

Abstract
The syntheses of three fragments, 2, 3, and 4, of amphidinolide B1 have been accomplished. The 1,3-isomerization of allylic alcohol 10 was accomplished via rhenium oxo catalysis and has been applied successfully in the synthesis. (–)-MIB-catalyzed asymmetric vinylzinc addition to aldehyde 31 and the regio- and stereoselective epoxidation of unsymmetrical divinyl methanol 32 were key steps.
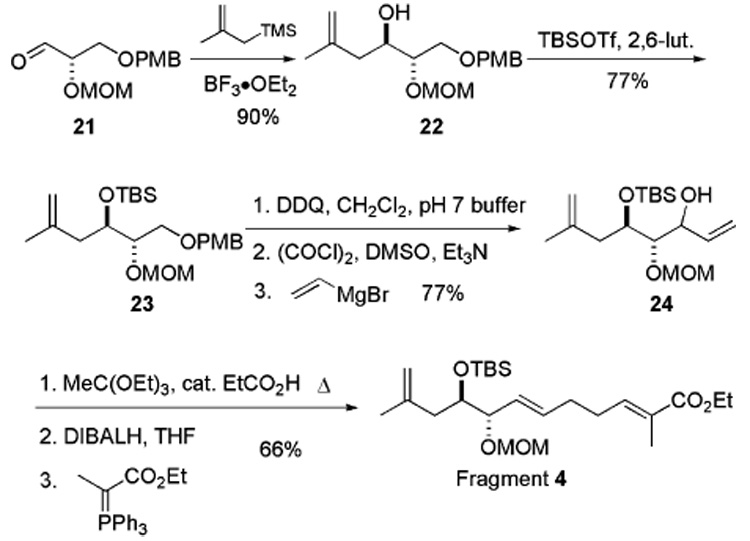
Amphidinolide B1 (1), a polyketide-based 26-membered macrolide, was isolated from a culture of the symbiotic marine dinoflagellate Amphidinium sp. (strain Y-5) in 1987 by Kobayashi et al.1,2 There have been several reports of partial syntheses toward 1; however, no completed total synthesis has been disclosed yet.3
In our laboratory, we undertook a multipronged approach toward the synthesis of amphidinolide B1 (1) (Scheme 1). One strategy was communicated earlier,4 whereas this paper represents an alternative strategy. Our previous strategy involved three major C–C bond-forming reactions and one C–O bond-forming reaction. This new retrosynthetic analysis involves two C–C and one C–O bond-forming coupling reactions: Suzuki–Miyaura coupling,5 an aldol reaction,6 and macrolactonization using the three fragments 2, 3, and 4. As C–C bond-forming reactions are traditionally more difficult than C–O bond-forming reactions, we hope that this new strategy will increase the efficiency of the synthesis.
Scheme 1.
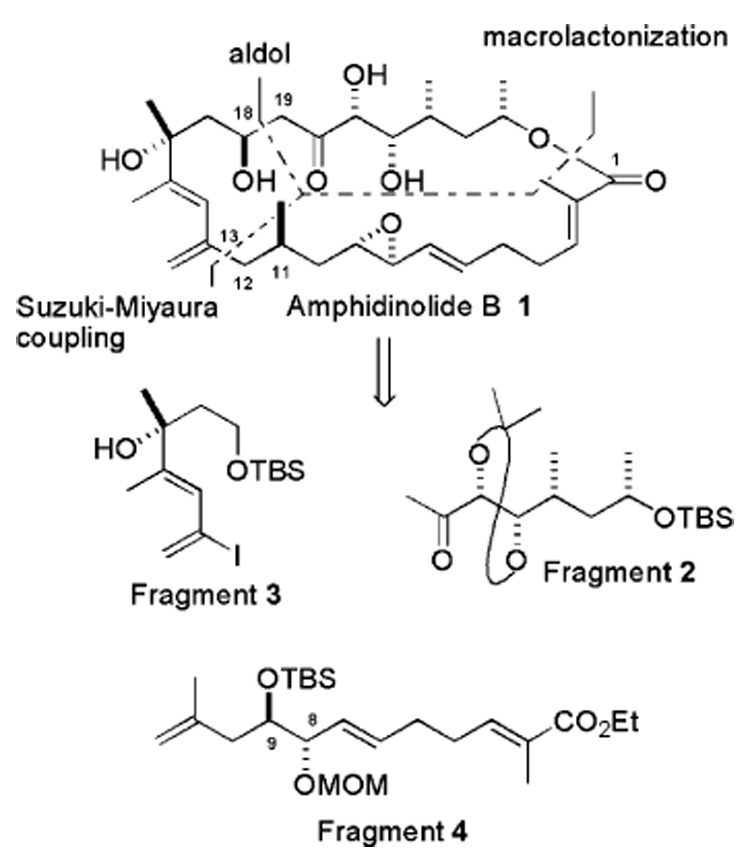
The synthesis of fragment 2 began with Brown’s crotylation reaction (Scheme 2).7 The reaction of trans-crotonaldehyde and (E)-crotyldiisopinocamphenylborane (prepared from trans-butene, n-BuLi, t-BuOK, and (+)-Ipc2BOMe) in the presence of BF3·OEt2 in THF-Et2O at −78 °C produced the allyl alcohol 6 in 50% yield and 14:1 anti/syn selectivity. Silyl protection of the hydroxyl group and a regioselective hydroboration–oxidation sequence furnished alcohol 7, which was oxidized to the corresponding aldehyde 8 using Dess–Martin periodinane. The asymmetric methylation of 8 with Me2Zn and Ti(OiPr)4 was examined with the chiral ligands BINOL and bissulfonamide.8 Unfortunately, the conversions were unacceptable in both cases (25% and 29%, respectively). However, diastereoselective methylation to obtain 9 was achieved with Seebach’s method9 [Me2Zn and Ti(OiPr)4 in the presence of (−)-TADDOL] in 92% yield and a 5–7:1 diastereomeric ratio (based on 1H NMR). Protection of the free alcohol as a TIPS ether (the minor isomer can be separated at this stage) and selective cleavage of the allylic TBS ether furnished 10 in 70% yield over two steps. At this point, the stage was set to manipulate allylic alcohol 10 to the requisite α,β-unsaturated ketone 12 and to install the remaining stereogenic centers. The 1,3-isomerization of 10 and subsequent oxidation promised to be an atom-efficient method to access ketone 12. Ph3SiOReO310 is currently the most effective catalyst for 1,3-isomerization.11 When we treated 10 in the presence of 1 mol % catalyst in ether at −60 °C, complete isomerization to regioisomeric allylic alcohol 11 was observed in 5 min. Oxidation of the alcohol 11 under Dess–Martin periodinane conditions furnished the α,β-unsaturated ketone 12. Swapping TIPS protection with TBS, Sharpless asymmetric dihydroxylation12 followed by acetonide formation completed the synthesis of fragment 2.
Scheme 2.
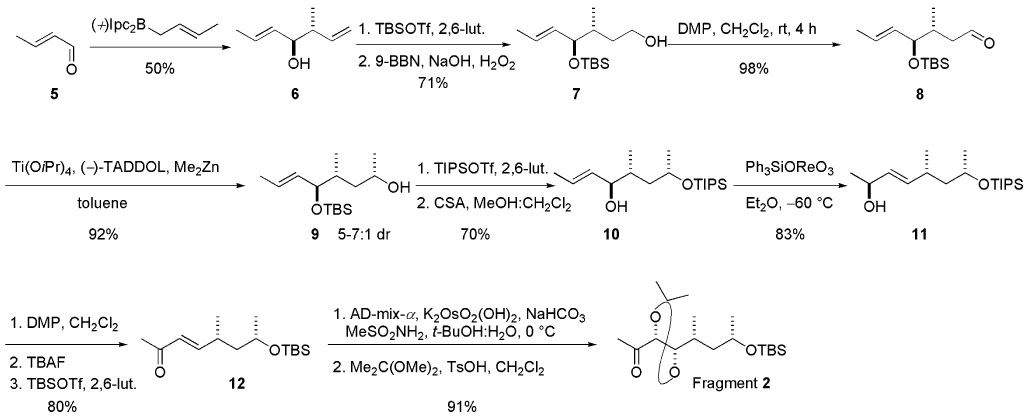
The synthesis of fragment 3 started with known vinyl iodide 1313 (Scheme 3). Transmetalation with t-BuLi in Et2O and capture of the resulting vinyllithium species with acetaldehyde yielded (±)-allylic alcohol 14. Sharpless kinetic resolution14 of (±)-14 gave the desired epoxide 15 in 85–90% enantiomeric excess (ee) and unreacted alcohol in 25–30% ee as a mixture that proved difficult to separate. The crude mixture was subjected to a Parikh–Doering oxidation to yield easily separable epoxy ketone 16 and the corresponding α,β-unsaturated ketone (not shown). The α,β-unsaturated ketone was subjected to a Luche reduction to produce (±)-allylic alcohol 14, which was recycled under Sharpless kinetic resolution conditions. The Horner–Wadsworth–Emmons condensation of epoxy ketone 16 with phosphonate in the presence of NaHMDS produced enyne 17 [82% yield, E/Z 6:1]. Simultaneous cleavage of TBS ether and the TMS group by TBAF followed by regioselective opening of the epoxide in the presence of LiAlH4 in Et2O furnished diol 18, which was separated from the minor (Z) isomer using column chromatography. Functionalization of the enyne 18 to the 1,3-diene iodide was quite problematic. After exploring several possibilities, we found that the triple bond could be silylstannylated regio- and stereoselectively employing n-Bu3SnSiMe2Ph/Pd(PPh3)4 to produce functionalized diene 19.15 TBAF-mediated removal of the PhMe2Si group produced stannane 20. Reaction with I2 and selective TBS protection completed the synthesis of fragment 3.
Scheme 3.
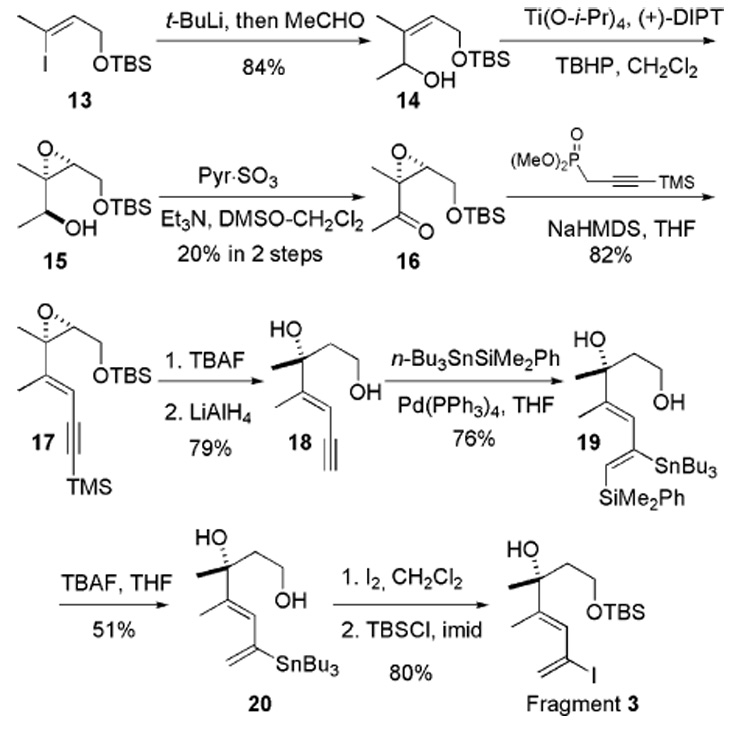
The synthesis of fragment 4 began with aldehyde 21 (Scheme 4). A nonchelation-controlled SE2′ reaction between aldehyde 21 and methallylsilane in the presence of BF3·OEt2 furnished homoallylic alcohol 22 in 90% yield and ≥95:5 diastereomeric ratio (dr).16 Hydroxyl group protection as a TBS ether 23, followed by oxidative deprotection of the PMB ether, revealed the primary alcohol. The alcohol was then converted to the aldehyde, which was subjected to vinyl-magnesium bromide addition to provide allyl alcohol 24 as a 1.5:1 diastereomeric mixture (based on 1H NMR). The allylic alcohol 24 underwent Johnson ortho ester Claisen rearrangement to provide the homologated ethyl ester exclusively as the (E) isomer. Reduction to the aldehyde using i-Bu2AlH in THF and the Wittig reaction furnished fragment 4 with exclusive (E) selectivity.
Scheme 4.
With all three fragments securely in hand, we sought to explore the possibility of setting up the C11 stereogenic centers by hydroboration and a subsequent (B)-alkyl Suzuki-Miyaura coupling reaction5 to assemble the 1,3-diene. We hoped to induce 1,3-stereocontrol on the basis of Evans’ alkyl-directed hydroboration model.17 We have been able to form the C12–C13 bond successfully using the (B)-alkyl Suzuki–Miyaura coupling reaction between (B)-alkylborane (derived from regioselective hydroboration of fragment 4) and (±)-1,3-vinyl iodide fragment 3 (synthesis not shown) to put together the C1–C18 portion of the molecule. However, investigations revealed that 1,3-stereocontrol cannot be achieved with this substrate, as hydroboration of fragment 4 with 9-BBN provided a 1:1 inseparable mixture of diastereoisomers at C11 (amphidinolide B1 numbering).
Considering A values, an alkyl group (i.e., Me = 1.74 kcal/mol) imparts more nonbonding interaction than an OSiR3 group (i.e., OSiMe3 = 0.74 kcal/mol), presumably because the SiR3 group can be turned away to avoid the steric interaction.18 This explains the loss of π-facial selectivity in our substrate. As separation of the diastereoisomers was not easy, we revised our strategy and decided to use compound 25 as a new fragment for the C1–C12 segment in which the C11 methyl stereogenic center is preinstalled (Scheme 5). Instead of coupling the 1,3-diene iodide fragment 3 to (B)-alkylborane (derived by hydroboration from fragment 4), we decided to couple it to the ate complex 26 derived from the corresponding iodide easily accessible from compound 25 in two steps.19 Preliminary studies involving (±)-alkenyliodide fragment 3 and a model alkyl boronate to assemble the 1,3-diene corroborate the feasibility of this strategy.20
Scheme 5.
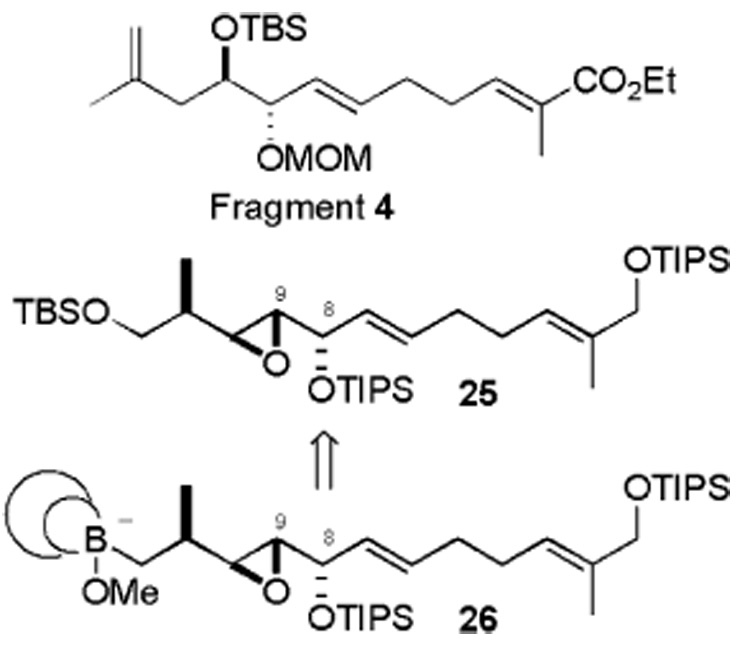
The synthesis of compound 25 commenced with isoprene monoxide (Scheme 6). TiCl4-mediated regioselective and stereoselective opening of the epoxide provided the alcohol, which was protected as a TIPS ether 28. It was then coupled with propargylmagnesium bromide in the presence of Pd-(PPh3)4 to give acetylene 29 in 83% yield. In parallel, aldehyde 31 was synthesized in two steps via the Wittig reaction, followed by reduction with i-Bu2AlH. The key strategy is to employ asymmetric addition of alkenylzinc to aldehyde 31 in the presence of Nugent’s21 isoborneol-based (−)-MIB ligand.22 Hydroboration of the terminal acetylene 29 with freshly prepared dicyclohexylborane proceeds regio-selectively to afford the alkenylborane. Transmetalation of this alkenylborane with Me2Zn generates the reactive alkenylzinc reagent in situ. Attempts to carry out the transmetalation at 0 °C, as described in the literature,23 led to decomposition of the substrate. After extensive experimentation, we found that the reaction could be performed successfully by adding Me2Zn followed by (−)-MIB at − 78 °C to alkenylborane. The mixture was then warmed to −20 °C over 10 min. The aldehyde 31 in hexane was added via syringe pump over 20 min while warming the mixture to 0 °C to furnish divinyl methanol 32 in 43% yield and ≥95% diastereomeric excess (de). This assembled the full carbon backbone of compound 25. The next challenge was to functionalize stereoselectively the C7–C8 double bond of this unsymmetrical divinyl methanol 32.
Scheme 6.
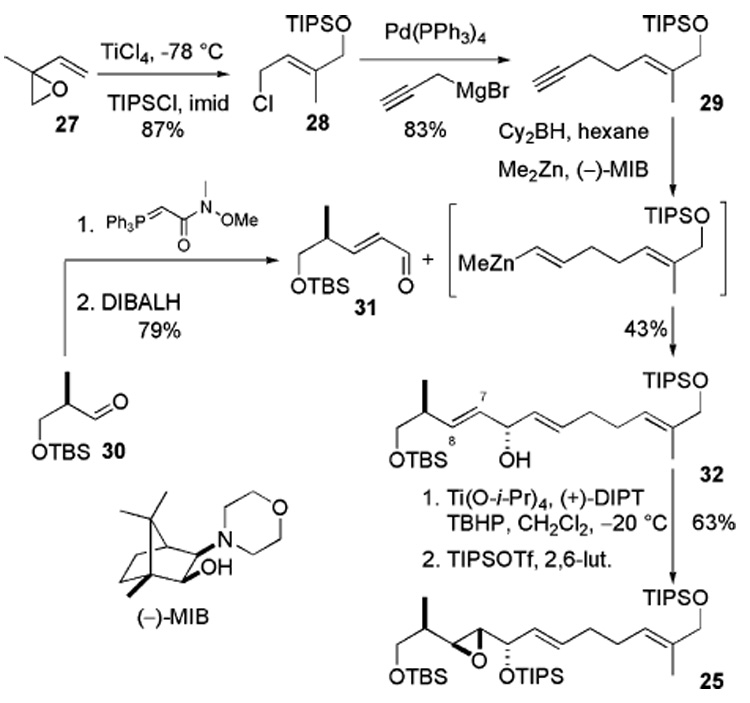
It is documented that the simple (±)-unsymmetrical divinyl methanols can be subjected to the Sharpless kinetic resolution conditions to synthesize the optically active monoepoxide.24 However, to the best of our knowledge, Sharpless asymmetric epoxidation has not been applied to desymmetrize a complex unsymmetrical divinyl methanol such as ours in the context of natural product synthesis. We were pleased to find that Sharpless’ asymmetric epoxidation provided the C7–C8 monoepoxide in 71% yield (6:1 dr). TIPS protection went uneventfully to furnish 25 in 90% yield.
In conclusion, we have accomplished the synthesis of three fragments, 2, 3 and, 4. Compound 25 will be used as the new fragment for the C1–C12 segment. 1,3-Isomerization of allylic alcohol via rhenium oxo catalysis has been applied successfully in our synthesis. (−)-MIB-catalyzed asymmetric vinylzinc addition to aldehyde 31, highly selective late-stage epoxidation of divinyl methanol 32, and regio- and stereo-selective silylstannylation to synthesize stannane 20 have been used as key steps. Continued advancement of these intermediates toward the eventual total synthesis of 1 are currently ongoing in our laboratory.
Acknowledgment
We gratefully acknowledge financial support from NIH (GM062120). J.S.S. acknowledges the American Chemical Society, Division of Medicinal Chemistry, and Aventis Pharmaceuticals for a pre-doctoral fellowship.
Footnotes
Supporting Information Available: Experimental procedures and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1. Ishibashi M, Ohizumi Y, Hamashima M, Nakamura H, Hirata Y, Sasaki T, Kobayashi J. J. Chem. Soc., Chem. Commun. 1987:1127.. For reviews: Kobayashi J, Ishibashi M. Chem. Rev. 1993;93:1753. Chakraborty T, Das S. Curr. Med. Chem.: Anti-Cancer Agents. 2001;1:131. doi: 10.2174/1568011013354660. Kobayashi J, Shimbo K, Kubota T, Tsuda M. Pure Appl. Chem. 2003;75:337. Kobayashi J, Tsuda M. Nat. Prod. Rep. 2004;21:77. doi: 10.1039/b310427n.
- 2.Kobayashi J, Ishibashi M, Nakamura H, Ohizumi Y, Yamasu T, Hirata Y, Sasaki T, Ohta T, Nozoe S. J. Nat. Prod. 1989;52:1036. doi: 10.1021/np50065a021. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kobayashi J, Tsuda M. Nat. Prod. Rep. 2004;21:77. doi: 10.1039/b310427n. [DOI] [PubMed] [Google Scholar]; (b) Cid MB, Pattenden G. Synlett. 1998:540. [Google Scholar]; (c) Cid MB, Pattenden G. Tetrahedron Lett. 2000;41:7373. [Google Scholar]; (d) Ishiyama H, Takemura T, Tsuda M, Kobayashi J. Tetrahedron. 1999;55:4583. [Google Scholar]; (e) Ishiyama H, Takemura T, Tsuda M, Kobayashi J. J. Chem. Soc., Perkin Trans. 1999;1:1163. [Google Scholar]; (f) Ohi K, Shima K, Hamada K, Saito Y, Yamada N, Ohba S, Nishiyama S. Bull. Chem. Soc. Jpn. 1998;71:2433. [Google Scholar]; (g) Ohi K, Nishiyama S. Synlett. 1999:573. [Google Scholar]; (h) Ohi K, Nishiyama S. Synlett. 1999:571. [Google Scholar]; (i) Chakraborty TK, Thippeswamy D, Suresh VR, Jayaprakash S. Chem. Lett. 1997:563. [Google Scholar]; (j) Chakraborty TK, Thippeswamy D. Synlett. 1999:150. [Google Scholar]; (k) Chakraborty TK, Thippeswamy D, Jayaprakash S. J. Ind. Chem. Soc. 1998;75:741. [Google Scholar]; (l) Chakraborty TK, Suresh VR, Vayalakkada R. Chem. Lett. 1997:565. [Google Scholar]; (m) Eng HM, Myles DC. Tetrahedron Lett. 1999;40:2275. [Google Scholar]; (n) Eng HM, Myles DC. Tetrahedron Lett. 1999;40:2279. [Google Scholar]; (o) Lee D-H, Lee S-W. Tetrahedron Lett. 1997;38:7909. [Google Scholar]; (p) Lee D-H, Rho M-D. Bull. Korean Chem. Soc. 1998;19:386. [Google Scholar]; (q) Lee D-H, Rho M-D. Tetrahedron Lett. 2000;41:2573. [Google Scholar]; (r) Zhang W, Carter RG, Yokochi AFT. J. Org. Chem. 2004;69:2569. doi: 10.1021/jo035829l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mandal AK, Schneekloth JS, Jr, Crews CM. Org. Lett. 2005;7:3645. doi: 10.1021/ol051175m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyaura N, Ishiyama T, Sasaki H, Ishikawa M, Satoh M, Suzuki A. J. Am. Chem. Soc. 1989;111:314. [Google Scholar]
- 6.Evans DA, Carter PH, Carreira EM, Charette AB, Prunet JA, Lautens M. J. Am. Chem. Soc. 1999;121:7540. [Google Scholar]
- 7.Brown HC, Bhat KS. J. Am. Chem. Soc. 1986;108:293. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]
- 8.(a) Takahashi H, Kawakita T, Ohno M, Yoshioka M, Kobayashi S. Tetrahedron. 1992;48:5691. [Google Scholar]; (b) Kitamoto D, Imma H, Nakai T. Tetrahedron Lett. 1995;36:1861. [Google Scholar]
- 9.(a) Schmidt B, Seebach D. Angew. Chem., Int. Ed. Engl. 1991;30:99. [Google Scholar]; (b) von dem Bussche-Hunnefeld JL, Seebach D. Tetrahedron. 1992;48:5719. [Google Scholar]
- 10.Schoop T, Roesky HW, Noltemeyer M, Schmidt H-G. Organometallics. 1993;12:571. [Google Scholar]
- 11.Bellemin-Laponnaz S, Gisie H, Le Ny J-P, Osborn JA. Angew Chem., Int. Ed. Engl. 1997;36:976. [Google Scholar]
- 12.Walsh PJ, Sharpless KB. Synlett. 1993:605. [Google Scholar]
- 13.Ashimori A, Bachand B, Calter MA, Govek SP, Overman LE, Poon DJ. J. Am. Chem. Soc. 1998;120:6488. [Google Scholar]
- 14.Gao Y, Hanson RM, Klunder JM, Ko SY, Masamune H, Sharpless KB. J. Am. Chem. Soc. 1987;109:5765. [Google Scholar]
- 15.(a) Mitchell TN, Wickenkamp R, Amamria A, Dieke R, Scheider U. J. Org. Chem. 1987;52:4868. [Google Scholar]; (b) Chenard BL, Van Zyl CM. J. Org. Chem. 1986;51:3561. [Google Scholar]
- 16.Reetz MT, Kesseler K. J. Org. Chem. 1985;50:5434. [Google Scholar]
- 17.Evans DA, Bartroli J, Godel T. Tetrahedron Lett. 1982;23:4577. [Google Scholar]
- 18.Eliel EL, Wilen SH, Mander LN. Stereochemistry of Organic Compounds. New York: John Wiley & Sons; 1993. p. 696. [Google Scholar]
- 19.Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]
- 20. Unpublished results. [Google Scholar]
- 21.Nugent WA. J. Chem. Commun. 1999:1369. [Google Scholar]
- 22.(a) Oppolzer W, Radinov RN. Hev. Chim. Acta. 1992;75:170. [Google Scholar]; (b) Pu L, Yu H-B. Chem. Rev. 2001;101:757. doi: 10.1021/cr000411y. Review: [DOI] [PubMed] [Google Scholar]
- 23.Chen YK, Lurain AE, Walsh PJ. J. Am. Chem. Soc. 2002;124:12225. doi: 10.1021/ja027271p. [DOI] [PubMed] [Google Scholar]
- 24.Honda T, Mizutani H, Kanai K. J. Chem. Soc., Perkin Trans. 1. 1996;14:1729. [Google Scholar]