

Abstract
Myriaporones are naturally occurring compounds which structurally resemble the southern hemisphere of the tedanolide family of macrolide antitumor agents. Despite the fact that myriaporone 3/4 represents only a portion of tedanolide, it nonetheless retains much of its biological activity. We show here that like tedanolide, myriaporone 3/4 inhibits protein synthesis and proliferation of mammalian cells with low nanomolar potencies but displays no prokaryotic growth inhibitory effect. Moreover, myriaporone 3/4 displays a very rapid, reversible and p21-independent activity to block S phase progression in mammalian cells. Structure–activity relationship studies revealed that the C18–C19 epoxide and the C14 hydroxymethyl group (tedanolide numbering) of myriaporone 3/4 are required for cell cycle inhibition. These constitute previously unidentified and/or novel pharmacophores for myriaporone 3/4. Our results show that the important biological activities associated with the structurally complex tedanolides are present and can be harnessed in the chemically much simpler myriaporones. This greatly increases the value of the latter as investigative tools for biochemical research as well as for development of potential therapeutics.
Introduction
For several years, the chemical diversity existing in marine sources has been shown to be comparable to that of terrestrial sources.1 Marine natural products such as the ion-channel blockers tetrodotoxin and saxitoxin and the phosphatase inhibitor okadaic acid have long been used extensively as research tools. More recent research into marine sources has led to the discovery of novel anti-cancer drug candidates such as ecteinascidin-743 (a transcription inhibitor from Ecteinascidia turbinata),2 didemnin B (a protein translation inhibitor from Trididemnum solidum)3 and discodermolide (a microtubule stabilizer/senescence accelerator from Discodermia sp.).4,5
Tedanolides are polyketide macrolides first identified by Schmitz and colleagues in isolates from the Caribbean fire sponge Tedania ignis.6 These compounds are cytotoxic, with potencies described from the sub-nanomolar6–8 to high nanomolar range.9 In murine in vivo tumor implantation studies, tedanolides have significantly slowed tumor growth.7,10 However, their mechanism of action has only recently been elucidated: Nishimura and colleagues11 demonstrated that 13-deoxytedanolide, a structurally-related macrolide isolated from the marine sponge Mycale adhaerens, binds to the 60S ribosomal subunit and can inhibit mRNA translation in a cell-free yeast extract. The resultant ribotoxic stress leads to the activation of SAPK/JNK and p38 MAP kinase,12 both well-established members of the stress-activated protein kinase family.
Following the discovery of the tedanolides, Rinehart and colleagues isolated myriaporones from the Mediterranean false coral Myriapora truncata.1 These simpler chemical entities strongly resemble the so-called “southern hemisphere” (C10–C23) of tedanolide. Myriaporone 3/4 is so named because the molecule exists as an equilibrium between open (myriaporone 4) and closed (myriaporone 3) structures. While tedanolide has yet to be prepared via total synthesis, two recent reports detail the successful total syntheses of 13-deoxytedanolide.13,14 Unfortunately, due to the structural complexity of the tedanolides, it is not clear that a total synthesis would play an important role in understanding their interesting biological activity and determining their therapeutic potential. In fact, there have been very few reports elaborating the biological activity of the tedanolides.11 Thus, the discovery of the myriaporones as a naturally-occurring highly related component of the more complex macrolide raised the possibility that the biological activity of the structurally more complicated tedanolides might be harnessed in a much smaller and simpler molecule. This could significantly expedite important structure–activity investigation and the development of biological probes for identification of partner proteins for these small molecules. Here we characterize the antiproliferative effects of myriaporone 3/4 and identify novel and important structural elements that contribute to its potent biological activity.
Results
Myriaporone 3/4 inhibits mammalian cell proliferation with low nanomolar potency
The biological activity of myriaporone 3/4 was examined against two different mammalian cells: P388 mouse leukemia cells and bovine aortic endothelial cells. The former is a transformed cell line which grows in suspension and the latter are adherent, primary cells. Despite significant differences in the growth patterns of these cells, myriaporone 3/4 effectively inhibited the proliferation of both (Fig. 1(a)) with IC50 values in the low nanomolar range (IC50 = 15.9 ± 1.0 nM for endothelial cells; IC50 = 15.4 ± 1.7 nM for P388 cells), which is approximately 18-fold more potent than was reported in the initial discovery of myriaporone.1 Compared in terms of potency to the known members of the tedanolide family, myriaporone 3/4 falls in the middle of them: it is more potent than the recently discovered tedanolide C,9 but less potent than 13-deoxytedanolide7 or tedanolide itself.6 Further, the biological activity of myriaporone 3/4 was not restricted to mammalian cells—in halo assays performed on Saccharomyces cerevisiae and Schizosaccharomyces pombe, myriaporone 3/4 inhibited the growth of both with S. pombe demonstrating slightly greater sensitivity (Fig. 1(b)). Sensitivity of yeast to this inhibitor is not as pronounced as in mammalian cells: use of dot titration assay to determine a well-defined concentration of myriaporone 3/4 that would inhibit S. cerevisiae proliferation showed that concentrations up to and including 2 µM failed to have any effect (Fig. 1(c)). Moreover, deletion of the pleiotropic drug resistance membrane transport protein (Δpdr5) in S. cerevisiae15 failed to alter the sensitivity of S. cerevisiae to myriaporone 3/4, although the mutation did have a general growth-retarding effect. This indicates that the yeast’s decreased sensitivity to myriaporone 3/4 relative to that of mammalian cells is not due to more effective drug efflux. The antiproliferative activity appears to be restricted to eukaryotic organisms, in that simultaneous halo assay performed on E. coli did not produce any myriaporone 3/4 growth inhibition zones (although E. coli were sensitive to the prokaryotic ribosome inhibitor, erythromycin) (Fig. 1(b)).
Fig. 1.
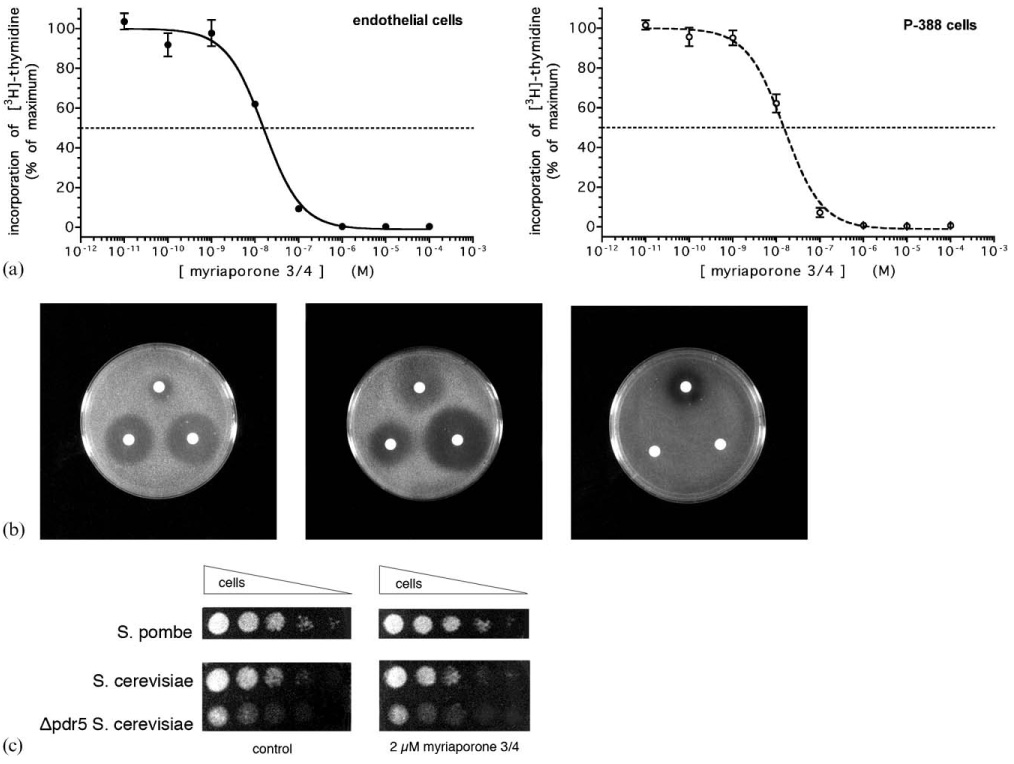
(a) Inhibition of mammalian cell proliferation by myriaporone 3/4. Cells were treated with the indicated concentrations of myriaporone 3/4 for 21 h, followed by 3 h incubation in the continued presence of drug and 15 µCi ml−1 [3H]-thymidine. Graphs represent the combined results of three independent experiments (error bars represent the standard error of the means). (b) Myriaporone 3/4 is selective for eukaryotes. Halo assays were performed on S. cerevisiae (left panel), S. pombe (center panel) and E. coli (right panel). Representative results are shown. Starting at the top of each plate photo and proceeding clockwise, the filters were dosed with 1 µmol erythromycin; 1 µmol myriaporone 3/4; and 1 µmol cycloheximide. (c) Yeast dot titration assay. Representative results are shown. S. pombe was spotted onto YES plates (without or with 2 µM myriaporone 3/4) and S. cerevisiae were spotted onto YPD plates (without or with 2 µM myriaporone 3/4).
Rapid-acting and reversible inhibition of cell growth
Some natural products exert their antiproliferative effect(s) rapidly, while others can have a more gradual onset as they may require target cells to be in a particular stage of the cell cycle or they may bind their target with relatively lower affinity. We found that despite their similar ultimate sensitivity to myriaporone 3/4, the two model cells showed significant kinetic differences in the onset of its effect. While P388 cells required nearly 20 h in order to achieve maximal growth inhibition, endothelial cells required less time and achieved maximal growth inhibition within 5 h—over four-fold more rapidly (Fig. 2(a)). This was surprising, in that P388 cells grow faster than the non-transformed endothelial cells and might be considered more metabolically susceptible to the adverse effect of an antiproliferative agent. In fact, a dose-response proliferation assay performed on endothelial cells treated with a 3 h pulse of myriaporone was identical to those performed following an overnight (>20 h) incubation (data not presented). Since incorporation of [3H]-thymidine is a direct measurement of cell progression through S phase, myriaporone proved to be a rapidly-acting S phase inhibitor. Interestingly, this inhibition of S phase was not mediated by p21 induction (Fig. 2(b)), in contrast to TNP 470—a structurally dissimilar, but well-studied, endothelial cell cycle inhibitor that does induce p21.16 As rapidly as myriaporone 3/4 acted on endothelial cells, its S phase arrest was just as rapidly reversed and the cells began to recover and proliferate immediately upon its withdrawal (Fig. 2(c)). By 3 h the cells had recovered 60%, and by 15 h the cells had nearly completely recovered. The presence of an epoxide at C18–C19 had made it attractive to speculate that myriaporone 3/4 might irreversibly inactivate its target protein via covalent adduct formation; however, the pattern of rapid growth recovery observed is inconsistent with an irreversible mechanism of action.
Fig. 2.
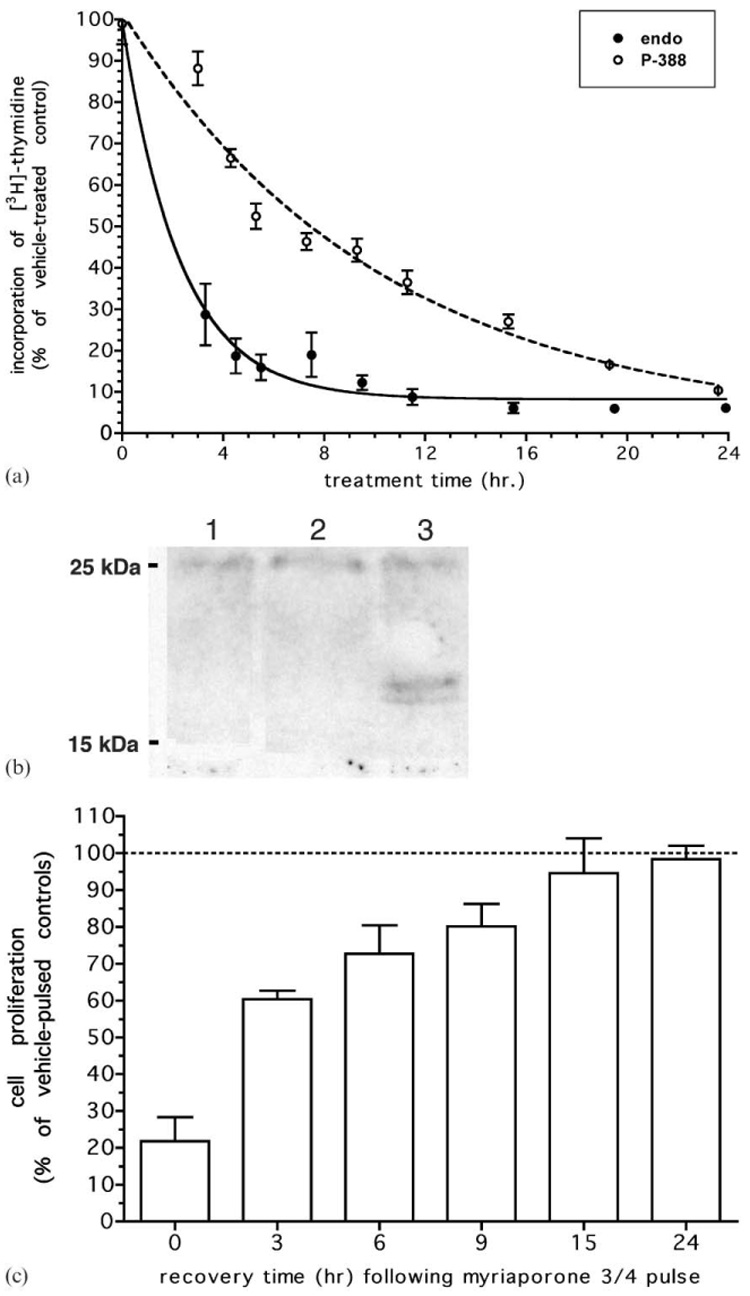
(a) Rate of onset of myriaporone 3/4. Cells were treated with 100 nM myriaporone 3/4 for the time indicated, with 15 µCi ml−1 [3H]-thymidine added over the final 3 h prior to harvest. Graph represents the averaged results of three independent experiments (error bars represent the standard error of the means). (b) Western blot for p21. Endothelial cells were treated for 24 h prior to harvest with either vehicle (0.1% DMSO, lane 1); 100 nM myriaporone 3/4 (lane 2); or 100 nM TNP 470 (lane 3). Whole cell proteins were probed with polyclonal antibody to p21 at 1 : 1000. Representative results are shown. (c) Recovery of cell proliferation following myriaporone 3/4 treatment. Endothelial cells were pulsed with 100 nM myriaporone 3/4 for 3 h, which was then removed and replaced with drug-free medium for the specified time. Over the final 3 h of each recovery, 15 µCi ml−1 [3H]-thymidine was added. Graph represents the averaged results of three independent experiments (error bars represent the standard error of the means).
Myriaporone 3/4 arrests cell growth via blockade of protein synthesis
A recent report11 has shown that 13-deoxytedanolide binds to the eukaryotic ribosome and can inhibit protein synthesis. In order to determine whether myriaporone 3/4 works through the same mechanism, [14C]-leucine labeled endothelial cells were incubated with varying concentrations of the drug and their ability to incorporate the isotope into cellular protein was measured. The results showed that myriaporone 3/4 was as effective at inhibiting protein synthesis as cycloheximide, a commonly-used eukaryotic translation inhibitor (Fig. 3(a)). Concentrations as low as 0.2 µM myriaporone 3/4 achieved a similar protein synthesis inhibition as a 100-fold greater concentration of cycloheximide. In fact, myriaporone 3/4 potently inhibited protein synthesis, with an IC50 = 11.3 ± 0.8 nM (Fig. 3(b)) virtually identical to that which was revealed in cell proliferation assays (Fig. 1(a)). This showed that the structurally less-complicated myriaporone was just as effective at inhibiting protein synthesis as structurally more-complicated molecules, and very likely works in the same way to block cell proliferation as do the tedanolides.
Fig. 3.
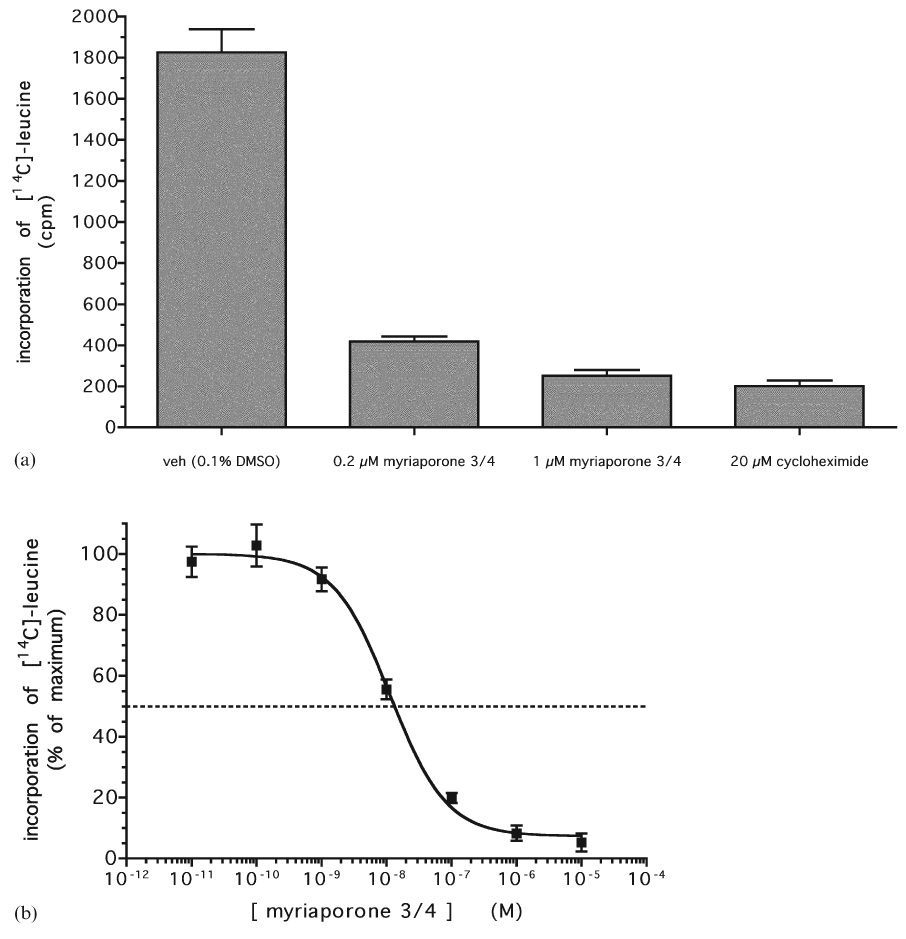
(a) Inhibition of in vivo protein synthesis by myriaporone 3/4. Endothelial cells were pre-treated with myriaporone 3/4 for 3 h, followed by another 3 h in the continued presence of the drug plus 1 µCi ml−1 [14C]-leucine. Graph represents the averaged results of three independent experiments (error bars represent the standard error of the means). (b) Dose–response of myriaporone 3/4 inhibition of protein synthesis. Graph represents the averaged results of three independent experiments (error bars represent the standard error of the means).
Novel pharmacophores determine myriaporone 3/4 biological activity
In order to identify those functional groups of myriaporone 3/4 which might be most responsible for its potent biological activity, a set of analogs was synthesized (Fig. 4). To make subsequent structural comparisons to the tedanolides more clear, the carbon atoms in myriaporone 3/4 and its analogs are numerically designated as in the macrolides themselves. Analogs lacking the C18–C19 epoxide (“MR-1-94”) or dehydroxylated at position C14 (“HC-1-195” and “HC-1-203”) were synthesized. HC-1-195 is identical to the southern hemisphere of tedanolide, while HC-1-203 is epimeric at the C13 position. These analogs were then tested for their activity against the model cell lines (Fig. 5). Given the evidence indicating a reversible interaction with its target, removal of the C18–C19 epoxide surprisingly had a profound effect on the activity of myriaporone 3/4, resulting in a 500- to 1000-fold decrease in activity (Table 1). This unexpectedly showed that the epoxide is indeed important for activity, albeit a readily reversible activity. Replacement of the C14 hydroxymethyl group with a methyl group in HC-1-195 had a similarly large detrimental effect on the activity; further shifting of the C13 hydroxyl group to the opposite epimer (in HC-1-203) nearly abolished the remaining activity. It is noteworthy that the tedanolide macrolide family already possesses a methyl group at position C14 and naturally lacks the hydroxymethyl group found in myriaporone 3/4. Thus, our finding concerning the importance of the C14 hydroxymethyl group is curious. Furthermore, the negative effect of the change in stereochemistry at the C13 hydroxyl group is equally somewhat curious, in that functional importance of this group has been equivocal in the past: tedanolide and 13-deoxytedanolide are reported to have comparable activities.8,9
Fig. 4.
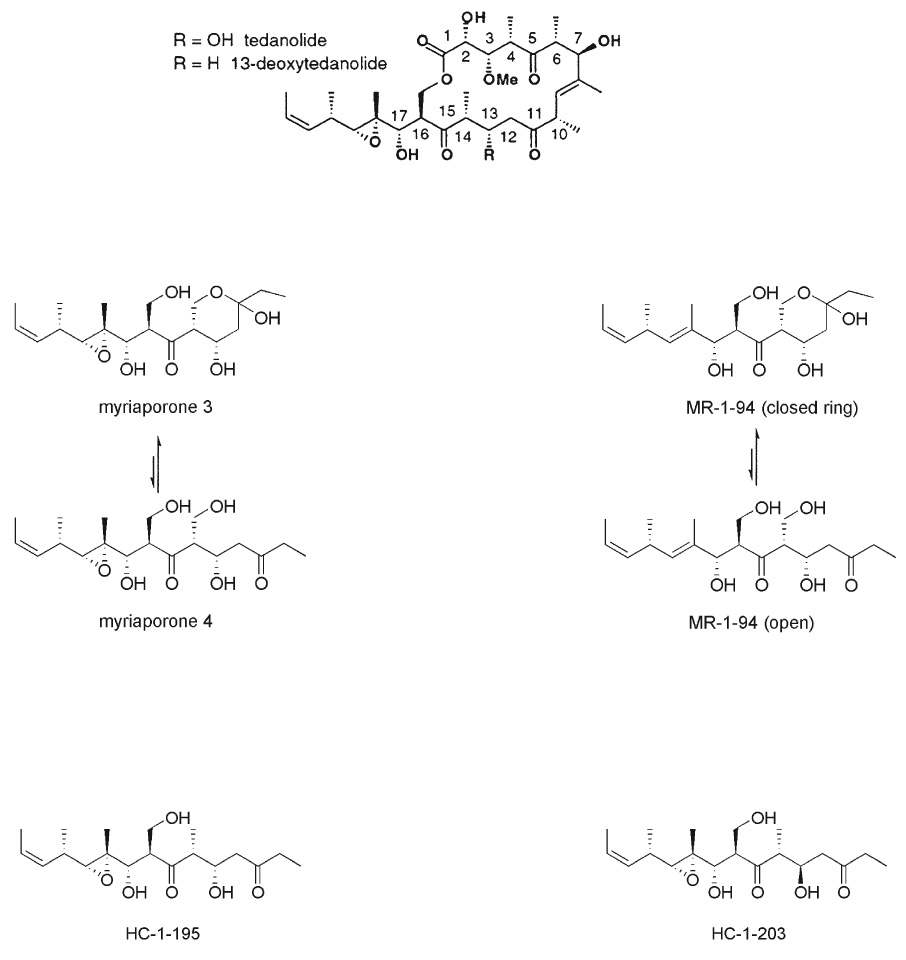
Chemical structures of the tedanolides, myriaporone 3/4, and the synthesized new analogs.
Fig. 5.
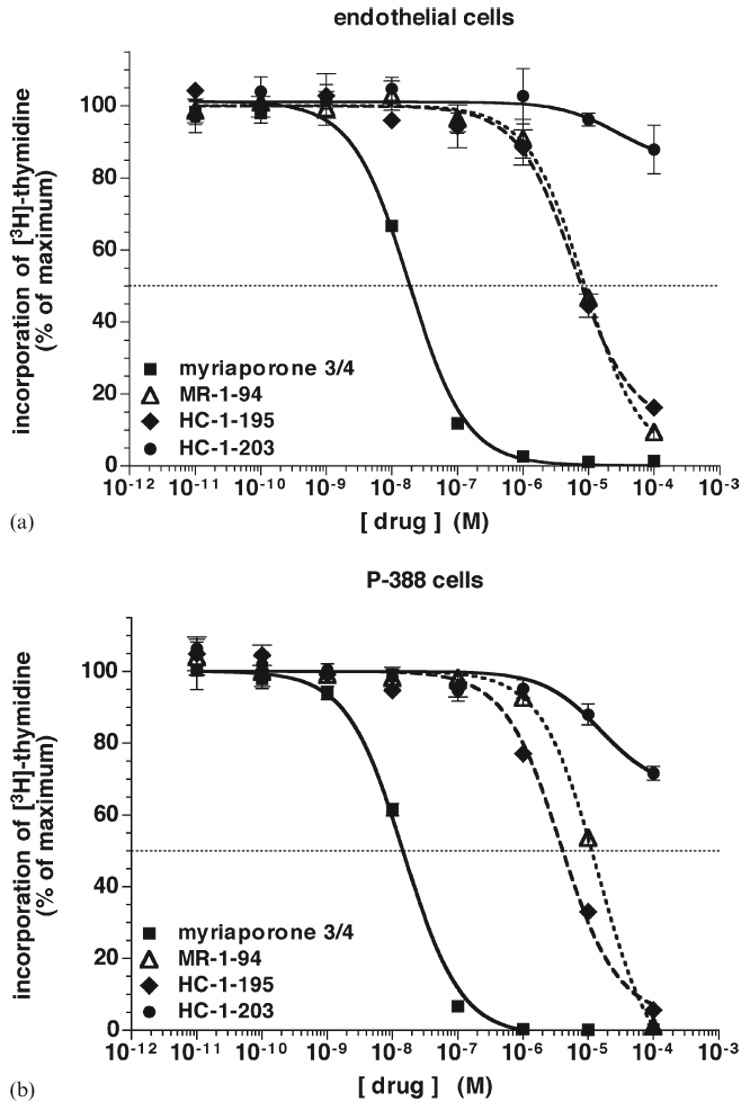
Activity of myriaporone 3/4 analogs to inhibit proliferation of mammalian cells: (a) endothelial cells, (b) P388 cells. Performed as in Fig. 1(a). Graphs represent the averaged results of three independent experiments (error bars represent the standard error of the means).
Table 1.
IC50 values of myriaporone 3/4 analogs at cultured mammalian cells
| Drug | Endothelial cells | P388 cells |
|---|---|---|
| Myriaporone 3/4 | 17.2 nM ± 1.1 nM | 14.9 ± 3.6 nM |
| MR-1-94 | 8.4 ± 1.2 µM | 14.4 ± 1.1 µM |
| HC-1-195 | 6.2 ± 2.3 µM | 3.8 ± 1.3 µM |
| HC-1-203 | >100 µM | >100 µM |
Discussion
In order to serve as a promising lead for the development of new therapeutics, a candidate small molecule should be large enough to allow for target specificity yet small enough to discourage spurious or harmful side effects mediated via structural elements unrelated to the pharmacophore. Thus, while the structurally complex tedanolide class of protein synthesis inhibitors provides researchers with a challenging exercise in chemical synthesis, the structurally simpler myriaporones could be pharmacologically more attractive provided they retain the important antiproliferative properties of the former. The present study supports the notion that they do: dose–response analysis reveals that myriaporone 3/4 remains a potent (low nanomolar) and rapid protein synthesis inhibitor and antiproliferative agent in mammalian cells. As with an earlier study6 describing S phase blockade at submicromolar concentrations of tedanolide, myriaporone 3/4 inhibited [3H]-thymidine incorporation into nuclei of the cultured cells tested. The high correlation of the IC50 values for growth inhibition and protein synthesis blockade by myriaporone 3/4 suggests that the latter is the mechanism by which the former is accomplished. Therefore, the lack of p21 induction coincident with S phase arrest by myriaporone 3/4 is not surprising given the inhibition of mRNA translation. It is a more likely scenario that myriaporone 3/4 blocks cyclin and histone synthesis leading to inhibition of S phase progression, which swiftly resumes once the drug is removed (Fig. 2(c)).
The spectrum of activity of myriaporone 3/4 clearly shows it to be selective for eukaryotes—halo assay revealed that growth of the budding yeast S. cerevisiae and the fission yeast S. pombe was also inhibited, albeit not as potently as were mammalian cells. However, parallel halo assays performed on E. coli revealed no growth inhibition. This is consistent with our observation that repeated attempts to co-crystallize myriaporone 3/4 with archaebacterial ribosomes (purified from Haloarcula marismortui) in order to obtain highly detailed structural information failed to reveal any interaction. Nevertheless, it is obvious that the simpler structure of myriaporone 3/4 (it lacks the “northern hemisphere” present in the tedanolides) does not result in a promiscuous spectrum of activity. The same insensitivity of prokaryotes to tedanolides is observed with myriaporone 3/4, suggesting that the larger ring size of the tedanolide macrolides (two to six carbons larger compared to prokaryotic antibiotics) is not the basis for their eukaryotic specificity.11 It must rather reside in the southern hemisphere.
In general, there exists a paucity of data in the literature regarding the structure–activity relationships of tedanolides and myriaporones. A lone previous report8 had identified structural features of 13-deoxytedanolide that were key to its biological activity. These included: the C8–C9 olefin in the northern hemisphere, the reduction of which reduced anti-proliferative activity over 700-fold; the hydrophobic end of the side chain (C21–C22), the conversion of which to an aldehyde abolished antiproliferative activity; and the C17 hydroxyl group in the southern hemisphere, the acetylation of which reduced activity over 100-fold. Taken together, the authors concluded that all three regions—the northern and southern hemispheres, and the side chain—contribute meaningfully to the activity of 13-deoxytedanolide. Since myriaporone 3/4 lacks a region analogous to the northern hemisphere of tedanolides, our study cannot directly address its importance for activity. Yet, it can be noted that 13-deoxytedanolide was reported to inhibit P388 cells with an IC50 of 0.16 nM,7 while our study shows that myriaporone 3/4 inhibits the same cell line with an IC50 of 14.9 nM—a 100-fold difference in potency. While this difference is less dramatic than would be anticipated based on the earlier structure–activity report,8 it nonetheless supports the notion that the northern hemisphere moderately contributes to drug activity. On the other hand, a similar comparison of our protein synthesis data (IC50 = 11.3 nM) with that reported for 13-deoxytedanolide (IC50 = 150 nM)11 at first suggests that myriaporone 3/4 is much more potent than the larger macrolide at inhibiting translation. However, this difference is more likely due to the use of a yeast system in the previous study, whereas our study used mammalian cells. As we have shown herein, yeast are not as sensitive to these drugs as are mammalian cells.
Our results have now identified an independent set of structural elements which are important for the activity of myriaporone 3/4. The presence of the C14 hydroxymethyl group is crucial for activity, as is the C18–C19 epoxide. Removal of either of these functionalities greatly diminishes activity anywhere from a few hundred-fold to nearly a thousand-fold. This is the first demonstration of the importance of this epoxide for the activity of myriaporones (and perhaps, by extension, the tedanolides). The epoxide result is curious, in that epoxides commonly inactivate target proteins irreversibly by formation of covalent adducts. However, the rapidly reversible effect observed strongly argues in favor of reversible binding of myriaporone 3/4 to its target protein. Thus, in this instance, the epoxide provides an important three-dimensional determinant that promotes drug activity without the need for irreversible modification of the target. Whether the interaction between myriaporone 3/4 is non-covalent or reversibly-covalent is not clear and remains to be conclusively determined.
Among the most obvious of consequences of dehydroxylation of the C14 hydroxymethyl group is that it prevents the intramolecular re-arrangement of the molecule into the closed ring structure of myriaporone 3, thereby permanently constraining HC-1-195 and HC-1-203 into the open structure of myriaporone 4. This may support the notion that the closed form of myriaporone 3/4 may be the more biologically active. Regardless, the finding that both the C18–19 epoxide and the C14 hydroxyl group are important for myriaporone 3/4 activity are very interesting in that neither structural determinant has been shown to play a significant role in the activity of tedanolides. Indeed, the C14 hydroxymethyl group is absent in the macrolides. Despite literature reports indicating the presence of the C13 hydroxyl group to be of nominal functional importance for tedanolide activity,8,9 we found that changing the C13 hydroxyl group to the opposite stereochemistry profoundly reduced biological activity. We reconcile our result with those of previous studies by proposing that, in the unnatural diastereomer, the C13 hydroxyl group now likely interferes with myriaporone binding to the ribosome, while in the natural stereochemistry the C13 hydroxyl group makes little to no meaningful contribution towards activity. While there is an overall structural similarity of myriaporone 3/4 to the southern hemisphere of tedanolide, our results clearly differentiate the presence of functionalities that either may be important to both classes of ribosome inhibitor (e.g. the C18–19 epoxide) or that appear to be uniquely important to myriaporone 3/4 (the C14 hydroxymethyl group). Efforts are ongoing to obtain a crystal structure of myriaporone 3/4 complexed with the eukaryotic ribosome to clarify the curious nature with which these groups contribute to target interaction to inhibit protein synthesis.
In summary, much of the cell growth and protein synthesis inhibitory activities possessed by the tedanolide class of macrolides is retained in myriaporone 3/4 while remaining selective for eukaryotes. Due to its comparatively simpler chemical structure, the task of evaluating the structural determinants that lead to myriaporone activity has been made easier. We have shown herein that the presence of the epoxide as well as the C14 hydroxymethyl group are crucial for its activity. This information will be useful not only in the ongoing design of candidate cancer therapeutics but also in the synthesis of affinity-based reagents that will allow for a more complete biochemical examination of the mechanism of action of these small molecules.
Experimental
Cell culture
All mammalian cells and cell lines were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% O2. P388 cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 50 units ml−1 penicillin and 50 µg ml−1 streptomycin. Bovine aortic endothelial cells were isolated and cultured as follows: (i) under aseptic conditions, freshly procured calf aorta was cut open lengthwise with a sterile surgical scissors; (ii) the intimal surface rinsed thoroughly with PBS supplemented with antibiotics (as described above); (iii) and the intimal surface gently scraped with sterile razors to collect the endothelial cells. The collected material was dispersed into PBS with antibiotics and pelleted by spinning at 800 rpm for 3 min in a tabletop centrifuge. The pellet was resuspended in 5 ml of 0.25% trypsin·EDTA for 2 min at room temperature to disaggregate the sheets of endothelial cells. Following centrifugation and removal of the trypsin·EDTA, the cells were plated out onto a gelatin-coated dish in a specialized formulation of d-MEM containing exclusively d-valine in place of l-valine (Invitrogen), and further supplemented with 10% FBS, 50 units ml−1 penicillin, 50 µg ml−1 streptomycin and 1% MEM non-essential amino acids. The supplemented d-valine growth medium was changed daily until the endothelial cells reached confluency (3 to 4 days), at which point the endothelial cells were subcultured in d-MEM containing l-valine.
[3H]-thymidine incorporation
Cells were seeded into 96 well plates at a density of 2000 to 3500 cells per well. Plates used for endothelial cells were pre-coated with gelatin to improve adherence. The medium was removed the next day and replaced with fresh medium containing the compound of interest at the specified concentration. After 20 h (or other specified incubation interval), each well of cells received another 20 µl of medium containing 2 µCi of [3H]-thymidine (Perkin-Elmer). Following another 3 h, the cells were harvested from the wells and passed through glass fibre filters using a Ska-Tron cell harvestor (Molecular Devices). The filters were transferred to vials, scintillant was added and the amount of radioactivity incorporated into the cells in the filters was quantified by scintillation counting. The resulting data was analyzed using PRISM software (GraphPad Software).
Microbial halo assay
E. coli were grown at 37 °C in LB broth or on LB plates (1.5% agar). S. pombe were grown at 30 °C in YES broth or on YES plates (1.5% agar). S. cerevisiae were grown at 30 °C in YPD broth or on YPD plates (1.5% agar). The Δpdr5 mutant was kindly provided by Joseph Heitman at Duke University (Durham, NC). For halo assays, actively growing yeast or bacteria liquid cultures were diluted into fresh medium to an A600 of 0.1. At that point 50–100 µl of diluted culture was removed to inoculate 7 ml of pre-warmed (52 °C) molten agar (aka “top” agar), quickly vortexed and then immediately poured out over a matching solidified agar plate (“bottom” agar). The top agar layer was allowed to congeal for 20 min after which time 6 mm diameter paper discs (BD Biosciences), pre-dosed with drug, were placed onto the dish using sterile forceps. The dishes were then incubated at appropriate temperature for 48 h, after which time halo development was evaluated.
Yeast dot titration
Yeast strains were inoculated into the appropriate medium and grown overnight at 30 °C with shaking. Cultures were then diluted to an A600 of 0.2, from which five serial dilutions (1 : 10 each time) were prepared. Using a multichannel pipettor, 4 µl aliquots of each set of yeast dilutions were simultaneously spotted in a row onto the appropriate agar medium plate already containing up to 2 µM myriaporone 3/4 (or vehicle-control). The liquid dots were allowed to absorb into the agar before being transferred to 30 °C for 48 h, at which time they were evaluated for drug effect.
p21 Western blot
Endothelial cells at 60–70% confluency were treated overnight with drug or vehicle. Following a quick rinse in ice-cold PBS, the cells were harvested in boiling hot 1% NP-40 lysis buffer (20 mM HEPES pH 7.9, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10 mM sodium fluoride, 10 mM sodium pyrophosphate and 1% NP-40). Samples were chilled on ice for 5 min at which time they were supplemented with protease inhibitors to a final concentration of 1 mM PMSF, 10 µg ml−1 pepstatin A, 10 µg ml−1 leupeptin and 0.3 TIU ml−1 aprotinin. Sample proteins were resolved by SDS gel electrophoresis and transferred to nitrocellulose. Following membrane block in 5% non-fat milk, samples were probed for p21 using rabbit polyclonal antibody C-19 (Santa Cruz Biotechnology) and developed using chemiluminescence.
Protein translation assay
Endothelial cells were plated into 24 well plates at 75 000 cells per well. The following day, the medium was removed and replaced with fresh medium containing myriaporone 3/4 at the specified concentrations (each concentration done in triplicate wells). After 3 h, medium was replaced with fresh medium containing myriaporone and 1 µCi ml−1 [3H]-leucine (Amersham Biosciences). After another 3 h, medium was removed and cells were rinsed twice with ice-cold PBS and then each well lysed in 450 µl of 0.1% SDS. Duplicate 200 µl aliquots from each well were each mixed with 25 µl of 20 mg ml−1 bovine serum albumin and 2 ml of ice-cold 10% trichloroacetic acid. The protein was allowed to precipitate during incubation on ice for 60 min, after which time the precipitate was collected on pre-wetted Whatman GF/B filters using a vacuum manifold (Millipore). Each filter was rinsed once with another 2 ml of ice-cold 10% trichloroacetic acid, and then once with 3 ml of ice-cold 95% ethanol. Filters were transferred to vials, scintillant was added and the amount of radioactivity incorporated into the cells in the filters was quantified by scintillation counting. The resulting data was analyzed using PRISM software (GraphPad Software, San Diego, CA).
Myriaporone 3/4 and analogs chemical syntheses
Myriaporone 3/4 was prepared via total synthesis utilizing the synthetic route developed in our laboratories.17 Deoxymyriaporones MR-1-94, HC-1-195 and HC-1-203 were prepared by related synthetic routes. Complete experimental details and full characterization for all new compounds are included in the supporting information.
Acknowledgements
The authors are grateful to members of the Crews lab for their helpful comments on the manuscript. This work was supported by NIH grants GM062120 (CMC) and CA81128 (RET). The synthetic aspects of this work were accomplished in collaboration with the Walther Cancer Research Center, University of Notre Dame.
Footnotes
Electronic supplementary information (ESI) available: Myriaporone analog synthesis.
References
- 1.Rinehart KL, Tachibana K. J. Nat. Prod. 1995;58:344–358. [Google Scholar]
- 2.Friedman D, Hu Z, Kolb EA, Gorfajn B, Scotto KW. Cancer Res. 2002;62:3377–3381. [PubMed] [Google Scholar]
- 3.Crews CM, Collins JL, Lane WS, Snapper ML, Schreiber SL. J. Biol. Chem. 1994;269:15411–15414. [PubMed] [Google Scholar]
- 4.Hung DT, Chen J, Schreiber SL. Chem. Biol. 1996;3:287–293. doi: 10.1016/s1074-5521(96)90108-8. [DOI] [PubMed] [Google Scholar]
- 5.Klein LE, Freeze BS, Smith AB, Horwitz SB. Cell Cycle. 2005;4:501–507. doi: 10.4161/cc.4.3.1550. [DOI] [PubMed] [Google Scholar]
- 6.Schmitz FJ, Gunasekera SP, Yalamanchili G, Hossain BM, van der Helm D. J. Am. Chem. Soc. 1984;106:7251–7252. [Google Scholar]
- 7.Fusetani N, Sugawara T, Matsunaga S, Hirota H. J. Org. Chem. 1991;56:4971–4974. [Google Scholar]
- 8.Nishimura S, Matsunaga S, Yoshida S, Nakao Y, Hirota H, Fusetani N. Bioorg. Med. Chem. 2005;13:455–462. doi: 10.1016/j.bmc.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 9.Chevallier C, Bugni TS, Feng X, Harper MK, Orendt AM, Ireland CM. J. Org. Chem. 2006 [Google Scholar]
- 10.Schmitz FJ, Gunasekera SP, Hossain BM, van der Helm D, Yalamanchili G. 87-7347. US Pat. 1988
- 11.Nishimura S, Matsunaga S, Yoshida M, Hirota H, Yokoyama S, Fusetani N. Bioorg. Med. Chem. 2005;13:449–454. doi: 10.1016/j.bmc.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 12.Lee K-H, Nishimura S, Matsunaga S, Fusetani N, Ichijo H, Horinouchi S, Yoshida M. Biosci. Biotechnol. Biochem. 2006;70:161–171. doi: 10.1271/bbb.70.161. [DOI] [PubMed] [Google Scholar]
- 13.Smith AB, Adams CM, Lodise-Barbarosa SA, Degnan AP. J. Am. Chem. Soc. 2003;125:350–351. doi: 10.1021/ja0289649. [DOI] [PubMed] [Google Scholar]
- 14.Julian LD, Newcom JS, Roush WR. J. Am. Chem. Soc. 2005;127:6186–6187. doi: 10.1021/ja050729d. [DOI] [PubMed] [Google Scholar]
- 15.Mahe Y, Lemoine Y, Kuchler K. J. Biol. Chem. 1996;271:167–172. doi: 10.1074/jbc.271.41.25167. [DOI] [PubMed] [Google Scholar]
- 16.Yeh J-R, Mohan R, Crews CM. Proc. Natl. Acad. Sci. U. S. A. 2000;97:12782–12787. doi: 10.1073/pnas.97.23.12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fleming KN, Taylor RE. Angew. Chem., Int. Ed. 2004;43:1728–1730. doi: 10.1002/anie.200353348. [DOI] [PubMed] [Google Scholar]