

Abstract
Thymidine dinucleotide (pTpT) stimulates melanogenesis in mammalian pigment cells and intact skin, mimicking the effects of UV irradiation and UV-mimetic DNA damage. Here it is shown that, in addition to tanning, pTpT induces a second photoprotective response, enhanced repair of UV-induced DNA damage. This enhanced repair results in a 2-fold increase in expression of a UV-damaged chloramphenicol acetyltransferase expression vector transfected into pTpT-treated skin fibroblasts and keratinocytes, compared with diluent-treated cells. Direct measurement of thymine dimers and (6–4) photoproducts by immunoassay demonstrates faster repair of both of these UV-induced photoproducts in pTpT-treated fibroblasts. This enhanced repair capacity also improves cell survival and colony-forming ability after irradiation. These effects of pTpT are accomplished, at least in part, by the up-regulation of a set of genes involved in DNA repair (ERCC3 and GADD45) and cell cycle inhibition (SDI1). At least two of these genes (GADD45 and SDI1) are known to be transcriptionally regulated by the p53 tumor suppressor protein. Here we show that pTpT activates p53, leading to nuclear accumulation of this protein, and also increases the specific binding of this transcription factor to its DNA consensus sequence.
UV irradiation of eukaryotic and prokaryotic cells leads to the formation of a variety of DNA photoproducts, predominately cyclobutane pyrimidine dimers. Their repair is accomplished largely through the excision repair system. Although well characterized in prokaryotes as part of the SOS response inducible by UV light and DNA damage (1–3), excision repair in eukaryotic cells only recently has begun to be defined at the biochemical and genetic levels (1–4). In particular, the existence of a coordinately regulated SOS response in eukaryotes has not been demonstrated. However, a variety of DNA-damage responsive genes have been identified (5–8), and at least one report demonstrates that excision repair is enhanced in a fibroblast cell line previously exposed to UV light or DNA-damaging chemicals (9).
Human skin exhibits a dual response to UV irradiation: initial repair of DNA photoproducts, followed by a variety of changes such as enhanced melanogenesis (tanning), known to protect against subsequent UV exposure. We previously have shown that the small DNA fragment thymidine dinucleotide (pTpT) enhances melanogenesis in pigment cells and intact skin (10, 11), mimicking the effects of UV-induced DNA damage. Here we report that pTpT also enhances the DNA repair capacity of human skin-derived cells, at least in part by activation of the p53 tumor suppressor protein and by induction of genes involved in DNA repair and/or cell cycle regulation after UV irradiation. These findings suggest the existence of an SOS response in mammalian tissues analogous to that described in prokaryotes, which is stimulated by the binding of single-stranded DNA to the Rec A protein with subsequent derepression of regulated genes (1–3). We postulate that pTpT may initiate this response and stimulate protective cellular responses without first inducing damage in the genomic DNA.
MATERIALS AND METHODS
Cell Culture.
Newborn keratinocytes were established as described (12), by using a modification of the method of Rheinwald and Green (13). First-passage keratinocytes were maintained in a nondifferentiating low Ca2+ medium (K-Stim, Collaborative Biomedical Products, Bedford, MA). Fibroblasts were established from dermal explants as described (14) and maintained in DMEM (GIBCO/BRL) supplemented with 10% bovine serum (HyClone).
SCC12F cells (15) were maintained in a DMEM-based keratinocyte growth medium supplemented with 10% fetal bovine serum and epidermal growth factor as described (16). The p53-null H1299 lung carcinoma cell line (17) was maintained in DMEM with 10% calf serum.
UV-Irradiation.
Cells were irradiated with a 1 KW Xenon arc solar simulator (XMN 1000–21, Optical Radiation, Azuza, CA) metered at 285 ± 5 nm by using a research radiometer (model IL1700A, International Light, Newburyport, MA), as described (18). Irradiations and metering were done filtered through a plastic culture dish lid, which effectively removes all wavelengths below 280 nm (19). Therefore, cells were exposed to a continuous spectrum of irradiation, including UVB (280–320 nm), UVA (320–400 nm), and visible and infrared wavelengths with the great majority of biologic activity in the UVB range. Accordingly, these cellular irradiation doses are designated as UVB. To UV-damage the pCAT reporter vector, the DNA dissolved in water was exposed to the solar simulator unfiltered by a plastic lid, allowing transmission of some UVC wavelengths (19). These irradiation doses also were quantified by metering at 285 ± 5 nm, but differ from the doses delivered to the cell cultures in that DNA-damaging UVC wavelengths were present and accounted for much of the biologic activity. Accordingly, the vector irradiation doses are designated as UVC. Of note, these UVC doses cannot be compared quantitatively to the UVB doses described above.
Chloramphenicol Acetyltransferase (CAT) Assay for DNA Repair.
Cells in serum-supplemented medium were treated with either 100 μM pTpT (Midland Certified Reagent, Midland, TX) or an equal volume of diluent (DMEM) alone for 5 days before transfection. Duplicate cultures of each condition were transfected by using the Lipofectin Reagent Kit (GIBCO/BRL) as suggested by the manufacturer and 5 μg reporter DNA, pCAT-Control vector (Promega,). This nonreplicating vector contains the CAT gene under control of simian virus 40 promoter and enhancer sequences. Before transfection, the vector DNA was either sham-irradiated or irradiated as described above. Dose response experiments showed that irradiation of the pCAT-control vector with 70, 100 and 140 mJ/cm2 UVC resulted in 65, 75, and 88% inhibition, respectively, of CAT activity in transfected fibroblasts and keratinocytes. For the repair experiments, the 100 mJ/cm2 dose was used to give an effective, but nonsaturating, degree of DNA damage to the pCAT expression vector. Cells were collected 24 hr after transfection in a lysis buffer provided in the CAT Enzyme Assay System (Promega) by using a protocol provided by the manufacturer. CAT enzyme activity was determined by using the liquid scintillation counting protocol and components of the assay system kit. 14C-labeled chloramphenicol [50–60 mCi (1.85–2.22 GBq) mmol] was purchased from New England Nuclear. Protein concentration in the cell extracts was determined by the method of Bradford (20).
Immunoassay for Repair of Thymine Dimers and (6–4) Photoproducts.
Preconfluent cell cultures were supplemented with either 100 μM pTpT or an equal volume of diluent for 5 days before irradiation with 50 mJ/cm2 UVB. The cells then were collected at various times, and the genomic DNA was isolated by using DNAzol (Molecular Research Center, Cincinnati) as described by the manufacturer. DNA (100 ng) from the UV-irradiated as well as sham-irradiated fibroblasts was spotted onto Hybond-N (Amersham) by using a slot-blot apparatus (Schleicher & Schuell). The filter then was baked at 80°C for 3 hr. Quantitation of thymine dimers and (6–4) photoproducts was carried out as described (21) by using mAbs MC-062 for thymine dimers and MC-082 for (6–4) photoproducts (Kamiya Biomedical, Tukwila, WA). Antibody binding was determined by using an ECL Western blot detection kit (Amersham), autoradiography, and densitometry. To evaluate the uniformity of DNA sample loading, each slot blot also was hybridized with 32P-labeled human genomic DNA (Promega) (22). The labeled DNA (50 ng) was added to 3–5 μg unlabeled DNA for the hybridization reaction to assure an excess of probe DNA. Slot blots of different amounts of human DNA (10–200 ng) also were hybridized to demonstrate a linear relationship between DNA on the filter and autoradiographic signal as determined by densitometry (data not shown).
Cell Survival After UV Irradiation.
Cultures of newborn dermal fibroblasts were treated for 5 days with either diluent or 100 μM pTpT. Duplicate cultures then were rinsed and then either untreated or exposed to 10, 20, 30 or 40 mJ/cm2 UVB (18), given fresh medium, cultured for an additional 1 or 3 days, collected by trypsinization after two washes in buffered saline, and counted by Coulter counter. The cell numbers from duplicate cultures were calculated and averaged.
To determine the colony-forming efficiency of keratinocytes, first passage cultures were supplemented with either 100 μM pTpT or an equal amount of diluent for 5 days before UV irradiation. Cells were either sham-irradiated or exposed to 30 mJ/cm2 or 60 mJ/cm2 UVB as described (18), trypsinized, and immediately replated onto a 3T3 feeder layer (12) at various densities. The cultures were monitored daily, and when colonies were easily visible (typically >30 cells/colony), the 3T3 cells were removed by mild EDTA treatment, and the keratinocytes were stained with Rhodanile blue. Culture dishes then were coded, and the colonies on each were counted in a blinded fashion by using a dissecting microscope.
Northern Blot Analysis.
Preconfluent cultures were given fresh medium supplemented with either 100 μM pTpT or an equal volume of diluent (DMEM). Cells were collected 24, 48, and 72 hr after additions and processed for total RNA isolation by using the Tri-Reagent extraction method (Molecular Research Center) following the manufacturer’s protocol. Ten micrograms of RNA from each sample were gel-electrophoresed, transferred to a nylon filter, and probed with 32P-labeled cDNA as described (22). The cDNA for GADD45 was generated by PCR by using primers based on the human GADD45 gene sequence (5). The cDNA for ERCC3 was purchased from the American Type Culture Collection. The SDI1 cDNA was the kind gift of J. Smith (Baylor, Houston) and has been described (21).
Staining of Normal Fibroblasts for p53 Localization.
Preconfluent cultures were treated with either diluent or 100 μM pTpT for 24 hr before cell staining. Cells first were fixed for 1 min in Histochoice fixative (Amresco, Solon, OH) followed by a 5-min rinse in PBS. p53 was detected by using the Vectastain Elite ABC kit (Vector Laboratories) and the p53-specific mAb 421 (Ab-1) (Oncogene). The protocol used for staining was that provided by the manufacturer. Photographic slides of stained cells were scanned by using a Scan Maker 35T (Microtek Lab, Redondo Beach, CA) and a Macintosh IIsi computer, adjusting brightness and contrast to maximize differences.
Transfection of H1299 Cells with a p53 Expression Vector.
Preconfluent cultures of p53-null H1299 cells were transfected with an expression vector containing the wild-type human p53 cDNA under the control of the human cytomegalovirus promoter/enhancer (Bert Vogelstein, The Johns Hopkins Oncology Center, Baltimore). Control transfections were performed by using the vector from which the p53 cDNA was removed. Transfections were carried out as described (7). One day after transfection, cells were collected for Western blot analysis by using 20 μg total protein as described (23). p53 was detected by using mAb421 (Ab-1) (Oncogene), anti-mouse Ig linked to horseradish peroxidase (Amersham) and an ECL kit (Amersham) following the manufacturer’s directions. At the time of protein collection, duplicate cultures of H1299 cells transfected with the p53 expression vector or control vector were given either diluent (DMEM) or 100 μM pTpT. After 24 hr, the cells were collected and processed for RNA isolation and Northern blot analysis with the SDI1 cDNA probe. The autoradiograph was scanned by using a Macintosh IIsi computer and Macintosh One Scanner with brightness and contrast adjusted to maximize differences in autoradiographic signals.
Electromobility Shift Assay for p53 Activity.
Cultures of H1299 cells were transfected with either control vector or the p53 expression vector. Twenty-four hours after transfection, both control and p53-expressing cells were collected by trypsinization and pelleted by centrifugation, then resuspended in either 2 ml of DMEM or 2 ml of DMEM containing 100 μM pTpT. The cells then were vortexed and incubated in suspension for 4–5 hr at 37°C, 5% CO2, with occasional vortexing. The cells again were pelleted by centrifugation, and nuclear extract was prepared as described by Tishler et al. (24). The amount of p53 in each nuclear extract then was determined by Western blot analysis and densitometry. The p53/consensus sequence-binding gel retardation assay was carried out essentially as described by Tishler et al. (24) and Hupp et al. (25). The wild-type p53-consensus sequence was 5′TTAAGGACATGCCCGGGCATGTCC3′. The mutated p53 consensus sequence was 5′TTAAGGACATGGCCGGCCATGTCC3′. Basic binding reactions were carried out on ice for 30 min and contained approximately 20 μg of nuclear extract (corrected so that each reaction contained the same amount of p53), 1 μg salmon sperm DNA, and 0.1 pmol 32P-labeled (GIBCO/BRL 5′ DNA terminus labeling system) wild-type consensus sequence in a final volume of 20 μl. After incubation, the reaction mix was separated on a 4% polyacrylamide gel (200 volts for 1.5 hr), and the p53/consensus sequence complex was detected by autoradiography.
RESULTS
DNA Repair in Control and pTpT-Treated Cells.
To measure the DNA repair capacity of normal human skin-derived fibroblasts and keratinocytes, we used an assay system in which a nonreplicating UV-damaged reporter plasmid containing the bacterial CAT gene under the control of simian virus 40 promoter and enhancer sequences is repaired by host cell repair enzymes after transfection (12). This system previously was shown to detect decreases of approximately 15% in DNA repair capacity in human cells associated with aging and early-onset skin cancers (26, 27) and was used to measure the DNA repair capacity of lymphocytes from workers exposed to benzene (28) and 1,3-butadiene (29).
Preliminary experiments were performed to determine the dose-response curve between pretransfection UV irradiation dose of the plasmid and reduction in CAT activity, assayed in keratinocyte and fibroblast culture lysates 16–24 hr after transfection. Because the reporter plasmid is nonreplicating, CAT activity is a direct measure of the extent of host cell enzyme-mediated DNA repair and restoration of biological activity. Compared with plasmids sham-irradiated before transfection, plasmids exposed to the selected dose of 100 mJ/cm2 UVC radiation generated only 23% and 24% as much CAT activity in keratinocytes and fibroblasts, respectively. However, lysates of keratinocytes and fibroblasts pretreated with 100 μM pTpT for 5 days before transfection with the irradiated plasmid displayed CAT activity that was 50% and 55%, respectively, that of controls (cultures transfected with sham-irradiated plasmid), indicating that pTpT treatment markedly increases the capacity of normal human cells to repair UV-induced DNA damage over the first 24 hr. Results were very consistent, with actual CAT values for duplicate cultures within 8% and 6% of the mean for keratinocytes and fibroblasts, respectively. The expression of the sham-irradiated plasmid was not higher in pTpT-treated cells than in non-pTpT-treated cells, establishing that the enhanced expression of UV-irradiated plasmid in pTpT-treated cells did not result from a general increase in plasmid transcription in these cells. Furthermore, the observed doubling of CAT readout is comparable to that reported by using the same assay system in CV-1 cells exposed to mitomycin C or UV-irradiation, from 25% to 50%–55% that in paired cultures transfected with sham-irradiated plasmid (9), and nearly twice the difference reported on average between cells derived from cancer-prone versus control subjects in a large population study in which low CAT plasmid repair capacity was suggested to be causally related to development of skin cancer (26).
To directly assess the rate of repair of UV-induced photoproducts in pTpT-treated cells vs. controls, paired cultures were UV-irradiated and DNA was harvested at multiple time points, then reacted with mAbs specific for thymine dimers or (6–4) photoproducts. Slot-blot analysis of DNA from cells harvested immediately after irradiation (time 0) with 50 mJ/cm2 UVB showed strong binding of the thymine dimer and (6–4) photoproduct antibodies. Neither diluent-treated nor pTpT-treated fibroblasts showed a decrease in dimer content within 6 hr after irradiation. However, by 12- and 24-hr postirradiation, pTpT-treated cells had removed 40% and 65% of dimers, respectively, whereas diluent-treated cells showed no decrease at 12 hr and had removed only 25% of dimers by 24 hr (Fig. 1A), a repair rate consistent with previous reports for mouse skin as well as cultured mouse and human fibroblasts (30, 31). As also previously reported (30, 31), the rate of repair of (6–4) photoproducts was considerably faster, with an approximately 50% reduction after 1 hr for controls (Fig. 1B). As for dimers, however, pTpT-treated cells removed (6–4) photoproducts far faster than diluent-treated cells, approximately 50% within 20 min.
Figure 1.

The effect of pTpT on the repair rate of thymine dimers and (6–4) photoproducts. Preconfluent cultures of newborn fibroblasts were supplemented with either 100 μM pTpT or an equal volume of diluent for 5 days before exposure to 50 mJ/cm2 UVB. The cells then were collected at various time points, and the genomic DNA was isolated. Thymine dimer (A) and (6–4) photoproduct (B) content were determined as described in Materials and Methods. Samples harvested immediately after irradiation (time 0) were set at 100%, and all time points were calculated as a percent of these values. No consistent effect of pTpT on the degree of initial DNA damage (time 0 values) was seen. Each normalized time point is the average of duplicate blots. Data for a representative donor (one of three separate experiments) are shown. The difference between diluent- and pTpT-treated cells was statistically significant [P < 0.013 for thymine dimers and P < 0.028 for (6–4) photoproducts; one-tailed t test].
Cell Survival After UV Irradiation.
Because enhanced DNA repair would be expected to increase cell survival after irradiation, we examined the effect of pretreatment with pTpT on fibroblast yield after UV irradiation. Cells treated for 5 days with diluent alone showed, as expected, continued exponential growth in serum-supplemented medium over the next 3 days in the absence of irradiation and a progressive decrease in the ratio of day 3 to day 1 postirradiation cell yields at UVB doses of 20, 30, and 40 mJ/cm2 (Fig. 2A), because of progressive cell death and detachment (Fig. 2B). Cells pretreated with pTpT also continued to grow exponentially in the absence of irradiation, but in contrast to controls exhibited continued slow net growth even after irradiation, maintaining day 3 cell yields above day 1 yields even at the highest UV doses tested (Fig. 2A). Furthermore, in contrast to controls (Fig. 2B), cells pretreated with pTpT appeared healthy after irradiation (Fig. 2C).
Figure 2.
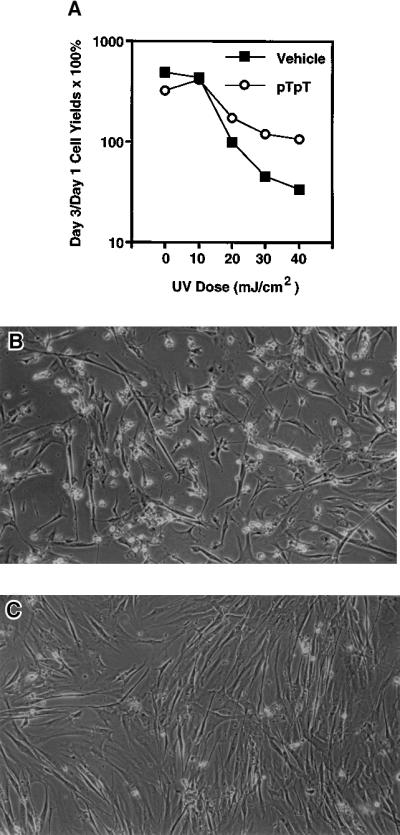
Effect of pTpT on the survival of fibroblasts after UV irradiation. The cell numbers from duplicate cultures were calculated and averaged as described in Materials and Methods. The ratio of cell yields (day 3/day 1) × 100% is presented for each condition (A), where 100% represents no change in cell yield between days 1 and 3. Day 1 cell yields were 140,000 ± 9,000 and 177,000 ± 21,000 for sham-irradiated pTpT-treated and diluent-treated cultures, respectively. After increasing UVB doses between 10 mJ/cm2 and 40 mJ/cm2, cell yields on day 1 were similar (70% to 120% of sham-irradiated day 1 cell yields). However, by day 3 cell yields progressively decreased to 78,200 ± 1,700 (or 34% sham-irradiated day 3 cultures) in diluent-treated cultures, implying cell death, whereas in pTpT-treated cultures, day 3 cell yields remained at or above the day 1 values, implying growth arrest without cell loss. Representative fields from diluent-treated control cells (B) and pTpT-treated cells (C), photographed 2 days after 30 mJ/cm2 UVB irradiation, are presented.
To determine the proportion of cells surviving the UV irradiation that were capable of mitosis, the colony-forming efficiency of normal human keratinocytes also was determined after UV irradiation. Preconfluent dishes of cells were either unirradiated or exposed to 30 mJ/cm2 or 60 mJ/cm2 UVB radiation and replated at various densities. Pretreatment of cells with pTpT reduced the colony-forming efficiency of unirradiated keratinocytes by 30% (data not shown), likely reflecting the effects of pTpT on slowing keratinocyte proliferation and hence promoting terminal differentiation (13). However, after exposure to 30 mJ/cm2 or 60 mJ/cm2 UVB radiation, pTpT-treated cells gave rise to more colonies than did diluent-treated cells (an average of 2,522 vs. 2,176 colonies per 200,000 cells plated after exposure to 30 mJ/cm2 and 344 vs. 275 colonies per 200,000 cells plated after exposure to 60 mJ/cm2). Representative colonies derived from replated pTpT-treated or diluent-treated keratinocytes after 30 mJ/cm2 irradiation are shown in Fig. 3A. Furthermore, when expressed as the percent of colonies formed from unirradiated cells, pTpT-treated and irradiated keratinocytes formed roughly twice as many colonies compared with diluent-treated and irradiated cells (Fig. 3B).
Figure 3.

Effect of pTpT on colony-forming efficiency of keratinocytes after UV irradiation. First passage cultures were supplemented with either 100 μM pTpT or an equal amount of diluent for 5 days before irradiation. Cells were either sham-irradiated or exposed to 30 mJ/cm2 or 60 mJ/cm2 UVB, trypsinized, and immediately replated onto a 3T3 feeder layer (12) at various densities as described. (A) Representative dishes of diluent- or pTpT-treated cells exposed to 30 mJ/cm2 UVB radiation. (B) Colony yield is presented as the percent of colonies formed from unirradiated keratinocytes. Nonpaired t test analysis found that the number of colonies formed between diluent- and pTpT-treated cells was statistically significant with P < 0.01 for 30 mJ/cm2 and P < 0.03 for 60 mJ/cm2.
Effect of pTpT on Gene Expression and Cell Proliferation.
To elucidate the mechanism of this pTpT effect, we examined the expression of three genes involved in DNA repair and in the cellular response to DNA damage: GADD45, ERCC3, and SDI1. GADD45 is induced by genotoxic stress (32) and recently was shown to interact with proliferating cell nuclear antigen to stimulate excision repair as well as inhibit progression of the cell cycle into the S phase (33). The ERCC3 gene product contains protein motifs similar to those of DNA and RNA helicases (34) and has been shown to participate in excision repair (35). This protein is also part of the basic transcription factor complex BTF2, suggesting it may function as a helicase during transcription and in excision repair (36). A third gene, SDI1, also called p21, WAF1, or CIP1, encodes a protein that inhibits cyclin-dependent kinases and prevents the G1 to S transition in the cell cycle (37) and also can inhibit DNA replication by repressing the activation of DNA polymerase δ by proliferating cell nuclear antigen (38, 39), an event believed to block chromosome replication until after DNA repair has occurred. Paired cultures of a human squamous cell carcinoma line SCC12F (15) or normal human fibroblasts were supplemented with either diluent or 100 μM pTpT, and samples were collected for total RNA isolation and Northern analysis. The mRNAs for GADD45, ERCC3, and SDI1 were up-regulated in pTpT-treated cells as early as 24 hr and remained elevated for at least 3 days (Fig. 4).
Figure 4.

Expression of DNA repair and cell cycle regulatory genes. Preconfluent cultures of SCC12F cells (15) were given fresh medium supplemented with either 100 μM pTpT (+) or an equal volume of diluent (DMEM) (−). Cells were collected 24, 48, and 72 hr after additions and processed for total RNA. Ten micrograms of RNA from each sample were gel-electrophoresed, transferred to a nylon filter, and probed as described (21) (A–C). The ethidium bromide-stained gel is shown to demonstrate relative loading of the RNA samples (D). Similar results were obtained using normal human fibroblasts (data not shown). Densitometric analysis showed a 2.5- to 3.5-fold increase at one or more time points for all three transcripts in pTpT-treated cells compared with diluent-treated controls.
Because GADD45 and SDI1 function in cell cycle arrest after DNA damage, we also examined the effect of pTpT on cell proliferation. Cultures of normal keratinocytes and fibroblasts as well as SCC12F cells were treated with either 100 μM pTpT or diluent as a control and harvested for cell counts. The cell yields in keratinocyte cultures treated with pTpT were reduced by 63% compared with those of paired control cultures after 3 days (Fig. 5A) and corresponded to complete growth arrest versus approximately one population doubling for control cells maintained in serum-free defined medium. Fibroblasts and SCC12F cells showed significant similar reductions in pTpT-treated cell yield over 3–5 days due to slowed or arrested growth (Fig. 5 B and C). This growth retardation was comparable at 50 μM and rapidly and completely reversible after removal of pTpT from the medium (data not shown).
Figure 5.

pTpT effects on cell proliferation. Normal human keratinocytes (A) and fibroblasts (B) and SCC12F cells (C) were maintained as described. Preconfluent cultures were given fresh medium supplemented with either 100 μM pTpT or diluent. Cells were collected by trypsinization daily and counted by Coulter counter. Values represent averages ± SD of duplicate cultures. Dose response experiments with fibroblasts and SCC12F cells found similar degrees of growth inhibition with pTpT concentrations between 50 μM and 150 μM and lesser inhibition at 25 μM. (D) Cultures of the p53-null H1299 lung carcinoma cell line (17) were given fresh medium supplemented with either 100 μM pTpT or diluent, and cells were collected on consecutive days by trypsinization and counted by Coulter counter. Values represent averages ± SD of duplicate cultures.
Activation of p53.
Both the GADD45 and SDI1 genes are known to be transcriptionally regulated by the tumor suppressor protein p53 (40, 41). After UV- or γ-irradiation, or treatment of cells with DNA-damaging chemical agents, a rapid stabilization and nuclear accumulation of p53 (42–44) results, after which this protein binds to specific promoter consensus sequences and modulates the transcription of regulated genes (44). Recent data suggest that p53 also can be activated by the binding of small single-stranded DNAs, as well as certain peptides and antibodies, to its carboxyl-terminal domain (45, 46). To determine whether the inhibitory effect of the dinucleotide pTpT on cell proliferation is mediated through p53, we examined the growth response of a p53 null lung carcinoma cell line, H1299. No inhibition of proliferation of pTpT-treated H1299 cells was seen compared with diluent-treated controls (Fig. 5D).
The effect of pTpT on the level and intracellular distribution of p53 then was examined in normal neonatal fibroblasts by immunoperoxidase staining using the p53-specific mAb421. Within 24 hr, an increase in intranuclear p53 was detected in many of the pTpT-treated cells but not in diluent-treated cells (Fig. 6), a pattern similar to that seen after UV- and X-irradiation (42–44) and as consistent with the induction by pTpT of the p53-regulated genes GADD45 and SDI1 in these cells and in SCC12F cells.
Figure 6.

Nuclear accumulation of p53 in normal human fibroblasts after pTpT addition. Preconfluent cultures of fibroblasts were treated with either diluent or 100 μM pTpT for 24 hr before cell staining. Photographic slides of stained cells were scanned, with brightness and contrast adjusted to maximize differences.
To further confirm that pTpT effects are mediated through p53, induction of SDI1 mRNA was examined by Northern analysis in p53-null H1299 cells. Western analysis confirmed that H1299 cells normally express no p53 and that H1299 cells transfected with a wild-type p53 expression vector express high levels of p53 (Fig. 7A). H1299 cells express a very low level of the SDI1 transcript, and this level is not affected by the addition of pTpT (Fig. 7B). Transfection of these cells with a wild-type p53 expression vector increased the level of SDI1 (Fig. 7 B and C) and rendered this transcript inducible by addition of pTpT (Fig. 7C). Moreover, pTpT treatment had no effect on the growth of H1299 cells (Fig. 5D), but inhibited growth of transfected H1299 cells (data not shown). These data strongly suggest that pTpT increases the transcription of selected genes via p53 and that these gene inductions alter cell behavior.
Figure 7.

pTpT up-regulates the expression of SDI1 mRNA through p53. Transfections of p53-null H1299 cells were carried out as described in Materials and Methods. (A) One day after transfection, cells were collected for Western blot analysis using 20 μg total protein as described (23). (B–D) Duplicate cultures of H1299 cells were transfected with the p53 expression vector (designated p53) or control vector (Ctrl) were given either diluent (DMEM) (designated −) or 100 μM pTpT (designated +). Northern blot analysis was performed with the SDI1 cDNA probe. The autoradiograph was scanned, and the brightness and contrast were adjusted to maximize differences in autoradiographic signals (B and C). (D) The ethidium bromide-stained gel is shown to demonstrate relative RNA loading. Densitometric analysis of the hybridization signals showed a >2-fold increase in the level of SDI1 mRNA by pTpT in the p53-transfected cells after correction for slight underloading of the +p53/+pTpT vs. +p53/−pTpT (D vs. C) lanes.
We next used the electromobility shift assay to measure the effect of pTpT on the binding of p53 to its specific wild-type DNA consensus sequence, using nuclear extracts from p53-transfected H1299 cells. Cells transfected with the p53 expression vector displayed low, but detectable, activity (Fig. 8, lane 1). In contrast, when binding reactions were performed with nuclear extract from pTpT-treated cells, the DNA binding activity was greatly enhanced (Fig. 8, compare lanes 1 and 2). In vivo exposure to pTpT followed by in vitro reaction with the p53-specific mAb421 resulted in strong DNA binding and also supershifted the band on the gel, identifying p53 as part of the complex (Fig. 8, lane 3). Excess unlabeled wild-type consensus sequence oligonucleotide (Fig. 8, lane 4), but not mutant consensus sequence oligonucleotide (Fig. 8, lane 5), competed in complex formation, demonstrating the specificity of this p53 binding. Thus, consistent with increasing transcription of p53-regulated genes, pTpT enhances the binding of p53 to its specific DNA consensus sequence.
Figure 8.

Activation of p53 by pTpT. Cultures of H1299 cells were transfected with either control vector or the p53 expression vector, treated with pTpT and processed for the electromobility shift assay as described in Materials and Methods. Lane 1, extract from cells transfected with the p53 expression vector. Lane 2, cells were transfected with the p53 expression vector and incubated with 100 μM pTpT. Lane 3, same as lane 2 but the binding reaction contained 0.1 μg mAb421. Lane 4, same as lane 3 but with a 100-fold excess (10 pmol) unlabeled wild-type p53 consensus sequence. Lane 5, same as lane 3 but with a 100-fold express (10 pmol) unlabeled mutant p53 consensus sequence. The consensus sequence DNA/p53 complex (C.S. DNA/p53) and the supershifted complex (C.S. DNA/p53/Ab421) are indicated by arrows.
DISCUSSION
We reported previously that pTpT induces melanogenesis (tanning), a UV-protective response, mimicking a signal generated by DNA damage, and/or the repair of this damage (10, 11). Here we show that pTpT induces a second UV-protective response, the repair of UV-damaged DNA. This repair was detected by two well established techniques. The first, the restoration of expression of a UV-damaged CAT reporter vector, has been used extensively to measure DNA repair capacity as a function of age (26) and exposure to environmental mutagens (28, 29). This assay also was used to show that pretreatment of normal human cells with a chemical carcinogen or UV light enhances the DNA repair capacity of these cells (9). The 100% increase in damaged CAT vector repair noted by these authors after pretreatment of cells with 1 μg/ml mitomycin C or 10 J/m2 UVC light is very similar to our results with 100 μM pTpT. Treatment of mammalian cells with DNA damaging agents also has been shown to enhance the survival of viruses carrying DNA damage (47, 48), supposedly through an enhanced DNA repair capacity of these cells. The effect of pTpT on virus survival remains to be determined.
The second technique measures the DNA photoproducts directly. The data from these experiments show a rapid removal of (6–4) photoproducts from irradiated DNA compared with a more gradual removal of thymine dimers. These relative rates of repair are consistent with those previously reported (49–51). Pretreatment of fibroblasts with pTpT enhanced the rate of removal of both of these photoproducts (Fig. 1).
Our data strongly suggest that the pTpT enhanced DNA repair results, at least in part, through the activation of the p53 tumor suppressor protein. p53, in fact, has been strongly implicated in the DNA repair process. In addition to transcriptionally activating repair-associated genes such as GADD45 (40, 41) and proliferating cell nuclear antigen (52), p53 also can bind single-stranded DNA and facilitate strand renaturation (53), perhaps reflecting a direct role in DNA repair. Furthermore, overall DNA repair is deficient in cells from Li–Fraumeni patients that express only mutant p53 (54).
Jayaraman and Prives (45) recently have shown that the sequence-specific DNA binding activity of p53 is stimulated by the binding of short single-stranded DNA fragments to a C-terminal domain of the protein. pTpT may similarly interact with p53, directly stimulating p53-mediated transcription. Alternatively, pTpT might act indirectly through upstream effectors of p53 function, such as the product of the ataxia telangiectasia mutated (ATM) gene or human homolog of the yeast RAD53 gene (55, 56). It should be noted, however, that the effect of pTpT on p53 activity is independent of p53 protein levels because equal amounts of protein, as determined by Western blot, were used for each reaction in the electromobility shift assay (Fig. 8). Although the exact mechanisms regulating p53 stability and activity are not fully understood, increases in p53 specific activity have been reported after exposure of cells to low levels of UV irradiation that did not effect p53 protein levels (46).
Regardless of pTpT’s precise mechanism of action, the present data demonstrate that pTpT induces multiple UV-protective responses in human skin, not only increased melanization, but also transient growth arrest and enhanced DNA repair. These findings suggest that mammalian cells can generate an SOS-like response to DNA damage, similar to that previously described in prokaryotic cells (1–3) and that this response can be evoked by exposure to small DNA fragments without the requirement for previous DNA damage.
Acknowledgments
We thank Dr. Faten Gad for assistance with the mAb assay for DNA damage and Kristin Allen and Ian Penner for overall technical assistance. Dr. S. Linn provided helpful discussions and comments on this work.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: pTpT, thymidine dinucleotide; CAT, chloramphenicol acetyltransferase.
A commentary on this article begins on page 12255.
References
- 1.Eller M S. Photodamage. Cambridge, MA: Blackwell; 1995. pp. 26–50. [Google Scholar]
- 2.Sancar A, Sancar G B. Annu Rev Biochem. 1988;57:29–67. doi: 10.1146/annurev.bi.57.070188.000333. [DOI] [PubMed] [Google Scholar]
- 3.Peterson K R, Ossanna N, Thliveris A T, Ennis D G, Mount D W. J Bacteriol. 1988;170:1–4. doi: 10.1128/jb.170.1.1-4.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sancar A. Science. 1994;266:1954–1956. doi: 10.1126/science.7801120. [DOI] [PubMed] [Google Scholar]
- 5.Hollander M C, Alamo I, Jackman J, Wang M G, McBride O W, Fornace A J. J Biol Chem. 1993;268:24385–24393. [PubMed] [Google Scholar]
- 6.Luethy J D, Holbrook N J. Cancer Res. 1992;52:5–10. [PubMed] [Google Scholar]
- 7.Fornace A J, Alamo I, Hollander M C. Proc Natl Acad Sci USA. 1988;85:8800–8804. doi: 10.1073/pnas.85.23.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sebastian J, Sancar G B. Proc Natl Acad Sci USA. 1991;88:11251–11255. doi: 10.1073/pnas.88.24.11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Protic M, Roilides E, Levine A S, Dixon K. Somatic Cell Mol Genet. 1988;14:351–357. doi: 10.1007/BF01534643. [DOI] [PubMed] [Google Scholar]
- 10.Eller M S, Yaar M, Gilchrest B A. Nature (London) 1994;372:413–414. doi: 10.1038/372413a0. [DOI] [PubMed] [Google Scholar]
- 11.Eller M S, Ostrom K, Gilchrest B A. Proc Natl Acad Sci USA. 1996;93:1087–1092. doi: 10.1073/pnas.93.3.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanulis-Praeger B M, Yaar M, Redziniak G, Meybeck A, Gilchrest B A. J Invest Dermatol. 1988;90:749–754. doi: 10.1111/1523-1747.ep12560950. [DOI] [PubMed] [Google Scholar]
- 13.Rheinwald J G, Green H. Cell. 1975;6:331–343. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- 14.Stanulis-Praeger B M, Gilchrest B A. J Cell Physiol. 1989;139:116–124. doi: 10.1002/jcp.1041390117. [DOI] [PubMed] [Google Scholar]
- 15.Rheinwald J G, Jermaine E, Beckell M A. Human Carcinogenesis. New York: Academic; 1983. pp. 86–96. [Google Scholar]
- 16.Yaar M, Karassik R, Schnipper L, Gilchrest B A. J Invest Dermatol. 1985;85:70–74. doi: 10.1111/1523-1747.ep12275353. [DOI] [PubMed] [Google Scholar]
- 17.Mitsudomi T, Steinberg S M, Nau M M, Carbone D, D’Amico D, Bodner S, Oie H K, Linnoila R I, Mulshine J L, Minna J D, Gazdar A F. Oncogene. 1992;7:171–180. [PubMed] [Google Scholar]
- 18.Gilchrest B A, Zhai S, Eller M S, Yarosh D B, Yaar M. J Invest Dermatol. 1993;101:666–672. doi: 10.1111/1523-1747.ep12371673. [DOI] [PubMed] [Google Scholar]
- 19.Werninghaus K, Handjani R-M, Gilchrest B A. Photodermatol Photoimmunol Photomed. 1991;8:236–242. [PubMed] [Google Scholar]
- 20.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Noda A, Ning Y, Venable S F, Pereira-Smith O M, Smith J R. Exp Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 22.Eller M S, Oleksiak M F, McQuaid T J, McAfee S G, Gilchrest B A. Exp Cell Res. 1992;199:328–336. doi: 10.1016/0014-4827(92)90387-n. [DOI] [PubMed] [Google Scholar]
- 23.Yaar M, Eller M S, DiBenedetto P, Reenstra W R, Zhai S, McQuaid T, Archambault M, Gilchrest R A. J Clin Invest. 1994;94:1550–1562. doi: 10.1172/JCI117496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tishler R B, Calderwood S K, Coleman C N, Price B D. Cancer Res. 1993;53:2212–2216. [PubMed] [Google Scholar]
- 25.Hupp T R, Meek D W, Midgley C A, Lane D P. Cell. 1992;71:875–886. doi: 10.1016/0092-8674(92)90562-q. [DOI] [PubMed] [Google Scholar]
- 26.Wei Q, Matanoski G M, Farmer E R, Hedayati M A, Grossman L. Proc Natl Acad Sci USA. 1993;90:1614–1618. doi: 10.1073/pnas.90.4.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moriwaki S-I, Ray S, Tarone R E, Kraemer K H, Grossman L. Mut Res. 1996;364:117–123. doi: 10.1016/0921-8777(96)00029-8. [DOI] [PubMed] [Google Scholar]
- 28.Hallberg, L. M., El Zein, R., Grossman, L. & Au, W. W. (1996) Environ. Health Perspect. 104, Suppl. 3, 529–534. [DOI] [PMC free article] [PubMed]
- 29.Hallberg, L. M., Bechtold, W. E., Grody, J., Legator, M. S. & Au, W. W. (1997) Mut. Res. 213–221. [DOI] [PubMed]
- 30.Mitchell D L, Karentz D. Environmental UV Photobiology. New York: Plenum; 1993. pp. 345–377. [Google Scholar]
- 31.Mitchell D L, Cleaver J E, Epstein J H. J Invest Dermatol. 1990;95:55–59. doi: 10.1111/1523-1747.ep12873312. [DOI] [PubMed] [Google Scholar]
- 32.Zhan Q, Lord K A, Alamo I, Hollander M C, Carrier F, Ron D, Kohn K W, Hoffman B, Lieberman D A, Fornace A. Mol Cell Biol. 1994;14:2361–2371. doi: 10.1128/mcb.14.4.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith M L, Chen I-T, Zhan Q, Bae I, Chen C-Y, Gilmer T M, Kastan M B, O’Conner P M, Fornace A J. Science. 1994;266:1376–1380. doi: 10.1126/science.7973727. [DOI] [PubMed] [Google Scholar]
- 34.Weeda G, Van Ham R C A, Vermeuler W, Bootsma D, van der Eb A J, Hoeijmakers H J. Cell. 1990;62:777–791. doi: 10.1016/0092-8674(90)90122-u. [DOI] [PubMed] [Google Scholar]
- 35.Weeda G, Van Ham R C A, Masurel R, Westerveld A, Odijk H, de Wit J, Bootsma D, van der Eb A J, Hoeijmakers H J. Mol Cell Biol. 1990;10:2570–2581. doi: 10.1128/mcb.10.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaeffer L, Roy R, Humbert S, Moncollin V, Vermeulen W, Hoeijmakers H J, Chambon P, Egly J-M. Science. 1993;260:58–63. doi: 10.1126/science.8465201. [DOI] [PubMed] [Google Scholar]
- 37.Harper J W, Adami G R, Wei N, Keyomarsi K, Elledge S J. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 38.Li R, Waga S, Hannon G J, Beach D, Stillman B. Nature (London) 1994;371:534–537. doi: 10.1038/371534a0. [DOI] [PubMed] [Google Scholar]
- 39.Waga S, Hannon G J, Beach D, Stillman B. Nature (London) 1994;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 40.Kastan M B, Zhan Q, El-Deiry W S, Carrier F, Jacks T, Walsh W V, Plunkett B S, Vogelstein B, Fornace A J. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 41.El-Diery W S, Tokino T, Velculescu V E, Levy D B, Parsons R, Trent J M, Lin D, Mercer W E, Kinzler K W, Vogelstein B. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 42.Fritsche M, Haessler C, Brandner G. Oncogene. 1993;8:307–318. [PubMed] [Google Scholar]
- 43.Nelson W G, Kastan M D. Mol Cell Biol. 1994;14:1815–1823. doi: 10.1128/mcb.14.3.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu X, Lane D P. Cell. 1993;75:765–778. doi: 10.1016/0092-8674(93)90496-d. [DOI] [PubMed] [Google Scholar]
- 45.Jayaraman J, Prives C. Cell. 1995;81:1021–1029. doi: 10.1016/s0092-8674(05)80007-8. [DOI] [PubMed] [Google Scholar]
- 46.Hupp T R, Sparks A, Lane D. Cell. 1995;83:237–245. doi: 10.1016/0092-8674(95)90165-5. [DOI] [PubMed] [Google Scholar]
- 47.Bockstahler L E. Prog Nucleic Acid Res Mol Biol. 1981;26:303–313. doi: 10.1016/s0079-6603(08)60413-4. [DOI] [PubMed] [Google Scholar]
- 48.Radman M. Photochem Photobiol. 1980;32:823–830. doi: 10.1111/j.1751-1097.1980.tb04062.x. [DOI] [PubMed] [Google Scholar]
- 49.Mitchell D L, Nairn P S. Photodermatology. 1988;5:61–64. [Google Scholar]
- 50.Roza L, van der Wulp K J M, MacFarlane J J, Lohman P H M, Baan R A. Photochem Photobiol. 1988;48:627–633. doi: 10.1111/j.1751-1097.1988.tb02873.x. [DOI] [PubMed] [Google Scholar]
- 51.Wani A F, D’Ambrosia S M, Alvi N K. Photochem Photobiol. 1987;46:477–482. doi: 10.1111/j.1751-1097.1987.tb04798.x. [DOI] [PubMed] [Google Scholar]
- 52.Morris G F, Bischoff J R, Mathews M B. Proc Natl Acad Sci USA. 1996;93:895–899. doi: 10.1073/pnas.93.2.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bakalkin G, Yakovleva T, Selivanova G, Magnusson K P, Szekely L, Kiseleva E, Klein G, Terenius L, Wiman K G. Proc Natl Acad Sci USA. 1994;91:413–417. doi: 10.1073/pnas.91.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ford J, Hanawalt P C. Proc Natl Acad Sci USA. 1996;92:8876–8880. doi: 10.1073/pnas.92.19.8876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walworth N C, Bernards R. Science. 1996;271:353–356. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- 56.Sanchez Y, Desany B A, Jones W J, Liu Q, Wand B, Elledge S J. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]