pathway diverges to an alternate pathway that may depend upon the availability of alternative repair factors or the position in the cell cycle.208
The notion of a cell-cycle-dependent response is echoed in the work of Akkari and colleagues, in which human fibroblasts, synchronized at different times in the cell cycle, were pulsed with nonfatal doses of psoralen and observed for evidence of arrest and repair.209 A response to psoralen-induced DNA interstrand cross-links was not evident until cells entered S phase and began replication. This study supports the idea that a stalled replication fork acts as a distress signal for the initiation of repair but does not explain the response to a potential block of transcription.
3.2.3. Mammalian Cells: Mechanistic Studies
The studies described in the preceding section focused on evaluating the effects of cross-linking agents on cell survival and the fate of the cross-linked DNA. Because these agents, in addition to producing DNA interstrand cross-links, are also capable of producing other lesions in both DNA and proteins, it is not always clear if the effects observed are due solely to the interstrand cross-link. A number of groups have prepared DNA substrates that carry a single, well-defined interstrand cross-link. These have been primarily interstrand cross-links of platinum compounds, mitomycin C, and psoralen. The interaction of purified repair proteins with such substrates has been examined using the electrophoretic mobility shift assay (EMSA) or, in some cases, surface plasmon resonance (SPR). These substrates have also been used in studies to monitor the fate of the cross-link in mammalian cells and mammalian-cell extracts.
Studies on platinum cross-links have been mainly limited to comparisons between cis- and trans-DDP and their novel counterparts, the trans-bis/tris platinum compounds (6a and 6b).210,211 While it is well-known that 1,2 (GG) and 1,3 (GNG) intrastrand adducts can be recognized and excised by the NER pathway,212,213 no incision is seen for cis- or trans-DDP interstrand cross-links nor the interstrand cross-links formed by the multinuclear class of platinum compounds.214,215 In addition, the protein-binding profile of platinum interstrand cross-links is quite different. For example, the high mobility group (HMG1) protein binds to cis-DDP intrastrand adducts by a different mode than to interstrand cross-links. It has been shown that the severe structural distortion of the intrastrand cross-link elicits HMG1 protein binding, whereas binding to a cis-DDP interstrand cross-link was achieved via intercalation of aromatic residues of the protein into the previously occupied cytosine site across from adducted guanines.216 Conversely, there is very little recognition by the HMG1 protein of DNA containing trans-DDP or trans-bis/tris-Pt interstrand cross-links.217 HMG1 proteins are believed to contribute to the effectiveness of cis-DDP as an antitumor agent because HMG1 shields intrastrand adducts from processing by NER while concomitantly diverting the p53 tumor-suppressing protein from its intended binding substrate.218 RPA, a protein involved in NER, has a lower affinity for an interstrand cross-linked cis-DDP compared to a nonplatinated duplex,219 as does MutS, a protein involved in mismatch repair.220 Proteins from the NHEJ pathway, Ku70 and 86, exhibit a 50–80% decrease in binding to cis-DDP interstrand versus intrastrand cross-linked DNA. The lack of binding inhibits Ku-dependent DNA-PKcs activation by 75%, as measured by an activity assay. The differences in structure, i.e., more unwinding and less bending, are deemed responsible for these differences.221
Mechanistic studies on mitomycin C interstrand cross-link processing are quite limited. A gel-shift study by Warren et al. demonstrated that there are proteins in human and hamster cell nuclear extracts that bind specifically to oligonucleotide duplexes that contain a 5′-CG-3′ mitomycin C interstrand cross-link. Extracts from various cell lines deficient in NER proteins were tested. The characteristic DNA/protein complex disappeared when a hamster cell extract deficient in ERCCI was incubated with the cross-linked duplex, indicating ERCC1, and by implication XPF, binds to mitomycin C interstrand cross-link.222 In a separate study, Mustra and coworkers found a 2.2–2.7-fold higher affinity of the XPA protein for a mitomycin C interstrand cross-linked duplex compared to a noncross-linked substrate.223 XPA is believed to be involved in the early damage recognition/validation step of NER.
A host-cell reactivation assay was used to study the processing of a mitomycin C interstrand cross-link in human and Chinese hamster cells.224,225 Plasmid DNA was prepared that contained a single 5′-CG-3′ mitomycin C interstrand cross-link. The cross-link was positioned between the transcription initiation site of a cytomegalovirus promoter and the translation initiation site of a firefly luciferase reporter gene. Cells were transfected with site-specific cross-linked or noncross-linked plasmid controls. Transcription and subsequent expression of the luciferase gene, which is easily monitored with a luminometer, is therefore dependent upon the ability of the cell to repair the interstrand cross-link. Human and CHO mutants that were defective in the NER proteins XPA, XPC, XPD, XPF, ERCC1, and XPG were unable to repair the cross-link, as indicated by the very low levels of luciferase expression. Chinese hamster lung fibro-blast homologous recombination mutants, XRCC2 and XRCC3, on the other hand, were active. Because the plasmid DNA used in these studies lacked any significant homology to cellular DNA and because the XRCC2 and XRCC3 mutants were able to repair the cross-link, the results suggested that a recombination independent repair pathway involving NER was responsible. An important role for lesion-bypass Pol η in this pathway was also suggested, because XPV mutant cell lines deficient in this enzyme showed a decrease in the reactivation of the luciferase reporter. This pathway appears to be error-prone, because repaired plasmids recovered from the transfected cells contained point mutations at the site of the mitomycin C interstrand cross-link.
The majority of mechanistic studies on interstrand cross-link repair have been carried out using DNA substrates that contain psoralen interstrand cross-links. Jones and Wood found that DNA containing multiple 4′-hydroxymethyl-4,5′,6-trimethylpsoralen interstrand cross-links was a poor substrate for XPA, a NER protein that binds preferentially to UV-induced 6–4 photoproducts and is believed to be involved in damage recognition.226 Early studies by Lambert and co-workers showed the existence of a psoralen inter-strand cross-link-specific binding protein in chromatin-associated protein extracts derived from normal human lymphoblastoid cell lines.227 Similar extracts from a FA complementation group A cell line did not contain this binding protein, suggesting that the protein was involved in interstrand cross-link recognition and possibly repair. Later studies by this group employed a biotinylated 105-bp DNA duplex that contained a single 4,5′,8-trimethylpsoralen interstrand cross-link.228 The biotin group allowed the capture of the duplex by streptavidin-coated polyacrylamide beads. Experiments with chromatin-associated proteins derived from HeLa cell extracts showed that α-spectrin (α-SpIIΣ*), a component of a nuclear protein complex, bound specifically to the cross-linked duplex. Proteins from FA complementation groups A, C, and G were also shown to bind to the 105-base-pair DNA substrate. It was not clear if these latter proteins bound directly to the DNA or were bound to α-spectrin.228
Lambert and co-workers also examined the ability of cell extracts to make incisions in a 132-base-pair DNA duplex that contained a single 4,5′,8-trimethylpsoralen cross-link located near the center of the duplex.229 The duplex was incubated with chromatin-associated protein extracts derived from a human lymphoblastoid cell line. Incisions were observed at the 4th and 5th phosphodiester bonds on the 3′ side of the furan and pyrone adducts, respectively, and at the 5th and 6th, and 13th and 14th phosphodiester bonds on the 5′ side of the furan and pyrone adducts, respectively.
Further studies were carried out in extracts derived from Fanconi anemia cell lines to determine if incision deficiency contributed to the hypersensitivity of Fanconi anemia cell lines to interstrand cross-linking agents.230 Extracts from Fanconi’s anemia complementation A (FA-A) deficient cells exhibited a 70% decrease in incision on the 3′ side of the furan adduct and an 85% decrease in incision on the 5′ side of the pyrone adduct of the cross-link. Similar decreases in the 5′-side incision efficiency were observed in XPF-deficient extracts. Because FA-A extracts contain XPF, it was concluded that other factors must be involved in the incision of the psoralen interstrand cross-link. Protein-mapping experiments showed that ERCC1 was a necessary cofactor for the 5′ incision, which involved the N and C termini of the XPF protein, and that the extreme N and C termini of XPF were involved to only a limited extent in the 3′ incision.231
To determine if other factors were involved in the incision reaction, experiments were carried out in HeLa cell chromatin-associated protein extracts in the absence or presence of anti-α-SpIIΣ* antibody.228 α-SpIIΣ* is a scaffolding protein that may be regulated by FA proteins and is involved in the repair of DNA interstrand cross-links.232 As described above, binding experiments showed that α-SpIIΣ* had a higher preference for interstrand cross-linked DNA over noncross-linked DNA.228 Experiments carried out in the presence of α-SpIIΣ* antibody showed a decrease in the levels of incision at sites of a psoralen furan-side interstrand cross-link. These results suggested a model in which FA complementation groups A, C, and G proteins modulate α-SpIIΣ* protein, either directly or indirectly, by regulating the expression or stability of the protein. In the case of FA cells, reduced levels of α-SpIIΣ* would provide less structural support around the cross-link and decrease the recruitment of repair factors, which in turn would result in a decreased efficiency of interstrand cross-link DNA repair.228
The studies of Lambert and co-workers described above showed that incisions were made on both sides of a 4,5′,8-trimethylpsoralen interstrand cross-link. Quite different results were obtained by Sancar and co-workers when they examined incision reactions in whole-cell extracts derived from CHO cells using a 140-base-pair linear duplex or a closed circular plasmid DNA substrate that contained a single 4′-hydroxymethyl-4,5′,6-trimethylpsoralen interstrand cross-link.233 In extracts obtained from wild-type AA8 cells, dual incisions were observed on the 5′ side of either the furan or pyrone adduct, producing a series of oligonucleotides 22–28 bases in length. No incisions were observed on the 3′ side of the interstrand cross-link. Thus, the interstrand cross-link remained essentially intact in these DNA substrates. Extracts deficient in XPF/ERCC1 or XPG, the NER endonucleases that are responsible, respectively, for 5′ and 3′ incisions, failed to create the dual incisions in these substrates. The same incisions were observed when the psoralen cross-linked linear duplex was incubated with a reconstituted NER system containing purified XPA, XPC/ hHR23B, TFIIH, XPF/ERCC1, XPG, and RPA. Omission of any of these proteins resulted in the loss of incision activity.
The dual incisions can potentially create a 22–28-nucleotide gap 5′ to the cross-link on one strand of the cross-linked DNA. To study the fate of this gap and the cross-link, experiments were conducted in hamster whole-cell extracts that allowed the repair synthesis to occur.41 This was accomplished by monitoring incorporation of α-[32P]-dCTP into the cross-linked plasmid DNA substrate. Incorporation of nucleotides into the gap was observed in a whole-cell extract from wild-type cells, but ligation of the resulting nick and removal of the cross-link were not observed. This result suggested that the cross-linked DNA underwent “futile” repair synthesis, resulting in the formation of a nick on the 5′ side of the unremoved cross-link. Consistent with the requirement for NER, futile repair synthesis was not observed in extracts from a XPG mutant cell line. However, repair synthesis was observed in XRCC2- and XRCC3-deficient extracts, suggesting that homologous recombination was not involved in this process. The role of futile repair synthesis in the processing of psoralen interstrand cross-links is unclear at this time, although it may serve as a signal that further processing of the cross-link is required.41
Incision/repair experiments were also carried out using purified XPF/ERCC1 heterodimer.41 No incisions on either side of the cross-link were observed with either the cross-linked linear duplex or the cross-linked closed circular plasmid DNA. However, the addition of RPA, a single-strand binding protein, resulted in the processing of the linear duplex. Two types of products were observed. One of these resulted from 3′ → 5′ exonucleolytic degradation, which stopped immediately after passing the cross-link. The other product resulted from complete exonucleolytic degradation of one of the strands of the cross-linked duplex. Similar results were obtained by Kuraoka et al.234 for a linear duplex that contained a single psoralen interstrand cross-link. No incisions were seen when the cross-linked duplex was incubated with XPF/ERCC1. However, these workers also tested a Y-shaped DNA substrate that contained a psoralen interstrand cross-link. This substrate, which to some extent mimics the stalled replication fork shown in Figure 20, was cleaved on both the 3′ and 5′ side of the furan adduct by XPF/ERCC1.
Interstrand cross-links have been postulated to induce strand breaks at stalled replication forks during DNA replication. A recent report by Bessho has demonstrated the formation of CIDs in a cell-free system.235 Closed circular plasmid DNA was prepared that contained a single psoralen interstrand cross-link located 205 nucleotides away from an SV40 origin of replication. When incubated with a nuclear extract derived from HeLa cells, the cross-link caused the termination of the replication at 1–50 nucleotides before the cross-link site. This resulted in the formation of a strand break near the site of the cross-link. Systems of this type could provide new insights into the role of replication and possibly recombination in the processing of interstrand cross-links.
Legerski and co-workers have devised an interesting cell-free system, called the cross-link repair synthesis (CRS) assay, to study interstrand cross-link repair.236 The assay, which is similar to the original DNA repair synthesis assay developed by Wood,213 utilizes closed circular plasmid DNA that contains a single 4,5′,8-trimethylpsoralen interstrand cross-link, a noncrossed-linked control plasmid whose sequence is identical to the cross-linked plasmid, and a donor plasmid whose size is larger than that of the cross-linked or control plasmids. The steps in the assay involve incubating the donor plasmid together with either the control or cross-linked plasmid in whole-cell extracts that contain α-[32 P]-dNTP. The plasmid DNAs are then deproteinized, linearized, and separated by agarose gel electrophoresis. The DNA is detected by ethidium bromide staining, and the incorporation of labeled dNTP is detected by autoradiography.
Incorporation of radioactivity into the cross-linked plasmid was stimulated by the presence of homologous donor plasmid. Surprisingly, radioactivity was also incorporated into the donor plasmid itself. This suggests that some form of recombination event had occurred, although the incorporation into only partially homologous donor plasmid was also observed. The incorporation of radioactivity into the donor plasmid was specifically stimulated by the presence of the interstrand cross-linked plasmid. Thus, no repair synthesis was observed when the plasmid contained a psoralen monoadduct or a double-strand break. Extracts from human lymphoid cells and hamster cells deficient in NER proteins XPA, XPC, and XPG gave essentially the same level of incorporation as those from wild-type cells. In contrast, extracts from hamster UV20 and UV41 (both of which are deficient in XPF/ERCC1), irs1 (deficient in XRCC2), or irs1SF (deficient in XRCC3) cells gave highly reduced levels of incorporation. In agreement with the results of Lambert and those of Sancar, these results suggest that XPF and ERCC1 are required for the repair synthesis. Consistent with this idea was the observation that the repair synthesis in XPF-and ERCC1-deficient extracts could be restored by the addition of purified XPF/ERRC1. The results further suggest that some type of recombination event was responsible for the repair synthesis. However, homologous recombination was ruled out because extracts immunodepleted of Rad51, the eukaryotic homologue of the E. coli homologous recombination protein RecA, showed no decrease in the repair synthesis. Further studies were carried out using extracts from an XPF human patient cell line.237 This extract was unable to support the repair synthesis of a plasmid that contained an N-acetoxy-N-acetylaminofluorene, a UV damage mimetic adduct, a result that is consistent with the inability of the extract to carry out NER. However, the extract did support the repair synthesis stimulated by the psoralen interstrand cross-linked plasmid. Similar results were also obtained using an extract from a hamster UV40 cell line that is deficient in XPF activity. These experiments suggested that some mutations in the XPF protein can impair NER activity but not the ability to participate in pathways that involve recombinational repair.
In subsequent studies conducted by Li and Zhang, the CRS assay was used to determine the role of other related proteins in the repair of a psoralen interstrand cross-link: PCNA protein, which is involved in the gap-filling step of NER, MutSβ, which is involved in mismatch repair, and the multifunctional RPA protein were all found to be required for repair synthesis.238,239 A model was proposed that involves these factors for the early processing of interstrand cross-links in vitro. However, this model does not describe the events that follow the unhooking of the cross-link, but according to supporting work, some form of nonhomologous recombination event must be involved, which may even include the formation of double-strand breaks.240
The fate of psoralen interstrand cross-links in cells was also examined using a host-cell reactivation system, similar to that described above for the mitomycin C interstrand cross-link.187 Plasmids were prepared that contained a single 4,5′,8-trimethylpsoralen interstrand cross-link inserted between a cytomegalovirus promoter and a gene coding for green fluorescent protein or luciferase. Transcription of either gene required the removal of the psoralen interstrand cross-link. The plasmids were transfected into human or hamster cell lines that were proficient or defective in various DNA repair pathways, and the efficiency of gene expression was determined relative to a control plasmid that did not contain an interstrand cross-link. Wild-type cells were capable of expressing the genes at approximately 50% of the level of the noncross-linked control plasmid, thus indicating that repair of the cross-link had occurred. NER mutants lacking XPA, XPB, XPC, XPD, XPF, XPG, or ERCC1 were all incapable of removing the cross-link. In contrast, homologous recombination mutants, XRCC2 or XRCC3, showed the same level of gene expression as wild-type cells. Repaired plasmids that were recovered and sequenced showed base substitutions at or near the site of the cross-link. Together, these results suggested that psoralen interstrand cross-link repair required the components of the NER pathway but that repair occurred in an error-prone and recombination-independent manner. The results are thus very similar to those seen with the mitomycin C interstrand cross-link.224,225 This would fit a model in which the cross-link is removed by the NER machinery and the resulting gap filled in by translesion bypass synthesis. Interestingly, an XPV mutant, which is defective in the translesion polymerase Pol η gene, was only moderately less efficient in repairing the cross-link compared to wild-type cells, suggesting that Pol η was not involved in the translesion step of the repair.
4. Conclusions
The formation of DNA interstrand cross-links plays a key role in the mechanism of action of a number of important cancer chemotherapeutic agents. Emerging evidence suggests that they may also be formed by environmental agents and as unwanted byproducts of cellular metabolic processes. A better understanding of the manner in which these lesions are dealt with by the cell could lead to the development of more effective therapeutic agents and strategies.
It should be evident from the preceding discussion that, despite the efforts of a considerable number of investigators, the mechanism by which DNA interstrand cross-links are repaired still remains an enigma. At the genetic level, protein members of a number of repair pathways have been identified to play a role and other proteins have been implicated. However, the details of how and under what circumstances they are called into action remains unclear. What is clear is that the repair of interstrand cross-links in eukaryotes involves factors from the NER and homologous recombination pathways but probably not the NHEJ pathway. Not clear, however, is the sequence of events, the mechanism of repair, nor the possible involvement of novel proteins not found in already established pathways. What is responsible for the recognition of damage: is it only a replicative block or is there some type of global damage recognition as is found in NER? At the molecular level, next to nothing is known about how the repair proteins interact with the cross-link and/or each other. Given this state of affairs, it is apparent that much more work lies ahead before we have a clear picture of this important repair process.
Acknowledgments
Work cited from our laboratory on interstrand cross-link synthesis and repair was supported by a grant from the National Cancer Institute (CA82785). The authors thank the reviewers of the original manuscript for their helpful comments and suggestions.
Biographies

Tracey McGregor Mason received her B.S. in chemistry from Longwood University (1991). She was employed as a technologist by Roche Biomedical Laboratories until 1994. Tracey subsequently joined the United States Peace Corps and served as a teacher of chemistry, biology, physics, and basic sciences to secondary school students in Fiji. Upon her return to Virginia, she obtained her Ph.D. in inorganic chemistry (2001) from Virginia Commonwealth University under the guidance of Nicholas P. Farrell. Her research concerned the interactions of di- and trinuclear platinum antitumor compounds with DNA. She went on to a 1 year postdoctoral position at University of Maryland Baltimore County in the lab of Richard Karpel where she studied the intra- and intermolecular interactions of T4 Phage gene 32 protein. Tracey is now in a second postdoctoral position at the Johns Hopkins Bloomberg School of Public Health in the Department of Biochemistry and Molecular Biology. She is supported in the laboratory of Paul S. Miller by an individual Ruth L. Kirschstein National Research Service Award. She is presently focusing her research on the repair process of synthetic DNA interstrand cross-links.

David Michael Noll received his B.A. in biochemistry from Boston College (1991). He received a Ph.D. in biochemistry from the Johns Hopkins University School of Public Health (1997). His research was conducted under Paul S. Miller and focused on the structural and biophysical properties of three-stranded DNA complexes. Postdoctoral research was carried out in the laboratory of Neil D. Clarke in the Department of Biophysics and Biophysical Chemistry at the Johns Hopkins University. David is currently a Research Associate in the Department of Biophysics and Biophysical Chemistry at Johns Hopkins University.

Paul S. Miller received his B.S. in chemistry from the State University of New York at Buffalo in 1965. He obtained a Ph.D. in chemistry from Northwestern University in 1969 under the guidance of Professor Robert L. Letsinger. He then carried out postdoctoral work in the laboratory of Dr. Paul O. P. Ts’o at Johns Hopkins University, where he was an American Cancer Society Postdoctoral Fellow from 1969 to 1971. He joined the faculty of the Department of Biochemistry in the School of Public Health at Johns Hopkins University in 1971 and is currently a full professor in the Department of Biochemistry and Molecular Biology. His research interests include the development of novel oligonucleotide analogues and derivatives for use as antisense and antigene agents and the study of DNA repair mechanisms. In addition to his research activities, he is also an Associate Editor of Bioconjugate Chemistry.
References
- 1.Lawley PD, Phillips DH. Mutat Res. 1996;355:13–40. doi: 10.1016/0027-5107(96)00020-6. [DOI] [PubMed] [Google Scholar]
- 2.Murnane JP, Byfield JE. Chem Biol Interact. 1981;38:75–86. doi: 10.1016/0009-2797(81)90154-x. [DOI] [PubMed] [Google Scholar]
- 3.Erickson LC, Bradley MO, Ducore JM, Ewig RAG, Kohn KW. Proc Natl Acad Sci USA. 1980;77:467–471. doi: 10.1073/pnas.77.1.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia ST, McQuillan A, Panasci L. Biochem Pharmacol. 1988;37:3189–3192. doi: 10.1016/0006-2952(88)90319-x. [DOI] [PubMed] [Google Scholar]
- 5.O’Connor PM, Kohn KW. Cancer Commun. 1990;2:387–394. doi: 10.3727/095535490820873949. [DOI] [PubMed] [Google Scholar]
- 6.Sunters A, Springer CJ, Bagshawe KD, Souhami RL, Hartley JA. Biochem Pharmacol. 1992;44:59–64. doi: 10.1016/0006-2952(92)90038-k. [DOI] [PubMed] [Google Scholar]
- 7.McHugh P, Spanswick V, Hartley J. Lancet Oncol. 2001;2:483–490. doi: 10.1016/S1470-2045(01)00454-5. [DOI] [PubMed] [Google Scholar]
- 8.Friedberg EC, Walker GC, Siede W. DNA Repair and Mutagenesis. ASM Press; Washington, DC: 1995. [Google Scholar]
- 9.Sancar A. Chem ReV. 2003;103:2203–2237. doi: 10.1021/cr0204348. [DOI] [PubMed] [Google Scholar]
- 10.Pegg AE. Mutat Res. 2000;462:83–100. doi: 10.1016/s1383-5742(00)00017-x. [DOI] [PubMed] [Google Scholar]
- 11.Falnes PO, Johansen RF, Seeberg E. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- 12.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 13.Falnes PO, Rognes T. Res Microbiol. 2003;154:531–538. doi: 10.1016/S0923-2508(03)00150-5. [DOI] [PubMed] [Google Scholar]
- 14.Mendelsohn ML, Moore DH, II, Lohman PH. Mutat Res. 1992;266:43–60. doi: 10.1016/0027-5107(92)90220-v. [DOI] [PubMed] [Google Scholar]
- 15.Lohman PH. Mutat Res. 1999;428:237–254. doi: 10.1016/s1383-5742(99)00051-4. [DOI] [PubMed] [Google Scholar]
- 16.Vogel EW, Barbin A, Nivard MJ, Stack HF, Waters MD, Lohman PH. Mutat Res. 1998;400:509–540. doi: 10.1016/s0027-5107(98)00060-8. [DOI] [PubMed] [Google Scholar]
- 17.Vogel EW, Nivard MJ, Ballering LA, Bartsch H, Barbin A, Nair J, Comendador MA, Sierra LM, Aguirrezabalaga I, Tosal L, Ehrenberg L, Fuchs RP, Janel-Bintz R, Maenhaut-Michel G, Montesano R, Hall J, Kang H, Miele M, Thomale J, Bender K, Engelbergs J, Rajewsky MF. Mutat Res. 1996;353:177–218. doi: 10.1016/0027-5107(96)00032-2. [DOI] [PubMed] [Google Scholar]
- 18.Yaghi BM, Turner PM, Denny WA, Turner PR, O’Connor CJ, Ferguson LR. Mutat Res. 1998;401:153–164. doi: 10.1016/s0027-5107(98)00005-0. [DOI] [PubMed] [Google Scholar]
- 19.Wijen JP, Nivard MJ, Vogel EW. Carcinogenesis. 2000;21:1859–1867. doi: 10.1093/carcin/21.10.1859. [DOI] [PubMed] [Google Scholar]
- 20.Vogel EW, Nivard MJ. Mutagenesis. 1993;8:57–81. doi: 10.1093/mutage/8.1.57. [DOI] [PubMed] [Google Scholar]
- 21.Tokuda K, Bodell WJ. Carcinogenesis. 1987;8:1697–1701. doi: 10.1093/carcin/8.11.1697. [DOI] [PubMed] [Google Scholar]
- 22.Bodell WJ, Aida T, Rasmussen J. Mutat Res. 1985;149:95–100. doi: 10.1016/0027-5107(85)90013-2. [DOI] [PubMed] [Google Scholar]
- 23.Bodell WJ, Aida T, Berger MS, Rosenblum ML. Environ Health Perspect. 1985;62:119–126. doi: 10.1289/ehp.8562119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colvin M. The Alkylating Agents. W B Saunders Co; Philadelphia, PA: 1982. [Google Scholar]
- 25.Colvin M. Alkylating Agents and Platinum Antitumor Compounds. Lea and Febiger; Philadelphia, PA: 1993. [Google Scholar]
- 26.Pratt WB, Ruddon RW, Ensminger WD, Maybaum J. Covalent DNA Binding Drugs. Oxford Press; New York: 1994. [Google Scholar]
- 27.Rajski SR, Williams RM. Chem ReV. 1998;98:2723–2795. doi: 10.1021/cr9800199. [DOI] [PubMed] [Google Scholar]
- 28.Hearst JE. Annu ReV Biophys Bioeng. 1981;10:69–86. doi: 10.1146/annurev.bb.10.060181.000441. [DOI] [PubMed] [Google Scholar]
- 29.Cimino GD, Gamper HD, Isaacs ST, Hearst JE. Annu Rev Biochem. 1985;54:1151–1193. doi: 10.1146/annurev.bi.54.070185.005443. [DOI] [PubMed] [Google Scholar]
- 30.Edelson RL, Berger CL, Gasparro FP. N Engl J Med. 1987;316:297–303. doi: 10.1056/NEJM198702053160603. [DOI] [PubMed] [Google Scholar]
- 31.Gasparro FP. Photochem Photobiol. 1996;63:553–557. doi: 10.1111/j.1751-1097.1996.tb05654.x. [DOI] [PubMed] [Google Scholar]
- 32.Dalla Via L, Marciani Magno S. Curr Med Chem. 2001;8:1405–1418. doi: 10.2174/0929867013372076. [DOI] [PubMed] [Google Scholar]
- 33.Baron ED, Stevens SR. Dermatol Ther. 2003;16:303–310. doi: 10.1111/j.1396-0296.2003.01642.x. [DOI] [PubMed] [Google Scholar]
- 34.Morison WL. Photodermatol Photoimmunol Photomed. 2004;20:315–320. doi: 10.1111/j.1600-0781.2004.00125.x. [DOI] [PubMed] [Google Scholar]
- 35.Zanolli M. Dermatol Clin. 2004;22:397–406. doi: 10.1016/j.det.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 36.Wilson VL, Foiles PG, Chung FL, Povey AC, Frank AA, Harris CC. Carcinogenesis. 2003;12:1483–1490. doi: 10.1093/carcin/12.8.1483. [DOI] [PubMed] [Google Scholar]
- 37.Niedernhofer LJ, Daniels JS, Rouzer CA, Greene RE, Marnett LJ. J Biol Chem. 2003;278:31426–31433. doi: 10.1074/jbc.M212549200. [DOI] [PubMed] [Google Scholar]
- 38.Dooley PA, Tsarouhtsis D, Korbel GA, Nechev LV, Shearer J, Zegar IS, Harris CM, Stone MP, Harris TM. J Am Chem Soc. 2001;123:1730–1739. doi: 10.1021/ja003163w. [DOI] [PubMed] [Google Scholar]
- 39.Dooley PA, Zhang M, Korbel GA, Nechev LV, Harris CM, Stone MP, Harris TM. J Am Chem Soc. 2003;125:62–72. doi: 10.1021/ja0207798. [DOI] [PubMed] [Google Scholar]
- 40.Kozekov D, Nechev LV, Moseley MS, Harris CM, Rizzo CJ, Stone MP, Harris TM. J Am Chem Soc. 2003;125:50–61. doi: 10.1021/ja020778f. [DOI] [PubMed] [Google Scholar]
- 41.Mu D, Bessho T, Nechev LV, Chen DJ, Harris TM, Hearst JE, Sancar A. Mol Cell Biol. 2000;20:2446–2454. doi: 10.1128/mcb.20.7.2446-2454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friedman HS, Colvin OM, Kaufmann SH, Ludeman SM, Bullock N, Bigner DD, Griffith OW. Cancer Res. 1992;52:5373–5378. [PubMed] [Google Scholar]
- 43.Bramson J, McQuillan A, Aubin R, Alaoui-Jamali M, Batist G, Christodoulopoulos G, Panasci LC. Mutat Res. 1995;336:269–278. doi: 10.1016/0921-8777(94)00063-c. [DOI] [PubMed] [Google Scholar]
- 44.Dong Q, Bullock N, Aliosman F, Colvin OM, Bigner DD, Friedman HS. Cancer Chemother Pharmacol. 1996;37:242–246. doi: 10.1007/BF00688323. [DOI] [PubMed] [Google Scholar]
- 45.Chaney SG, Sancar A. J Natl Cancer Inst. 1996;88:1346–1360. doi: 10.1093/jnci/88.19.1346. [DOI] [PubMed] [Google Scholar]
- 46.Dong Q, Johnson SP, Colvin OM, Bullock N, Kilborn C, Runyon G, Sullivan DM, Easton J, Bigner DD, Nahta R, Marks J, Modrich P, Friedman HS. Cancer Chemother Pharmacol. 1999;43:73–79. doi: 10.1007/s002800050865. [DOI] [PubMed] [Google Scholar]
- 47.Panasci L, Xu ZY, Bello V, Aloyz R. Anticancer Drugs. 2002;13:211–220. doi: 10.1097/00001813-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Noll DM, Noronha AM, Wilds CJ, Miller PS. Front Biosci. 2004;9:421–437. doi: 10.2741/1246. [DOI] [PubMed] [Google Scholar]
- 49.Ojwang JO, Grueneberg DA, Loechler EL. Cancer Res. 1989;49:6529–6537. [PubMed] [Google Scholar]
- 50.Millard JT, Raucher S, Hopkins PB. J Am Chem Soc. 1990;112:2459–2460. [Google Scholar]
- 51.Cullis PM, Merson-Davies L, Weaver R. J Am Chem Soc. 1995;117:8033–8034. [Google Scholar]
- 52.Dong Q, Barsky D, Colvin ME, Melius DF, Ludeman SM, Moravek JF, Colvin OM, Bigner DD, Modrich P, Friedman HS. Proc Natl Acad Sci USA. 1995;92:12170–12174. doi: 10.1073/pnas.92.26.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rink SM, Solomon MS, Taylor MJ, Rajur SB, McLaughlin LW, Hopkins PB. J Am Chem Soc. 1993;115:2551–2557. [Google Scholar]
- 54.Rink SM, Hopkins PB. Biochemistry. 1995;34:1439–1445. doi: 10.1021/bi00004a039. [DOI] [PubMed] [Google Scholar]
- 55.Rink SM, Lipman R, Alley SC, Hopkins PB, Tomasz M. Chem Res Toxicol. 1996;9:382–389. doi: 10.1021/tx950156q. [DOI] [PubMed] [Google Scholar]
- 56.Norman D, Live D, Sastry M, Lipman R, Hingerty BE, Tomasz M, Broyde S, Patel DJ. Biochemistry. 1990;29:2861–2875. doi: 10.1021/bi00463a032. [DOI] [PubMed] [Google Scholar]
- 57.Reishus JW, Martin DS. J Am Chem Soc. 1961;83:2457–2462. [Google Scholar]
- 58.Kartalou M, Essigmann JM. Mutat Res. 2001;478:1–21. doi: 10.1016/s0027-5107(01)00142-7. [DOI] [PubMed] [Google Scholar]
- 59.Baik MH, Friesner RA, Lippard SJ. J Am Chem Soc. 2002;124:4495–4503. doi: 10.1021/ja017588+. [DOI] [PubMed] [Google Scholar]
- 60.Malinge JM, Perez C, Leng M. Nucleic Acids Res. 1994;22:3834–3839. doi: 10.1093/nar/22.19.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang H, Zhu L, Reid BR, Drobny GP, Hopkins PB. Science. 1995;270:1842–1845. doi: 10.1126/science.270.5243.1842. [DOI] [PubMed] [Google Scholar]
- 62.Paquet F, Perez C, Leng M, Lancelot G, Malinge JM. J Biomol Struct Dyn. 1996;14:67–77. doi: 10.1080/07391102.1996.10508930. [DOI] [PubMed] [Google Scholar]
- 63.Coste F, Malinge JM, Serre L, Shepard W, Roth M, Leng M, Zelwer C. Nucleic Acids Res. 1999;27:1837–1846. doi: 10.1093/nar/27.8.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eastman A, Barry MA. Biochemistry. 1987;26:3303–3307. doi: 10.1021/bi00386a009. [DOI] [PubMed] [Google Scholar]
- 65.Brabec V, Leng M. Proc Natl Acad Sci USA. 1993;90:5345–5349. doi: 10.1073/pnas.90.11.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pinto AL, Lippard SJ. Proc Natl Acad Sci USA. 1985;82:4616–4619. doi: 10.1073/pnas.82.14.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bancroft DP, Lepre CA, Lippard SJ. J Am Chem Soc. 1990;112:6860–6871. [Google Scholar]
- 68.Dalbies R, Payet D, Leng M. Proc Natl Acad Sci USA. 1994;91:8147–8151. doi: 10.1073/pnas.91.17.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Paquet F, Boudvillain M, Lancelot G, Leng M. Nucleic Acids Res. 1999;27:4261–4268. doi: 10.1093/nar/27.21.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boudvillain M, Dalbies R, Aussourd C, Leng M. Nucleic Acids Res. 1995;23:2381–2388. doi: 10.1093/nar/23.13.2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lippert B. Met Ions Biol Syst. 1996;33:105–141. [PubMed] [Google Scholar]
- 72.Gupta AK, Anderson TF. J Am Acad Dermatol. 1987;17:703–734. doi: 10.1016/s0190-9622(87)70255-2. [DOI] [PubMed] [Google Scholar]
- 73.Isaacs ST, Shen C-kJ, Hearst JE, Rapoport H. Biochemistry. 1977;16:1058–1064. doi: 10.1021/bi00625a005. [DOI] [PubMed] [Google Scholar]
- 74.Spielmann HP, Dwyer TJ, Sastry SS, Hearst JE, Wemmer DE. Proc Natl Acad Sci USA. 1995;92:2345–2349. doi: 10.1073/pnas.92.6.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Speilmann HP, Dwyer TJ, Hearst JE, Wemmer DE. Biochemistry. 1995;34:12937–12953. [PubMed] [Google Scholar]
- 76.Hwang GS, Kim JK, Choi BS. Biochem Biophys Res Commun. 1996;219:191–197. doi: 10.1006/bbrc.1996.0204. [DOI] [PubMed] [Google Scholar]
- 77.Haran TE, Crothers DM. Biochemistry. 1988;27:6967–6971. doi: 10.1021/bi00418a044. [DOI] [PubMed] [Google Scholar]
- 78.Kumar S, Johnson WS, Tomasz M. Biochemistry. 1993;32:1364–1372. doi: 10.1021/bi00056a023. [DOI] [PubMed] [Google Scholar]
- 79.Spielmann HP, Sastry SS, Hearst JE. Proc Natl Acad Sci USA. 1992;89:4514–4518. doi: 10.1073/pnas.89.10.4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kobertz WR, Essigmann JM. J Am Chem Soc. 1997;119:5960–5961. [Google Scholar]
- 81.Nechev LV, Kozekov I, Harris CM, Harris TM. Chem Res Toxicol. 2001;14:1506–1512. doi: 10.1021/tx0100690. [DOI] [PubMed] [Google Scholar]
- 82.Nechev LV, Harris CM, Harris TM. Chem Res Toxicol. 2000;13:421–429. doi: 10.1021/tx990167+. [DOI] [PubMed] [Google Scholar]
- 83.Noll DM, Noronha AM, Miller PS. J Am Chem Soc. 2001;123:3405–3411. doi: 10.1021/ja003340t. [DOI] [PubMed] [Google Scholar]
- 84.Noronha AM, Noll DM, Miller PS. Nucleosides Nucleotides Nucleic Acids. 2001;20:1303–1307. doi: 10.1081/NCN-100002542. [DOI] [PubMed] [Google Scholar]
- 85.Noronha AM, Noll DM, Wilds CJ, Miller PS. Biochemistry. 2002;41:760–771. doi: 10.1021/bi011610u. [DOI] [PubMed] [Google Scholar]
- 86.Noronha AM, Wilds CJ, Miller PS. Biochemistry. 2002;41:8605–8612. doi: 10.1021/bi025671j. [DOI] [PubMed] [Google Scholar]
- 87.Wilds CJ, Noronha AM, Robidoux S, Miller PS. J Am Chem Soc. 2004;126:9257–9265. doi: 10.1021/ja0498540. [DOI] [PubMed] [Google Scholar]
- 88.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 89.Sancar A. J Biol Chem. 1995;270:15915–15918. doi: 10.1074/jbc.270.27.15915. [DOI] [PubMed] [Google Scholar]
- 90.Prakash S, Prakash L. Mutat Res. 2000;451:13–24. doi: 10.1016/s0027-5107(00)00037-3. [DOI] [PubMed] [Google Scholar]
- 91.Krogh BO, Symington LS. Annu Rev Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 92.Aylon Y, Kupiec M. DNA Repair. 2004;3:797–815. doi: 10.1016/j.dnarep.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 93.Aylon Y, Kupiec M. Mutat Res. 2004;566:231–248. doi: 10.1016/j.mrrev.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 94.Thacker J, Zdzienicka MZ. DNA Repair. 2004;3:1081–1090. doi: 10.1016/j.dnarep.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 95.Helleday T. Mutat Res. 2003;532:103–115. doi: 10.1016/j.mrfmmm.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 96.Valerie K, Povirk LF. Oncogene. 2003;22:5792–5812. doi: 10.1038/sj.onc.1206679. [DOI] [PubMed] [Google Scholar]
- 97.Kuzminov A. Microbiol Mol Biol Rev. 1999;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Courcelle J, Belle JJ, Courcelle CT. Res Microbiol. 2004;155:231–237. doi: 10.1016/j.resmic.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 99.Boudsocq F, Ling H, Yang W, Woodgate R. DNA Repair. 2002;1:343–358. doi: 10.1016/s1568-7864(02)00019-8. [DOI] [PubMed] [Google Scholar]
- 100.Pham P, Rangarajan S, Woodgate R, Goodman MF. Proc Natl Acad Sci USA. 2001;98:8350–8354. doi: 10.1073/pnas.111007198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sutton MD, Walker GC. Proc Natl Acad Sci USA. 2001;98:8342–8349. doi: 10.1073/pnas.111036998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Plosky BS, Woodgate R. Curr Opin Genet Dev. 2004;74:317–353. doi: 10.1016/j.gde.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 103.Yang W. Curr Opin Struct Biol. 2003;13:23–30. doi: 10.1016/s0959-440x(02)00003-9. [DOI] [PubMed] [Google Scholar]
- 104.Broomfield S, Hryciw T, Xiao W. Mutat Res. 2001;486:167–184. doi: 10.1016/s0921-8777(01)00091-x. [DOI] [PubMed] [Google Scholar]
- 105.Lehmann AR. Mutat Res. 2002;509:23–34. doi: 10.1016/s0027-5107(02)00227-0. [DOI] [PubMed] [Google Scholar]
- 106.Prakash S, Johnson RE, Prakash L. Annu ReV Biochem. 2004 doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 107.Dronkert ML, Kannar R. Mutat Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- 108.Scharer OD. ChemBioChem. 2005;6:27–32. doi: 10.1002/cbic.200400287. [DOI] [PubMed] [Google Scholar]
- 109.Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, Pena-Diaz J, Otterlei M, Slupphuag G, Krokan HE. DNA Repair. 2004;3:1389–1407. doi: 10.1016/j.dnarep.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 110.Lawley PD, Brookes P. Nature. 1965;206:480–483. doi: 10.1038/206480a0. [DOI] [PubMed] [Google Scholar]
- 111.Cole RS. J Bacteriol. 1971;107:846–852. doi: 10.1128/jb.107.3.846-852.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cole RS. Proc Natl Acad Sci USA. 1973;70:1064–1068. doi: 10.1073/pnas.70.4.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sancar A, Franklin KA, Sancar G, Tang MS. J Mol Biol. 1985;184:725–734. doi: 10.1016/0022-2836(85)90316-x. [DOI] [PubMed] [Google Scholar]
- 114.van Houten B, Gamper H, Hearst JE, Sancar A. J Biol Chem. 1986;261:14135–14141. [PubMed] [Google Scholar]
- 115.van Houten B, Gamper H, Holbrook SR, Hearst JE, Sancar A. Proc Natl Acad Sci USA. 1986;83:8077–8081. doi: 10.1073/pnas.83.21.8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cheng S, Houten BV, Gamper HB, Sancar A, Hearst JE. J Biol Chem. 1988;263:15110–15117. [PubMed] [Google Scholar]
- 117.Cheng S, Sancar A, Hearst JE. Nucleic Acids Res. 1991;19:657–663. doi: 10.1093/nar/19.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sladek FM, Munn MM, Rupp WD, Howard-Flanders P. J Biol Chem. 1989;264:6755–6765. [PubMed] [Google Scholar]
- 119.Jones BK, Yeung AT. Proc Natl Acad Sci USA. 1988;85:8410–8414. doi: 10.1073/pnas.85.22.8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jones BK, Yeung AT. J Biol Chem. 1990;265:3489–3496. [PubMed] [Google Scholar]
- 121.Munn MM, Rupp WD. J Biol Chem. 1991;266:24748–24756. [PubMed] [Google Scholar]
- 122.Pu WT, Kahn R, Munn MM, Rupp WD. J Biol Chem. 1989;264:20697–20704. [PubMed] [Google Scholar]
- 123.Snowden A, Kow YW, van Houten B. Biochemistry. 1990;29:7251–7259. doi: 10.1021/bi00483a013. [DOI] [PubMed] [Google Scholar]
- 124.van Houten B, Snowden A. Bioessays. 1993;15:51–59. doi: 10.1002/bies.950150108. [DOI] [PubMed] [Google Scholar]
- 125.Oh EY, Grossman L. Nucleic Acids Res. 1986;14:8557–8571. doi: 10.1093/nar/14.21.8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Oh EY, Grossman L. Proc Natl Acad Sci USA. 1987;84:3638–3642. doi: 10.1073/pnas.84.11.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lage C, de Padula M, de Alencar TA, da Fonseca Goncalves SR, da Silva Vidal L, Cabral-Neto J, Leitao AC. Mutat Res. 2003;544:143–157. doi: 10.1016/j.mrrev.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 128.Grueneberg DA, Ojwang JO, Benasutti M, Hartman S, Loechler EL. Cancer Res. 1991;51:2268–2272. [PubMed] [Google Scholar]
- 129.Berardini M, Mackay W, Loechler EL. Biochemistry. 1997;36:3506–3513. doi: 10.1021/bi962778w. [DOI] [PubMed] [Google Scholar]
- 130.Berardini M, Foster PL, Loechler EL. J Bacteriol. 1999;181:2878–2882. doi: 10.1093/gao/9781884446054.article.t031385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Piette J, Gamper HB, van de Vorst A, Hearst JE. Nucleic Acids Res. 1988;16:9961–9977. doi: 10.1093/nar/16.21.9961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Noll DM, de Silva MW, Noronha AM, Wilds CJ, Colvin OM, Gamcsik MP, Miller PS. Biochemistry. 2005;44:6764–6775. doi: 10.1021/bi050014n. [DOI] [PubMed] [Google Scholar]
- 133.Henriques JA, Moustacchi E. J Bacteriol. 1981;148:248–256. doi: 10.1128/jb.148.1.248-256.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sung P, Trujillo KM, van Komen S. Mutat Res. 2000;451:257–275. doi: 10.1016/s0027-5107(00)00054-3. [DOI] [PubMed] [Google Scholar]
- 135.Cejka P, Vondrejs V, Storchova Z. Genetics. 2001;159:953–963. doi: 10.1093/genetics/159.3.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Henriques JA, Andrade HH, Bankmann M, Brendel M. Curr Genet. 1989;16:75–80. doi: 10.1007/BF00393398. [DOI] [PubMed] [Google Scholar]
- 137.Bankmann M, Brendel M. J Photochem Photobiol B. 1989;3:33–52. doi: 10.1016/1011-1344(89)80019-3. [DOI] [PubMed] [Google Scholar]
- 138.Bankmann M, Brendel M. J Photochem Photobiol B. 1989;4:57–74. doi: 10.1016/1011-1344(89)80102-2. [DOI] [PubMed] [Google Scholar]
- 139.Magana-Schwencke N, Henriques JA, Chanet R, Moustacchi E. Proc Natl Acad Sci USA. 1982;79:1722–1726. doi: 10.1073/pnas.79.6.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Grossmann KF, Ward AM, Matkovic ME, Folias AE, Moses RE. Mutat Res. 2001;487:73–83. doi: 10.1016/s0921-8777(01)00106-9. [DOI] [PubMed] [Google Scholar]
- 141.Nelson JR, Lawrence CW, Hinkle DC. Science. 1996;272:1646–1649. doi: 10.1126/science.272.5268.1646. [DOI] [PubMed] [Google Scholar]
- 142.Morrison A, Christensen RB, Alley J, Beck AK, Bernstine EG, Lemontt JF, Lawrence CW. J Bacteriol. 1989;171:5659–5667. doi: 10.1128/jb.171.10.5659-5667.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Henriques JA, Moustacchi E. Genetics. 1980;95:273–288. doi: 10.1093/genetics/95.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cassier C, Chanet R, Henriques JA, Moustacchi E. Genetics. 1980;96:841–857. doi: 10.1093/genetics/96.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Henriques JA, Brozmanova J, Brendel M. J Photochem Photobiol B. 1997;39:185–196. doi: 10.1016/s1011-1344(97)00020-1. [DOI] [PubMed] [Google Scholar]
- 146.Barber LJ, Ward TA, Hartley JA, McHugh PJ. Mol Cell Biol. 2005;25:2297–2309. doi: 10.1128/MCB.25.6.2297-2309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Beljanski V, Marzilli LG, Doetsch PW. Mol Pharmacol. 2004;65:1496–1506. doi: 10.1124/mol.65.6.1496. [DOI] [PubMed] [Google Scholar]
- 148.Saffran WA, Ahmed S, Bellevue S, Pereira G, Patrick T, Sanchez W, Thomas S, Alberti M, Hearst JE. J Biol Chem. 2004;279:36462–36469. doi: 10.1074/jbc.M402323200. [DOI] [PubMed] [Google Scholar]
- 149.Greenberg RB, Alberti M, Hearst JE, Chua MA, Saffran WA. J Biol Chem. 2001;276:31551–31560. doi: 10.1074/jbc.M103588200. [DOI] [PubMed] [Google Scholar]
- 150.Miller RD, Prakash L, Prakash S. Mol Cell Biol. 1982;2:939–948. doi: 10.1128/mcb.2.8.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Meniel V, Magana-Schwencke N, Averbeck D. Mutagenesis. 1995;10:543–548. doi: 10.1093/mutage/10.6.543. [DOI] [PubMed] [Google Scholar]
- 152.Jachymczyk WJ, von Borstel RC, Mowat MR, Hastings PJ. Mol Gen Genet. 1981;182:196–205. doi: 10.1007/BF00269658. [DOI] [PubMed] [Google Scholar]
- 153.Dardalhon M, de Massy B, Nicolas A, Averbeck D. Curr Genet. 1998;34:30–42. doi: 10.1007/s002940050363. [DOI] [PubMed] [Google Scholar]
- 154.Dardalhon M, Averbeck D. Mutat Res. 1995;336:49–60. doi: 10.1016/0921-8777(94)00037-7. [DOI] [PubMed] [Google Scholar]
- 155.McHugh PJ, Sones WR, Hartley JA. Mol Cell Biol. 2000;20:3425–3433. doi: 10.1128/mcb.20.10.3425-3433.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.McHugh PJ, Gill RD, Waters R, Hartley JA. Nucleic Acids Res. 1999;27:3259–3266. doi: 10.1093/nar/27.16.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.de Silva IU, McHugh PJ, Clingen PH, Hartley JA. Mol Cell Biol. 2000;20:7980–7990. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, Kanaar R. Mol Cell Biol. 2004;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wilborn F, Brendel M. Curr Genet. 1989;16:331–338. doi: 10.1007/BF00340711. [DOI] [PubMed] [Google Scholar]
- 160.Li X, Moses RE. DNA Repair. 2003;2:121–129. doi: 10.1016/s1568-7864(02)00192-1. [DOI] [PubMed] [Google Scholar]
- 161.Ruhland A, Kircher M, Wilborn F, Brendel M. Mutat Res. 1981;91:457–462. doi: 10.1016/0165-7992(81)90052-x. [DOI] [PubMed] [Google Scholar]
- 162.Ruhland A, Haase E, Siede W, Brendel M. Mol Gen Genet. 1981;181:346–351. doi: 10.1007/BF00425609. [DOI] [PubMed] [Google Scholar]
- 163.Callebaut I, Moshous D, Mornon JP, de Villartay JP. Nucleic Acids Res. 2002;30:3592–3601. doi: 10.1093/nar/gkf470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, Fischer A, de Villartay JP. Cell. 2001;105:177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 165.Ma Y, Pannicke U, Schwarz K, Lieber MR. Cell. 2002;108:781–794. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 166.Tran PT, Erdeniz N, Dudley S, Liskay RM. DNA Repair. 2002;1:895–912. doi: 10.1016/s1568-7864(02)00114-3. [DOI] [PubMed] [Google Scholar]
- 167.Antony E, Hingorani MM. Biochemistry. 2003;42:7682–7693. doi: 10.1021/bi034602h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chanet R, Cassier C, Moustacchi E. Mutat Res. 1985;145:145–155. doi: 10.1016/0167-8817(85)90021-5. [DOI] [PubMed] [Google Scholar]
- 169.Watkins JF, Sung P, Prakash S, Prakash L. Genes Dev. 1993;7:250–261. doi: 10.1101/gad.7.2.250. [DOI] [PubMed] [Google Scholar]
- 170.Dohmen RJ, Madura K, Bartel B, Varshavsky A. Proc Natl Acad Sci USA. 1991;88:7351–7355. doi: 10.1073/pnas.88.16.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Robzyk K, Recht J, Osley MA. Science. 2000;287:501–504. doi: 10.1126/science.287.5452.501. [DOI] [PubMed] [Google Scholar]
- 172.Bailly V, Prakash S, Prakash L. Mol Cell Biol. 1997;17:4536–4543. doi: 10.1128/mcb.17.8.4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bailly V, Lauder S, Prakash S, Prakash L. J Biol Chem. 1997;272:23360–23365. doi: 10.1074/jbc.272.37.23360. [DOI] [PubMed] [Google Scholar]
- 174.Bailly V, Lamb J, Sung P, Prakash S, Prakash L. Genes Dev. 1994;8:811–820. doi: 10.1101/gad.8.7.811. [DOI] [PubMed] [Google Scholar]
- 175.Ulrich HD, Jentsch S. EMBO J. 2000;19:3388–3397. doi: 10.1093/emboj/19.13.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Morey NJ, Doetsch PW, Jinks-Robertson S. Genetics. 2003;164:443–455. doi: 10.1093/genetics/164.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Morrison A, Sugino A. Mol Gen Genet. 1994;242:289–296. doi: 10.1007/BF00280418. [DOI] [PubMed] [Google Scholar]
- 178.Morrison A, Johnson AL, Johnston LH, Sugino A. EMBO J. 1993;12:1467–1473. doi: 10.1002/j.1460-2075.1993.tb05790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Saeki T, Machida I, Nakai S. Mutat Res. 1980;73:251–265. doi: 10.1016/0027-5107(80)90192-x. [DOI] [PubMed] [Google Scholar]
- 180.Mondon P, Shahin MM. Mutat Res. 1992;279:121–128. doi: 10.1016/0165-1218(92)90254-w. [DOI] [PubMed] [Google Scholar]
- 181.Arman IP, Dutova TA. Genetika. 1975;11:80–89. [PubMed] [Google Scholar]
- 182.Legerski RJ, Gray HB, Jr, Robberson DL. J Biol Chem. 1977;252:8740–8746. [PubMed] [Google Scholar]
- 183.Edfeldt NB, Harwood EA, Sigurdsson ST, Hopkins PB, Reid BR. Nucleic Acids Res. 2004;32:2785–2794. doi: 10.1093/nar/gkh606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Edfeldt NB, Harwood EA, Sigurdsson ST, Hopkins PB, Reid BR. Nucleic Acids Res. 2004;32:2795–2801. doi: 10.1093/nar/gkh607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Caulfield JL, Wishnok JS, Tannenbaum SR. Chem Res Toxicol. 2003;16:571–574. doi: 10.1021/tx020117w. [DOI] [PubMed] [Google Scholar]
- 186.Aloyz R, Xu ZY, Bello V, Bergeron J, Han FY, Yan Y, Malapetsa A, Alaoui-Jamali MA, Duncan AM, Panasci L. Cancer Res. 2002;62:5457–5462. [PubMed] [Google Scholar]
- 187.Wang X, Peterson CA, Zheng H, Nairn RS, Legerski RJ, Li L. Mol Cell Biol. 2001;21:713–720. doi: 10.1128/MCB.21.3.713-720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Liu N. J Biomed Biotechnol. 2002;2:106–113. doi: 10.1155/S1110724302204040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Thompson LH. Mutat Res. 1996;363:77–88. doi: 10.1016/0921-8777(96)00008-0. [DOI] [PubMed] [Google Scholar]
- 190.Thacker J. Biochimie. 1999;81:77–85. doi: 10.1016/s0300-9084(99)80041-8. [DOI] [PubMed] [Google Scholar]
- 191.Pichierri P, Averbeck D, Rosselli F. Hum Mol Genet. 2002;11:2531–2546. doi: 10.1093/hmg/11.21.2531. [DOI] [PubMed] [Google Scholar]
- 192.Lee JH, Paull TT. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 193.Wang X, D’Andrea AD. DNA Repair. 2004;3:1063–1069. doi: 10.1016/j.dnarep.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 194.Pichierri P, Rosselli F. Cell Cycle. 2004;3:698–700. [PubMed] [Google Scholar]
- 195.Venkitaraman AR. Nat Rev Cancer. 2004;4:266–276. doi: 10.1038/nrc1321. [DOI] [PubMed] [Google Scholar]
- 196.Jansen JG, de Wind N. DNA Repair. 2003;2:1075–1085. doi: 10.1016/s1568-7864(03)00119-8. [DOI] [PubMed] [Google Scholar]
- 197.Lehmann AR. Cell Cycle. 2003;2:300–302. [PubMed] [Google Scholar]
- 198.Bergoglio V, Bavoux C, Verbiest V, Hoffmann JS, Cazaux C. J Cell Sci. 2002;115:4413–4418. doi: 10.1242/jcs.00162. [DOI] [PubMed] [Google Scholar]
- 199.Ohashi E, Ogi T, Kusumoto R, Iwai S, Masutani C, Hanaoka F, Ohmori H. Genes Dev. 2000;14:1589–1594. [PMC free article] [PubMed] [Google Scholar]
- 200.Vos JM, Hanawalt PC. Cell. 1987;50:789–799. doi: 10.1016/0092-8674(87)90337-0. [DOI] [PubMed] [Google Scholar]
- 201.Larminat F, Bohr VA. Nucleic Acids Res. 1994;22:3005–3010. doi: 10.1093/nar/22.15.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Perego P, Gatti L, Caserini C, Supino R, Colangelo D, Leone R, Spinelli S, Farrell N, Zunino F. J Inorg Biochem. 1999;77:59–64. doi: 10.1016/s0162-0134(99)00142-7. [DOI] [PubMed] [Google Scholar]
- 203.Colella G, Pennati M, Bearzatto A, Leone R, Colangelo D, Manzotti C, Daidone MG, Zaffaroni N. Br J Cancer. 2001;84:1387–1390. doi: 10.1054/bjoc.2001.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu JC, Sasaki YF. Environ Mol Mutagen. 2000;35:206–221. doi: 10.1002/(sici)1098-2280(2000)35:3<206::aid-em8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 205.de Silva IU, McHugh PJ, Clingen PH, Hartley JA. Nucleic Acids Res. 2002;30:3848–3856. doi: 10.1093/nar/gkf479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Simpson LJ, Sale JE. EMBO J. 2003;22:1654–1664. doi: 10.1093/emboj/cdg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sonoda E, Sasaki MS, Morrison C, Yamaguchi-Iwai Y, Takata M, Takeda S. Mol Cell Biol. 1999;19:5166–5169. doi: 10.1128/mcb.19.7.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sasaki MS, Takata M, Sonoda E, Tachibana A, Takeda S. Cytogenet Genome Res. 2004;104:28–34. doi: 10.1159/000077463. [DOI] [PubMed] [Google Scholar]
- 209.Akkari YM, Bateman RL, Reifsteck CA, Olson SB, Grompe M. Mol Cell Biol. 2000;20:8283–8289. doi: 10.1128/mcb.20.21.8283-8289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zaludova R, Zakovska A, Kasparkova J, Balcarova Z, Kleinwachter V, Vrana O, Farrell N, Brabec V. Eur J Biochem. 1997;246:508–517. doi: 10.1111/j.1432-1033.1997.00508.x. [DOI] [PubMed] [Google Scholar]
- 211.Hegmans A, Berners-Price SJ, Davies MS, Thomas DS, Humphreys AS, Farrell N. J Am Chem Soc. 2004;126:2166–2180. doi: 10.1021/ja036105u. [DOI] [PubMed] [Google Scholar]
- 212.Szymkowski DE, Yarema K, Essigmann JM, Lippard SJ, Wood RD. Proc Natl Acad Sci USA. 1992;89:10772–10776. doi: 10.1073/pnas.89.22.10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wood RD. Ann N Y Acad Sci. 1994;726:274–279. doi: 10.1111/j.1749-6632.1994.tb52827.x. [DOI] [PubMed] [Google Scholar]
- 214.Kasparkova J, Zehnulova J, Farrell N, Brabec V. J Biol Chem. 2002;277:48076–48086. doi: 10.1074/jbc.M208016200. [DOI] [PubMed] [Google Scholar]
- 215.Zamble DB, Mu D, Reardon JT, Sancar A, Lippard SJ. Biochemistry. 1996;35:10004–10013. doi: 10.1021/bi960453+. [DOI] [PubMed] [Google Scholar]
- 216.Kartalou M, Essigmann JM. Mutat Res. 2001;478:1–21. doi: 10.1016/s0027-5107(01)00142-7. [DOI] [PubMed] [Google Scholar]
- 217.Kasparkova J, Farrell N, Brabec V. J Biol Chem. 2000;275:15789–15798. doi: 10.1074/jbc.M000777200. [DOI] [PubMed] [Google Scholar]
- 218.Kasparkova J, Pospisilova S, Brabec V. J Biol Chem. 2001;276:16064–16069. doi: 10.1074/jbc.M101224200. [DOI] [PubMed] [Google Scholar]
- 219.Patrick SM, Turchi JJ. J Biol Chem. 1999;274:14972–14978. doi: 10.1074/jbc.274.21.14972. [DOI] [PubMed] [Google Scholar]
- 220.Fourrier L, Brooks P, Malinge JM. J Biol Chem. 2003;278:21267–21275. doi: 10.1074/jbc.M301390200. [DOI] [PubMed] [Google Scholar]
- 221.Turchi JJ, Patrick SM, Henkels KM. Biochemistry. 1997;36:7586–7593. doi: 10.1021/bi963124q. [DOI] [PubMed] [Google Scholar]
- 222.Warren AJ, Ihnat MA, Ogdon SE, Rowell EE, Hamilton JW. Environ Mol Mutagen. 1998;31:70–81. [PubMed] [Google Scholar]
- 223.Mustra DJ, Warren AJ, Hamilton JW. Biochemistry. 2001;40:7158–7164. doi: 10.1021/bi002820u. [DOI] [PubMed] [Google Scholar]
- 224.Zheng H, Wang X, Warren AJ, Legerski RJ, Nairn RS, Hamilton JW, Li L. Mol Cell Biol. 2003;23:754–761. doi: 10.1128/MCB.23.2.754-761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Ahn B, Kang D, Kim H, Wei Q. Mol Cells. 2004;18:249–255. [PubMed] [Google Scholar]
- 226.Jones CJ, Wood RD. Biochemistry. 1993;32:12096–12104. doi: 10.1021/bi00096a021. [DOI] [PubMed] [Google Scholar]
- 227.Hang B, Yeung AT, Lambert MW. Nucleic Acids Res. 1993;21:4187–4192. doi: 10.1093/nar/21.18.4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.McMahon LW, Sangerman J, Goodman SR, Kumaresan K, Lambert MW. Biochemistry. 2001;40:7025–7034. doi: 10.1021/bi002917g. [DOI] [PubMed] [Google Scholar]
- 229.Kumaresan KR, Hang B, Lambert MW. J Biol Chem. 1995;270:30709–30716. doi: 10.1074/jbc.270.51.30709. [DOI] [PubMed] [Google Scholar]
- 230.Kumaresan KR, Lambert MW. Carcinogenesis. 2000;21:741–751. doi: 10.1093/carcin/21.4.741. [DOI] [PubMed] [Google Scholar]
- 231.Kumaresan KR, Hwang M, Thelen MP, Lambert MW. Biochemistry. 2002;41:890–896. doi: 10.1021/bi011614z. [DOI] [PubMed] [Google Scholar]
- 232.McMahon LW, Walsh CE, Lambert MW. J Biol Chem. 1999;274:32904–32908. doi: 10.1074/jbc.274.46.32904. [DOI] [PubMed] [Google Scholar]
- 233.Bessho T, Mu D, Sancar A. Mol Cell Biol. 1997;17:6822–6830. doi: 10.1128/mcb.17.12.6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, Wood RD. J Biol Chem. 2000;275:26632–26636. doi: 10.1074/jbc.C000337200. [DOI] [PubMed] [Google Scholar]
- 235.Bessho T. J Biol Chem. 2003;278:5250–5254. doi: 10.1074/jbc.M212323200. [DOI] [PubMed] [Google Scholar]
- 236.Li L, Peterson CA, Lu X, Wei P, Legerski RJ. Mol Cell Biol. 1999;19:5619–5630. doi: 10.1128/mcb.19.8.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zhang N, Zhang X, Peterson C, Li L, Legerski R. Nucleic Acids Res. 2000;28:4800–4804. doi: 10.1093/nar/28.23.4800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Li L, Peterson CA, Zhang X, Legerski RJ. Nucleic Acids Res. 2000;28:1424–1427. doi: 10.1093/nar/28.6.1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Zhang N, Lu X, Zhang X, Peterson CA, Legerski RJ. Mol Cell Biol. 2002;22:2388–2397. doi: 10.1128/MCB.22.7.2388-2397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Zhang N, Lu X, Legerski RJ. Biochem Biophys Res Commun. 2003;309:71–78. doi: 10.1016/s0006-291x(03)01535-3. [DOI] [PubMed] [Google Scholar]