

Abstract
Three microcolin A and B analogs have been synthesized. Their biological activity profiles were evaluated against several cell lines, revealing the existence of a structural determinant whose role in mediating the antiproliferative effect of the microcolins has heretofore gone unrecognized. While previously described as potent immunosuppressive natural products, we found that these microcolin analogs possessed no selective cytotoxicity when comparing immune cell versus nonimmune cell proliferation. In addition, the amino-terminus of microcolin A has been modified to incorporate a biotinylated polyethylene glycol linker. The biological activity of this biotinylated microcolin A analog was determined. This affinity reagent was shown to retain limited biological activity and thus can serve as a useful probe for elucidating the mechanism of action of microcolin A.
Keywords: Natural products, Immunosuppressant, Synthesis
Pharmacological intervention aimed at suppressing the immune system plays an important role in the management of autoimmune diseases, the prevention of rejection after organ transplantation, and treatment of graft versus host diseases. In recent years, many new immunosuppressive drugs have been discovered and developed for clinical use in organ transplantation. The commonly used immunosuppressive drugs fall into five groups with different mechanisms of action: (a) regulators of gene expression; (b) alkylating agents; (c) inhibitors of de novo purine synthesis; (d) inhibitors of de novo pyrimidine synthesis; and (e) inhibitors of kinases and phosphatases. Despite phenomenal success in this disease area, many of these drugs have troublesome side effects and high toxicity profiles. More effective and specific immunosuppressive therapy is needed to further reduce the high morbidity due to infections, malignancies, and graft loss due to chronic rejection after organ transplantation. Thus, the search for more efficacious and tolerant immunosuppressive drugs is vigorously pursued in both academia and pharmaceutical industry. In our chemical biology program, we are interested in developing natural products as biological probes to identify novel proteins for therapeutic intervention (target validation) and the exploration of cell biology (chemical genetics).1
Microcolins A and B are potent antiproliferative and immunosuppressive natural products discovered by Koehn and co-workers from the Venezuelan blue-green algae Lyngbya majuscule (Scheme 1).2–4 Microcolins A and B were found to be potent in the human two-way mixed lymphocyte response (MLR) assay with EC50 values reported in the subnanomolar to nanomolar range.3,5 Despite this potent activity and potential clinical use, the mechanism of action for the microcolins remains unknown. Furthermore, microcolins are distinctive in structure relative to other immunosuppressive drugs. This suggests that the immunosuppressive properties of microcolins A and B might be mediated by a novel mechanism.
Scheme 1.

Retrosynthetic analysis.
Microcolin A has been synthesized by Decicco and Grover6 as well as by Andrus et al.7 Koehn et al.5 prepared several analogs by semisynthetic modification and/or degradation of the natural product. They showed that the C-10 free hydroxy functionality, as opposed to general oxygen functionality, is important for the biological activity. Striking loss of activity occurs upon reduction of the pyrrolenone ring C2–C3 olefin. The EC50 value for 2,3-dihydromicrocolin A is 10,000-fold less potent than native microcolin A in both murine and human MLR. However, the C22 OAc can be replaced with OH without any significant loss of activity. It suggests that the pyrrolenone function is an important structural element for immunosuppressive activity. Despite considerable research to determine a structure–activity profile, there have been no reported studies to determine the role of C4 methyl group and 34,36-dimethyloctanoyl side chain. We were particularly interested to know the role of 34,36-dimethyloctanoyl side chain because we would like to design a molecular probe based on microcolin A without impairing biological activity for our biochemical studies. In this communication, we report the synthesis of three microcolin analogs. We also describe the design and synthesis of biotinylated microcolin A for use as a molecular probe. The pharmacological activity profiles of each analog against different cell lines are also discussed.
A very expedient and highly efficient synthesis of tripeptide was developed (Scheme 2). Commercially available Boc-threonine 5 was coupled in the presence of BOP-Cl/Et3N to commercially available valine derivative 6, followed by acetylation (Ac2O/DMAP) to produce the known dipeptide 76 in 62% yield. Boc deprotection of 7 and coupling of the resulting hydrochloride salt (BOP-Cl/Et3N) to commercially available N-methyl Boc-leucine 9 and subsequent hydrogenation furnished the tripeptide carboxylic acid 3 in 38% yield.6 The tri-peptide 3 was accessed in five steps in 24% yield from commercially available starting material.
Scheme 2.

Synthesis of the tripeptide. Reagents and conditions: (a) BOP-Cl, Et3N, CH2Cl2, rt, 12 h, 67%; (b) Ac2O, DMAP, EtOAc, rt, 6 h, 93%; (c) 4 N HCl, dioxane, 0 °C, 4 h, 89%; (d) BOP-Cl, 9, Et3N, CH2Cl2, 8 h, 45%; (e) 10% Pd/C, H2, 96%.
Our route to synthesize the pyrrolenone fragment was based on a modified version of pentafluorophenyl ester-based protocol reported by Andrus et al. (Scheme 3).7 Following Silverman’s protocol, lactone 11 was opened up in the presence of NaN3/MeOH to give Cbz-protected cis-4-hydroxyproline methyl ester 12 in 99% yield.8 Silylation and hydrolysis gave carboxylic acid, which was reacted with pentafluorophenol to produce 13.
Scheme 3.

Synthesis of the pyrrolenone. Reagents and conditions: (a) Ph3P, DEAD, THF, rt, 3 h, 55%; (b) NaN3, MeOH, 45 °C, 24 h, 99%; (c) TBSCl, imidazole, DMF, rt, 36 h, 94%; (d) LiOH, THF/MeOH/H2O, 0 °C, 24 h, 81%; (e) pentafluorophenol, DCC, EtOAc, 90%; (f) (±) 5-methyl-2-pyrrolidinone, BuLi, THF, 15 min, 13, 3 h, 15 = 40%, 16 = 45%; (g) 10% Pd/C, Boc2O, H2, MeOH, 1 h; (h) LDA, −78 °C, 15 min, PhSeBr, H2O2, AcOH, H2O, 0 °C, 30 min; (i) 4 N HCl, dioxane, 0 °C, 4 h, 4a = 57%, 4b = 51% over last three steps.
This stable activated ester 137 was reacted with imidate 14 derived from commercially available racemic 5-methyl- 2-pyrrolidinone to produce mixed imide 157 and 16 in 40% and 45% yields, respectively. Both diastereoisomers were separated by flash column chromatography. Each diastereoisomer was then converted to pyrrolenone 4a7 and 4b following the three-step sequence (Pd/C, BOC2O, H2; LDA, PhSeBr, H2O2,9 and HCl). Pyrrolenone 4c was synthesized from Cbz-protected proline following Andrus et al. protocol (Scheme 4). Enantiomerically pure 5-methyl-2-pyrrolidinone was easily synthesized from commercially available 18 according to the conditions of Amstutz et al.10
Scheme 4.
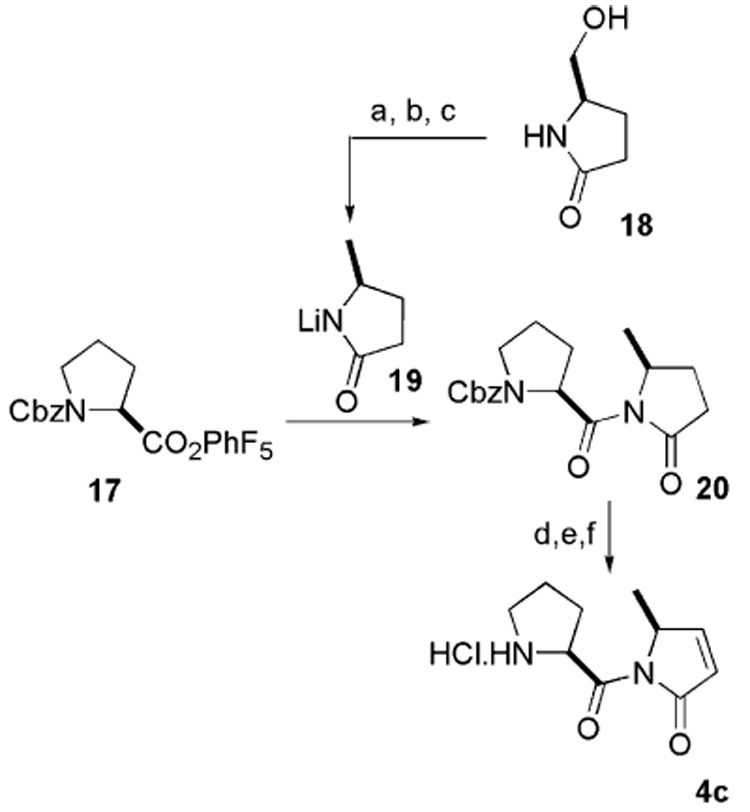
Synthesis of pyrrolenone. Reagents and conditions: (a) Ph3P, CBr4, MeCN, 4 h, 73%; (b) Bu3SnH, AIBN, PhH, 70 °C, 3 h, 61%; (c) BuLi, THF, 15 min; (d) 10% Pd/C, Boc2O, H2, MeOH, 1 h, 90%; (e) LDA, −78 °C, 15 min, PhSeBr, H2O2, AcOH, H2O, 0 °C, 30 min, 60%; (f) 4 N HCl, dioxane, 0 °C, 4 h, 50%.
The tripeptide carboxylic acid 3 was coupled with 4a, 4b, and 4c separately to produce 21, 22, and 23, respectively, in 67%, 73%, and 56% yields (Scheme 5).6 Hydrochloric acid treatment produced the salt, which was then coupled with octanoic acid in the presence of BOP-Cl/Et3N to yield 24,11 25,12 and 26.13
Scheme 5.

Synthesis of microcolin A analogs. Reagents and conditions: (a) BOP-Cl, Et3N, CH2Cl2, rt, 12 h, 21 = 67%, 22 = 73%, 23 = 56%; (b) 4 N HCl, dioxane, 0 °C, 4 h; (c) BOP-Cl, octanoic acid, Et3N, CH2Cl2, 12 h, 24 = 63%, 25 = 50%, 26 = 43% over two steps.
Once these compounds were in hand, we next investigated their activity on different cell lines (Table 1). We have found that the microcolin A analog 24 potently inhibits both primary lymphocytes and immune cell lines with EC50 values in the low nanomolar range. In fact, 24 is similarly potent at inhibiting the proliferation of all cell lines tested in this study, including nontransformed (mouse) C2C12 myoblast cells and even primary (bovine) endothelial cells. Rather curiously, this is the first study ever to test whether microcolin A actually is selective for immune cells (all previous studies have relied heavily on the mixed lymphocyte reaction and exclusively on immune-derived cells and cell lines to assess microcolin A activity). This finding raises significant doubts about the therapeutic potential of microcolins as immunosuppressants, and rather points toward their more likely usefulness as antineoplastic agents.
Table 1.
Biological activity profiles of microcolin A analogs 24, 25, and 26
| Entry | Cell name | IC50 |
|---|---|---|
| 24 | Mouse mixed lymphocyte reaction | 32.8 nM |
| Mouse PMA ionomycin-stimulated lymphocytes | 30.3 nM | |
| EL-4 (mouse thymoma) | 45.6 nM | |
| Jurkat (human T-cell leukemia) | 39.4 nM | |
| P-388 (mouse T-cell leukemia) | 66.7 nM | |
| N1E-115 (mouse neuroblastoma) | 35.9 nM | |
| C2C12 (mouse myoblast) | 43.4 nM | |
| BAE (bovine aortic endothelial) | 36.7 nM | |
| 25 | EL-4 (mouse thymoma) | 6 µM |
| P-388 (mouse T-cell leukemia) | 7 µM | |
| 26 | EL-4 (mouse thymoma) | 6 µM |
| P-388 (mouse T-cell leukemia) | 5 µM |
Analogs 25 and 26 displayed striking loss of activity: 26, which lacks the C10 prolylhydroxyl group, was 100-fold less potent than 24, thereby confirming the importance of this functional group in mediating the antiproliferative effects of the microcolins. Interestingly, 25, which differs from 24 in the stereochemistry of the C4 methyl group, had a 100-fold reduced potency as well. Moreover, in a competition assay, the potency of 24 to inhibit cell proliferation was unaffected by the presence of molar excess 25, indicating that the proper stereochemistry of the C4 methyl group is important for target protein binding. This finding, therefore, has identified a novel structural determinant important for the antiproliferative activity of the microcolins, and further corroborates the notion that the pyrrolylproline functionality is the active pharmacophore.
Biotinylated derivatives of the epoxide containing natural products fumagillin, parthenolide, epoxomicin, and eponemycin have proven useful in the identification of their intracellular targets.14–17 Structure–activity relationship studies by Koehn et al. and our studies involving the C4 methyl group of microcolin A established that the pyrrolylproline C-terminus is the pharmacophore. Our studies with the octanoyl analog demonstrated that 34,36-dimethyl side chain plays an anciliary role in imparting pharmacological activity. Therefore, we decided to attach a biotin handle to the amino-terminus of microcolin A (Scheme 6). The hydrochloride salt derived from 21 was coupled with commercially available PEG-biotin carboxylic acid to furnish biotinylated microcolin A 27 in modest 40% yield.18
Scheme 6.

Synthesis of biotinylated microcolin A derivative. Reagents and conditions: (a) dPEG4™ -biotin acid, BOP-Cl, Et3N, CH2Cl2, rt, 12 h, 40%.
Once the synthesis was complete, we measured the inhibitory potency of the biotinylated microcolin A 27 (Table 2). Of all the analogs tested in this study, 27 had the least biological activity, with IC50 values between 200- and 300-fold less potent than 24 when tested against P-388 cells and EL-4 cells, respectively. Nevertheless, at these readily achievable higher concentrations, this reagent may prove useful in identifying the target protein of these small molecules.
Table 2.
Biological activity of biotinylated microcolin A
| Entry | Cell name | IC50 (µM) |
|---|---|---|
| 27 | EL-4 (mouse thymoma) | 10 |
| P-388 (mouse T-cell leukemia) | 10 |
In conclusion, we have synthesized microcolin analogs as well as designed and synthesized biotinylated microcolin A as a biological probe. It was also demonstrated that microcolin A analog 24 is comparably potent to the native microcolin A 1 and that, unexpectedly, shows no immune cell selectivity in its antiproliferative activity when tested on a wide panel of cell types. Our study has revealed the importance of the C4 methyl group stereochemistry in mediating the activity of these compounds. Despite the impaired biological activity of biotinylated microcolin A, it can still be useful as molecular probe for biochemical studies. We now aim to identify and characterize the molecular target of microcolin A by employing affinity chromatographic techniques. We will report our progress in this regard in due course.
Acknowledgments
This work was supported by NIH Grant GM62120 (C.M.C.) and a Leukemia Lymphoma Society Award (J.H.).
References and notes
- 1.Crews CM, Splittgerber U. Trends Biochem. Sci. 1999;24:317. doi: 10.1016/s0968-0004(99)01425-5. [DOI] [PubMed] [Google Scholar]
- 2.Koehn FE, Longley RE, Reed JK. J. Nat. Prod. 1992;55:613. doi: 10.1021/np50083a009. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L-H, Longley RE, Koehn FE. Life Sci. 1997;60:751. doi: 10.1016/s0024-3205(96)00645-5. [DOI] [PubMed] [Google Scholar]
- 4.Zhang L-H, Longley RE. Life Sci. 1999;64:1013. doi: 10.1016/s0024-3205(99)00028-4. [DOI] [PubMed] [Google Scholar]
- 5.Koehn FK, McConnell OJ, Longley RE, Sennett SH, Reed JK. J. Med. Chem. 1994;37:3181. doi: 10.1021/jm00045a024. [DOI] [PubMed] [Google Scholar]
- 6.Decicco CP, Grover P. J. Org. Chem. 1996;61:3534. [Google Scholar]
- 7.Andrus MB, Li W, Keyes RF. J. Org. Chem. 1997;62:5542. [Google Scholar]
- 8.Gomez-Vidal JA, Silverman RB. Org. Lett. 2001;3:2481. doi: 10.1021/ol0161054. [DOI] [PubMed] [Google Scholar]
- 9.Reich HJ, Renga JM, Reich IL. J. Am. Chem. Soc. 1975;97:5434. [Google Scholar]
- 10.Amstutz R, Ringdahl B, Karlen B, Roch M, Jenden DJ. J. Med. Chem. 1985;28:1760. doi: 10.1021/jm00150a004. [DOI] [PubMed] [Google Scholar]
- 11.Compound 24. The substrate 21 (5.3 mg, 7.80 µmol) was treated with 4 N HCl in dioxane (0.23 ml, 0.92 mmol) at 0 °C and stirred for 4 h. The solvent was evaporated under reduced pressure to give crude hydrochloride salt, which was carried to the next step without further purification. A solution of hydrochloride salt and octanoic acid (1.6 µl, 10 µmol) in CH2Cl2 (1 ml) was treated with BOP-Cl (2.55 mg, 10 µl) and Et3N (1.4 µl) at 0 °C. It was then warmed to room temperature and stirred for 12 h. The reaction was quenched by the addition of aqueous saturated NaHCO3. The aqueous layer was extracted with ethyl acetate (2×2 ml). The organic layers were combined, washed with brine, dried (Na2SO4), filtered and evaporated under reduced pressure. The residue was purified by preparative TLC (90% EtOAc in hexane) to give compound 24 (3.5 mg, 63%). ; 1H (500 MHz, CDCl3) δH: 7.29 (1H, dd, J = 8.1 and 2.2), 6.89 (1H, d, J = 10.0), 6.10 (1H, dd, J = 6.0 and 1.5), 5.68 (1H, dd, J = 10 and 2.0), 5.25 (3H, m), 5.05 (1H, d, J = 11.0), 4.97 (1H, dd, J = 9.5 and 3.5), 4.83 (1H, m), 4.40 (1H, br s), 3.85 (2H, m), 3.13 (3H, s), 2.94 (3H, s), 2.49 (1H, m), 2.37 (2H, m), 2.22 (1H, m), 2.03 (3H, s), 2.01 (1H, m), 1.67 (4H, m), 1.49 (3H, d, J = 6.5), 1.31 (10H, br m), 1.18 (3H, d, J = 7.0), 1.01 (3H, d, J = 6.5), 0.95 (3H, d, J = 6.5), 0.90 (6H, m) and 0.83 (3H, d, J = 7.0); 13C NMR (125 MHz, CDCl3) δc: 175.1, 174.6, 171.6, 170.2, 170.2, 154.5, 125.7, 72.3, 69.0, 59.2, 58.6, 58.0, 56.8, 54.2, 51.9, 37.1, 36.3, 33.9, 30.8, 30.5, 29.7, 29.3, 29.6, 27.2, 25.5, 24.0, 23.1, 23.6, 21.9, 22.1, 19.0, 18.3, 17.8, 17.4, 14.3. IR (neat) 3420, 3011; HRMS (FAB+): calcd for C37H62N5O9 (M+H)+ 720.4548; found 720.4550.
- 12.Compound 25. Compound 22 (12.2 mg, 21 µmol) was treated with 4 N HCl in dioxane (0.5 ml, 2.0 mmol) and allowed to stir at 0 °C. After 4 h, the solvent was removed to give the crude. To a solution of octanoic acid (5 µl, 32 µmol) and the crude amine in CH2Cl2 (3 ml) triethylamine (4 µl, 29 mmol) was added followed by BOP-Cl (6.7 mg, 26 µmol) at room temperature. After being stirred for 16 h, the mixture was quenched by the addition of H2O. The mixture was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with sodium sulfate, filtrated, and evaporated under reduced pressure. The residue was purified by preparative TLC (17% hexanes in ethyl acetate) to yield 25 (7.5 mg, 50%) as white foam. ; 1H NMR (400 MHz, CDCl3) δH: 7.28 (1H, dd, J = 6.1 and 2.0), 6.90 (1H, d, J = 8.9), 6.10 (1H, dd, J = 6.1 and 1.5), 5.78 (1H, dd, J = 10.1 and 2.0), 5.25 (1H, m), 5.22 (1H, m), 5.02 (1H, d, J = 11.1), 4.95 (1H, dd, J = 8.8 and 3.4), 4.85 (1H, qt, J = 6.7 and 1.8), 4.40 (1H, br s), 3.85 (2H, m), 3.11 (3H, s), 2.92 (3H, s), 2.50 (1H, m), 2.35 (2H, m), 2.26 (1H, m), 2.01 (3H, s), 1.98 (1H, m), 1.65 (4H, m), 1.44 (3H, d, J = 6.7), 1.25 (10H, br s), 1.17 (3H, d, J = 6.5), 1.03 (3H, d, J = 6.6), 0.94 (3H, d, J = 6.6), 0.89 (3H, d, J = 6.6), 0.88 (3H, t, J = 6.0), and 0.82 (3H, d, J = 6.6). 13C NMR (125 MHz, CDCl3) δc: 174.3, 173.6, 171.2, 170.0, 169.9, 169.1, 154.5, 125.3, 71.7, 68.6, 59.2, 58.6, 58.0, 56.8, 54.2, 51.9, 37.1, 36.3, 33.9, 30.8, 30.5, 29.7, 29.4, 29.1, 27.2, 25.1, 25.0, 23.1, 22.6, 21.9, 21.1, 19.0, 18.4, 17.7, 17.4, 14.1. IR (neat) 3420, 3011; HRMS (FAB+): calcd for C37H62N5O9 (M+H)+ 720.4548; found 720.4539.
- 13.Compound 26. Compound 23 (20.0 mg, 30 µmol) was treated with 4 N HCl (1.0 ml, 4.0 mmol) and allowed to stir at 0 °C. After 3 h, the solvent was removed to give the crude. To a solution of octanoic acid (8 µl, 50 µmol) and the crude amine in CH2Cl2 (1.5 ml) triethylamine (7 µl, 50 mmol) was added followed by BOP-Cl (15.2 mg, 60 µmol) at room temperature. After being stirred for 18.5 h, the mixture was quenched by the addition of H2O. The mixture was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with sodium sulfate, filtrated, and evaporated under reduced pressure. The residue was purified by preparative TLC (17% hexanes in ethyl acetate) to yield 26 (9 mg, 43%) as colorless oil. ; 1H NMR (500 MHz, CDCl3) δH: 7.25 (1H, dd, J = 6.0 and 2.0), 6.87 (1H, d, J = 8.5), 6.08 (1H, dd, J = 6.0 and 1.5), 5.48 (1H, dd, J = 8.5 and 5.5), 5.26 (1H, dq, J = 6.5 and 2.0), 5.23 (1H, dd, J = 9.0 and 6.5), 5.06 (1H, d, J = 11.0), 4.98 (1H, dd, J = 9.0 and 6.5), 4.79 (1H, qt, J = 7.0 and 1.5), 3.83 (1H, m), 3.74 (1H, m), 3.14 (3H, s), 2.93 (3H, s), 2.45 (1H, m), 2.37 (2H, m), 2.29 (1H, m), 2.03 (3H, s), 2.01 (1H, m), 1.97 (1H, m), 1.88 (1H, m), 1.69 (4H, m), 1.48 (3H, d, J = 6.5), 1.33 (9H, m), 1.18 (3H, d, J = 6.5), 1.01 (3H, d, J = 7.0), 0.95 (3H, d, J = 6.5), 0.91 (3H, d, J = 6.0), 0.89 (3H, t, J = 6.0), 0.82 Hz (3H, d, J = 6.5); 13C NMR (125 MHz, CDCl3) δc: 174.6, 172.4, 171.6, 170.2, 168.7, 154.2, 125.9, 69.3, 60.4, 59.7, 58.5, 54.6, 52.6, 48.4, 36.8, 34.4, 32.1, 32.2, 31.0, 30.1, 29.8, 29.5, 29.3, 27.7, 25.5, 25.4, 25.0, 23.5, 23.0, 23.0, 22.4, 21.4, 19.3, 18.8, 17.6, 17.5, 14.5. IR (neat) 3417, 3015; HRMS(FAB+): calcd for C37H62N5O8 (M+H)+ 704.4598; found 704.4590.
- 14.Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, et al. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6099. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwok HB, Koh B, Ndubuisi M, Elofsson M, Crews CM. Chem. Biol. 2001;8:759. doi: 10.1016/s1074-5521(01)00049-7. [DOI] [PubMed] [Google Scholar]
- 16.Meng L, Mohan R, Kwok BHK, Elofsson M, Sin N, et al. Proc. Natl. Acad. Sci. U.S.A. 1999;96:10403. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng L, Kwok BHB, Sin N, Crews CM. Cancer Res. 1999;59:2798. [PubMed] [Google Scholar]
- 18.Compound 27. Compound 21 (26.7 mg, 38 µmol) was treated with 4 N HCl (1.5 ml, 6.0 mmol) and allowed to stir at 0 °C. After 4 h, the solvent was removed to give the crude product. To a solution of dPEG4-biotin acid (28.4 mg, 58 µmol) and the crude amine in CH2Cl2 (0.8 ml) triethylamine (20 µl, 144 µmol) was added followed by BOP-Cl (15.2 mg, 60 µmol) at room temperature. After the mixture was stirred for 12.5 h, the solvent was removed under reduced pressure. The residue was purified by preparative TLC (14% methanol in chloroform) to yield 27 (16.3 mg, 40%) as white foam. ; 1H NMR (400 MHz, CDCl3) δH: 7.27 (1H, dd, J = 6.0 and 2.0), 7.17 (1H, d, J = 8.6), 6.80 (1H, br s), 6.73 (1H, br s), 6.21 (1H, br s), 6.09 (1H, dd, J = 6.0 and 1.5), 5.62 (1H, dd, J = 9.7 and 2.9), 5.23 (2H, m), 5.01 (1H, d, J = 11.1), 4.96 (1H, m), 4.81 (1H, m), 4.51 (1H, m), 4.38 (1H, br s), 4.33 (1H, m), 3.82 (2H, m), 3.73 (2H, m), 3.71–3.63 (14H, m), 3.57 (2H, br t, J = 7.8), 3.44 (2H, m), 3.16 (1H, m), 3.11 (3H, s), 2.93 (3H, s), 2.90 (1H, dd, J = 12.8 and 3.8), 2.75 (1H, d, J = 12.8), 2.70 (1H, m), 2.61 (1H, m), 2.53 (1H, m), 2.27 (1H, m), 2.23 (1H, t, J = 7.4), 1.99 (3H, s), 1.97 (1H, m), 1.76–1.62 (6H, m), 1.47 (3H, d, J = 6.7), 1.24 (2H, m), 1.17 (3H, d, J = 6.4), 0.99 (3H, d, J = 6.4), 0.93 (3H, d, J = 6.6), 0.87 (3H, d, J = 6.5), and 0.81 Hz (3H, d, J = 6.7). 13C NMR (125 MHz, CDCl3) δc: 175.5, 174.1, 173.4, 172.0, 171.1, 170.1, 170.0, 169.9, 168.8, 154.2, 125.4, 71.4, 70.6, 70.5, 70.2, 69.9, 68.9, 67.3, 66.6, 59.3, 58.6, 58.2, 56.6, 54.4, 52.3, 51.7, 39.2, 36.6, 35.8, 34.9, 34.1, 31.9, 31.0, 30.6, 29.7, 28.2, 27.2, 25.5, 25.0, 23.1, 21.9, 21.8, 21.1, 19.2, 18.4, 17.4, 17.0. IR (neat) 3420, 3054; HRMS (FAB+): calcd for C50H83N5O15 (M+H)+ 1067.5699; found 1067.5718.