

Abstract
The metal-dependent deacetylase LpxC catalyzes the first committed step of lipid A biosynthesis in Gram-negative bacteria. Accordingly, LpxC is an attractive target for the development of inhibitors that may serve as potential new antibiotics for the treatment of Gram-negative bacterial infections. Here, we report the 2.7 Å resolution X-ray crystal structure of LpxC complexed with the substrate analogue inhibitor, TU-514, and the 2.0 Å resolution structure of LpxC complexed with imidazole. The X-ray crystal structure of LpxC complexed with TU-514 allows for a detailed examination of the coordination geometry of the catalytic zinc ion and other enzyme-inhibitor interactions in the active site. The hydroxamate group of TU-514 forms a bidentate chelate complex with the zinc ion and makes hydrogen bond interactions with conserved active site residues E78, H265, and T191. The inhibitor C-4 hydroxyl group makes direct hydrogen bond interactions with E197 and H58. Finally, the C-3 myristate moiety of the inhibitor binds in the hydrophobic tunnel of the active site. These intermolecular interactions provide a foundation for understanding structural aspects of enzyme-substrate and enzyme-inhibitor affinity. Comparison of the TU-514 complex with cacodylate and imidazole complexes suggests a possible substrate diphosphate binding site and highlights residues that may stabilize the tetrahedral intermediate and its flanking transition states in catalysis. Evidence is also presented for a catalytic zinc ion in the native zinc enzyme coordinated by H79, H238, D242, and two water molecules with square pyramidal geometry. These results suggest that the native state of this metallohydrolase may contain a pentacoordinate zinc ion, which contrasts with the native states of archetypical zinc hydrolases such as thermolysin and carboxypeptidase A.
The outer leaflet of the outer membrane of a Gram-negative bacterium is composed of lipopolysaccharide (LPS), which serves as a permeability barrier that protects the bacterium against erythromycin and other antibiotics (1-6).1 Each LPS molecule contains O-antigen polysaccharide and core oligosaccharide domains covalently attached to lipid A, a glucosamine-based phospholipid that is the immunologically active component of LPS (4-6). Lipid A is required for the viability of Gram-negative bacteria and its biosynthesis has accordingly attracted attention as a therapeutic target for the treatment of Gram-negative bacterial infections (5-10).
The first committed step in lipid A biosynthesis is catalyzed by a metal-dependent deacetylase, UDP-(3-O-((R)-3-hydroxymyristoyl))-N-acetylglucosamine deacetylase (LpxC), which hydrolyzes UDP-(3-O-((R)-3-hydroxymyristoyl))-N-acetylglucosamine to form acetate and UDP-(3-O-((R)-3-hydroxymyristoyl))glucosamine (11-14). The three-dimensional structure of LpxC from Aquifex aeolicus has been determined by X-ray crystallography (15) and NMR spectroscopy (16, 17) and reveals a novel α + β fold and the new zinc-binding motif, HKX(L,F)D. In the mechanism first proposed based on analysis of the X-ray crystal structure (15), E78 serves as a general base by abstracting a proton from the zinc-bound water molecule to promote nucleophilic attack at the substrate, and this aspect of the catalytic mechanism has been probed in detailed structural and enzymological studies (17-19).2 The results of site-directed mutagenesis studies are consistent with this role for E78 in catalysis (18, 19). The oxyanion of the resulting tetrahedral intermediate is stabilized by zinc coordination and hydrogen bonding with T191 based on intermolecular interactions observed in the LpxC-cacodylate and LpxC-palmitate complexes (19). Cacodylate and palmitate also hydrogen bond with E78 and H265, implicating these residues in the stabilization of the tetrahedral intermediate and its flanking transition states (19), which is consistent with the results of site-directed mutagenesis studies (18, 19) and the pKa values of ∼6 and ∼8 measured for E78 (18, 19) and H265, respectively (17). Site-directed mutagenesis experiments and analysis of the NMR structure of LpxC also implicate K239 as an electrostatic catalyst in the stabilization of the tetrahedral intermediate (16, 18). Although E78 has been considered a potential general acid catalyst for the protonation of the leaving amino group in the collapse of the tetrahedral intermediate (15, 18), analysis of the LpxC-cacodylate complex suggests that H265 is more appropriately positioned to protonate the leaving group (19). Accordingly, E78 and H265 may function as a general acid-base catalyst pair (Figure 1) (19, 20).
Figure 1.
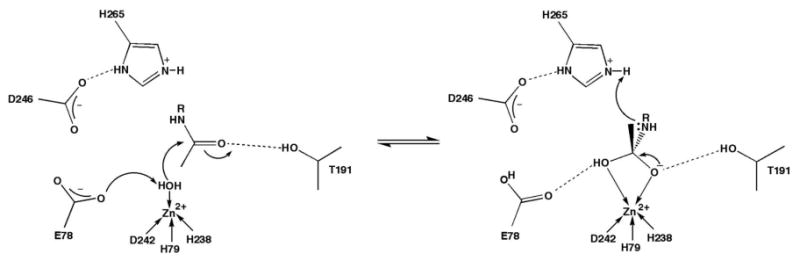
Proposed mechanism of LpxC. E78 serves as a general base to abstract a proton from a zinc-bound water molecule, which attacks the substrate carbonyl to form a tetrahedral intermediate stabilized by interactions with zinc and T191. The imidazolium side chain of H265 serves as a general acid and protonates the amine leaving group to facilitate collapse of the tetrahedral intermediate.
Recently, the structure of LpxC from A. aeolicus complexed with the substrate analogue inhibitor TU-514 (1,5-anhydro-2-C-(carboxymethyl-N-hydroxamide)-2-deoxy-3-O-myristoyl-d-glucitol) (21-23) has been determined by NMR spectroscopy (16, 17). TU-514 lacks the UDP moiety and the 3-hydroxy group of the myristoyl ester chain of the actual substrate (Figure 2). Additionally, in place of the scissile amide linkage of the substrate, TU-514 contains a hydroxamate “warhead” that targets the catalytic zinc ion. Although the hydroxamate group is a key feature responsible for high affinity inhibitor binding to LpxC, the NMR structure of the enzyme-inhibitor complex does not reveal the characteristic five-membered ring zinc-chelate interaction expected for the inhibitor hydroxamate group; instead, zinc coordination is described as tetracoordinate (16). Although the NMR structure of the LpxC–TU-514 complex yields important information regarding other features of inhibitor binding (16, 17), e. g., the binding of the O-3 myristic acid of TU-514 in the active site hydrophobic tunnel (where fatty acid binding is observed in the X-ray crystal structure of the zinc-inhibited enzyme (15)), X-ray crystallography is the definitive technique for the examination of precise enzyme-inhibitor zinc coordination and hydrogen bond interactions in the active site.
Figure 2.
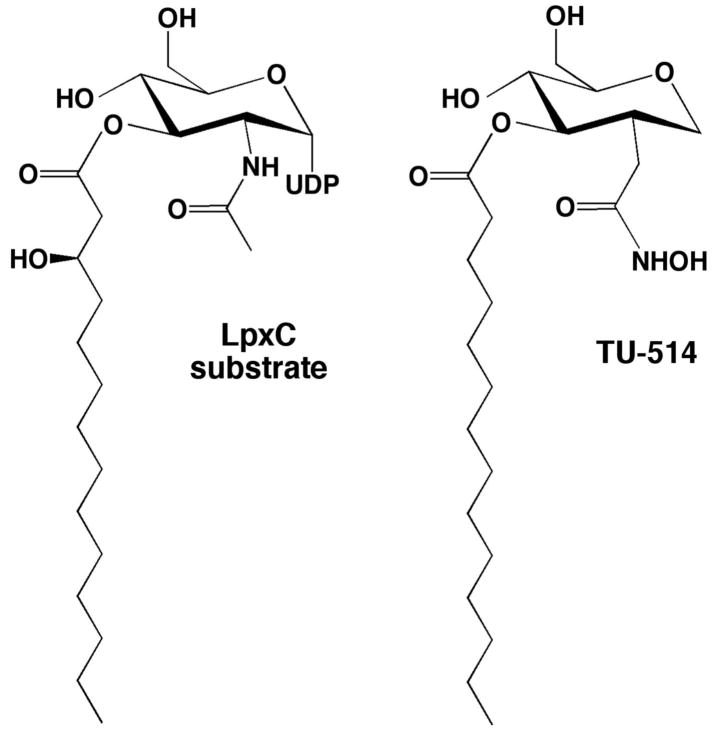
The biological substrate of LpxC, UDP-(3-O-((R)-3-hydroxymyristoyl))-N-acetylglucosamine, and the substrate analogue inhibitor 1,5-anhydro-2-C-(carboxymethyl-N-hydroxamide)-2-deoxy-3-O-myristoyl-d-glucitol (TU-514; IC50 ∼ 7 μM and Ki ∼ 650 nM against LpxC enzymes from A. aeolicus and E. coli) (21, 23).
Here, we report the 2.7 Å-resolution X-ray crystal structure of A. aeolicus LpxC complexed with TU-514, which reveals a pentacoordinate zinc ion. We also present structural evidence for two zinc-bound water molecules in the structure of wild-type LpxC determined in the presence of 5 mM imidazole. These structures provide key inferences on enzyme-substrate recognition and changes in the zinc coordination polyhedron that may accompany catalysis.
Materials and Methods
Structure determination of LpxC complexed with TU-514
C193A/ΔD284-L294 LpxC from A. aeolicus (henceforth “LpxC”) was overexpressed in E. coli and purified as described (15, 24). Crystallization of the zinc-inhibited enzyme was achieved as previously reported (19) and crystals were cross-linked with glutaraldehyde using the technique described by Lusty (25), in which LpxC crystals from hanging drops were placed over a microbridge containing a sitting droplet of 2 μL of 25% glutaraldehyde (pH 3). The microbridge rested within a reservoir containing 500 μL of stabilization buffer (100 mM HEPES (pH 7.5), 180 mM NaCl, 16% PEG 3350, and 5 mM ZnSO4). The cross-linking reaction was allowed to proceed for 45 minutes, after which the coverslip containing the hanging drop of crystals was placed over a well containing 500 μL of stabilization buffer lacking ZnSO4. An inhibitor buffer solution (100 mM HEPES (pH 7.5), 180 mM NaCl, 16% PEG 3350, 1% glycerol, and 2 mM TU-514) was slowly added to the hanging drop to a final volume of 20 μL, and crystals were equilibrated for 16 hours. Crystals were prepared for data collection by cryoprotection with 25% glycerol followed by flash-cooling in liquid nitrogen. Diffraction data were measured to 2.7 Å at Cornell High Energy Synchrotron Source (CHESS, beamline F1, Ithaca, NY). Crystals were isomorphous with those of the zinc-inhibited enzyme and belonged to space group P61 with unit cell parameters a = b = 100.7 Å, c = 121.3 Å, with two monomers in the asymmetric unit. Data were indexed and merged using the program HKL2000 (26). The structure was solved by the difference Fourier technique. It was evident in the initial electron density maps that the inhibitory zinc ion had been displaced by TU-514, which was coordinated to the catalytic zinc ion (Zn2+A) with its O-3 myristoyl ester chain extending into the hydrophobic tunnel. Poor electron density for the C-11–C-14 atoms of the myristoyl ester was observed, presumably due to the increasing molecular disorder of the aliphatic tail as it nears the end of the hydrophobic tunnel. Additionally, the C-11–C-14 segment adopts different conformations in monomers A and B in order to avoid steric clashes in the crystal lattice. Loss of the inhibitory zinc ion, Zn2+B, was confirmed in anomalous scattering difference maps (data not shown). Iterative cycles of refinement and model building were performed with the programs CNS (27) and O (28), respectively, to improve the structure as monitored by Rfree. Restrained noncrystallographic symmetry (NCS) was employed in refinement with the appropriate weights and slightly relaxed as refinement progressed as guided by Rfree. Atomic coordinates of solvent molecules and TU-514 were added during the last stages of refinement. Data collection and refinement statistics are reported in Table 1.
Table 1. Data Collection and Refinement Statistics.
| Complex | LpxC–TU-514 | LpxC-imidazole |
|---|---|---|
| Resolution range (Å) | 50.0-2.7 | 30-2.0 |
| Reflections (measured/unique) | 142655/19244 | 330287/47936 |
| Completeness (%) (overall/outer shell) | 97.7/97.2 | 99.8/99.7 |
| Rmergea (overall/outer shell) | 0.069/0.538 | 0.081/0.319 |
| <I/σ> (overall/outer shell) | 12.6/2.2 | 21.4/3.9 |
| Protein atoms (no.) b | 4298 | 4298 |
| Solvent atoms (no.) b | 64 | 419 |
| Metal ions (no.) b | 5 | 5 |
| Ligand atoms (no.) b | 60 | 88 |
| Reflections used in refinement (work/ free) | 16755/846 | 45400/2393 |
| R/Rfree c | 0.210/0.241 | 0.195/0.235 |
| r.m.s. deviations | ||
| bonds (Å) | 0.007 | 0.006 |
| angles (deg.) | 1.3 | 1.2 |
| proper dihedral angles (deg.) | 23.6 | 23.5 |
| Improper dihedral angles (deg.) | 0.8 | 0.7 |
Rmerge = Σ|Ij - <Ij>|/ΣIj, where Ij is the observed intensity for reflection j and <Ij> is the average intensity calculated for reflection j from replicate data.
Per asymmetric unit
R = Σ| |Fo| - |Fc| | / ΣFo, where R and Rfree are calculated using the working and test reflection sets, respectively.
Structure determination of LpxC complexed with imidazole
Crystals of zinc-inhibited LpxC were first cross-linked by transferring crystals to a hanging drop containing 10 μL of stabilization buffer (100 mM HEPES (pH 8.0), 180 mM NaCl, 12% PEG 3350, 1% glycerol, and 5 mM ZnSO4) over a well containing 500 μL of stabilizing buffer and 40 μL of 25% glutaraldehyde for approximately 3.5 hours. Subsequently, crystals were soaked in stabilizing buffer plus 5 mM imidazole for 15 hours. Crystals were cryoprotected in 22% glycerol and flash-frozen in liquid nitrogen. Data were collected at National Synchrotron Light Source at Brookhaven National Laboratory (beamline X4A, Upton, NY). Crystals belonged to space group P61 with unit cell parameters a = b = 101.2 Å, c = 122.2 Å. Data were indexed and merged using the program HKL2000 (26). The structure was solved using the difference Fourier technique. Initial electron density maps showed that the inhibitory zinc ion had been displaced by imidazole. In addition, other imidazole-zinc coordination interactions were evident at the Zn2+C site, and a fatty acid remnant from heterologous expression in E. coli was observed bound in the hydrophobic tunnel. In monomer A, the fatty acid was observed bound in a flipped orientation with its carboxylate head group extending from the solvent-exposed end of the tunnel. In monomer B, the fatty acid was observed bound with its carboxylate head group extending into the active site, accepting hydrogen bonds from water molecules #292 and #214. Another fatty acid remnant was bound at the interface between the two monomers. Refinement and model building were performed with the programs CNS (27) and O (28), respectively. With further refinement, the electron density for imidazole at the catalytic zinc ion was interpretable as a 50%-occupied zinc-bound imidazole and a pair of 50%-occupied zinc-bound water molecules. Additional solvent molecules were added during the last stages of refinement. Data collection and refinement statistics are listed in Table 1.
In the figures, molecular structures are illustrated using the programs Bobscript (29) and PyMOL (30). All least squares plane calculations for pentacoordinate complexes were made using routines implemented in the program SHELX (31).
Results
LpxC–TU-514 complex
The transfer of zinc-inhibited LpxC crystals into a solution containing 2 mM TU-514 results in the loss of the inhibitory zinc ion (Zn2+B) and the binding of TU-514 in the enzyme active site. The structures of enzyme monomers A and B in the asymmetric unit of the crystal are essentially identical, with an r.m.s. deviation of 0.019 Å for 267 protein Cα atoms. An electron density map of the LpxC–TU-514 complex is displayed in Figure 3. The structure of the enzyme-inhibitor complex is essentially identical to that of the zinc-inhibited enzyme, with an r.m.s. deviation of 0.241 Å for 267 Cα atoms. Both oxygens of the TU-514 hydroxamate group coordinate to the catalytic zinc ion (ZnA2+) such that the hydroxamate forms a 5-membered ring chelate complex. In addition to ZnA2+ coordination, the hydroxamate OH group is within hydrogen bonding distance to E78, H265, and the noncoordinating carboxylate oxygen of the zinc ligand D242. The hydroxamate C=O group accepts a hydrogen bond from T191. It is possible that zinc coordination facilitates ionization of the hydroxamate, as considered by Holmes and Matthews in the first X-ray crystal structures of hydroxamate inhibitors complexed with a zinc enzyme (32). The nitrogen atom of the hydroxamate is within hydrogen bonding distance to H265, which is presumed to be a hydrogen bond donor due to its elevated pKa of ∼8 (17).
Figure 3.
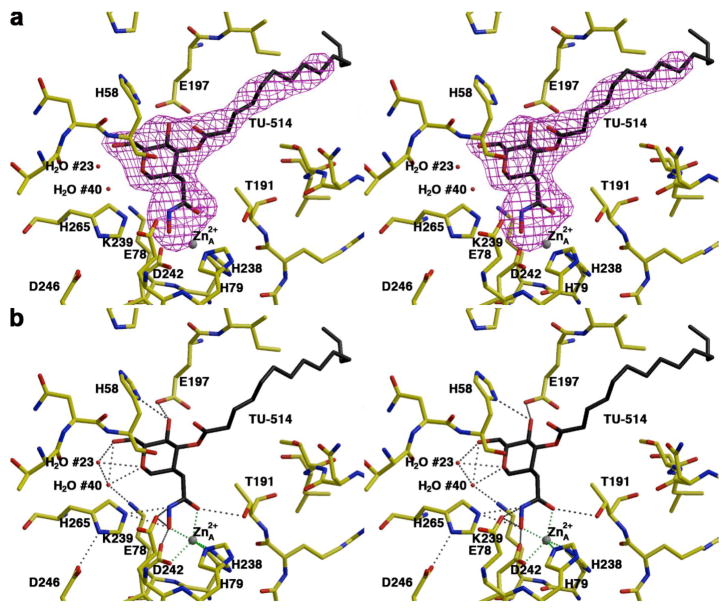
(a) Simulated annealing omit electron density map of LpxC complexed with TU-514 (monomer A, contoured at 4σ). Atoms are color-coded as follows: C = yellow (protein) or black (TU-514), O = red, N = blue; zinc appears as a gray sphere; water molecules appear as red spheres. (b) Dashed lines indicate zinc coordination (green) and hydrogen bond (gray) interactions in the LpxC–TU-514 complex.
In the LpxC–TU-514 complex, the ZnA2+ coordination polyhedron is pentacoordinate with approximate square pyramidal geometry (average deviation = 15° from ideal geometry), with ZnA2+ ∼0.5 Å out of the least-squares plane defined by H238, D242, and the hydroxamate C=O and OH groups; H79 is the apical ligand. The average trigonality index τ = 0.13 for the two enzyme molecules in the asymmetric unit, which corresponds to square pyramidal coordination geometry with very slight trigonalization (τ = 0 for ideal square pyramidal and τ = 0 for ideal trigonal bypyramidal geometry) (33). The calculation of the trigonality index τ as an angular structural parameter is analogous to the approach of Muetterties and Guggenberger in using certain shape-determining dihedral angles to characterize the reaction coordinate of square pyramidal⇔trigonal bipyramidal ligand rearrangements in Berry pseudorotation processes (34). The distances of zinc coordination interactions are recorded in Table 2. In Figure 4, the square pyramidal ZnA2+ coordination polyhedron in the LpxC–TU-514 complex is compared with ZnA2+ coordination polyhedra observed in other LpxC complexes.
Table 2. ZnA2+ Coordination Interactions (Å)*.
| Ligand | LpxC–TU-514
Complex |
LpxC-Imidazole
Complex |
|---|---|---|
| H79 Nε2 | 2.1/2.1 | 2.1/2.1 |
| H238 Nε2 | 2.0/2.1 | 2.1/2.1 |
| D242 Oδ1 | 2.2/2.0 | 2.1/2.1 |
| Hydroxamate C=O | 2.2/2.1 | — |
| Hydroxamate OH | 2.0/1.9 | — |
| Imidazole N1 | — | 2.1/2.0 |
| H2O #416 / #414 | — | 2.3/2.3 |
| H2O #417 / #415 | — | 2.3/2.3 |
monomer A/B
Figure 4.
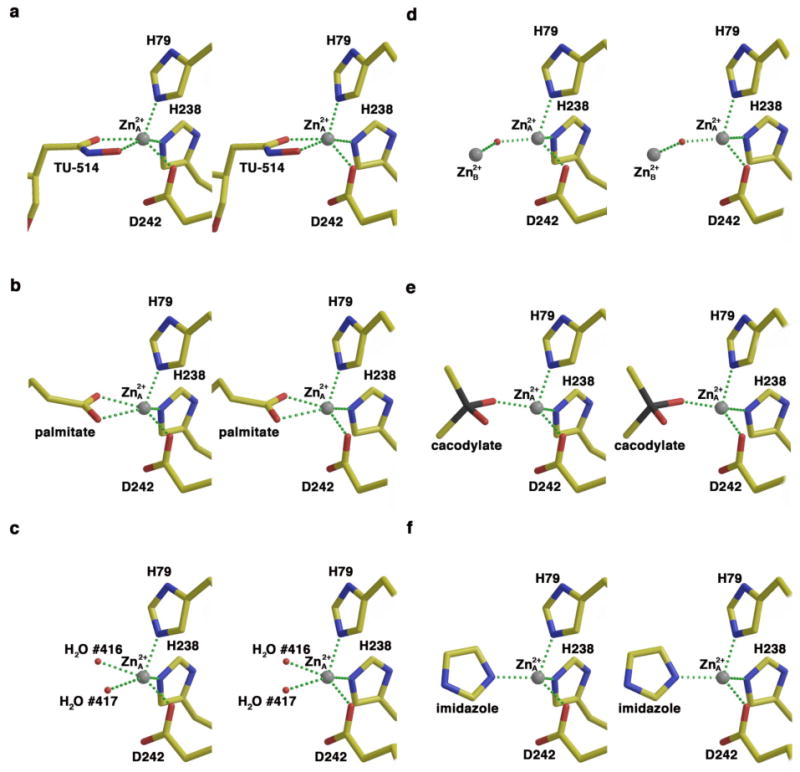
Square pyramidal zinc coordination geometry in the (a) LpxC–TU-514 complex, (b) LpxC-palmitate complex (PDB accession code 1YH8) (19), and (c) LpxC-imidazole complex (50%-occupied zinc-bound water molecules); tetrahedral zinc coordination geometry in the (d) zinc-inhibited enzyme (PDB accession code 1P42) (15), (e) LpxC-cacodylate complex (PDB accession code 1YHC) (19), and (f) LpxC-imidazole complex (zinc-bound imidazole). Atoms are color-coded as follows: C = yellow, O = red, N = blue As = black; zinc appears as a gray sphere; zinc-bound water molecules appear as small red spheres in (c), and the metal-bridging water molecule appears as a small red sphere in (d).
The hexose ring of TU-514 is in the chair conformation and makes extensive hydrogen bond interactions in the active site, as expected from the results of chemical mapping experiments (35). In monomer A, the hexose ring oxygen accepts hydrogen bonds from water molecules #23 and #40, and these water molecules also hydrogen bond to the C-6 hydroxyl group of the hexose ring. Water molecule #40 is hydrogen bonded to water molecule #23 and K239 (Figure 3). In monomer B, the hexose ring oxygen and the C-6 hydroxyl group are hydrogen bonded to water molecule #41. In both monomers A and B, the C-4 hydroxyl group of the hexose ring hydrogen bonds with E197 and H58. Residue K239 is strictly conserved among 61 currently sequenced LpxC enzymes, but E197 is only partially conserved (16/61 as glutamate, 4/61 as glutamine, and 41/61 as aspartate), and H58 is not conserved (1/61; this residue usually appears as methionine or leucine, and sometimes as arginine).
Although the r.m.s. deviations of the TU-514 coordinates between the X-ray crystal structure (monomer A) and the ensemble of NMR structures (PDB accession code 1XXE) (17) are in the range of 1.45 – 2.31 Å, the crystal structure of the LpxC–TU-514 complex is in general agreement with the NMR structure (16, 17) with regard to the binding of the O-3 myristate moiety in the active site hydrophobic tunnel. This binding is comparable to that observed for myristate in the crystal structure determination of the zinc-inhibited enzyme (15) as well as that observed for palmitate binding to the native enzyme (19). A superposition of the NMR structure ensemble and the X-ray crystal structure is found in Figure 5.
Figure 5.
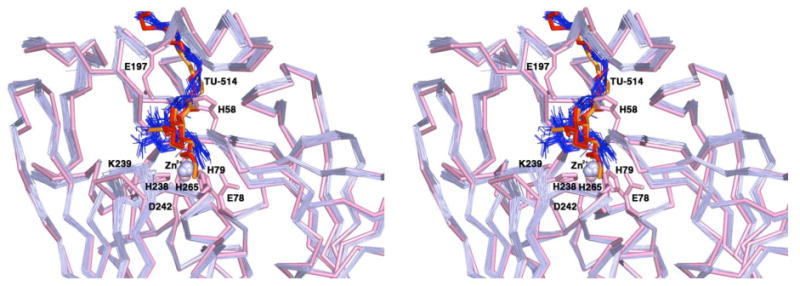
Superposition of the X-ray crystal structure of the LpxC–TU-514 complex (monomers A and B) with the ensemble of 25 NMR structures determined by Coggins and colleagues (17) (PDB code 1XXE), oriented so that the viewer is looking directly into the main active site cleft. Atoms from the X-ray crystal structure are color-coded as follows: main chain carbon atoms = pink; zinc appears as a pink sphere; TU-514 appears in red (monomer A) and orange (monomer B). Selected side chains are indicated and are colored pink. Atoms from the ensemble of 25 NMR strutures are color-coded as follows: main chain carbon atoms = light blue; Zn2+ ions = light blue spheres; TU-514 molecules = blue.
LpxC-imidazole complex
The transfer of zinc-inhibited LpxC crystals into a solution containing 5 mM imidazole results in loss of the inhibitory zinc ion (Zn2+B) and the binding of imidazole to the catalytic zinc ion (Zn2+A) and to another zinc ion (Zn2+C) identified in the zinc-inhibited structure (15). Electron density for Zn2+A-bound imidazole is characterized by additional features interpretable as a pair of Zn2+A-bound water molecules (#417/#415 and #416/#414 in monomer A/B), mutually exclusive with imidazole at 50% occupancy (Figure 6). The pentacoordinate water-bound complex represents the unliganded, native state of LpxC, and is similar to that observed for the native states of MshB, a zinc deacetylase from Mycobacterium tuberculosis (36), and PgdA, a zinc deacetylase from Streptococcus pneumoniae (37). The structure of the LpxC-imidazole complex is otherwise identical to that of the zinc-inhibited enzyme (15), with an r.m.s. deviation of 0.249 Å for 267 Cα atoms.
Figure 6.
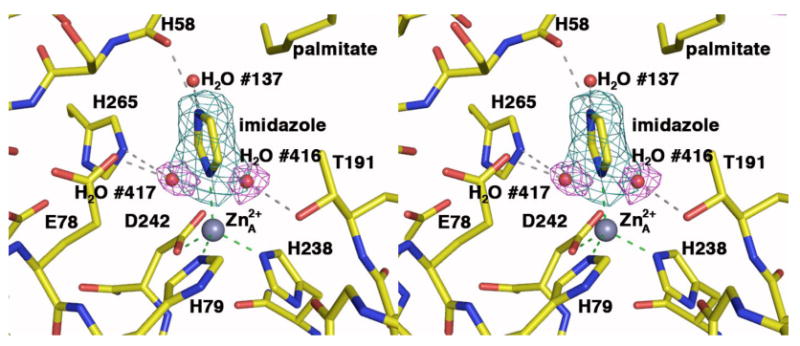
Simulated annealing omit electron density maps of LpxC (monomer A) complexed with imidazole (cyan, contoured at 4σ) and water molecules #416 and 417 (magenta, contoured at 4σ). Atoms are color-coded as follows: C = yellow, O = red, N = blue; zinc appears as a gray sphere and water molecules appear as red spheres. Dashed lines indicate zinc coordination (green) and hydrogen bond (gray) interactions.
The Zn2+A coordination polyhedron is tetrahedral in the complex with imidazole (average deviation = 9° from ideal geometry). In monomer A, the imidazole N1 atom coordinates to Zn2+A and N3–H donates a hydrogen bond to the backbone carbonyl of H58. In monomer B, N3–H donates a hydrogen bond to the backbone carbonyl of H58 and also to water molecule #214.
In the unliganded enzyme structure, water molecules #417/#415 and #416/#414 (monomer A/B) form a pentacoordinate complex with Zn2+A that adopts approximate square pyramidal geometry (average deviation = 13° from ideal geometry) with Zn2+A ∼0.4 Å out of the least-squares plane defined by H238, D242, and water molecules #417/#415 and #416/#414; H79 is the apical ligand. The average trigonality index τ = 0.12 for the two enzyme molecules in the asymmetric unit, which corresponds to very slightly trigonalized square pyramidal coordination geometry (33). Water molecule #417/#415 is within hydrogen bonding distance to E78 and H265, and water molecule #416/#414 is within hydrogen bonding distance to T191. Zinc coordination polyhedra in LpxC complexes are illustrated and compared in Figure 4.
Discussion
Structural basis of inhibitor affinity
The X-ray crystal structure of the LpxC–TU-514 complex provides important information that will aid in the future design of inhibitors capable of targeting enzymes from a broad spectrum of Gram-negative bacteria. To date, it has been demonstrated that TU-514 exhibits modest inhibition against LpxC enzymes from A. aeolicus and E. coli with IC50 values of ∼7 μM against each (21) and Ki values of ∼650 nM against each (23). It is notable that apart from a water-mediated hydrogen bond interaction between the hexose ring oxygen of TU-514 and strictly conserved K239, all of the intermolecular hydrogen bond interactions of the hexose ring occur with LpxC residues that are at best only partially conserved across the species. Accordingly, the hexose ring may be dispensable in inhibitor design. In this regard, it is interesting that fatty acid derivatives containing functional groups such as CO2-, SO3- and PO32-, but completely lacking a hexose ring moiety, exhibit dissociation constants as low as 300 nM against LpxC (15). Additionally, a slow-binding LpxC inhibitor lacking the hexose moiety, CHIR-090, exhibits a Ki value of ∼1 nM (23).
Despite the significant differences between the X-ray crystal structure and the NMR structure of the LpxC–TU-514 complex in the vicinity of the catalytic zinc ion, a feature common to both structures is the insertion of the C3-myristoyl tail into the conserved, ∼15-Å-long hydrophobic tunnel. As previously suggested, this may stabilize the conformation of the βαβ subdomain that frames the hydrophobic tunnel and forms one wall of the active site cleft (15, 16). The NMR spectra indicate increased mobility for this region of the protein structure in the absence of inhibitor binding, so the insertion of the C-3 myristoyl tail into the hydrophobic tunnel is likely required to stabilize the active site in the proper conformation for catalysis (16). This hypothesis is supported by more than a 5 × 106-fold decrease in activity observed for the catalysis of the deacetylation of UDP-N-acetylglucosamine, which lacks a 3-O-(R-3 hydroxymyristoyl) substituent to bind in the hydrophobic tunnel (24).
Thus, as first noted by Whittington and colleagues (15), two key, minimal components of an LpxC inhibitor are (1) a functional group to coordinate to Zn2+A and (2) a long hydrophobic moiety to occupy the hydrophobic tunnel. Additional affinity determinants outlined by Coggins and colleagues (17) include interactions with the hydrophobic patch defined by F161, F192, and F194, and the “basic patch” defined by K239, R143, R262, and H265. Among the 61 currently sequenced LpxC enzymes, residues within the hydrophobic patch are either fully conserved (F192) or highly conserved (F161 occasionally appears as tyrosine, and F194 occasionally appears as leucine). However, residues R143 and R262 within the basic patch are poorly conserved, with only K239 and H265 appearing as strictly conserved residues. The basic patch is proposed as a possible binding site for the substrate diphosphate group (17), and analysis of the X-ray crystal structure of the LpxC-UDP complex suggests that additional intermolecular interactions can be exploited in the basic patch to enhance enzyme-inhibitor affinity (unpublished results).
Inferences on substrate binding
The X-ray crystal structures of the LpxC–TU-514 complex and the LpxC-cacodylate complex (19) yield important inferences on substrate binding in the active site. The UDP group is an axial substituent of the C-1 carbon of the hexose ring of the substrate and is proposed to interact with positively charged residues, such as those in the basic patch (17), for stabilization of the negatively charged diphosphate group. Interestingly, in the previously reported structure of LpxC complexed with cacodylate (19) (PDB accession code 1YHC), a sulfate ion binds in the basic patch. This sulfate ion makes an electrostatic interaction with K239 (3.5 Å) and accepts a hydrogen bond from the backbone NH of H265, the carboxylic acid head group of palmitate, and a water molecule. The sulfate ion is positioned approximately 2 Å away from the hexose C-1 atom of TU-514 when the structures of the LpxC–TU-514 and LpxC-cacodylate complexes are superimposed (Figure 7a). Indeed, the crystal structure of the LpxC-UDP complex confirms that the bound sulfate ion mimics the binding of a UDP phosphate group (unpublished results).
Figure 7.
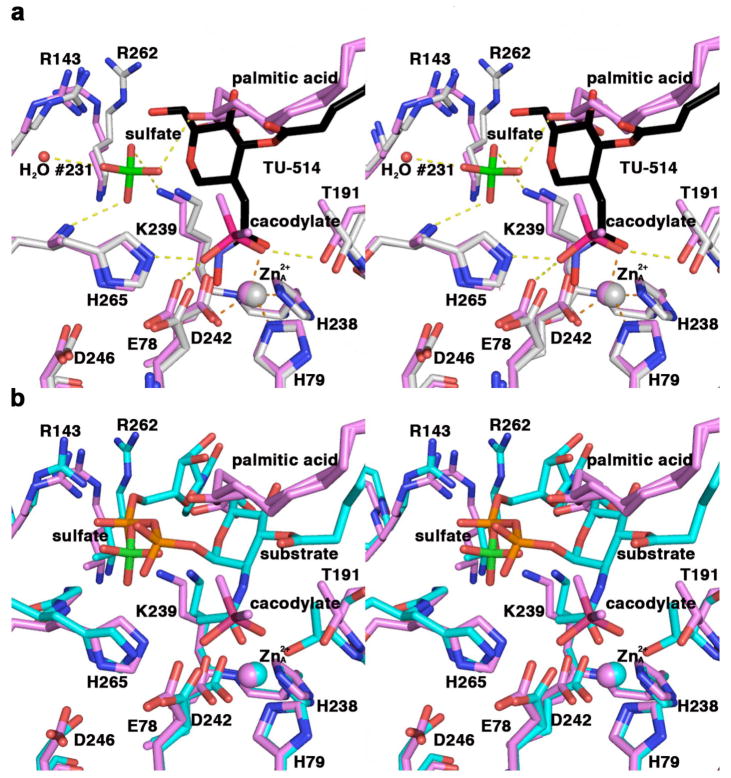
(a) Superposition of LpxC-cacodylate (violet) and LpxC–TU-514 (gray; TU-514 is shown in black) complexes. Hydrogen bond interactions (yellow) and metal coordination interactions (orange) in the LpxC-cacodylate complex are indicated with dashed lines. All non-carbon atoms are color coded as follows: O = red; N = blue; S = green; As = magenta; zinc ions are violet (LpxC-cacodylate complex) or gray (LpxC–TU-514 complex) spheres; water molecule #231 appears as a small red sphere. (b) Superposition of the LpxC-cacodylate complex (violet) and a model of the LpxC-substrate complex (cyan). All non-carbon atoms are color coded as follows: O = red; N = blue; S = green; As = magenta; P = orange; zinc ions are violet (LpxC-cacodylate complex) or cyan (model of the LpxC–substrate complex) spheres.
Superposition of the LpxC-cacodylate complex with the LpxC–TU-514 complex also provides insight regarding the position and orientation of the tetrahedral intermediate in the proposed catalytic mechanism (17-19). The arsenic atom of cacodylate is 0.7 Å away from the carbonyl carbon of the hydroxamate head group of TU-514. The As-O2 bond closely aligns with the hydroxamate C=O group and may thus represent the position of the substrate C=O group in the enzyme-substrate complex. The structural inferences emanating from the comparison of the two complexes in Figure 7a suggest the model for substrate binding illustrated in Figure 7b, which updates the model first presented by Whittington and colleagues (15).
Zinc coordination geometry and catalysis
Both tetrahedral and square pyramidal Zn2+A coordination geometries are observed in X-ray crystal structures of LpxC complexes, and interconversions between these two geometries may accompany catalysis. The proposed catalytic mechanism involves ionization of a water molecule bound to a tetrahedrally-coordinated Zn2+A with the subsequent formation of an oxyanion intermediate stabilized by Zn2+A coordination and electrostatic interactions with E78, H265, and T191 (Figure 1) (17-19). Unexpectedly, the imidazole-LpxC complex provides structural evidence for a native Zn2+A ion coordinated by H79, H238, D242, and two water molecules with square pyramidal geometry. This could possibly indicate that substrate binding requires the displacement of one zinc-bound water molecule to allow for some degree of substrate C=O---Zn2+A interaction and the retention of the second zinc-bound water molecule as the catalytic nucleophile. This would be similar to the mechanisms proposed for the zinc deacetylases MshB from M. tuberculosis (36) and PgdA from S. pneumoniae (37), both of which contain pentacoordinate zinc ions in their native states.
However, extended X-ray absorption fine structure (EXAFS) spectroscopic studies of LpxC from A. aeolicus and P. aeruginosa suggest a 4-coordinate zinc ion and a 5-coordinate Co2+ ion (38), so it seems that the equilibrium between a 4- and 5-coordinate metal ion in LpxC is sensitive to changes in the protein environment. In fact, the conditions utilized in the EXAFS experiments are quite different from those employed in the X-ray crystallographic studies: EXAFS experiments were performed with protein prepared under conditions of low ionic strength (samples were desalted prior to analysis and stabilized in 25 mM HEPES (pH 7.0), 1.5 mM tris-2-carboxyethylphosphine, and 10% glycerol), whereas the crystals of LpxC with two zinc-bound water molecules were prepared under conditions of higher ionic strength and stabilized in 100 mM HEPES (pH 8.0), 180 mM NaCl, 12% PEG 3350, 22% glycerol, 5 mM ZnSO4, and 5 mM imidazole. Differences in protein preparations may contribute to the apparent discrepancy between EXAFS and X-ray crystallography results.
The interesting mechanistic feature that the current work highlights is that the native zinc ion in LpxC has a propensity for pentacoordination with square pyramidal coordination geometry under some conditions, which could modify extant mechanistic proposals (15-20). The possibility of a pentacoordinate rather than a tetracoordinate catalytic zinc ion in the native state of LpxC is consistent with native pentacoordinate zinc ions in other zinc-dependent bacterial deacetylases (36, 37) and contrasts with the zinc coordination observed in the native states of the archetypical zinc hydrolases thermolysin (39) and carboxypeptidase A (40). Future structural studies of LpxC will allow us to clarify aspects of Zn2+A coordination changes in the catalytic mechanism.
Acknowledgments
We thank the Cornell High Energy Synchrotron Source (CHESS, Ithaca, NY) and the National Synchrotron Light Source at Brookhaven National Laboratory (NSLS, Upton, NY) for beamline access. Also, we thank Dr. Luigi Di Costanzo for assistance with data collection and Drs. Marjorie Harding and Anthony Addison for helpful scientific discussions.
Footnotes
This work was supported by National Institutes of Health grants GM49758 (D. W. C.) and GM40602 (C. A. F.)
The atomic coordinates for LpxC complexed with TU-514 and imidazole have been deposited in the Protein Data Bank, www.rcsb.org, with accession codes 2GO4 and 2GO3, respectively.
Abbreviations: LPS, lipopolysaccharide; TU-514, 1,5-anhydro-2-C-(carboxymethyl-N-hydroxamide)-2-deoxy-3-O-myristoyl-d-glucitol; UDP, uridine 5′-diphosphate; NCS, noncrystallographic symmetry; HEPES, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid; PEG, polyethylene glycol; EXAFS, extended X-ray absorption fine structure.
Here, the numbering system for the well-studied LpxC enzyme from E. coli is adapted for the A. aeolicus enzyme. Based on an alignment performed with Clustal W, important residues conserved between E. coli/A. aeolicus correspond as follows: R58/H58, E78/E73, H79/H74, K143/R137, F161/F155, T191/T179, F192/F180, F194/F182, E197/D185, H238/H226, K239/K227, D242/D230, D246/D234, K262/R250, and H265/H253.
References
- 1.Vaara M. Agents that increase the permeability of the outer membrane. Microbiol Rev. 1992;56:395–411. doi: 10.1128/mr.56.3.395-411.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vuorio R, Vaara M. The lipid A biosynthesis mutation lpxA2 of Escherichia coli results in drastic antibiotic supersusceptibility. Antimicrob Agents Chemother. 1992;36:826–829. doi: 10.1128/aac.36.4.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaara M. Outermembrane permeability barrier to azithromycin, clarithromycin, and roxithromycin in Gram-negative enteric bacteria. Antimicrob Agents Chemother. 1993;37:354–356. doi: 10.1128/aac.37.2.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nikaido H, Vaara M. Molecular basis of bacterial outer membrane permeability. Microbiol Rev. 1985;49:1–32. doi: 10.1128/mr.49.1.1-32.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raetz CRH. Biochemistry of endotoxins. Annu Rev Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 6.Raetz CRH. Bacterial endotoxins: extraordinary lipids that activate eukaryotic signal transduction. J Bacteriol. 1993;175:5745–5753. doi: 10.1128/jb.175.18.5745-5753.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen MH, Patchett AA, Galloway SM, Hyland SA, Anderson MS, Raetz CRH. Antibacterial agents that inhibit lipid A biosynthesis. Science. 1996;274:980–982. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 8.Wyckoff TJ, Raetz CRH, Jackman JE. Antibacterial and anti-inflammatory agents that target endotoxin. Trends Microbiol. 1998;6:154–159. doi: 10.1016/s0966-842x(98)01230-x. [DOI] [PubMed] [Google Scholar]
- 9.Vaara M. Endotoxin in health and disease. Marcel Dekker, Inc.; New York, NY: 1999. pp. 31–32. [Google Scholar]
- 10.Rick PD, Raetz CRH. Endotoxin in health and disease. Marcel Dekker, Inc.; New York, NY: 1999. pp. 283–304. [Google Scholar]
- 11.Anderson MS, Bulawa CE, Raetz CR. The biosynthesis of Gram-negative endotoxin. Formation of lipid A precursors from UDP-GlcNAc in extracts of Escherichia coli. J Biol Chem. 1985;260:15536–15541. [PubMed] [Google Scholar]
- 12.Anderson MS, Robertson AD, Macher I, Raetz CRH. Biosynthesis of lipid A in Escherichia coli: identification of UDP-3-O-[(R)-3-hydroxymyristoyl]-alpha-D-glucosamine. Biochemistry. 1988;27:1908–1917. doi: 10.1021/bi00406a017. [DOI] [PubMed] [Google Scholar]
- 13.Anderson MS, Bull HG, Galloway SM, Kelly TM, Mohan S, Radika K, Raetz CRH. UDP-N-acetylglucosamine acyltransferase of Escherichia coli. The first step of endotoxin biosynthesis is thermodynamically unfavorable. J Biol Chem. 1993;268:19858–19865. [PubMed] [Google Scholar]
- 14.Young K, Silver LL, Bramhill D, Cameron P, Eveland SS, Raetz CRH, Hyland SA, Anderson MS. The envA permeability/cell division gene of Escherichia coli encodes the second enzyme of lipid A biosynthesis. UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase. J Biol Chem. 1995;270:30384–30391. doi: 10.1074/jbc.270.51.30384. [DOI] [PubMed] [Google Scholar]
- 15.Whittington DA, Rusche KM, Shin H, Fierke CA, Christianson DW. Crystal structure of LpxC, a zinc-dependent deacetylase essential for endotoxin biosynthesis. Proc Natl Acad Sci U S A. 2003;100:8146–8150. doi: 10.1073/pnas.1432990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coggins BE, Li X, McClerrin AL, Hindsgaul O, Raetz CRH, Zhou P. Structure of the LpxC deacetylase with a bound substrate-analog inhibitor. Nat Struct Biol. 2003;10:645–651. doi: 10.1038/nsb948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coggins BE, McClerrin AL, Jiang L, Li X, Rudolph J, Hindsgaul O, Raetz CRH, Zhou P. Refined solution structure of the LpxC–TU-514 complex and pKa anlaysis of an active site histidine: insights into the mechanism and inhibitor design. Biochemistry. 2005;44:1114–1126. doi: 10.1021/bi047820z. [DOI] [PubMed] [Google Scholar]
- 18.McClerren AL, Zhou P, Guan Z, Raetz CRH, Rudolph J. Kinetic analysis of the zinc-dependent deacetylase in the lipid A biosynthetic pathway. Biochemistry. 2005;44:1106–1113. doi: 10.1021/bi048001h. [DOI] [PubMed] [Google Scholar]
- 19.Hernick M, Gennadios HA, Whittington DA, Rusche KM, Christianson DW, Fierke CA. UDP-(3-O-((R)-3-hydroxymyristoyl))-N-acetylglucosamine deacetylase functions through a general acid-base catalyst pair mechanism. J Biol Chem. 2005;280:16969–16978. doi: 10.1074/jbc.M413560200. [DOI] [PubMed] [Google Scholar]
- 20.Hernick M, Fierke CA. Zinc hydrolases: the mechanisms of zinc-dependent deactylases. Arch Biochem Biophys. 2004;433:71–84. doi: 10.1016/j.abb.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 21.Jackman JE, Fierke CA, Tumey LN, Pirrung M, Uchiyama T, Tahir SH, Hindsgaul O, Raetz CRH. Antibacterial agents that target lipid A biosynthesis in Gram-negative bacteria. J Biol Chem. 2000;275:11002–11009. doi: 10.1074/jbc.275.15.11002. [DOI] [PubMed] [Google Scholar]
- 22.Li X, Uchiyama T, Raetz CRH, Hindsgaul O. Synthesis of a carbohydrate-derived hydroxamic acid inhibitor of the bacterial enzyme (LpxC) involved in lipid A biosythesis. Org Lett. 2003;5:539–541. doi: 10.1021/ol027458l. [DOI] [PubMed] [Google Scholar]
- 23.McClerren AL, Endsley S, Bowman JL, Andersen NH, Guan Z, Rudolph J, Raetz CRH. A slow, tight-binding inhibitor of the zinc-dependent deacetylase LpxC of lipid A biosynthesis with antibiotic activity comparable to ciproflaxin. Biochemistry. 2005;44:16574–16583. doi: 10.1021/bi0518186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackman JE, Raetz CRH, Fierke CA. UDP-(3-O-(R-3-hydroxymyristoyl))-N-acetylglucosamine deacetylase of Escherichia coli is a zinc metalloenzyme. Biochemistry. 1999;38:1902–1911. doi: 10.1021/bi982339s. [DOI] [PubMed] [Google Scholar]
- 25.Lusty CJ. A gentle vapor-diffusion technique for cross-linking of protein crystals for cryocrystallography. Applied Crystallogr. 1999;32:106–112. [Google Scholar]
- 26.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 27.Brünger AT, Adams PD, Clore GM, De Lano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 28.Jones TA, Zou JY, Cowan SW, Kjeldjaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 29.Esnouf RM. An extensively modified version of MOLSCRIPT that includes greatly enhanced coloring capabilities. J Graph Model. 1997;15:132–134. 112–113. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 30.DeLano WL. The PyMOL molecular graphics system. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 31.Sheldrick GM, Schneider TR. SHELXL: High resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 32.Holmes MA, Matthews BW. Binding of hydroxamic acid inhibitors to crystalline thermolysin suggests a pentacoordinate zinc intermediate in catalysis. Biochemistry. 1981;20:6912–6920. doi: 10.1021/bi00527a026. [DOI] [PubMed] [Google Scholar]
- 33.Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC. Synthesis, structure, and spectroscopic properties of copper (II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper (II) perchlorate. J Chem Soc Dalton Trans. 1984:1349–1356. [Google Scholar]
- 34.Muetterties EL, Guggenberger LJ. Idealized polytopal forms. Description of real molecules referenced to idealized polygons or polyhedra in geometric reaction path form. J Am Chem Soc. 1974;96:1748–1756. [Google Scholar]
- 35.Li X, McClerren AL, Raetz CRH, Hindsgaul O. Mapping the active site of the bacterial enzyme LpxC using novel carbohydrate-based hydroxamic acid inhibitors. J Carb Chem. 2005;24:583–609. [Google Scholar]
- 36.Maynes JT, Garen C, Cherney MM, Newton G, Arad D, Av-Gay Y, Fahey RC, James MNG. The crystal structure of 1-D-myo-inosityl-2-acetamido-2-deoxy-alpha-D-gucopyranoside deacetylase (MshB) from Mycobacterium tuberculosis reveals a zinc hydrolase with a lactate dehydrogenase fold. J Biol Chem. 2003;278:47166–47170. doi: 10.1074/jbc.M308914200. [DOI] [PubMed] [Google Scholar]
- 37.Blair DE, Schuttelkopf AW, McRae JI, van Aalten DMF. Structure and metal-dependent mechanism of peptidoglycan deactylase, a streptococcal virulence factor. Proc Natl Acad Sci U S A. 2005;102:15429–15434. doi: 10.1073/pnas.0504339102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McClure CP, Rusche KM, Peariso K, Jackman JE, Fierke CA, Penner-Hahn JE. EXAFS studies of the zinc sites of UDP-(3-O-acyl)-N-acetylglucosamine deacetylase (LpxC) J Inorg Biochem. 2003;94:78–85. doi: 10.1016/s0162-0134(02)00611-6. [DOI] [PubMed] [Google Scholar]
- 39.Matthews BW. Structural basis of the action of thermolysin and related zinc peptidases. Acc Chem Res. 1988;21:333–340. [Google Scholar]
- 40.Christianson DW, Lipscomb WN. Carboxypeptidase A. Acc Chem Res. 1989;22:62–69. [Google Scholar]