

Abstract
A new design for DNA bases and base pairs is described in which the pyrimidine bases are widened by naphtho-homologation. Two naphtho-homologated deoxyribosides, dyyT (1) and dyyC (2) were synthesized and could be incorporated into oligonucleotides as suitably protected phosphoramidite derivatives. The deoxyribosides were found to be fluorescent, with emission maxima at 446 and 433 nm, respectively. Studies with single substitutions of 1 and 2 in the natural DNA context revealed exceptionally strong base stacking propensity for both. Sequences containing multiple substitutions of 1 and 2 paired opposite adenine and guanine were subsequently mixed and studied by several analytical methods. Data from UV mixing experiments, FRET measurements, fluorescence quenching experiments, and hybridizations on beads suggest that complementary “doublewide DNA” (yyDNA) strands may self-assemble into helical complexes with 1:1 stoichiometry. Data from thermal denaturation plots and CD spectra were less conclusive. Control experiments in one sequence context gave evidence that yyDNA helices, if formed, are preferentially antiparallel and are sequence selective. Hypothesized base pairing schemes are analogous to Watson-Crick pairing, but with glycosidic C1′-C1′ distances widened by over 45%, to ca. 15.2 Å. The possible self-assembly of the double-wide DNA helix establishes a new limit for the size of information-encoding, DNA-like molecules, and the fluorescence of yyDNA bases suggests uses as reporters in monomeric and oligomeric forms.
Keywords: naphthalene, homolog, steric effects, expanded helix, base pair, fluorescence
Introduction
The sizes and shapes of DNA bases can have powerful effects in their roles as encoders of genetic information. For example, in the DNA helix, the purine-pyrimidine architecture of natural DNA pairs works in four different variations because this conserved structure gives virtually the same geometry for all four pairs.1 This allows for a regular backbone conformation in the helix. Although mismatched base pairs can stack and hydrogen bond in ways similar to correct pairs, the relative rigidity of the DNA backbone helps enforce the correct geometry and elicits an energetic cost if the geometry is wrong.2 In enzyme active sites, DNA bases and pair sizes can be even more tightly constrained by the surrounding protein,3 imposing very high selectivity that is crucial in biological processes such as DNA replication.
We have undertaken a program to test the limits of size and steric constraints in the DNA backbone and in proteins that process or bind DNA. This has been pursued by two strategies: by development of a series of nonpolar DNA nucleoside mimics of increasing size,4 and by constructing DNA base analogues that retain hydrogen bonding capacity, but are rendered larger than the natural bases by benzo-homologation.5,6 In the latter vein, we recently described a set of benzo-homologous nucleosides, called “xDNA” (short for “expanded DNA”) nucleosides.7 Using the natural DNA backbone but possessing an expanded base pair size, xDNA pairs can be assembled into helices that are more stable than natural DNA owing to their enhanced base stacking proficiency. More recently, we described a second geometric nucleobase expansion, also employing benzo-homologation, termed “yDNA” for “wide DNA”.8 Like xDNA, this second variant also expands the base pair length by approximately 2.4 Å (the width of a benzene ring), but with a different vector and geometry from that of xDNA. Experiments have shown that, also like xDNA, yDNA helices are more thermally stable than natural DNA of the analogous sequence, most likely because the added base surface area and hydrophobicity contributes to greater stacking affinity.7d,7f,8a,8c In addition to their high affinity for natural sequences of DNA, xDNA and yDNA oligomers are also of interest because they are fluorescent,7a,7b,7f,8a,8b which suggests their possible use in detection or imaging of natural nucleic acid sequences.
Other researchers have also begun to investigate the effects of increased DNA base pair sizes on the stability of the double helix. For example, a number of examples of isolated purine-purine base pairs within natural DNA have been reported.9 Those experiments have shown that single pairs of non-canonical width within a natural-sized DNA context is destabilizing to the helix. However, when all the pairs take on such expanded width, the local energy cost of conformational distortion is lessened or lost altogether.7,8 In this vein, a few cases of helices having apparently all-purine-purine pairs have been reported,10 although structural information is not yet available on those systems. In related experiments, Matsuda has examined the possibility of expanding DNA base pairs in their width and/or length, with designs for pairs having four rather than the canonical three hydrogen bonding groups.11 Matteucci, Moreau, Seley, and Saito all have reported DNA base analogs larger than natural ones, although they were not designed to expand the size of the helix.6,12
To date, the greatest expansion of the helix reported is that associated with the yDNA and xDNA systems, and the xDNA system is the only case for which there is high-resolution structural information.7c The added base pair width in that 10-base-pair example results in a right handed helix with more base pairs per turn than the natural helix. Despite the difference in size, the phosphodiester backbone conformation resembles the natural B-form, with small adjustments in dihedral angles made to account for the larger helix diameter.
The successful self-assembly of the large xDNA and yDNA helices raises the interesting question as to how far the DNA base pair size can be extended and still retain the iconic helix-forming ability. Here we report a more extreme test of this question, by development of a new, doubly widened base structure, which we abbreviate yyDNA (for “doublewide DNA”). Nucleosides containing these highly fluorescent base analogs may be useful as reporters in biochemical systems. In addition, some lines of evidence suggest that this greater geometric displacement, amounting to approximately 4.8 Å, may still allow for sequence-selective double helix assembly.
Results
Design and synthesis
The previous yDNA base design started with the pyrimidine bases C and T(U), and extended them by insertion of benzene rings, with an extension vector 2.4 Å in length (for one ring) and an orientation −120° from the glycosidic bond.8a,8b Thus yyDNA, with a second benzene ring relative to yDNA (see Figure 1, and Figure S1 in Supporting Information), simply extends that earlier design further in the same direction. For example, yyC, a naphtho-homologated variant of cytosine, could in principle pair with G to yield a base pair extended by 4.8 Å (the width of naphthalene). When compared with the natural glycosidic distance in a base pair (10.4 Å) this yields an increase of over 45%, assuming that such a 4.8 Å distortion of the DNA backbone is not too energetically costly. Of course, yyT might be expected to pair with A in the analogous manner. Both the doubly widened deoxyribosides were previously unknown; the closest literature precedents are a naphtho-homologated ribonucleoside of adenine reported by Leonard in 1984,5b and a naphtho-uracil N-nucleoside described by Moreau eight years ago.6b
Figure 1.
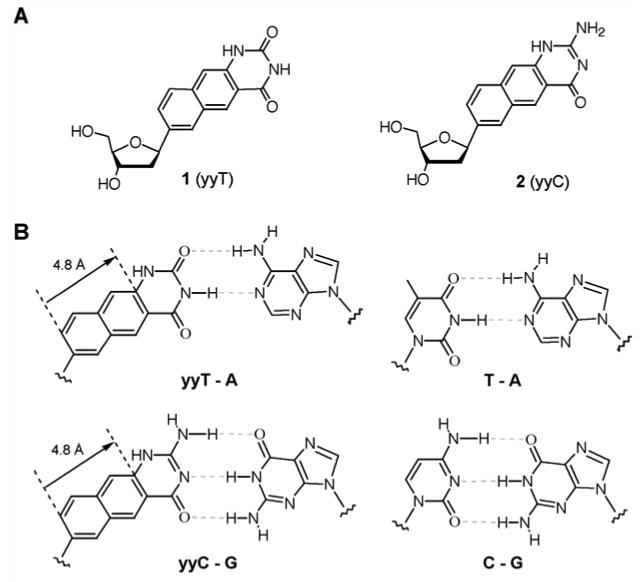
The widened nucleoside components of double-wide DNA (yyDNA). A. Nucleosides 1 and 2, which are naphtho-homologated forms of deoxyuridine and deoxycytidine. B. Proposed structures of yyDNA base pairs (left), with analogous natural pairs at right. The distance from glycosidic C1′ to the opposite one in a pair is expected to be ca. 15.2 Å (compared with 10.4 Å in natural DNA).
The structural designs for yyT and yyC bases leave open the possibility of structural variations. Their designs make them homologous to the natural pyrimidines (uracil and cytosine, respectively) and formally, their structures are most closely related to pseudouracil and pseudoisocytosine. The change in electronic structure due to naphtho-substitution might in principle alter tautomeric equilibria. As a preliminary check on this we calculated heats of formation of several possible tautomers of both yyT and yyC (see the Supporting file). The results suggest that the desired yyT tautomer 1 is the most stable one, while yyC may have one tautomer lower in energy than the structure in 2, which is predicted to be second-lowest in energy. It is not yet known how solvation would affect these relative energies. In addition to tautomeric variation, it should be pointed out that the point of glycosidic attachment in yyT and yyC make it likely that both syn and anti rotamers would have similar energies in the free nucleosides. Similar tautomer and syn/anti situations are also present for the previously described yDNA nucleosides, which have been shown to hybridize selectively and stably.8b
The new doubly widened nucleoside analogues were prepared in an approach analogous to the syntheses of the earlier yDNA benzopyrimidine nucleosides,8b using iodinated base precursors and Heck chemistry13 to form the key glycosidic C-C bonds with beta-anomeric selectivity (Schemes 1 and 2). Elaboration of the final doublewide nucleosides required 5-7 steps from the simple aromatic precursors (see Experimental section for details and Supporting Information for spectra). The overall yield of the free nucleoside dyyT (1) was 19% from 3-amino-2-naphthoic acid, while the yield of dyyC (2) was 20% from the same commercial starting material. The beta anomeric geometry of the nucleosides, which was anticipated from the precedents,8b,13 was confirmed by 1H-NOE experiments (see Supporting Information). Like the earlier yDNA nucleosides, the dyyT and dyyC nucleosides were observed to be fluorescent (Figure 2 and Table 1), with emission maxima in water at 446 and 433 nm, respectively, which is shifted to the red of those earlier compounds by the greater conjugation (see Table 1).
Scheme 1.
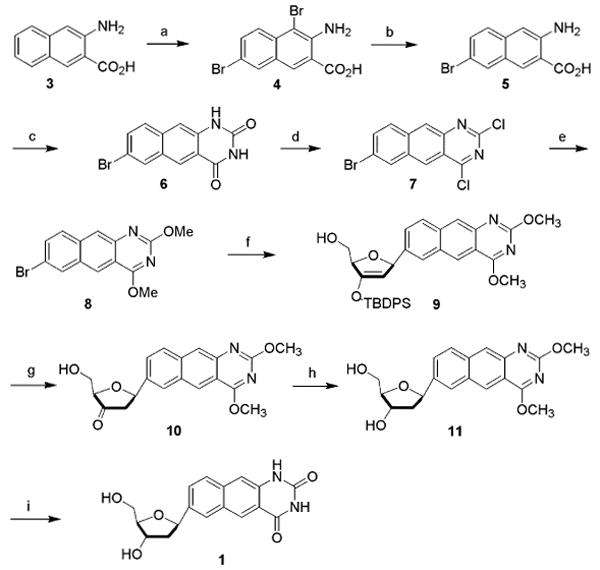
Reagents and conditions: (a) Br2, HOAc (94%); (b) Sn, HCl, HOAc (89%); (c) urea, phenol (99%); (d) POCl3 (44%) (e) NaOMe/MeOH (91%); (f) 3′-O-TBDPS-1,2-dehydro-2-deoxy-d-ribofuranose, Pd2(dba)3, P(tBu)3, Cy2NMe, dioxane, 70-80°C (68%); (g) TBAF, HOAc (cat), THF; (h) Na(OAc)3BH, CH3CN-HOAc, -10°C (97%); (i) Nal, HOAc, 60°C (78%).
Scheme 2.
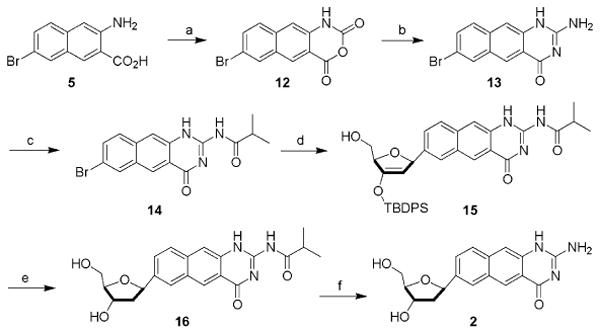
Reagents and conditions: (a) ethyl chloroformate, PBr3 (83%); (b) S-methylisothiouronium sulfate, Na2CO3 (48%); (c) isobutyric anhydride, DIPEA (85%); (d) 3′-O-TBDPS-1,2-dehydro(d)ribofuranose, Pd2(dba)3, P(tol)3, Bu3N, dioxane, 60-80°C (67%); (e) (i) HOAc, TBAF; (ii) Na(OAc)3BH, -10°C, (88%); (f) NH3/MeOH, rt (90%).
Figure 2.

Fluorescence of yyDNA bases. A. Excitation and emission spectra of dyyT (1) in methanol. B. Excitation and emission spectra of dyyC (2) in methanol. Excitation spectra are shown with dashed lines and were measured at the emission maxima; emission spectra are shown with solid lines, and were taken with excitation at the long-wavelength absorption maxima (Table 1).
Table 1.
Absorption absorption and fluorescence data for compounds 1 and 2, with comparison to smaller analogues. R = deoxyribose.
| compound | Absorption maximum
(nm) |
Molar absorptivity
(M-1cm) |
Emission maximum
(nm) |
Quantum yield
(φ) |
|---|---|---|---|---|
|
262a | 30100 | ||
| 362 | 1510 | 446 | 0.67 | |
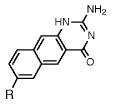
|
256a | 51500 | ||
| 371 | 1690 | 433 | 0.35 | |
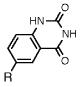
|
221 b | 38100 | ||
| 315 | 3040 | 379 | 0.54 | |
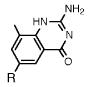
|
231 b | 33900 | ||
| 312 | 2780 | 394 | 0.40 | |
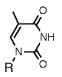
|
267a | 9900 | -- | -- |
|
271 a | 7300 | -- | -- |
Data measured in water.
Data measured in methanol.
Incorporation into natural DNA helices
To test their helix-forming ability, the yyT and yyC nucleosides were then incorporated into DNA by preparing suitably protected nucleoside phosphoramidite precursors (see Supporting Information), and combining these phosphoramidites with the commercially available phosphoramidite derivatives of the natural nucleosides. Intact incorporation of the new nucleosides was confirmed by MALDI-TOF mass spectrometry of several cases (see the Supporting file). A number of sequences containing yyT and yyC in various contexts was prepared to test stacking and pairing properties in the natural DNA context, and to test effects of sequence and structure on stability of any putative widened helices that might be formed in some sequences (Table 2 and Table S1). Helix-formation was then measured by UV-monitored thermal denaturation experiments; buffers typically contained 10 mM Mg2+ and 100 mM Na+ (see Experimental section) and were maintained at pH 7.0.
Table 2.
Stacking proficiency of base analogues, as measured by thermodynamic data for self-complementary duplexes containing a 5′ dangling modified base. Sequence is 5′-dXCGCGCG.a
| dangling base | Tmb (°C) |
ΔG°37c (kcal/mol) |
ΔΔG°37d (kcal/mol) |
|---|---|---|---|
| (none) | 41.7 ± 0.5 | -8.1 ± 0.2 | --- |
| yyT | 66.3 ± 0.5 | -12.5 ± 0.3 | -4.4 ± 0.5 |
| yyC | 60.8 ± 0.5 | -11.1 ± 0.7 | -3.0 ± 0.9 |
| yTe | 56.6 ± 0.5 | -10.9 ± 0.1 | -2.8 ± 0.3 |
| yCe | 53.4 ± 0.5 | -9.7 ± 0.2 | -1.6 ± 0.4 |
| Te | 50.0 ± 0.5 | -9.2 ± 0.5 | -1.1 ± 0.7 |
| Ce | 47.8 ± 0.5 | -9.3 ± 0.2 | -1.2 ± 0.4 |
Conditions: 1 M NaCI, 10 mM phosphate (pH 7.0) with 0.1 mM EDTA.
Tm values are for 5.0 μM oligonucleotide.
Averages of values from van't Hoff and curve fitting methods.
Values per each dangling base, relative to the core duplex.
Data from ref. 8b.
Studies with yyDNA bases in a 5′ dangling position at the end of the hexameric self-complementary sequence context 5′-dCGCGCG14 provided a measure of the relative stacking propensity of yyT and yyC in the DNA context, as compared with their smaller homologs. The data are shown in Table 2. The doubly widened bases were found to stabilize the duplex markedly, and did so with greater proficiency than the smaller yDNA bases, by margins of 1.4-1.6 kcal/mol (37 °C) in this context. Indeed, the yyT base showed stabilization rivaling the most strongly stacking bases known, including benzoadenines,7d,8a 5-nitroindole,14 and N-substituted adenines.15
Additional oligonucleotides containing single substitutions of dyyT and dyyC were prepared to test their effects in a pairing position in the center of a 12 base-pair natural DNA duplex. The data are shown in the Supporting Information file (Table S1). When paired opposite natural DNA bases, yyT and yyC were moderately destabilizing as compared to the case with an A-T base pair at the same position, as was previously observed for the smaller yC and yT bases.8b Unlike those smaller analogues, however, yyT and yyC showed little or no sequence selectivity among the four natural bases, suggesting that their extra size prevents a Watson-Crick-like pairing configuration in the context of the natural DNA backbone.
Testing the doubly widened helical context
Several new oligonucleotides containing 1 and/or 2 were prepared to examine possible assembly properties of such a DNA-homologous system, where every base pair is widened by naphtha-homologation. Table 3 lists sequences that were investigated. Two classes of sequences were prepared: non-self-complementary cases in which yyDNA nucleosides were intermixed with natural nucleosides on both strands (entries 1-4); and cases in which 1 and 2 were segregated onto one strand (entries 5-8), to be paired with a complementary strand containing only natural dA and dG.
Table 3.
yyDNA sequences in this study. Naphtho-widened nucleotides are underlined.
| entry | putative duplex sequencea | comment |
|---|---|---|
| 1 | 5′- T A C A T G
3′- A T G T A C |
hexameric mixed sequence |
| 2 | 5′- T A T T A T A
3′- A T A A T A T |
all yyT - A pairs |
| 3 | 5′- C G C C G C G
3′- G C G G C G C |
all yyC - G pairs |
| 4 | 5′- C C T G A C G
3′- G G A C T G C |
heptameric mixed sequence |
| 5 | 5′- C C T T C T C C
3′- G G A A G A G G |
all-yyDNA strand; natural DNA complement |
| 6 | 5′- C C T T C T C C
5′- G G A A G A G G |
“, parallel orientation |
| 7 | 5′- C C T T C T C C
3′- G G A  G A G G G A G G |
“, yyT-G mismatch |
| 8 | 5′- C C T T C T C C
3′- G G A G  A G G A G G |
“, yyC-A mismatch |
AII oligomers have a 3′ phosphate group.
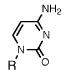
Initial results of UV-monitored thermal melting experiments with pairs of yyDNA sequences that were expected to be complementary yielded unusual changes in absorbance that suggested more than two structural states in the equilibria (Figure 3, solid lines). In addition, changes in absorbance with temperature were quite small, suggesting either that no complex was formed, or that the higher-temperature states (putatively “denatured”) were characterized by little change in stacking from the lower-temperature “complexed” states. Significantly, experiments with the individual strands alone showed in some cases increasing absorbance with temperature (data not shown), also consistent with the strong stacking propensity measured for these extended aromatic bases.
Figure 3.
Changes in thermal denaturation behavior on mixing of complementary yyDNA strands. Shown are UV-monitored denaturation plots of mixed strands (sequences shown) involving yyDNA bases (solid lines), with comparison to the mathematically added data for denaturation of the component single strands (dashed lines). See Figure S3 (Supporting file) for controls.
This non-two-state denaturation behavior required us to adopt a number of methods aside from simple melting curves to determine whether, in fact, intermolecular complexes were formed between selected pairs of strands containing 1 and/or 2. First we examined data in which the component single strands were denatured separately, compared to those in which they were both present in the same vessel. We plotted curves of the mixed melting data along with curves representing the mathematical addition of separate strands (Figure 3). The experiments showed that for five complementary sequence combinations (Table 3, entries 1-5), the data with both strands together gave clearly different curve shapes than the mathematically combined individual component curves, strongly suggesting noncovalent complexes for these sequences. For comparison, three control cases were examined in the same way, but using clearly noncomplementary strands containing 1, 2 (Figure S2 in Supporting Information). In those cases the shapes of the denaturation curves with strands combined were virtually identical to those generated by mathematically adding the data for separate strands, as expected if the strands were not interacting.
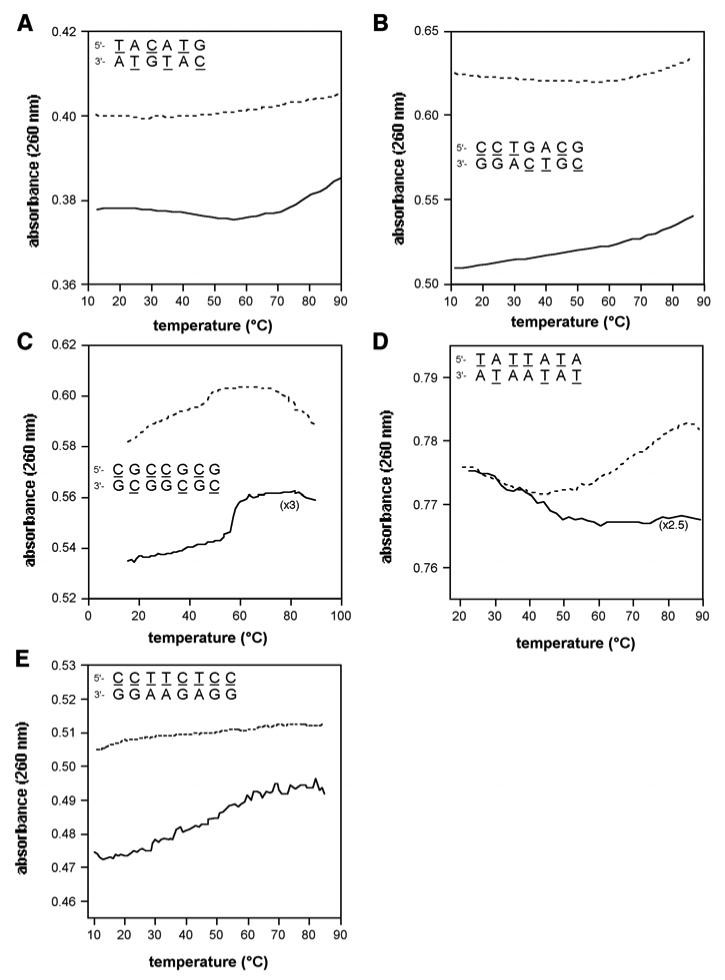
As a second test of noncovalent complexation in the yyDNA system, we performed UV-monitored mixing experiments at varied stoichiometry (Figure 4). If the absorption data in such an experiment deviate from monotonic, single-line behavior, this gives evidence for complexation with associated hyper- or hypo-chromicity arising from changes in stacking when binding occurs. Although the magnitudes of chromicity changes ranged from small to very small (an effect previously observed for the smaller yDNA,8c and consistent with strong stacking in the single-stranded state), the results for four cases were consistent with possible formation of intermolecular complexes (Figure 4). In these four cases, the data were better fit with two lines than with a single line, and the intersections of the lines at near 0.5 mole fraction were consistent with 1:1 stoichiometry in the four sequence contexts. For the fifth case (sequence entry 4 in Table 3), no significant hyper- or hypo-chromicity was observed (data not shown), thus providing no evidence for a complex from this mixing experiment.
Figure 4.
UV-monitored mixing data, showing evidence for 1:1 yyDNA complexes. (A-D), data for entries 1,2,3,5 in Table 3. Conditions: 100 mM NaCl, 10 mM MgCl2, 10 mM Na•PIPES (pH 7.0), 25 °C, with a total DNA concentration of 2 μM. See Figure S4 (Supporting file) for controls.
Stoichiometric mixing experiments were also carried out with three variants of the fifth entry sequence (Table 3); these three corresponded to a parallel-oriented complement (entry 6), and two singly-mismatched antiparallel variants (entries 7 and 8). While the fully complementary case had shown mixing data consistent with a 1:1 complex (Figure 4A), the three variants all showed no such hypo- or hyper-chromicity (Figure S3), although the sequences were closely related. The data suggest, therefore, that this putative yyDNA complex may be preferentially oriented antiparallel, and that it pairs with sequence selectivity, discriminating against a single mismatched base opposite yyT or yyC. However, since the hypochromicity amounts were small in all cases, more data were needed to evaluate whether yyDNA complexes were indeed formed.
To gain further evidence for any complexation and associated helical structure, CD spectra were measured for the putative duplex-forming sequences in Table 3 as well as for the individual component strands. The spectra of the complementary mixtures showed two classes of spectral behavior: one for the two simple, related sequences containing only one type of base pair (entries 2 and 3 in Table 3), and somewhat different spectral shape for the more complex mixed sequences (entries 1, 4, 5). The spectra are shown in Fig. S4, Supporting Information. For both of the simple sequences a prominent negative peak appeared at ∼220 nm, and a positive peak at 280 nm. For the mixed sequences, there was a negative peak at 240-250 nm, and a positive peak at 270 nm. Spectra of the component single strands alone showed similar overall shapes, but with significant differences (Fig. S4). The presence of the induced CDs in the chromophores of the single strands alone provided further support for the presence of helical stacked structure even in the absence of a complement. For four of five cases, the CD spectra of the mixed oligomers showed differences from those of the single strands, providing additional evidence of interaction between the strands. For example, the spectra for the entry 1 sequence show clear differences in the region 330-420 nm when strands were mixed as compared when they were measured alone, and spectra from entries 2 and 5 also showed differences. The spectra of the simple sequences (entries 3 and 4 in Table 3), however, showed less change, with a small difference at 245 nm for entry 3 sequence, and no observable difference with the entry 4 sequence. Overall, for the three mixed-sequence cases, the appearance of a negative peak at ∼240 nm and a positive one at ∼270 nm is similar to that seen for B-DNA, suggesting the presence of a right-handed helical turn.
As a fourth approach to evaluating bimolecular helix formation in the yyDNA context, we took advantage of the inherent fluorescence of the yyDNA bases (shown in Fig. 2). Sequence entry 5 (Table 3) provides a special opportunity, since all the bases of the yyDNA strand are inherently fluorescent. We considered the possibility that these bases, which are excited at 370 nm and emit at 440 nm, might be able to undergo fluorescence resonance energy transfer (FRET) with an acceptor dye of appropriate absorption characteristics. Fluorescein (FAM) was chosen as a candidate, since its absorption maximum (470 nm) overlaps well with the emission of yyDNA bases. We prepared a natural DNA complement of this yyDNA strand carrying a FAM label; the label was added at the end of a hexathymidine spacer to increase the distance from donor to acceptor to a rough distance more typically favorable for FRET. We then measured the fluorescence of the FAM-labeled DNA alone, of the yyDNA strand alone (both at 2 μM), and of the mixture with excitation at 370 nm, where yyDNA maximally absorbs. The data are shown in Fig. 5. The FAM-labeled strand showed a small emission peak at 525 nm, representing the small FAM emission due to low absorption of FAM at 370 nm. The yyDNA strand alone showed the expected emission of yyDNA bases centered at 430 nm. The mixture showed both peaks; significantly, the mixture showed a greater FAM:yyDNA peak intensity ratio for the mixture than for the mathematical addition of the spectra for the two separate strands. This is as would be expected with energy transfer from yyDNA chromophores to the FAM chromophore, and is consistent with formation of a noncovalent complex between the two strands. A control experiment with a FAM-labeled target DNA having scrambled sequence showed no change in the FAM:yyDNA emission ratios on mixing (data not shown).
Figure 5.

Fluorescence resonance energy transfer (FRET) experiment showing evidence for complex involving yyDNA sequence (entry 5, Table 3). Conditions: 100 mM NaCl, 10 mM MgCl2, 10 mM Na•PIPES (pH 7.0), 25 °C, with a total DNA concentration of 2 μM. The probe sequence was 5′-dGGAGAAGGTTTTTT-FAM, where FAM is 2-(6-carboxyfluorescein)-4-aminobutyl)-propyl-3-O-succinoyl-long chain alkylamino.
To gain more evidence for or against a putative yyDNA double-stranded complex, we carried out two more lines of experiments, again making use of the inherent fluorescence of yyDNA bases. The first set of experiments made use of a common fluorescence quencher, dabcyl, conjugated to complementary or non-complementary probes, to be mixed with possibly complementary yyDNA strands (see structures in the Supporting file). We found that in experiments when a quencher-containing complementary strand was admixed with the fluorescent yyDNA, a small decrease in fluorescence intensity was consistently seen (refer to spectra in Fig. 6), and a greater degree of quenching (19% decrease) was observed for the same sequence with 2 dabcyl groups. However, a similar quencher-conjugated DNA having a scrambled sequence did not affect emission intensity. Aside from these experiments, it is not yet known whether dabcyl is inherently a weak or strong quencher of yyDNA bases; however, we did observe a similar effect with the previously described and structurally related “yDNA” (data not shown), where dabcyl was only able to quench the analogous sequence by a similarly small amount. yDNA double helix formation has been previously confirmed by several methods.8
Figure 6.

Effects of adding quencher-conjugated DNA complements to the fluorescence of a yyDNA strand. The yyDNA strand sequence was 5′-CCTTCTCC; DNA complement is 5′- GGAGAAGG with one or two dabcyl conjugates at the ends (see Supporting file (Fig. S5) for structures). Scrambled sequence: 5′-AGAGAGAG. Conditions: 100 mM NaCl, 10 mM MgCl2, 10 mM Na•PIPES (pH 7.0), 25 °C, with a total oligomer concentration of 2 μM.
The final experiments involved attempting to visualize the fluorescence of yyDNA by carrying out potential hybridizations on PEG-polystyrene beads under an epifluorescence microscope. We prepared two sets of beads with different DNA oligonucleotides covalently conjugated. One oligonucleotide sequence was putatively complementary (in the antiparallel sense) to the yyDNA oligomer 5′-CCTTCTCC, while the other oligonucleotide contained a scrambled control sequence. These were allowed to swell in hybridization buffer; then the yyDNA oligomer was incubated with the beads, and then the beads were washed with buffer twice. Digital fluorescence images were obtained under an epifluorescence microscope. Results (Figure 7) show significantly increased fluorescence for the sequence-complementary beads (images D,F) relative to background (B) and relative to scrambled-sequence beads (C,E). This result is consistent with formation of a sequence-selective noncovalent complex between the yyDNA and the complementary DNA sequence on the polymer beads.
Figure 7.
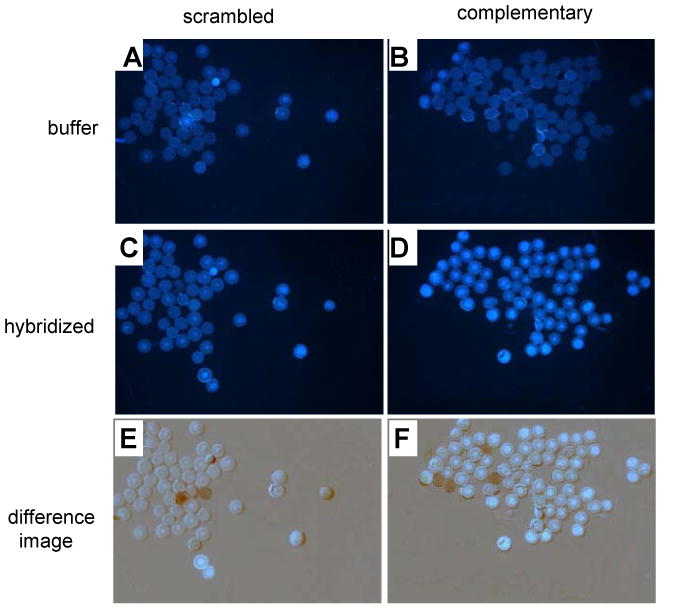
Hybridization studies on 60-micron PEG-polystyrene beads. Images show DNA-conjugated beads with buffer alone (A,B); beads after hybridization with yyDNA (5′-dyyCCTTCTCC) and washing (C,D); and digitally subtracted difference images (E,F). A,C,E: scrambled control DNA sequence 5′- GAGAGAGA on beads; B,D,F: complementary sequence (5′- GGAGAAGG) on beads. Hybridization and wash buffer contained 100 mM NaCl, 10 mM MgCl2, 10 mM Na•PIPES (pH 7.0), 25 °C. Increased fluorescence with complementary beads vs. scrambled controls (compare D to C and F to E) is consistent with sequence-selective hybridization of yyDNA. Anomalous dark and light spots in E (2 spots) and F (4 spots) arose from beads that moved between background and hybridization images.
Discussion
We have described the efficient synthesis of two new naphtho-homologous nucleosides (1 and 2), and their incorporation into oligonucleotides. The nucleosides are readily available in preparative quantities; the overall synthesis yields are comparable to those achieved previously for the smaller yT and yC benzo-homologated analogues.8b In addition to their ability to be employed in widened helix self assembly (see below), we anticipate that these compounds may have useful applications either as mononucleotides or as single substitutions in DNA. Strongly stacking large nucleobase analogues have been employed recently as tools to study base-flipping mechanisms in DNA-modifying enzymes;16 the inherent fluorescence of 1 and 2 might also be useful in that respect. Beyond this, the visible-wavelength fluorescence of these compounds may have other uses in reporting and sensing, adding to a growing literature of fluorescent DNA base replacements.17
The unusually strong stacking ability of yyT and yyC bases suggests their use in generically stabilizing helices (such as in arrays and diagnostics) by substitution at the ends; such “end-capping” has been studied recently as a means to stabilizing hybridization irrespective of sequence.18 Indeed, we find that 1 and 2 each stabilize a duplex more than addition of a full A-T base pair.7d,14 The end-stacking studies show that yyT and yyC stabilize the duplex by greater factors than earlier (and smaller) yT and yC bases. We hypothesize that the added stacking proficiency arises from increased size (which favors van der Waals contributions) and from increased hydrophobicity, which can strongly influence stacking in modified DNAs.14 The observance of strong induced CDs in these bases suggests that the stacking is substantial in the single-stranded state, which is also consistent with our observation of only small changes in chromicity on binding a complementary strand. Similar behavior (although not as pronounced) was previously observed in the smaller yDNA system.8c
Unlike the smaller yDNA system, the yyDNA sequences did not display simple two-state melting behavior, and this fact made it necessary to evaluate the possibility of fully naphtha-homologated helix formation by several different analytical methods. Taken together, several lines of evidence point to possible formation of helical widened DNA complexes involving naphtho-homologated base pairing involving 1 and 2. Differences in UV-monitored thermal denaturation, and changes in CD spectra, on mixing complementary yyDNA strands indicate that at least in some cases the strands interact to yield differences in base interactions. Absorbance-monitored mixing experiments also add evidence to this interaction, and they further suggest that complexes are 1:1 in stoichiometry, as expected for a DNA-like pairing model. Controls carried out with mismatched and with parallel-oriented strands show no such evidence of strand-strand interactions, consistent with sequence selective binding (analogous to DNA) and consistent with preferentially antiparallel orientation (again, similar to DNA). These controls also lend confidence in the mixing experiments, since they show data that are well-fit by straight lines, whereas the experimental data with fully complementary strands are better fit by two lines.
Data from a number of fluorescence studies were more strongly suggestive of the formation of fully-widened yyDNA complexes. These were carried out in a sequence context wherein one strand was fully substituted with yyDNA bases, and the putative complement was natural DNA. Results of mixing a yyDNA strand with complementary DNA having a fluorescein FRET acceptor showed evidence for energy transfer between the yyDNA and the fluorescein. Studies were also carried out with dabcyl quencher-conjugated DNAs; these showed successively increased quenching of yyDNA fluorescence as one or two dabcyl groups were attached to a DNA complement, but showed no quenching when the DNA sequence was noncomplementary. Finally, hybridization studies on beads, monitored by fluorescence microscopy, were also supportive of a sequence-selective complexation between a yyDNA strand and a DNA sequence. Thus the fluorescence data are suggestive of an antiparallel, sequence-selective complex having fully naphtha-homologated base pair size. These studies also demonstrate the general utility of the fluorescence of the yyT and yyC chromophores.
The observations of possible 1:1 strand complexation, with apparent preference for antiparallel strand orientation and sequence selectivity, is strongly suggestive of a pairing model for yyDNA that is related to that of natural DNA. Model building suggests (Figure 8) that yyT – A and yyC – G base pairs can be assembled with Watson-Crick-like hydrogen-bonded pairing (Fig. 1B) to form a right-handed helix bearing similarity to B-form DNA. A right-handed model is also supported by the B-DNA-like resemblance of CD spectra (Fig. S4). In the modeling, the sugar conformations and phosphodiester backbone needed only small adjustments (chiefly to δ and ζ dihedral angles in the preliminary models) to adapt to the longer chain-chain distance. The main differences from natural DNA in this hypothetical model are the increased distance across a pair, the increased major and minor groove widths, and a lowered degree of helical turn per pair. Similar adjustments (although smaller in magnitude) are proposed for the earlier benzo-homologous yDNA system, and new structural evidence supports that model (H. Lu, S. R. Lynch, E. T. Kool, unpublished results). Clearly, more structural data are needed for the more geometrically extreme yyDNA system before confident conclusions can be made about its pairing and topography.
Figure 8.
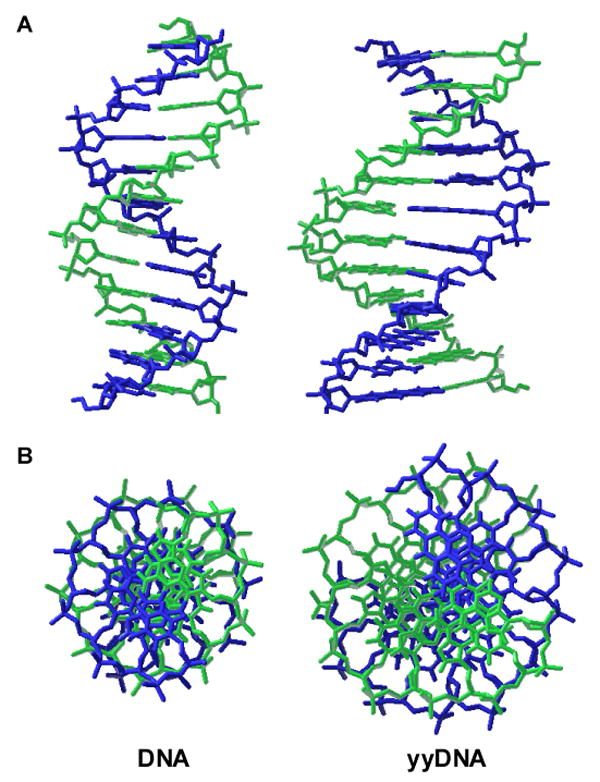
Molecular models comparing natural B-form DNA dodecamer (left) with a plausible model of yyDNA composed of yyT-A and yyC-G pairs (right). Side views (A) and end views (B) are shown. Both modeled structures (AMBER force field; continuum water) are right-handed and antiparallel double helices, with similar sugar conformations. The chief differences predicted for yyDNA are widened grooves and a lower degree of twisting, resulting in ca. 14 base pairs per 360° turn of the helix.
The experiments suggest that strands fully composed of yyDNA bases may be able to recognize natural DNA. Since yyDNA bases are fluorescent, this raises the possibility that yyDNAs might have applications in detection or imaging of natural nucleic acids. Of course, it remains to be seen whether analogous naphtho-homologous variants of adenine and guanine could be prepared, and whether they could be combined with the doubly-widened pyrimidines. In addition, future studies will be needed to evaluate how the fluorescence of yyDNA changes with sequence. The earlier (and smaller) yDNA system was shown to bind to natural DNAs and to RNAs;8a,8c the present larger system, if it has this capability, has one possible advantage, in that its absorption occurs at longer wavelength, and its fluorescence emission appears well into the visible range. However, the apparently strong single-stranded stacking of yyDNA bases also complicates the measurement of binding: whereas standard nucleic acid probes can easily be monitored using thermal denaturation, the present yyDNAs do not give simple denaturation curves. This also prevents the measurement of the affinity of yyDNAs for their complements using standard thermal denaturation curve-fitting methods. More experiments involving other equilibrium binding techniques and/or calorimetry will be needed to evaluate the thermodynamics of these complexes.
Overall, the results suggest that the combination of yyT, yyC, A, and G nucleotides may make up the basis for a new four-base genetic self-recognition system having unprecedented size. If this is the case, then it establishes that a surprising degree of geometric plasticity is possible in the base pairs of designed genetic systems, as long as homologous structures for the base pairs are maintained.
Experimental section
General Experimental Methods
Reagents were purchased from Aldrich and used without further purification. All water-sensitive reactions were carried out in oven-dried glassware with stirring bar under argon atmosphere. Anhydrous solvents pyridine, CH3CN, and CH2Cl2 were distilled over CaH2 under a nitrogen atmosphere. THF was distilled from Na/benzophenone under a nitrogen atmosphere prior to use. All other anhydrous solvents were used directly without further distillation. Thin-layer chromatography was carried out using Silica Gel 60 F254 plates. Column chromatography was performed using Silica Gel, sizes 32-63. All 1H, 13C NMR spectra were recorded on Varian Inova 400 or 500 MHz instruments as solutions in CDCl3 unless otherwise indicated, and the chemical shifts are reported in parts per million (ppm, δ). Coupling constants are reported in hertz (Hz). Assignments of the anomeric stereochemistry for the compounds 1 and 2 were confirmed by NOE experiments. High-resolution mass spectra were recorded at UCR MS Facility in DEI or FAB modes.
Preparation of nucleosides 1 and 2
3-Amino-4,7-dibromo-naphthalene-2-carboxylic acid (4)
3-Amino-naphthalene-2-carboxylic acid 3 (5.45 g, 29.11 mmol) was dissolved in glacial acetic acid (100 mL) and cooled to 5 °C. To this vigorously stirred solution was added dropwise a solution of bromine (3.0 mL, 58.70 mmol) in glacial acetic acid (20 mL). After the addition at 5°C, the reaction mixture was refluxed for 5 h, cooled and poured into ice water. The solid was filtered with suction and washed with water, followed with small volume of methanol (ca. 25 mL). The dibrominated product 4 (9.44 g, 94%) was obtained after vacuum dried without further purification as a yellowish brown solid. 1H NMR (DMSO-d6, 400 MHz): δ 8.55 (s, 1H), 8.20 (d, 1H, J= 2.0 Hz), 7.77 (d, 1H, J = 9.2 Hz), 7.68 (dd, 1H, J = 9.2, 2.0 Hz). 13C NMR (DMSO-d6, 100 MHz): δ 169.22, 145.15, 134.09, 133.79, 133.31, 132.17, 127.02, 126.85, 116.72, 115.68, 103.77. HRDEIMS m/z: [M]+ calcd for C11H7NO2Br2, 342.8843 found 342.8826.
3-Amino-7-dibromo-naphthalene-2-carboxylic acid (5)
3-Amino-4,7-dibromo-naphthalene-2-carboxylic acid 4 (7.56 g, 21.80 mmol) was suspended in glacial acetic acid (40 mL) and 37% w/w HCl (30 mL) and 300 mesh tin (2.60 g, 21.80 mmol) were added. The reaction mixture was reflux overnight, cooled and diluted with ice water. The solid was suction filtered, washed with water and then small volume of methanol (ca. 20 mL), and air dried to afford 7-bromo product 5 (5.23 g, 89%) as an orange powder. 1H NMR (DMSO-d6, 400 MHz): δ 8.47 (s, 1H), 8.11 (s, 1H), 7.58 (d, 1H, J= 8.8 Hz), 7.50 (dd, 1H, J = 9.2, 2.0 Hz), 7.21 (s, 1H). 13C NMR (DMSO-d6, 100 MHz): δ 168.63, 144.30, 134.92, 132.47, 131.50, 130.80, 127.62, 127.06, 116.87, 115.19, 111.51. HRDEIMS m/z: [M]+ calcd for C11H8NO2Br, 264.9738 found 264.9749.
7-Bromobenzoquazoline-2,4-dione (6)
3-Amino-7-dibromo-naphthalene-2-carboxylic acid 5 (5.14 g, 19.30 mmol), urea (6.00 g, 100.0 mmol) and phenol (8.00 g) were heated to 150 °C in a round-bottom flask, and the melt was stirred overnight at that temperature. The mixture was then allowed to cool to 100 °C, and a volume equivalent of water and ethanol were added.2 The resulting mixture was stirred for 30 min to allow unreacted urea and phenol to dissolve. The precipitate was suction-filtered and the filter-cake was washed with water, then with methanol. The product 6 (5.47 g, 99%) was obtained after vacuum drying without further purification as a yellowish brown powder. 1H NMR (DMSO-d6, 400 MHz): δ 8.61 (s, 1H), 8.37 (s, 1H), 7.86 (d, 1H, J= 8.8 Hz), 7.66 (d, 1H, J = 8.8 Hz), 7.51 (s, 1H). 13C NMR (DMSO-d6, 100 MHz): δ 162.66, 150.28, 137.02, 134.77, 131.97, 131.10, 129.45, 128.99, 128.31, 117.66, 116.03, 110.53. HRDEIMS m/z: [M]+ calcd for C12H7N2O2Br, 289.9690 found 289.9679.
7-Bromo-2,4-dichlorobenzoquinazoline (7)
A mixture of bromobenzoquinazolinedione 6 (4.22 g, 14.50 mmol) and phosphorus oxychloride (25.0 mL, 270 mmol) was stirred in an oven-dried round-bottom flask fitted with a condenser. The mixture was heated at reflux for 12 h. Then the solution was allowed to cool to room temperature and volatiles were removed in vacuo. The residue was dissolved in CH2Cl2 (120 mL) and washed with water (2 × 60 mL). The organic layer was dried (Na2SO4) and concentrated in vacuo. The resulting residue was purified by silica column chromatography (hexane/ EtOAc 6:1) to yield 7 (2.08 g, 44%) as an orange powder. 1H NMR (CDCl3, 400 MHz): δ 8.79 (s, 1H), 8.52 (s, 1H), 8.33 (s, 1H), 7.98 (d, 1H, J = 8.8 Hz), 7.76 (dd, 1H, 8.0, 2.0 Hz). 13C NMR (CDCl3, 100 MHz): δ 165.61, 146.19, 135.63, 133.30, 131.03, 129.83, 126.55, 126.29, 122.37, 120.55. HRDEIMS m/z: [M]+ calcd for C12H5N2Cl2Br, 325.9013 found 325.9003.
7-Bromo-2,4-dimethoxybenzoquinazoline (8)
7-Bromo-2,4-dichlorobenzoquinazoline 7 (1.80 g, 5.52 mmol) was suspended in 0.5 M sodium methoxide in methanol (30.0 mL). The reaction mixture was heated at reflux under argon atmosphere for 24 h. The reaction mixture was then cooled to room temperature and the solvent was removed in vacuo. The orange residue was purified by silica column chromatography (CH2Cl2 initially and CH2Cl2 / EtOAc 5:1 subsequently) to yield 8 (1.60 g, 91%) as an orange solid. 1H NMR (CDCl3, 400 MHz): δ 8.50 (s, 1H), 8.10 (d, 2H, J = 4.8 Hz), 7.77 (d, 1H, J = 9.2 Hz), 7.56 (dd, 1H, J = 9.2, 1.6 Hz), 4.23 (s, 1H), 4.13 (s, 1H). 13C NMR (CDCl3, 100 MHz): δ 169.83, 161.64, 146.94, 134.87, 131.34, 130.78, 130.71, 129.24, 123.65, 122.83, 119.10, 114.14, 54.85, 54.82. HRDEIMS m/z: [M]+ calcd for C14H11N2O2Br, 318.0003 found 317.9998.
2,4-Dimethoxy-7-{2′R-cis-3-[2′3′-dehydro-3′-(tert-butyldiphenylsilyloxy)]-5′-hydromethyl-2′-furanyl}benzoquinazole (9)
The dimethoxybenzoquinazoline 8 (1.38 g, 4.33 mmol) and 1,2-Dehydro-3-O-(tert-butyldiphenylsilyl)-5-hydroxymethylfuran19 (1.47 g, 4.16 mmol) were charged into an oven-dried round bottom flask equipped with a septum. Dried dioxane (30 mL) was added to the flask to form a suspension and the mixture was bubbled with argon for 45 min. Pd2(dba)3 (60 mg, 0.07 mmol) and P(tBu)3 (61 mg, 0.3 mmol) were charged into a separated round-bottom flask, and suspended in dried dioxane (10 mL). The catalyst-ligand mixture was stirred at room temperature and degassed with argon bubbling before being transferred to the glycal-benzsoquinazoline suspension via hypodermic syringe under inert atmosphere. N-methyldicyclohexylamine (1.00 mL, 4.72 mmol) was added to the reaction mixture in a portion. The septum-sealed flask was heated to 70-80 °C for 12 h and allowed to cool to room temperature. After the removal of the volatiles, the residue was purified with silica column chromatography (hexanes/ EtOAc 5:1 initially and 2:1 subsequently) to afford 9 (1.66 g 68%) as slightly yellow foam. 1H NMR (CDCl3, 400 MHz): δ 8.50 (s, 1H), 8.10 (s, 1H), 7.87 (d, 2H, J = 7.2 Hz), 7.79 (m, 3H), 7.49 (m, 6 H), 7.30 (dd, 1H, J = 8.4, 1.2 Hz), 5.72 (d, 1H, J = 2.8 Hz), 4.84 (m, 1H), 4.52 (s, 1H), 4.24 (s, 3H), 4.12 (s, 3H), 3.90 (bs, 2H), 1.12 (s, 9H). 13C NMR (CDCl3, 100 MHz): δ 169.89, 161.44, 151.10, 146.83, 138.70, 136.53, 135.80, 135.34, 131.37, 131.11, 130.28, 130.20, 129.71, 127.99, 127.90, 127.67, 127.29, 124.81, 122.26, 113.52, 102.49, 84.77, 83.38, 62.77, 54.72, 54.68, 26.33, 19.18. HRFABMS m/z: [M + H]+ calcd for C35H37N2O5Si, 593.2472 found 593.2468.
1′-β-[7-(2,4-Dimethoxybenzoquinazoline)]-2′-deoxy-D-ribofuranose (11)
A solution of compound 9 (1.00 g, 1.69 mmol) in anhydrous THF (15 mL) was chilled in an ice-water bath. Glacial acetic acid (0.5 mL) and 1M TBAF solution in THF (2.2 mL) were added sequentially via hypodermic syringe. The reaction mixture was allowed to stir at 0 °C for 15 min and the volatiles were removed in vacuo. The crude intermediate of 2,4-dimethoxy-7-(β-D-glycero-pentofuran-3′-ulos-1′yl)-benzoquinazoline 10 was subjected to next step without purification and characterized by NMR: 1H NMR (CD3OD, 400 MHz): δ 8.65 (s, 1H), 8.12 (s, 1H), 7.87 (d, 1H, J = 8.4 Hz), 7.78 (dd, 1H, J = 8.4, 1.2 Hz), 5.38 (dd, 1H, J = 10.8, 6.0 Hz), 4.09 (m, 1H), 3.92 (m, 2H), 2.95 (dd, 1H, J = 17.6, 5.6 Hz), 2.52 (dd, 1H, J = 17.6, 10.8 Hz). 13C NMR (CD3OD, 100 MHz): δ 214.41, 162.88, 150.36, 137.52, 136.74, 136.14, 128.94, 128.07, 128.01, 127.14, 126.90, 115.40, 110.28, 82.78, 76.51, 60.56, 45.18. The crude product was then dissolved in a solution of CH3CN (15 mL) and glacial acetic acid (3 mL). The resulting mixture was chilled to -10 °C in an ice-salt bath followed by the addition of sodium triacetoxyboronhydride (0.90 g, 4.23 mmol) in one portion. The reaction mixture was allowed to stir at the low temperature for 5 min and was concentrated in vacuo. The residue was purified by silica column chromatography (EtOAc initially and EtOAc/ methanol 20:1 subsequently) to give 11 (0.58 g, 97%) as a yellow foam. 1H NMR (CD3OD, 400 MHz): δ 8.20 (s, 1H), 7.77 (s, 2H), 7.70 (d, 1H, J = 8.4 Hz), 7.48 (d, 1H, J = 8.8 Hz), 5.21 (dd, 1H, J = 10.4, 5.6 Hz), 4.35 (1H, m), 4.02 (s, 3H), 3.94 (s, 3H), 3.71 (d, 1H, J = 4.8 Hz), 2.25 (m, 1H), 2.00 (m, 1H). 13C NMR (CD3OD, 100 MHz): δ 171.12, 162.59, 147.40, 140.27, 137.61, 131.10, 128.75, 127.94, 126.65, 125.42, 122.64, 114.01, 89.32, 81.40, 74.46, 64.11, 55.37, 55.20, 44.57. HRDEIMS m/z: [M]+ calcd for C19H20N2O5, 356.1372 found 356.1380.
1′-β-[7-(Benzoquinazoline2,4-dione)]-2′-deoxy-D-ribofuranose (1)
To a solution of compound 11 (0.30 g, 0.84 mmol) in glacial acetic acid (15b mL) was added sodium iodide (0.50 g, 3.33 mmol). The reaction mixture was heated to 60 °C for 45 min., and then volatiles were removed in vacuo. The residue was dissolved in EtOAc (100 mL), extracted with saturated Na2SO3 (aq) (3 × 20 mL) and saturated sodium bicarbonate solution (2 × 15 mL). The aqueous layers were back extracted with EtOAc (2 × 15 mL). The combined organics were dried (Na2SO4) and concentrated in vacuo. The residue was purified by silica column chromatography (EtOAc/methanol 10:1) to yield 1 (0.22 g, 78%) as a yellow powder. 1H NMR (CD3OD, 400 MHz):δ 8.60 (s, 1H), 7.99 (s, 1H), 7.80 (d, 1H, J = 8.8 Hz), 7.65 (dd, 1H, J = 8.8, 2.0 Hz), 7.48 (s, 1H), 5.28 (dd, 1H, J = 10.4, 5.6 Hz), 4.37 (m, 1H), 4.02 (m, 1H), 3.73(d, 2H, J = 4.8 Hz), 2.28 (m, 1H))). 2.04 (m, 1H). 13C NMR (CD3OD, 100 MHz): δ 162.93, 150.39, 139.23, 136.43, 135.89, 128.78, 128.20, 128.01, 126.82, 126.07, 115.24, 110.22, 87.99, 79.22, 72.53, 62.50, 43.44. HRDEIMS m/z: [M]+ calcd for C17H16N2O5, 328.1055 found 328.1047.
7-Bromo-1H-naphtho-1,3oxazine-2,4-dione (12)
Intermediate 5 (6.11 g, 23.00 mmol) was dissolved in anhydrous THF (100 mL), to which was added ethyl chloroformate (5.4 mL, 56.73 mmol) dropwise at room temperature. After addition, the mixture was heated at reflux for 24 h under argon. The mixture was allowed to cool to room temperature and the volatiles were removed in vacuo to yield an orange solid. The residue was redissolved in anhydrous diethyl ether (65 mL), to which was added PBr3 (2.2 mL, 23.40 mmol) cautiously. After refluxing for 24 h under Ar, the brick red turbid reaction mixture was chilled in ice-water. The precipitate formed was filtered by suction filtration, washed with petroleum ether and CH2Cl2. The filter-cake was dried in vacuo to give 12 (5.57 g, 83%) as a reddish brown powder. 1H NMR (DMSO-d6, 400 MHz): δ 8.67 (s, 1H), 8.38 (s, 1H), 7.87 (d, 1H, J = 8.8 Hz), 7.71 (dd, 1H, J = 8.8, 2.0 Hz), 7.47 (s, 1H). 13C NMR (DMSO-d6, 100 MHz): δ 159.78, 146.84, 136.52, 135.42, 132.88, 131.28, 130.90, 129.89, 129.17, 118.34, 112.15, 110.66. HRDEIMS m/z: [M]+ calcd for C12H6NO3Br 290.9531, found 290.9541.
2-Amino-7-bromobenzoquinazolin-4-one (13)
A mixture of 12 (4.50 g, 15.00 mmol), sodium carbonate (1.60 g, 12.00 mmol) and S-methylisothiourea hemisulfate (1.40 g, 13.00 mmol) was dissolved in a solvent mixture of CH3CN-H2O (40mL/ 10 mL). The mixture was then heated at reflux for 18 h, then cooled to room temperature. The yellow precipitate was filtered with suction filtration and the filter-cake was washed with a solution of H2O and methanol. As the vacuum-dried yellow solid was poorly soluble in various solvents, the crude product 13 (4.35 g, 48%) was subjected to the next step without further purification. HRDEIMS m/z: [M]+ calcd for C12H8N3OBr 288.9851, found 288.9855.
N-(7-Bromo-4-oxo-1,4-dihydro-benzoquinazolin-2-yl)-isobutyramide (14)
Isobutyric anhydride (10.0 mL, 60.20 mmol) was added to a solution of 13 (2.29 g, 7.89 mmol) in distilled N,N-diisopropylethylamine (1 mL), and the solution was stirred at reflux for 16 h. The resulting mixture was concentrated in vacuo and purified by silica column chromatography (CH2Cl2/EtOAc/MeOH 6:1:1) to give 14 (2.41 g, 85%) a golden yellow powder. 1H NMR (DMSO-d6, 400 MHz): δ 8.74 (s, 1H), 8.45 (s, 1H), 7.99 (s, 1H), 7.98 (d, 1H, J = 11.2 Hz), 7.70 (dd, 1H, J = 8.8, 2.0 Hz), 2.78 (m, 1H), 1.12 (d, 6H, J = 7.2 Hz). 13C NMR (DMSO-d6, 100 MHz): δ 180.49, 156.09, 134.89, 131.46, 130.93, 130.77, 129.55, 127.04, 122.54, 120.21, 118.64, 34.89, 18.92. HRDEIMS m/z: [M]+ calcd for C16H14N3O2Br, 359.0269 found 359.0277.
{(2′R)-cis-3-[2′,3′-Dehydro-3′-(tert-butyldiphenylsilyloxy)]5′-hydroxymethyl-2′-furanyl}-7-(4-oxo-1,4-dihydroquinazolin-2-yl)]-isobutyramide (15)
Isobutyramide 14 (1.10 g, 2.80 mmol) and 1,2-Dehydro-3-O-(tert-butyldiphenylsilyl)-5-hydroxymethylfuran17 (1.10 g, 3.00 mmol) were charged into an oven-dried round bottom flask equipped with a septum. Dried dioxane (30 mL) was added to the flask to form a suspension and the mixture was bubbled with argon for 30 min. Pd2(dba)3 (70 mg, 0.07 mmol) and P(tol)3 (90 mg, 0.30 mmol) were charged into a separated round-bottom flask, and suspended in dried dioxane (15 mL). The catalyst-ligand mixture was stirred at room temperature and degassed with argon bubbling before being transferred to the glycal-isobutyramide suspension via hypodermic syringe under inert atmosphere. Tri(n-butyl)amine (1.0 mL, 4.21 mmol) was added to the reaction mixture in a portion. The septum-sealed flask was heated to 60 to 80 °C for 18 h and allowed to cool to room temperature. After the removal of the volatiles, the residue was purified with silica column chromatography (hexanes/ EtOAc 3:1) to afford 15 (1.14 g, 59%) as slightly yellow foam. 1H NMR (CDCl3, 400 MHz): δ 8.32 (s, 1H), 7.95 (d, 2H, J = 6.4 Hz), 7.82 (d, 2H, J = 6.0 Hz), 7.52 (m, 7H), 7.39 (s, 1H), 7.24 (dd, 2H, J = 8.4, 1.6 Hz), 5.71 (bs, 1H), 4.88 (bs, 1H), 4.42 (s, 1H), 4.03 (bs, 2H), 2.76 (m, 1H), 2.51 (m, 1 H), 1.47 (m, 1H), 1.28 (m, 6H), 1.14 (s, 9H). 13C NMR (CDCl3100 MHz): δ 151.33, 138.49, 136.04, 135.86, 135.30, 131.27, 130.87, 130.57, 130.17, 129.67, 128.48, 127.92, 127.86, 127.07, 101.64, 85.19, 83.95, 62.61, 60.28, 53.14, 36.27, 26.27, 20.94, 20.54, 19.26, 19.14, 18.68, 14.06, 13.90. HRFABMS m/z: [M + H]+ calcd for C37H40N3O5Si, 634.2737 found 634.2711.
1′-β-{7-[N-(4-oxo-1,4-Dihydrobenzoquinazolin-2-yl)]-isobutyramide}-2′-deoxy-D-ribofuranose (16)
A solution of 15 (1.15 g, 1.78 mmol) in anhydrous THF (12 mL) was chilled in an ice-water bath. Glacial acetic acid (0.5 mL) and 1M TBAF solution in THF (3.5 mL, 3.50 mmol)) were added sequentially via hypodermic syringe. The reaction mixture was allowed to stir at 0 °C for 20 min, then ammonium hydroxide (4 mL) was added to quench the reaction, and the volatiles were removed in vacuo. The intermediate was dissolved in a solution of CH3CN (15 mL) and glacial acetic acid (3 mL). The resulting mixture was chilled to -10 °C in an ice-salt bath followed by the addition of sodium triacetoxyborohydride (1.13 g, 5.34 mmol) in a portion. The reaction mixture was allowed to stir at the low temperature for 10 min and concentrated in vacuo. The residue was purified by silica column chromatography (EtOAc initially, EtOAc/methanol 20:1 subsequently) to afford 16 (0.60 g, 88%) as a slight yellow powder. 1H NMR (DMSO-d6, 400 MHz): δ 8.69 (s, 1H), 8.08 (s, 1H), 7.96 (d, 1H, J = 11.6 Hz), 7.94 (s, 1H), 7.62 (d, 1H, J = 8.8 Hz), 5.18 (dd, 1H, J = 10.4, 5.2 Hz), 4.25 (d, 1H, J = 5.2 Hz), 3.86 (bs, 1H), 3.53 (m, 2H), 2.79 (m, 1H), 2.16 (m, 1H), 1.90 (m, 1H), 1.53 (m, 1H), 1.11 (d, 6H, J = 6.8 Hz). 13C NMR (DMSO-d6, 100 MHz): δ 180.27, 139.99, 136.09, 129.68, 127.42, 125.75, 119.54, 88.04, 79.28, 72.53, 62.52, 60.76, 43.46, 19.24, 18.99. HRDEIMS m/z: [M]+ calcd for C21H23N3O5, 397.1637 found 397.1645.
1′-β-7-[2-Amino-(4-oxo-1,4-dihydroquinazolin-2-yl)]-2′-deoxy-D-ribofuranose (2)
A solution of compound 16 (0.36 g, 0.09 mmol) in 5 mL saturated ammonium/methanol solution was stirred at room temperature for overnight in a sealed tube. The volatiles were removed in vacuo, and the residue was purified by washing with EtOAc to afford pure free deoxyriboside dyyC (2) (0.26 g, 90%) as a white solid. 1H NMR (DMSO-d6, 400 MHz): δ 8.53 (s, 1H), 7.96 (s, 1H), 7.82 (d, 1H, J = 8.8 Hz), 7.60 (s, 1H), 7.52 (d, 1H, J = 8.8 Hz), 5.14 (m, 1H), 4.82 (bs, 1H), 4.22 (d, 1H, J = 4.0 Hz), 3.53 (m, 2H), 2.12 (m, 1H), 1.88 (m, 1H). 13C NMR (DMSO-d6, 100 MHz): δ 138.34, 128.22, 127.13, 126.95, 126.91, 125.85, 118.22, 87.96, 79.37, 72.55, 62.55, 43.40. HRFABMS m/z: [M + H]+ calcd for C17H18N3O4, 328.1293 found 328.1290.
Oligonucleotide synthesis and characterization
5′-O-tritylated phosphoramidite derivatives of dyyT and dyyC were prepared as described in the Supporting Information file. Oligonucleotides were synthesized on Applied Biosystems 394 and 392 synthesizers using standard β-cyanoethyl phosphoramidite chemistry. The coupling time for the modified nucleoside phosphoramidites was extended from 100 seconds to 900 seconds. Stepwise coupling yields for the extended bases were all greater than 98% as determined by trityl cation response. All widened oligonucleotides were synthesized in DMT-off mode on the 5′- end and deprotected from CPG (alkyl controlled pore glass) supports in concentrated ammonia overnight at 55 °C. All oligonucleoitdes were prepared with 3′ phosphate groups (phosphate-ON reagent, Glen Research) to avoid the need for modified CPG. Oligonucleotides were purified by preparative 20% denaturing polyacrylamide gel electrophoresis (PAGE), and isolated by excision and elution from gel. The recovered material was subsequently quantified by absorption at 260 nm with molar extinction coefficients determined by the nearest neighbor method. All synthesized oligodeoxynucleotides were confirmed by MALDI mass spectroscopy. Data are given in Supporting Information. Oligoribonucleotides were purchased from Dharmacon (Boulder, CO) and were used without further purification.
Thermal denaturation studies
Solutions for the thermal denaturation studies contained the indicated concentrations of self-complementary or non-self complementary strands containing nucleoside analogues. The buffer contained NaCl, MgCl2, and Na·PIPES (100 mM, 10 mM, and 10 mM respectively) at pH 7.0. After the solutions were prepared they were heated to 95°C and annealed in the UV-VIS spectrophotometer Varian Cary 1 equipped with a thermoprogrammer at the rate of 1.0 °C /min. Melting studies were carried out in Teflon-stoppered 1 cm path length quartz cuvettes under a steady flow of nitrogen current to prevent moisture condensation on quartz wall at low temperature. Absorbance was monitored while temperature was raised from 5 to 95 °C, or lowered from 95 to 5 °C at a rate of 1°C /min. Sequences were monitored at 260 nm except the indicated ones. Computer fitting of the melting data with MeltWin software generated melting temperature (Tm), Free energies (ΔG°) and other thermodynamic data for the “dangling end” duplexes and for the 12 bp duplexes in the Supporting file. The curve fit values were compared with those calculated from van't Hoff plots by plotting 1/Tm verse ln(CT/4) and matched with good agreement.
Circular Dichroism Measurements
CD spectra were recorded on an AVIV CD spectrometer (model: 62A DS) from 400 to 180 nm at 15 °C. Data were collected for 1 sec in 1 nm intervals in a 1 cm path-length quartz cell. All samples were in 3 μM total oligonucleotide concentration with 1:1 single strand ratios when paired. The buffer solution contained 100 mM NaCl, 10 mM MgCl2, 10 mM Na•PIPES, pH=7.0.
Fluorescence measurements
Fluorescence spectra were recorded on a Fluorolog-3 spectrometer (model: FL3-11) equipped with polarizer. Data were collected from sample in a 1 cm path-length quartz cell. All samples were in 2 μM total oligonucleotide concentration with 1:1 single strand ratios when paired. The buffer solution contained 100 mM NaCl, 10 mM MgCl2, 10 mM Na•PIPES, pH=7.0.
Hybridizations on PEG-polystyrene beads
Preparation of modified PEG-polystyrene beads and determination of loading were based on the reported procedures.20 DNA was synthesized on the beads using Pac-dA CE and iPr-Pac-dG CE phosphoramidites purchased from Glen Research. Deprotection of the Pac and cyanoethyl groups was done by the reactions of the beads with ammonium hydroxide at room temperature, after which the beads were dried. Using double-sided adhesive, beads were immobilized onto the surface of glass slides. They were then soaked in hybridization buffer (NaCl/MgCl2/Na•PIPES, 100 mM/10 mM/10 mM) for 12 h at room temperature, and background fluorescence images under the buffer were taken a Nikon Eclipse E800 epifluorescence microscope. 10 μL of the yyDNA solution at 90 μM concentration was then pipetted onto the immobilized beads. The hybridization solutions were allowed to stand 12 h at room temperature in the dark. Then the immobilized beads were washed with the hybridization buffer three times, and then a post-hybridization image was taken. Microscope settings were as follows: excitation filter 340-380 nm, emission observed at 450-510 nm. Real images of background and hybridization samples are shown in Fig. 7a-d. The images of loaded beads with buffer and the loaded beads after hybridization were also compared by overlaying an inverse image (Adobe Photoshop) of the background with the real image of the hybridization solution, using 50% transparency (Fig. 7e,f). In such an overlay, anything not appearing 50% gray shows optical changes between background image and experimental image.
Supplementary Material
Description of synthesis of trityl phosphoramidite derivatives of 1 and 2; 1H NMR spectra of synthetic intermediates; thermal denaturation and UV-monitored mixing data; circular dichroism spectra; oligonucleotide characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We acknowledge the National Institutes of Health (GM63587) for support. A.H. F. L. ackowledges a fellowship from the Croucher Foundation. We thank Samuel Tan for assistance with polymer-supported DNA synthesis.
References
- 1.Watson JD, Crick FHC. Cold Spr Harbor Symp Quant Biol. 1953;18:123–131. doi: 10.1101/sqb.1953.018.01.020. [DOI] [PubMed] [Google Scholar]
- 2.SantaLucia J, Jr, Hicks D. Annu Rev Biophys Biomol Struct. 2004;33:415–440. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- 3.(a) Petruska J, Goodman MF, Boosalis MS, Sowers LC, Cheong C, Tinoco I., Jr Proc Natl Acad Sci USA. 1988;85:6252–6256. doi: 10.1073/pnas.85.17.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kool ET. Annu Rev Biochem. 2002;71:191–219. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kim T, Kool ET. Org Lett. 2004;6:3949–3952. doi: 10.1021/ol048487u. [DOI] [PubMed] [Google Scholar]; (b) Kim T, Kool ET. J Org Chem. 2005;70:2048–2053. doi: 10.1021/jo048061t. [DOI] [PubMed] [Google Scholar]; (c) Kim TW, Delaney JC, Essigmann JM, Kool ET. Proc Natl Acad Sci USA. 2005;102:15803–15808. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Leonard NJ. Accounts Chem Res. 1982;15:128–135. [Google Scholar]; (b) Stevenson TM, Leonard NJ. J Org Chem. 1984;49:2158–2164. [Google Scholar]
- 6.(a) Godde F, Toulme JJ, Moreau S. Nucleic Acids Res. 2000;28:2977–2985. doi: 10.1093/nar/28.15.2977. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Godde F, Toulme JJ, Moreau S. Biochemistry. 1998;37:13765–13775. doi: 10.1021/bi9811967. [DOI] [PubMed] [Google Scholar]
- 7.(a) Liu H, Gao J, Lynch SR, Maynard L, Saito D, Kool ET. Science. 2003;302:868–871. doi: 10.1126/science.1088334. [DOI] [PubMed] [Google Scholar]; (b) Liu H, Gao J, Saito D, Maynard L, Kool ET. J Am Chem Soc. 2004;126:1102–1109. doi: 10.1021/ja038384r. [DOI] [PubMed] [Google Scholar]; (c) Liu H, Lynch SR, Kool ET. J Am Chem Soc. 2004;126:6900–6905. doi: 10.1021/ja0497835. [DOI] [PubMed] [Google Scholar]; (d) Gao J, Liu H, Kool ET. J Am Chem Soc. 2004;126:11826. doi: 10.1021/ja048499a. [DOI] [PubMed] [Google Scholar]; (e) Liu H, Gao J, Kool ET. J Am Chem Soc. 2005;127:1396–1402. doi: 10.1021/ja046305l. [DOI] [PubMed] [Google Scholar]; (f) Liu H, Gao J, Kool ET. J Org Chem. 2004;70:639–647. doi: 10.1021/jo048357z. [DOI] [PubMed] [Google Scholar]; (g) Gao J, Liu H, Kool ET. Angew Chem Int Ed. 2005;44:3118–3122. doi: 10.1002/anie.200500069. [DOI] [PubMed] [Google Scholar]
- 8.(a) Lu H, He K, Kool ET. Angew Chem Int Ed. 2004;43:5834–5836. doi: 10.1002/anie.200461036. [DOI] [PubMed] [Google Scholar]; (b) Lee AHF, Kool ET. J Org Chem. 2004;70:132–140. doi: 10.1021/jo0483973. [DOI] [PubMed] [Google Scholar]; (c) Lee AHF, Kool ET. J Am Chem Soc. 2005;127:3332–3338. doi: 10.1021/ja0430604. [DOI] [PubMed] [Google Scholar]
- 9.Geyer CR, Battersby TR, Benner SA. Structure. 2003;11:1485–1498. doi: 10.1016/j.str.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 10.(a) Seela F, Melenewski A, Wei C. Bioorg Med Chem Lett. 1997;17:2173–2176. [Google Scholar]; (b) Groebke K, Hunziker J, Fraser W, Peng L, Diederichsen U, Zimmermann K, Holzner A, Leumann C, Eschenmoser A. Helv Chim Acta. 1998;81:375–474. [Google Scholar]; (c) Seela F, Wei C, Melenewski A, Feiling E. Nucleosides Nucleotides. 1998;17:2045–2052. [Google Scholar]
- 11.(a) Minakawa N, Kojima N, Hikishima S, Sasaki T, Kiyosue A, Atsumi N, Ueno Y, Matsuda A. J Am Chem Soc. 2003;125:9970–9982. doi: 10.1021/ja0347686. [DOI] [PubMed] [Google Scholar]; (b) Hikishima S, Minakawa N, Kuramoto K, Fujisawa Y, Ogawa M, Matsuda A. Angew Chem Int Ed. 2005;44:596–598. doi: 10.1002/anie.200461857. [DOI] [PubMed] [Google Scholar]
- 12.(a) Flanagan WM, Wolf JJ, Olson P, Grant D, Lin KY, Wagner RW, Matteucci MD. Proc Natl Acad Sci USA. 1999;96:3513–3518. doi: 10.1073/pnas.96.7.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Seley KL, Januszczyk P, Hagos A, Zhang L, Dransfield DT. J Med Chem. 2000;43:4877–4883. doi: 10.1021/jm000326i. [DOI] [PubMed] [Google Scholar]; (c) Okamoto A, Tanaka K, Fukuta T, Saito I. Chembiochem. 2004;5:958–963. doi: 10.1002/cbic.200400010. [DOI] [PubMed] [Google Scholar]
- 13.(a) DeVries RA, Vosejpka PC, Ash ML. In: Catalysis of Organic Reactions. Herkes FE, editor. Chapter 37 Marcel Dekker; New York: 1998. [Google Scholar]; (b) Littke AF, Fu GC. J Org Chem. 1999;64:10–11. doi: 10.1021/jo9820059. [DOI] [PubMed] [Google Scholar]; (c) Littke AF, Fu GC. J Am Chem Soc. 2001;123:6989–7000. doi: 10.1021/ja010988c. [DOI] [PubMed] [Google Scholar]
- 14.Guckian KM, Schweitzer BA, Ren RXF, Sheils CJ, Tahmassebi DC, Kool ET. J Am Chem Soc. 2000;122:2213–2222. doi: 10.1021/ja9934854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakano S, Uotani Y, Nakashima S, Anno Y, Fujii M, Sugimoto N. J Am Chem Soc. 2003;125:8086–8087. doi: 10.1021/ja034465b. [DOI] [PubMed] [Google Scholar]
- 16.(a) Jiang YL, Stivers JT, Song F. Biochemistry. 2002;41:11248–11254. doi: 10.1021/bi026227j. [DOI] [PubMed] [Google Scholar]; (b) Beuck C, Singh I, Bhattacharya A, Hecker W, Parmar VS, Seitz O, Weinhold E. Angew Chem Int Ed. 2003;42:3958–3960. doi: 10.1002/anie.200219972. [DOI] [PubMed] [Google Scholar]; (c) Kwon K, Jiang YL, Stivers JT. Chem Biol. 2003;10:351–359. doi: 10.1016/s1074-5521(03)00077-2. [DOI] [PubMed] [Google Scholar]
- 17.(a) Ward DC, Reich E, Stryer L. J Biol Chem. 1969;244:1228–1237. [PubMed] [Google Scholar]; (b) Secrist JA, Barrio JR, Leonard NJ. Science. 1972;175:646–647. doi: 10.1126/science.175.4022.646. [DOI] [PubMed] [Google Scholar]; (c) Ren RXF, Chaudhuri NC, Paris PL, Rumney S, IV, Kool ET. J Am Chem Soc. 1996;118:7671–7678. doi: 10.1021/ja9612763. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Charubala R, Maurinsh J, Rosler A, Melguizo M, Jungmann O, Gottlieb M, Lehbauer J, Hawkins M, Pfleiderer W. Nucleosides Nucleotides. 1997;16:1369–1378. [Google Scholar]; (e) Paris PL, Langenhan J, Kool ET. Nucleic Acids Res. 1998;26:3789–3793. doi: 10.1093/nar/26.16.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Coleman RS, Madaras ML. J Org Chem. 1998;63:5700–5703. [Google Scholar]; (g) Strässler C, Davis NE, Kool ET. Helv Chim Acta. 1999;82:2160–2171. doi: 10.1002/(sici)1522-2675(19991215)82:12<2160::aid-hlca2160>3.0.co;2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Seela F, Becher G, Chen YM. Nucleosides Nucleotides & Nucleic Acids. 2000;19:1581–1598. doi: 10.1080/15257770008045448. [DOI] [PubMed] [Google Scholar]; (i) Rao P, Benner SA. J Org Chem. 2001;66:5012–5015. doi: 10.1021/jo005743h. [DOI] [PubMed] [Google Scholar]; (j) Chen D, Beuscher AE, IV, Stevens RC, Wirsching P, Lerner RA, Janda KD. J Org Chem. 2001;66:1725–1732. doi: 10.1021/jo001676f. [DOI] [PubMed] [Google Scholar]; (k) Lehbauer J, Pfleiderer W. Helv Chim Acta. 2001;84:2330–2342. [Google Scholar]; (l) Liu C, Martin CT. J Mol Biol. 2001;308:465–475. doi: 10.1006/jmbi.2001.4601. [DOI] [PubMed] [Google Scholar]; (m) Singleton SF, Shan F, Kanan MW, McIntosh CM, Stearman CJ, Helm JS, Webb KJ. Org Lett. 2001;3:3919–3922. doi: 10.1021/ol0167863. [DOI] [PubMed] [Google Scholar]; (n) Gao J, Strässler C, Tahmassebi DC, Kool ET. J Am Chem Soc. 2002;124:11590–11591. doi: 10.1021/ja027197a. [DOI] [PubMed] [Google Scholar]; (o) Tanaka K, Okamoto A, Saito I. Nucleic Acids Res Suppl. 2003:171–172. doi: 10.1093/nass/3.1.171. [DOI] [PubMed] [Google Scholar]; (p) Gao J, Watanabe S, Kool ET. J Am Chem Soc. 2004;126:12748–12749. doi: 10.1021/ja046910o. [DOI] [PubMed] [Google Scholar]; (q) Saito Y, Miyauchi Y, Okamoto A, Saito I. Chem Commun. 2004;7:1704–1705. doi: 10.1039/b405832a. [DOI] [PubMed] [Google Scholar]; (r) Okamoto A, Tanaka K, Fukuta T, Saito I. Chembiochem. 2004;5:958–963. doi: 10.1002/cbic.200400010. [DOI] [PubMed] [Google Scholar]; (s) Hawkins ME, Balis FM. Nucleic Acids Res. 2004;32:e62. doi: 10.1093/nar/gnh060. [DOI] [PMC free article] [PubMed] [Google Scholar]; (t) Cuppoletti A, Cho Y, Park JS, Strässler C, Kool ET. Bioconj Chem. 2005;16:528–534. doi: 10.1021/bc0497766. [DOI] [PubMed] [Google Scholar]; (u) Greco NJ, Tor Y. J Am Chem Soc. 2005;127:10784–10785. doi: 10.1021/ja052000a. [DOI] [PubMed] [Google Scholar]; (v) Sandin P, Wilhelmsson LM, Lincoln P, Powers VE, Brown T, Albinsson B. Nucleic Acids Res. 2005;33:5019–5025. doi: 10.1093/nar/gki790. [DOI] [PMC free article] [PubMed] [Google Scholar]; (w) Cho Y, Kool ET. ChemBioChem. 2005;6 in press. [Google Scholar]; (x) Kim SJ, Kool ET. J Am Chem Soc. 2006;128:6164–6171. doi: 10.1021/ja0581806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Matray TJ, Kool ET. J Am Chem Soc. 1998;120:6191–6192. doi: 10.1021/ja9803310. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Uotani Y, Nakano S, Nakashima S, Anno Y, Fujii M, Sugimoto N. Nucleic Acids Res Suppl. 2003:79–80. doi: 10.1093/nass/3.1.79. [DOI] [PubMed] [Google Scholar]; (c) Zhang L, Long H, Boldt GE, Janda KD, Schatz GC, Lewis FD. Org Biomol Chem. 2006;4:314–322. doi: 10.1039/b513694f. [DOI] [PubMed] [Google Scholar]
- 19.(a) Zhang HC, Daves GDJ. J Org Chem. 1992;57:4690–4696. [Google Scholar]; (b) Lan T, McLaughlin LW. Biochemistry. 2001;40:968–976. doi: 10.1021/bi0015212. [DOI] [PubMed] [Google Scholar]
- 20.Gao J, Watanabe S, Kool ET. J Am Chem Soc. 2004;126:12748–12749. doi: 10.1021/ja046910o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of synthesis of trityl phosphoramidite derivatives of 1 and 2; 1H NMR spectra of synthetic intermediates; thermal denaturation and UV-monitored mixing data; circular dichroism spectra; oligonucleotide characterization. This material is available free of charge via the Internet at http://pubs.acs.org.