

Abstract
Although glial cells are recognized for their roles in maintaining neuronal function, there is growing appreciation of the ability of resident CNS cells to initiate and/or augment inflammation following trauma or infection. The tachykinin, substance P (SP), is well known to augment inflammatory responses at peripheral sites and its presence throughout the CNS raises the possibility that this neuropeptide might serve a similar function within the brain. In support of this hypothesis, we have recently demonstrated the expression of high affinity receptors for SP (Neurokinin-1 (NK-1) receptors) on microglia and shown that this tachykinin can significantly elevate bacterially induced inflammatory prostanoid production by isolated cultures of these cells. In the present study, we demonstrate that endogenous SP/NK-1R interactions are an essential component in the initiation and/or progression of CNS inflammation in vivo following exposure to two clinically relevant bacterial CNS pathogens, Neisseria meningitidis and Borrelia burgdorferi. We show that in vivo elevations in inflammatory cytokine production and decreases in the production of an immunosuppressive cytokine are markedly attenuated in mice genetically deficient in the expression of the NK-1R or in mice treated with a specific NK-1R antagonist. Furthermore, we have used isolated cultures of microglia and astrocytes to demonstrate that SP can augment inflammatory cytokine production by these resident CNS cell types following exposure to either of these bacterial pathogens. Taken together, these studies indicate a potentially important role for neurogenic exacerbation of resident glial immune responses in CNS inflammatory diseases, such as bacterial meningitis.
Bacterial infections of the CNS are serious and often intractable conditions affecting the meninges and the brain parenchyma. Such infections are associated with high levels of inflammatory cytokines including IL-6 and TNF-α in the CNS that result in neurological dysfunction (1). Although the brain has traditionally been viewed as a “victim organ” of infiltrating leukocytes, it has become increasingly apparent that resident glial cells, such as microglia and astrocytes, play an important role in the initiation and/or progression of immune responses following pathogen invasion. These glial cells can perceive bacteria via microbial pattern recognition receptors, including members of the TLR and nucleotide-binding domain leucine rich repeat region containing (NLR) receptor families, to promote recruitment and Ag-specific activation of infiltrating leukocytes (2-7).
A compelling body of evidence has accumulated to show that the neuropeptide substance P (SP)3 plays an important role in augmenting inflammatory immune responses at the peripheral sites, such as the gastrointestinal tract and skin (as reviewed in Ref. 8). Central to the ability of SP to augment inflammation is the finding that this neuropeptide can modulate the function of myeloid cells, such as macrophages and dendritic cells, via high affinity neurokinin-1 (NK-1) receptors (9). Interaction of SP with NK-1R activates the transcription factor NF-κB and facilitates the production of key proinflammatory cytokines including IL-6 and TNF-α (8, 9). Furthermore, we have demonstrated that SP can also reduce immunosuppressive cytokine production by activated macrophages (10), there by providing another means by which this neuropeptide could exacerbate inflammation.
SP is the most abundant tachykinin in the brain (11) and we have documented the expression of NK-1Rs on microglia (12), a CNS cell type that shares a common myeloid lineage with macrophages and dendritic cells. Importantly, we have also recently shown that SP can synergistically augment Borrelia burgdorferi-induced expression of cyclooxygenase-2 in microglia and increase subsequent secretion of the potentially inflammatory prostanoid, PGE2 (13). Although these findings are consistent with the effects of SP in the periphery, the ability of this neuropeptide to exacerbate bacterially induced CNS inflammation has yet to be established. In the present study, we demonstrate that SP augments inflammatory cytokine production by resident glial cells following exposure to disparate and clinically relevant bacterial pathogens of the CNS. Moreover, we show that endogenous SP is an essential component in the initiation and/or progression of CNS inflammation in vivo following bacterial administration. Taken in concert, these data provide evidence of a role for this neuropeptide during inflammatory CNS disorders. Furthermore, the ability of a systemically delivered SP receptor antagonist to significantly reduce immune responses within the CNS may represent an important new therapeutic strategy to combat the potentially lethal consequences of inflammation within the brain.
Materials and Methods
SP receptor-deficient mice
SP receptor-deficient mice, bred for >10 generations onto a C57BL/6 background, were derived at the University of Iowa Medical Center (14). These mice were originally derived from induced mutations made by insertion of the lacZ gene into exon 1 of the SP receptor (15). SP receptor-deficient mice were routinely screened by PCR to confirm disruption of the SP receptor, as previously described (15, 16), using the positive and negative strand primers CCAACACCTCCAAGACTTCTG and GCCACAGCTATGGAGTAGAT for wild type, and TCCAGACTGCCTTGG GAAAA and GCCACAGCTGTCATGGAGTAGAT for SP receptor deficiency, respectively.
Isolation of primary murine microglia
Mouse neonatal brain microglia were isolated, as described previously by our laboratory (7, 12, 13, 17-19), and cultured in RPMI 1640 with 10% FBS and 20% conditioned medium from LADMAC cells (American Type Culture Collection number CRL-2420) as a source of CSF-1. Cells isolated in this manner are >95% authentic microglia as assessed by their characteristic morphology and expression of CD11b and F4/80.
Isolation and characterization of primary murine astrocytes
Mouse neonatal brain astrocytes were isolated, as described previously by our laboratory (6, 13, 20, 21), and cultured in RPMI 1640 containing 10% FBS. Cells isolated in this manner are >97% authentic astrocytes due to their characteristic morphology, expression of glial fibrillary acidic protein, and the absence of CD11b as determined by immunofluorescent microscopy.
Culture of B. burgdorferi and Neisseria meningitidis and preparation of Ag lysates
A clonal low passage of B. burgdorferi strain N40 was used throughout these studies and was grown in BSK II medium while N. meningitidis strain MC58 was grown overnight in 5 ml of GC medium plus hemoglobin with Isovitalex enrichment solution at 37°C, as previously described (6, 20). For cell lysates, bacteria were centrifuged at 10,000 × g and washed three times with PBS before pulsing three times with a cell sonicator. B. burgdorferi Ag isolates generated in this manner have previously been demonstrated to be free of detectable levels of LPS (6, 7, 17, 19). For in vitro exposure of isolated glia to N. meningitidis or B. burgdorferi, bacteria were harvested by centrifugation and washed twice in PBS. Confluent cell layers of glia were washed three times with 4 ml of PBS to remove growth media and then exposed to bacteria at M.O.I. of between 3:1 and 30:1 bacteria to cells in media without antibiotics for 120 min at 37°C. Following this period, cell cultures were washed and incubated in media with 10% FBS supplemented with 25 μg/ml gentamicin to kill remaining extracellular bacteria. At 12-24 h following this procedure, culture supernatants were collected. In other experiments, glial cells were exposed to 1-10 μg/ml B. burgdorferi or N. meningitidis cell lysates as indicated. These doses were selected based upon their documented ability to induce submaximal cytokine responses by glial cells (19, 20).
Intracerebral (i.c.) administration of bacteria
Viable N. meningitidis or B. burgdorferi (1 × 106 bacteria) were administered via i.c. injection as previously described by our laboratory (20) into NK1R-/-, wild type C57BL/6 mice (Jackson ImmunoResearch Laboratories), or wild type animals that received s.c. doses of l-703,606 (Sigma-Aldrich) at days -1, 0, and +1, or vehicle. Anesthetized animals were secured in a stereotaxic platform and stand and received an i.c. injection containing pathogens in Ringer’s solution (final volume of 1 μl), or vehicle only, 1 mm lateral and 1 mm posterior to the bregma using a Hamilton positive displacement syringe (7001 series) with a 25-gauge needle and a tubing guard to ensure constant depth of administration (3-3.5 mm). Eight animals per group were used, and animals displaying signs such as seizures, abnormal gait/ataxia, failure to open eyes, or other physical disabilities were euthanized immediately. At 3 days post infection, animals were euthanized and all brain tissue removed and weighed. The brain tissue was mechanically disrupted in a glass homogenizer in 3 ml PBS and then centrifuged. Supernatants were analyzed for cytokine content by specific capture ELISA and RNA extracted from the cell pellets for real-time PCR analysis.
All studies were performed in accordance with relevant federal guidelines and institutional policies regarding the use of animals for research purposes.
Isolation of RNA and real-time PCR
Isolation of total RNA was performed based on a single step, acid guanidium thiocyanate-phenol-chloroform extraction method as performed previously in our laboratory (7, 13, 19, 21). Real-time PCR amplification was performed on reverse transcribed cDNA using a light cycler (Roche). Positive and negative stranded primers used for the reactions, respectively, were GATGCTACCAAACTGGATATAATC and GGTCCTTAGCCAC TCCTTCTGTG for IL-6 amplification, GAGTGGTGCCAGCCGATGG GTTGT and GATGAGTTGGTCCCCCTTCTCCAG for TNF-α, GTAC AGCCGGGAAGACAATA and ATCACTCTTCACCTGCTCCA to amplify mRNA encoding IL-10 and CCATCACCATCTTCCAGGAGCG AG, and CACAGTCTTCTGGGTGGCAGTGAT to amplify mRNA encoding the housekeeping gene, G3PDH. Quantitech SYBR green PCR mix (Qiagen) was used to conduct the quantitative PCR as previously described by our laboratory (7). These primers were designed using Oligo 4.0 primer analysis software (National Biosciences) based on their location in different exons of the genomic sequences for each in addition to their lack of significant homology to sequences present in GenBank (MacVector Sequence analysis software; IBI). PCR amplification of the housekeeping gene, G3PDH, was performed on cDNA from each sample to insure similar input of RNA and efficiencies of reverse transcription. The identities of the PCR-amplified fragments were verified by size comparison with DNA standards (Promega).
Quantification of IL-6, TNF-α, and IL-10 secretion in glial culture supernatants
Specific capture ELISAs were performed to quantify IL-6, TNF-α, and IL-10 and secretion by glial cells as described previously by our laboratory (13, 19, 20). Cytokine levels in brain homogenates were normalized to total brain weight and reported as ng or pg/g of brain tissue.
Bacterial counts
Postinfection, 100 μl of the isolated brain homogenates were cultured on GC agar plates with hemoglobin plus Isovitalex enrichment solution (Becton Dickinson Microbiology system) for 16 h at 37°C and N. meningitidis colonies subsequently counted.
Histochemical and immunofluorescent analyses of murine brain sections
Infected and non-infected mice were euthanized 72 h postinfection and the brains were perfused with Prefer fixative (Anatech) via intra-atrial injection. Samples were embedded in paraffin and sectioned serially (5-7 um). Cellularity was assessed in axial sections stained with H&E by counting the total number of cell nuclei within three quadrants from two low power (×20 objective) microscopy fields of the same cortical locations in animals from each experimental group.
Demyelination was measured in coronal sections by Luxol fast blue-Cresyl violet staining according to the directions provided by the manufacturer (American MasterTech Scientific). In brief, paraffin embedded sections were heated and rehydrated with decreasing concentrations of ethyl alcohol before rinsing in distilled water and Luxol fast blue staining at 60°C for 18 h. Gray and white matter was further differentiated using 0.05% lithium carbonate and washing with 70% alcohol. Finally, the slides were placed in Cresyl Echt violet stain for 10 min and differentiated with alcohol. After dehydration, slides were washed in xylene and coverslips were mounted using permanent mounting media and myelination of the corpus callosum visually assessed in a low power (×10 objective) microscopy field in animals from each experimental group.
Astrogliosis was assessed in deparaffinized coronal sections blocked with goat serum (Zymed Laboratories) according to glial fibrillary acidic protein (GFAP) expression. GFAP was detected using a monoclonal mouse Ab directed against murine GFAP (Molecular Probes) and an AlexaFluor 488-conjugated chicken anti-mouse secondary Ab (Molecular Probes). Cell nuclei were visualized with 4′,6-diamidino-2-phenylindole, dihydrochloride (Molecular Probes) and the cover slip was mounted using ProLong Gold antifade reagent (Invitrogen). GFAP expression was assessed in multiple microscopy fields (×40 objective) of the same cortical locations in animals from each experimental group using an Olympus IX70 Fluoroview confocal microscopy system. Fluorescence intensity was quantified using Fluoview 500 Version 4.3 and National Institutes of Health Image software to acquire microscopic field section images and to determine mean gray values for multiple fluorescent fields.
Acute isolation and cytometric analysis of CNS cells
Mixed CNS cells were acutely isolated from infected and uninfected animals using a protocol modified from Campanella et al. (22). In brief, brains were rapidly removed and mechanically disrupted in a glass homogenizer, washed, and resuspended in PBS/30% Percoll (Fluka; Sigma-Aldrich) solution. This was overlaid on a gradient containing 37 and 70% Percoll solutions and centrifuged at 500 × g for 20 min at room temperature. Cells were then collected from the 37/70% Percoll interface and washed with PBS. Cells were permeabilized with CytoFix/CytoPerm Plus according to the directions provided by the manufacturers (BD Pharmingen) before staining with a AlexaFluor 488-conjugated monoclonal mouse Ab directed against murine GFAP (Molecular Probes). These cells were analyzed using a FACSCalibur flow cytometer (BD Biosciences) to determine the intensity of GFAP-associated fluorescence relative to the signal obtained with an Ab directed against an irrelevant Ag. In each experiment, a minimum of 50,000 cells were counted and data is reported as the geometric mean of the fluorescence signal.
Statistical analyses
All results are presented as the mean ± SEM and were tested statistically by Student’s t test or one-way ANOVA with Tukey’s posthoc test as appropriate using commercially available software (Sigma-Aldrich). Results were considered to be statistically significant at a probability of <0.05.
Results
Endogenous SP/NK-1R interactions are required for increased CNS cellularity, astrogliosis, and demyelination following in vivo bacterial administration
To begin to determine the role of SP in bacterially induced inflammatory glial responses, we have investigated the morphological changes associated with in vivo administration of N. meningitidis in the presence or absence of endogenous SP/NK-1R interactions. As shown in Fig. 1, i.c. injection of known numbers of N. meningiditis (1 × 106 organisms) elicits significant (p < 0.05) elevations in cellularity and numbers of astrocytes in the cerebral cortex (three quadrants/field counted from two fields/section from four animals per experimental group) with an average increase in cell number per quadrant from 71 ± 11 to 124 ± 34 cells (n = 4) and an increase in GFAP staining from 18 ± 3 to 98 ± 24 arbitrary intensity units (n = 4). In addition, administration of N. meningitidis elicits demyelination as evidenced by reduced Luxol blue staining of the corpus callosum as shown in Fig. 1 (n = 4).
FIGURE 1.
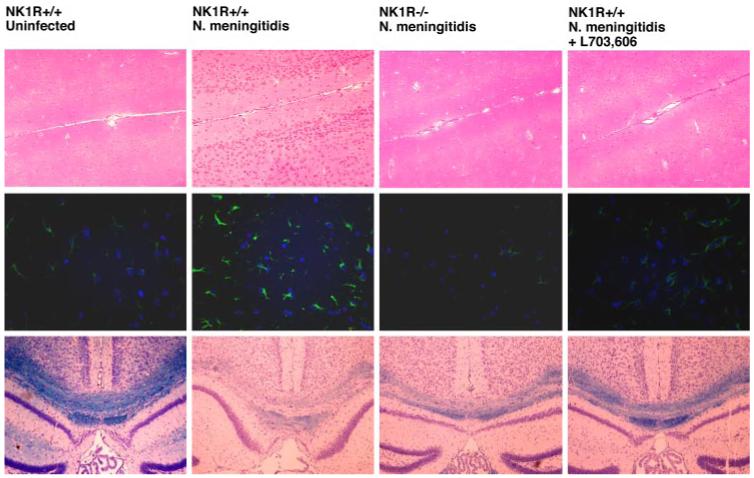
Bacterially induced increases in CNS cellularity, astrogliosis, and demyelination are markedly reduced in the absence of endogenous SP/NK-1R interactions. N. meningitidis (1 × 106) was administered via i.c. injection into C57BL/6 wild-type (NK1R+/+) and NK-1R-deficient (NK1R-/-) mice. In addition, a group of wild-type mice received s.c. injections (5 mg/kg) of the NK-1R antagonist l-703,606 at days -1, 0, and +1 relative to bacterial challenge. At 72 h postinfection, brain tissue was perfused in situ, isolated, and prepared for histochemical analysis by lightfield microscopy or immunofluorescent microscopy. Upper panels, Representative H&E-stained axial cortical fields (×20 objective) from one of the four animals in each group. Middle panels, Representative GFAP (green) and 4′,6-diamidino-2-phenylindole, dihydrochloride (blue) immunofluorescence (×40 objective) in coronal cortical fields from one of the four animals in each group. Bottom panels, Representative Luxol blue staining of the corpus callosum in coronal fields (×10 objective) from one of the four animals in each group.
Interestingly, similar administration of N. meningitidis into NK1R-/- animals failed to elicit a significant increase in cellularity, with an average cell number per quadrant of 65 ± 15 cells (n = 4), or astrogliosis, with an average GFAP-associated fluorescence of 14 ± 2 arbitrary intensity units (n = 4) (Fig. 1). NK1R-/- mice also exhibited less demyelination than their wild-type counterparts (Fig. 1). To further investigate the relative importance of endogenous SP/NK-1R interactions in host responses to in vivo CNS infections, we have determined the effect of s.c. administration of a specific NK-1R antagonist, L703,606 (5 mg/kg; days -1, 0, and + 1 to infection), on cerebral cortex cellularity and astrogliosis following N. meningitidis administration in wild-type animals. As shown in Fig. 1, N. meningitidis failed to elicit a significant increase in cellularity in L703,606-treated mice with an average cell number per quadrant of 72 ± 11 cells and exhibited significantly less astrogliosis than untreated animals with an average GFAP-associated fluorescence of 57 ± 6 arbitrary fluorescence units (n = 4). Furthermore, the degree of demyelination following bacterial administration was markedly reduced in L703,606-treated animals as shown in Fig. 1.
To confirm our immunofluorescence microscopy findings, parallel experiments were performed using flow cytometry to assess GFAP expression in acutely isolated glial cells. As shown in Fig. 2A, elevations GFAP-associated fluorescence following N. meningitidis infection were inhibited in L703,606-treated mice (n = 6). Interestingly, the absence or pharmacological inhibition of endogenous SP/NK-1R interactions did not significantly change the number of viable bacteria that could be isolated from infected brain tissue (1679 ± 36, 1852 ± 92, and 1817 ± 83 bacteria/ml in wild-type, NK1R-/-, and antagonist-treated NK1R+/+ animals, respectively).
FIGURE 2.
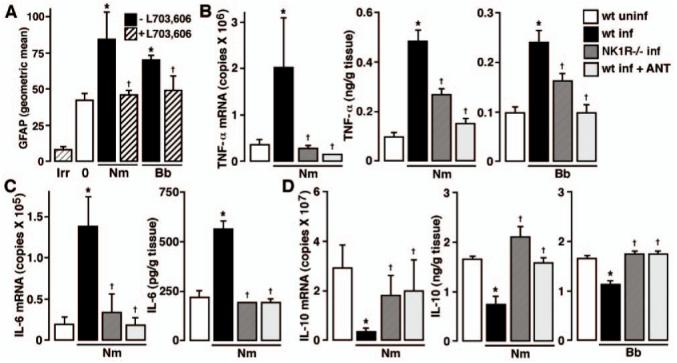
In vivo astrogliosis and inflammatory cytokine production are markedly reduced in the absence of endogenous SP/NK-1R interactions. A, Wild-type C57BL/6 mice were given vehicle or infected with N. meningitidis (Nm) or B. burgdorferi (Bb) (1 × 106) via i.c. injection and were untreated or received s.c. administration of the NK-1R antagonist l-703,606 (5 mg/kg) at days -1, 0, and +1 relative to bacterial challenge. At 72 h postinfection, glial cells were isolated, permeabilized, and GFAP expression determined by flow cytometry. n = 6; Asterisk indicates significant difference from uninfected (0) animals, and dagger indicates significant difference from infected animals that did not receive the NK-1R antagonist. Fluorescence signal obtained using an isotype and species matched Ab directed against an irrelevant Ag is indicated (Irr). B-D, N. meningitidis (1 × 106) (Nm) or B. burgdorferi (1 × 106) (Bb) was administered via i.c. injection into C57BL/6 wild-type (wt) or NK-1R-deficient (NK1R-/-) mice. In addition, groups of wild-type mice received s.c. injections (5 mg/kg) of the NK-1R antagonist l-703,606 at days -1, 0, and +1 to bacterial challenge (wt inf + ANT). At 72 h postinfection, mRNA or tissue homogenates were isolated for measurement of TNF-α (B), IL-6 (C), and IL-10 (D) mRNA and protein expression by real-time PCR and specific capture ELISA, respectively. n = 8; Asterisk indicates significant difference from uninfected (wt uninf) animals, and dagger indicates significant difference from infected wild-type animals (wt inf).
Finally, we have investigated the role of endogenous SP/NK-1R interactions in the changes in the astrogliosis evoked by a disparate bacterial pathogen, B. burgdorferi, the causative agent of Lymeneuroborreliosis. As shown in Fig. 2A, in vivo exposure to B. burgdorferi elicits a marked increase in GFAP-associated fluorescence in acutely isolated glial cells. Importantly, this astrogliosis is markedly attenuated by L703,606 treatment (n = 6). Similarly, this antagonist attenuated B. burgdorferi-induced demyelination as evidenced by reduced Luxol blue staining of the corpus callosum (data not shown). Taken together, these data indicate that endogenous SP/NK-1R interactions play an important role in the increase in CNS cellularity and/or astrogliosis following in vivo bacterial administration.
Endogenous SP/NK-1R interactions are required for increased inflammatory cytokine production following in vivo bacterial administration
To further determine the role of SP in bacterially induced inflammatory CNS responses in vivo, we have investigated the production of proinflammatory cytokines following in vivo administration of two disparate bacterial CNS pathogens in the presence or absence of endogenous SP/NK-1R interactions. As shown in Fig. 2B, i.c. injection of known numbers of N. meningitidis (1 × 106 organisms) elicits significant (p < 0.05) elevations in the expression of mRNA encoding TNF-α and release of this protein. Importantly, these elevations are significantly lower in NK1R-/- mice following N. meningitidis administration (Fig. 2B). Furthermore, increases in the expression of mRNA encoding TNF-α and secretion of this inflammatory cytokine following bacterial challenge were inhibited in wild-type animals that received s.c. administration of L703,606 (5 mg/kg; days -1, 0, and +1 to infection) (Fig. 2B).
In parallel experiments, animals were exposed to B. burgdorferi (1 × 106 organisms) administered via i.c. injection. As shown in Fig. 2B, this pathogen also elicited significant elevations in TNF-α levels. Similar to N. meningitidis, B. burgdorferi-mediated elevations in TNF-α protein levels were significantly lower in NK1R-/- mice (Fig. 2B). Furthermore, B. burgdorferi-induced elevations in TNF-α mRNA levels were inhibited following s.c. administration of L703,606 (5 mg/kg; days -1, 0, and +1toin-fection) (5.28 ± 0.23 × 104 vs 1.05 ± 0.60 × 104 copies in infected mice vs infected animals that received L703,606, respectively, from 0.38 ± 0.02 × 104 copies in uninfected mice) and bacterially induced elevations in TNF-α protein levels were abolished by this antagonist (Fig. 2B).
In addition, we have assessed the importance of SP interactions with its receptor in the production of an alternate inflammatory cytokine, IL-6, following in vivo bacterial challenge. As shown in Fig. 2C, i.c. injection of known numbers of N. meningitidis (1 × 106 organisms) elicits significant (p < 0.05) elevations in the expression of mRNA encoding IL-6 and release of this protein (n = 8). Again, these elevations were significantly reduced in NK1R-/- mice and wild-type animals that received s.c. administration of L703,606 (5 mg/kg; days -1, 0, and + 1 to infection) (Fig. 2C). Taken in concert, these data show that endogenous SP/NK-1R interactions play an essential role in the increased production of key inflammatory cytokines within the CNS following in vivo bacterial administration.
Endogenous SP/NK-1R interactions are required for decreased immunosuppressive cytokine production following in vivo bacterial administration
We have investigated the importance of endogenous SP/NK-1R interactions in the production of IL-10, an immunosuppressive cytokine that is thought to play an important role in the maintenance of the immunoquiescent environment of the CNS, following in vivo bacterial challenge. As shown in Fig. 2D, i.c. injection of N. meningitidis (1 × 106 organisms) elicits significant (p < 0.05) reductions in the expression of mRNA encoding IL-10 and decreased protein levels of this anti-inflammatory cytokine. Consistent with our findings for inflammatory cytokine production, this decrease was significantly reduced in NK1R-/- mice following N. meningitidis administration (Fig. 2D). Furthermore, the decreases in IL-10 mRNA and protein expression following bacterial challenge were inhibited in wild-type animals that received s.c. administration of L703,606 (5 mg/kg; days -1, 0, and +1toin-fection) (Fig. 2D).
In parallel experiments, animals were exposed to B. burgdorferi (1 × 106 organisms) administered via i.c. injection. As shown in Fig. 2D, this pathogen also elicited significant reductions in IL-10 levels. Similar to N. meningitidis, B. burgdorferi-mediated reductions in IL-10 protein levels were not apparent in NK1R-/- mice (Fig. 4C). Furthermore, B. burgdorferi-induced decreases in IL-10 mRNA levels were inhibited following s.c. administration of L703,606 (5 mg/kg; days -1, 0, and +1 to infection) (0.29 ± 0.11 × 106 vs 1.29 ± 0.78 × 106 copies in infected mice vs infected animals that received L703,606, respectively, from 1.25 ± 0.37 × 106 copies in uninfected mice) and bacterially induced reductions in IL-10 protein levels were abolished by this antagonist (Fig. 2D). Taken together, these data show that endogenous SP/NK-1R interactions within the CNS play an essential role in the decreased production of an important immunosuppressive cytokine following in vivo bacterial administration and provides an additional mechanism whereby this neuropeptide could exacerbate CNS inflammation associated with bacterial challenge.
FIGURE 4.
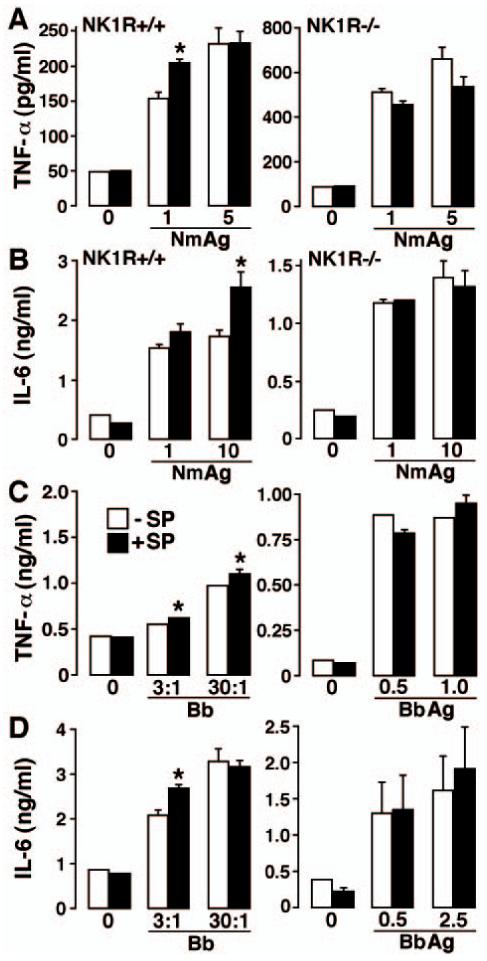
SP augments bacterially induced inflammatory cytokine production by primary astrocytes in a NK-1R-dependent manner. Cells (2 × 106) from wild-type (NK1R+/+) and NK-1R knockout (NK1R-/-) animals were untreated (0) or exposed to N. meningitidis (Nm: 1, 5, or 10 μg/ml), viable B. burgdorferi (Bb: M.O.I. of 3 or 30 bacteria to each astrocyte) or B. burgdorferi Ags (Bb Ag: 0.5, 1, or 2.5 μg/ml) in the absence (-SP) or presence (+SP) of SP (5 nM). At 12 (A and B) or 24 h (C and D) following bacterial challenge, culture supernatants were assayed for the presence of TNF-α (A and C) and IL-6 (B and D) by specific capture ELISA. Data are presented as the mean of triplicate determinations of samples from three separate experiments ± SEM. Asterisks indicate statistically significant differences between cytokine levels in the absence or presence of SP.
SP augments proinflammatory cytokine production by isolated microglia in response to disparate bacterial pathogens of the CNS
To determine the effects of SP on bacterially induced inflammatory cytokine production by resident myeloid cells of the CNS, isolated cultures of primary microglia were untreated or infected with viable N. meningitidis at varying M.O.I. (10 and 30 bacteria to each microglia) or exposed to whole cell N. meningitidis lysates (1-10 μg/ml) in the absence or presence of SP agonist (5 nM). As shown in Fig. 3A, SP alone fails to elicit elevations in TNF-α production. However, SP significantly augmented (p < 0.05, n = 3) production of TNF-α at 12 h after infection with viable N. meningitidis or exposure to bacterial lysates (Fig. 3A). To confirm that the effects of SP are mediated via specific interactions with the NK-1R, parallel experiments were performed using microglia derived from mice genetically deficient in the expression of the NK-1R (NK1R-/-). As shown in Fig. 3A, SP failed to augment N. meningitidis-induced TNF-α production by microglia derived from NK1R-/- animals. Furthermore, we performed additional experiments using L703,606 (10 nM) and verified that addition of this NK-1R antagonist inhibits the SP-mediated augmentation of TNF-α production by microglia derived from wild type following exposure to N. meningitidis lysates (data not shown).
FIGURE 3.
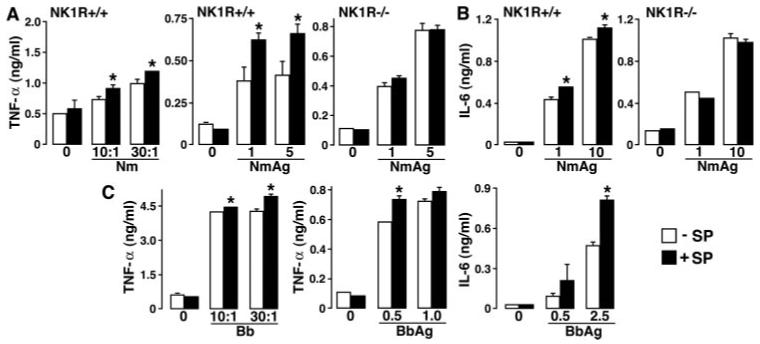
SP augments inflammatory cytokine production by primary microglia following exposure to N. meningitidis or B. burgdorferi in an NK-1R-dependent manner. Cells (2 × 106) from wild-type (NK1R+/+) and NK-1R knockout (NK1R-/-) animals were untreated or exposed to either viable N. meningitidis (Nm: M.O.I. of 10 or 30 bacteria to each glial cell), N. meningitidis Ags (Nm Ag: 1 and 5 μg/ml), viable B. burgdorferi (Bb: M.O.I. of 10 or 30 bacteria to each glial cell), or B. burgdorferi Ags (Bb Ag: 0.5, 1, and 2.5 μg/ml) in the absence (-SP) or presence (+SP) of SP (5 nM). At 12 h following bacterial challenge, culture supernatants were isolated and assayed for the presence of TNF-α (A and C) or IL-6 (B and C) by specific capture ELISA. Data are presented as the mean of triplicate determinations of samples from three separate experiments ± SEM. Asterisks indicate statistically significant differences between cytokine levels in the absence or presence of SP.
In addition, we have assessed the effects of SP on the production of an alternate inflammatory cytokine IL-6 following in vitro bacterial challenge. Primary microglia were untreated or exposed to whole cell N. meningitidis lysates (1-10 μg/ml) in the absence or presence of SP agonist (5 nM). As shown in Fig. 3B, SP alone fails to elicit elevations in IL-6 production but significantly augments (p < 0.05, n = 3) production of IL-6 at 12 h following exposure to bacterial lysates. To again confirm that the effects of SP are mediated via specific interactions with the NK-1R, we have verified that L703,606 (10 nM) inhibits SP-mediated augmentation of IL-6 production by microglia (data not shown), and performed parallel experiments using microglia derived from NK1R-/- mice. As shown in Fig. 3B, SP failed to augment N. meningitidis-induced IL-6 production by microglia derived from NK1R-/- animals. In contrast, SP failed to significantly alter N. meningitidis(5 μg/ml)-induced production of the anti-inflammatory cytokine IL-10 (266 ± 33 vs 272 ± 35 pg/ml in the absence or presence of SP, respectively).
In parallel experiments, primary microglia were untreated or infected with viable B. burgdorferi at varying M.O.I. (10 and 30 bacteria to each microglia) or exposed to whole cell B. burgdorferi lysates (0.5-2.5 μg/ml) that lack detectable levels of LPS (23) in the absence or presence of SP agonist (5 nM). As shown in Fig. 3C, SP alone fails to elicit elevations in TNF-α or IL-6 production. However, SP significantly augmented (p < 0.05, n = 3) production of TNF-α and IL-6 at 24 h following infection with viable B. burgdorferi or exposure to bacterial lysates (Fig. 3C) but did not alter IL-10 production elicited by 1 μg/ml B. burgdorferi (321 ± 67 vs 344 ± 68 ng/ml in the absence or presence of SP, respectively). Taken together, these data suggest that the ability of SP to augment bacterially induced inflammatory cytokine production by microglia is not limited to TLR4-mediated bacterial responses.
SP augments proinflammatory cytokine production by isolated astrocytes in response to N. meningitidis and B. burgdorferi
To further investigate the effects of SP on bacterially induced inflammatory cytokine production by resident cells of the CNS, isolated cultures of primary astrocytes were untreated or infected with viable N. meningitidis at varying M.O.I. (3 and 10 bacteria to each astrocyte) or exposed to whole cell N. meningitidis lysates (1-10 μg/ml) in the absence or presence of SP agonist (5 nM). SP alone fails to elicit elevations in either TNF-α (Fig. 4A) or IL-6 (Fig. 4B) production. However, SP significantly (p < 0.05, n = 3) augmented production of TNF-α 24 h after infection with viable N. meningitidis (10:1 bacteria to each astrocyte) (553 ± 32 vs 615 ± 23 pg/ml by infected cells vs infected cells plus SP, respectively, from 297 ± 29 pg/ml by uninfected cells) or 12 h exposure to bacterial lysates (Fig. 4A). Similarly, SP significantly augmented N. meningitidis lysate-mediated IL-6 secretion by astrocytes at 12 h following challenge (Fig. 4B) but failed to significantly alter production of IL-10 (116 ± 9 vs 105 ± 26 pg/ml in the absence or presence of SP, respectively). To again confirm that the effects of SP are mediated via specific interactions with the NK-1R, we have verified that L703,606 (10 nM) inhibits SP-mediated augmentation of IL-6 production by astrocytes (data not shown), and performed parallel experiments using astrocytes derived from NK1R-/- mice. As shown in Fig. 4, A and B, SP failed to significantly alter N. meningitidis-induced TNF-α or IL-6 production by astrocytes derived from these knockout animals.
In parallel experiments, primary astrocytes were untreated or infected with viable B. burgdorferi at varying M.O.I. (3 and 30 bacteria to each astrocyte) or exposed to whole cell B. burgdorferi lysates (0.5-2.5 μg/ml) in the absence or presence of SP agonist (5 nM). SP significantly augmented (p < 0.05, n = 3) production of TNF-α (Fig. 4C) and IL-6 (Fig. 4D) at 24 h following infection with viable B. burgdorferi but not B. burgdorferi lysates (0.5-2.5 μg/ml).
Discussion
Bacterial infections of the CNS constitute a group of highly damaging and often life-threatening diseases that have shown dramatic increases in incidence over the last two decades. What makes the etiology of these diseases so perplexing is that severe CNS inflammation can be initiated by bacterial species that are generally regarded to be of low virulence (as reviewed in Ref. 1). Although such responses may represent protective immune responses to certain pathogens, inflammation elicited by infectious agents often results in progressive damage to the CNS. A hallmark of developing immune responses is the synergistic interactions between cells and their products, which can amplify the response. Such amplification and positive feedback loops serve to recruit cells to the site of infection, while promoting activation signals that continue to expand the response.
It is now widely accepted that the neuropeptide SP plays an important role in exacerbating inflammatory immune responses at peripheral sites upon infection. In particular, it has been demonstrated that SP can promote the immune functions of peripheral myeloid cells, including macrophages and dendritic cells, via its high affinity NK-1R (9). SP is found throughout the CNS, with evidence for both neuronal and glial cells as potential sources of this neuropeptide (11). Such expression may be constitutive, or may be elicited following injury or exposure to CNS pathogens. Importantly, glial cells have been demonstrated to express NK-1Rs (11, 12, 14, 24-26) and this raises the possibility that SP similarly augments the immune functions of resident CNS cells during infection.
In the present study, we have demonstrated that endogenous SP/NK-1R interactions are required for maximal inflammatory responses to in vivo challenge with N. meningitidis or B. burgdorferi. We have shown that bacterially induced increases in cortical CNS cellularity and/or gliosis, and demyelination are markedly reduced in animals genetically deficient in the expression of the NK-1R. In addition, the absence of endogenous SP/NK-1R interactions significantly attenuates the production of the key inflammatory cytokines, IL-6 and TNF-α, while simultaneously preventing an infection-associated decrease in the immunosuppressive cytokine IL-10. Furthermore, we have verified our results using a specific nonpeptide NK-1R antagonist, L703,606. Previous studies have indicated that such nonpeptide NK-1R antagonists can cross the blood-brain barrier and exert central effects (27-29). We demonstrate that systemic administration of this agent significantly inhibits N. meningitidis and B. burgdorferi-induced CNS gliosis, demyelination, and associated inflammatory cytokine elevations while attenuating concomitant decreases in IL-10 levels. As such, endogenous SP may augment inflammation within the brain in two ways, first by elevating levels of inflammatory mediators, and second by lowering levels of a cytokine that are thought to contribute to the immunoquiescent environment of the healthy CNS (30, 31).
The present findings are consistent with previous studies showing that SP/NK-1R interactions are required for the maintenance of chronic EAE-associated inflammation (32). In addition, SP antagonists have been found to reduce in vivo CNS inflammation elicited by Trypanosoma brucei (33) and attenuate astrocyte activation in a murine model of post-treatment reactive encephalopathy that mimics late stage diminazene-treated African Trypanosomiasis (34). In contrast, the authors of the latter study documented an increased neuroinflammatory response in NK-1R-deficient animals (35). Furthermore, decreased levels of SP and its receptor were observed in postmortem Parkinson’s disease patients and some reported neuroprotective effects of SP appear to contradict a role for SP in augmenting neuroinflammation (36). However, it is important to note that the increased neuroinflammatory response in NK-1R-deficient animals observed in the post-treatment reactive encephalopathy model was found to be directly attributable to the actions of diminazene (37), and it is not clear whether the pathogenesis of bacterial CNS infections bears any resemblance to a non-infectious neurodegenerative disease, such as Parkinson’s disease.
Although it is highly likely that SP augments the inflammatory responses of infiltrating leukocytes due to its documented effects on macrophages and dendritic cells (8, 10, 11, 17, 19, 20), the acknowledged role of resident glia in CNS inflammation and the observed NK-1R-dependent astrogliosis has led us to investigate the direct effects of this neuropeptide on glial cell responses to clinically relevant bacterial pathogens. In this study, we have established that SP can significantly augment the production of IL-6 and TNF-α by isolated cultures of microglia and astrocytes in an NK-1R-dependent manner following challenge with N. meningitidis or B. burgdorferi. Interestingly, while SP augments inflammatory cytokine production by astrocytes following interaction with viable B. burgdorferi, this exacerbation was not seen with B. burgdorferi lysates and the reasons for this difference are presently unclear.
The present study is in agreement with previous work demonstrating that SP can exacerbate the immune responses of activated glial cells. Although some evidence exists that SP is a full and sufficient stimulus to induce microglial and astrocyte responses (38-45), several studies indicate that SP can only augment glial responses in a similar manner to that seen in macrophages. For example, SP was reported to be insufficient to induce IL-1 production by murine microglia, but synergistically augments LPS-induced IL-1 production (39). In addition, stereotaxic injection of SP into the brainstem has been reported to increase IFN-γ-mediated MHC class II up-regulation in parenchymal microglia (46). Furthermore, SP has been reported to augment LPS-induced production of TNF-α by astrocytes (25) and can potentiate the ability of IL-1β to induce the production of IL-6 and PGE2 by these cells (47). Finally, the present findings are in agreement with our recent demonstration that SP synergistically augments prostanoid synthesis by murine microglia in response to suboptimal doses of B. burgdorferi (19).
Although exogenous SP significantly augments glial cell responses to bacterial pathogens, it must be noted that these in vitro effects were less marked than the reductions in the in vivo inflammatory responses observed in the absence and/or reduction of SP/NK-1R interactions. This discrepancy may result, in part, from a direct effect of SP on infiltrating leukocytes, such as macrophages. This contention is supported by the apparent need for endogenous SP/NK-1R interactions in the reductions in IL-10 levels observed following in vivo infection, while no significant effect of SP was observed on in vitro IL-10 production by bacterially challenged microglia or astrocytes. However, it is also possible that our in vitro experiments underestimate the importance of SP, as such studies fail to model the chronic effects of this neuropeptide on glial cell function. Alternatively, the use of isolated cultures may negate the cumulative effect that SP could have on microglia and astrocyte responses, and the cross-talk that is likely to occur between these cell types and/or infiltrating leukocytes in vivo. These issues are the subjects of ongoing investigations by our laboratory.
Taken together, the present studies indicate that SP is an important component in the generation of bacterially induced CNS inflammation. The ability of SP to augment inflammatory responses in the CNS has important ramifications for the development of protective immune responses or progressive CNS damage. As shown in our proposed model in Fig. 5, SP could contribute to CNS inflammation that is disproportionate to the stimulus. Such an effect might explain previous observations in microbe-initiated CNS diseases, such as Lyme neuroborreliosis, in which robust inflammation occurs in the presence of relatively low numbers of spirochetes (48). Although future studies will be required to determine whether the net result of such exacerbation is helpful or harmful to the host, the present observation that prevention of SP/NK-1R interactions fails to significantly elevate the number of viable bacteria that can be isolated from the CNS would appear to support the latter conclusion. Accordingly, the ability of a systemically administered NK-1R antagonist to attenuate CNS inflammation may represent a novel therapeutic strategy for the treatment of bacterially induced CNS diseases. Such a notion is particularly attractive given the ongoing study of similar agents for the clinical treatment of depression and anxiety (49-51).
FIGURE 5.
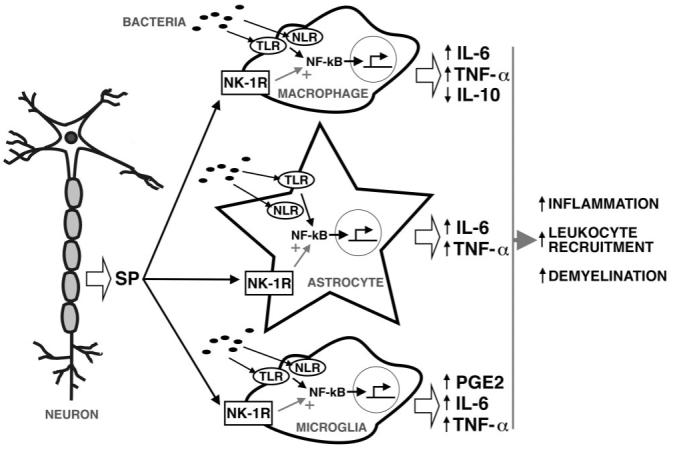
Proposed model in which SP exacerbates the early inflammatory responses of resident CNS cells following bacterial infection. Bacterial motifs are recognized by TLR and NLR pattern recognition receptors expressed by perivascular macrophages, microglia, and astrocytes leading to NF-κB activation and resulting in inflammatory cytokine production via NF-κB activation. SP released by neurons and perhaps activated glia acts on these NK-1R bearing CNS cells to alter NF-κB activation or function. In this manner, SP augments TNF-α and IL-6 production by astrocytes and microglia, and limits IL-10 production by perivascular macrophages and/or infiltrating leukocytes, thereby exacerbating CNS inflammation and damage.
Footnotes
This work was supported by Grants NS050325 and NS057434 to I.M. from the National Institutes of Health.
- SP
- substance P
- NK-1
- neurokinin-1
- i.c.
- intracerebral
- GFAP
- glial fibrillary acidic protein.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Chauhan VS, Marriott I. Bacterial infections of the central nervous system: a critical role for resident glial cells in the initiation and progression of inflammation. Curr. Immunol. Rev. 2007;3:133–143. [Google Scholar]
- 2.Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- 3.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 4.Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 2004;17:942–964. doi: 10.1128/CMR.17.4.942-964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kielian T, Esen N, Bearden ED. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia. 2005;49:567–576. doi: 10.1002/glia.20144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sterka D, Marriott I. Characterization of nucleotide-binding oligomerization domain (NOD) protein expression in primary murine microglia. J. Neuroimmunol. 2006;179:65–75. doi: 10.1016/j.jneuroim.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 7.Sterka D, Rati DM, Marriott I. Functional expression of NOD2, a novel pattern recognition receptor for bacterial motifs, in primary murine astrocytes. Glia. 2006;53:322–330. doi: 10.1002/glia.20286. [DOI] [PubMed] [Google Scholar]
- 8.Marriott I, Bost KL. Substance P. In: Oppenheim JJ, Feldmann M, editors. The Cytokine Reference. Academic Press; London: 2001. [Google Scholar]
- 9.Marriott I, Bost KL. Substance P receptor mediated macrophage responses. Adv. Exp. Med. Biol. 2001;493:247–254. doi: 10.1007/0-306-47611-8_30. [DOI] [PubMed] [Google Scholar]
- 10.Marriott I, Bost KL. Substance P diminishes lipopolysaccharide and interferon-γ-induced TGF-β 1 production by cultured murine macrophages. Cell. Immunol. 1998;183:113–120. doi: 10.1006/cimm.1998.1248. [DOI] [PubMed] [Google Scholar]
- 11.Marriott I. The role of tachykinins in central nervous system inflammatory responses. Front. Biosci. 2004;9:2153–2165. doi: 10.2741/1377. [DOI] [PubMed] [Google Scholar]
- 12.Rasley A, Bost KL, Olson JK, Miller SD, Marriott I. Expression of functional NK-1 receptors in murine microglia. Glia. 2002;37:258–267. doi: 10.1002/glia.10034. [DOI] [PubMed] [Google Scholar]
- 13.Rasley A, Marriott I, Halberstadt CR, Bost KL, Anguita J. Substance P augments Borrelia burgdorferi-induced prostaglandin E2 production by murine microglia. J. Immunol. 2004;172:5707–5713. doi: 10.4049/jimmunol.172.9.5707. [DOI] [PubMed] [Google Scholar]
- 14.Blum AM, Metwali A, Kim-Miller M, Li J, Qadir K, Elliott DE, Lu B, Fabry Z, Gerard N, Weinstock JV. The substance P receptor is necessary for a normal granulomatous response in murine schistosomiasis mansoni. J. Immunol. 1999;162:6080–6085. [PubMed] [Google Scholar]
- 15.Bozic CR, Lu B, Hopken UE, Gerard C, Gerard NP. Neurogenic amplification of immune complex inflammation. Science. 1996;273:1722–1725. doi: 10.1126/science.273.5282.1722. [DOI] [PubMed] [Google Scholar]
- 16.Elsawa SF, Taylor W, Petty CC, Marriott I, Weinstock JV, Bost KL. Reduced CTL response and increased viral burden in substance P receptor-deficient mice infected with murine γ-herpesvirus 68. J. Immunol. 2003;170:2605–2512. doi: 10.4049/jimmunol.170.5.2605. [DOI] [PubMed] [Google Scholar]
- 17.Rasley A, Anguita J, Marriott I. Borrelia burgdorferi induces inflammatory mediator production by murine microglia. J. Neuroimmunol. 2002;130:22–31. doi: 10.1016/s0165-5728(02)00187-x. [DOI] [PubMed] [Google Scholar]
- 18.Taylor WR, Rasley A, Bost KL, Marriott I. Murine γherpesvirus-68 infects microglia and induces high levels of pro-inflammatory cytokine production. J. Neuroimmunol. 2003;136:75–83. doi: 10.1016/s0165-5728(03)00011-0. [DOI] [PubMed] [Google Scholar]
- 19.Rasley A, Bost KL, Marriott I. Murine γherpesvirus-68 elicits robust levels of interleukin-12 p40, but not interleukin-12 p70 production, by murine microglia and astrocytes. J. Neurovirol. 2004;10:171–180. doi: 10.1080/13550280490444119. [DOI] [PubMed] [Google Scholar]
- 20.Rasley A, Tranguch SL, Rati DM, Marriott I. Murine glia express the immunosuppressive cytokine, interleukin-10, following exposure to Borrelia burgdorferi or Neisseria meningitidis. Glia. 2006;53:583–592. doi: 10.1002/glia.20314. [DOI] [PubMed] [Google Scholar]
- 21.Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia. 2003;43:281–291. doi: 10.1002/glia.10256. [DOI] [PubMed] [Google Scholar]
- 22.Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;33:586–592. doi: 10.1161/hs0202.103399. [DOI] [PubMed] [Google Scholar]
- 23.Anguita J, Barthold SW, Persinski R, Hedrick MN, Huy CA, Davis RJ, Flavell RA, Fikrig E. Murine lyme arthritis development mediated by p38-activated protein kinase activity. J. Immunol. 2002;168:6352–6357. doi: 10.4049/jimmunol.168.12.6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Too HP, Marriott DR, Wilkin GP. Preprotachykinin-A and substance P receptor (NK1) gene expression in rat astrocytes in vitro. Neurosci. Lett. 1994;182:185–187. doi: 10.1016/0304-3940(94)90793-5. [DOI] [PubMed] [Google Scholar]
- 25.Luber-Narod J, Kage R, Leeman SE. Substance P enhances the secretion of tumor necrosis factor-α from neuroglial cells stimulated with lipopolysaccharide. J. Immunol. 1994;152:819–824. [PubMed] [Google Scholar]
- 26.Van Ginkel FW, Pascual DW. Recognition of neurokinin 1 receptor (NK1-R): an antibody to a peptide sequence from the third extracellular region binds brain NK1-R. J. Neuroimmunol. 1996;67:49–58. doi: 10.1016/s0165-5728(96)00033-1. [DOI] [PubMed] [Google Scholar]
- 27.Watson JW, Gonsalves SF, Fossa AA, McLean S, Seeger T, Obach S, Andrews PL. The anti-emetic effects of CP-99,994 in the ferret and the dog: role of the NK1 receptor. Br. J. Pharmacol. 1995;115:84–94. doi: 10.1111/j.1476-5381.1995.tb16324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrews PL, Okada F, Woods AJ, Hagiwara H, Kakaimoto S, Toyoda M, Matsuki N. The emetic and anti-emetic effects of the capsaicin analogue resiniferatoxin in Suncus murinus, the house musk shrew. Br. J. Pharmacol. 2000;130:1247–1254. doi: 10.1038/sj.bjp.0703428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsuchiya M, Fujiwara Y, Kanai Y, Mizutani M, Shimada K, Suga O, Ueda S, Watson JW, Nagahisa A. Anti-emetic activity of the novel nonpeptide tachykinin NK1 receptor antagonist ezlopitant (CJ-11,974) against acute and delayed cisplatin-induced emesis in the ferret. Pharmacology. 2002;66:144–152. doi: 10.1159/000063796. [DOI] [PubMed] [Google Scholar]
- 30.Ledeboer A, Breve JJ, Wierinckx A, van der Jagt S, Bristow AF, Leysen JE, Tilders FJ, Van Dam AM. Expression and regulation of interleukin-10 and interleukin-10 receptor in rat astroglial and microglial cells. Eur. J. Neurosci. 2002;16:1175–1185. doi: 10.1046/j.1460-9568.2002.02200.x. [DOI] [PubMed] [Google Scholar]
- 31.Rozenfeld C, Martinez R, Figueiredo RT, Bozza MT, Lima FR, Pires AL, Silva PM, Bonomo A, Lannes-Vieira J, De Souza W, Moura-Neto V. Soluble factors released by Toxoplasma gondii-infected astrocytes down-modulate nitric oxide production by γ interferon-activated microglia and prevent neuronal degeneration. Infect. Immun. 2003;71:2047–2057. doi: 10.1128/IAI.71.4.2047-2057.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reinke EK, Johnson MJ, Ling C, Karman J, Lee J, Weinstock JV, Sandor M, Fabry Z. Substance P receptor mediated maintenance of chronic inflammation in EAE. J. Neuroimmunol. 2006;180:117–125. doi: 10.1016/j.jneuroim.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy PG, Rodgers J, Jennings FW, Murray M, Leeman SE, Burke JM. A substance P antagonist, RP-67,580, ameliorates a mouse meningoencephalitic response to Trypanosoma brucei brucei. Proc. Natl. Acad. Sci. USA. 1997;94:4167–4170. doi: 10.1073/pnas.94.8.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kennedy PG. The pathogenesis and modulation of the post-treatment reactive encephalopathy in a mouse model of Human African Trypanosomiasis. J. Neuroimmunol. 1999;100:36–41. doi: 10.1016/s0165-5728(99)00196-4. [DOI] [PubMed] [Google Scholar]
- 35.Chen LW, Yung KK, Chan YS. Neurokinin peptides and neurokinin receptors as potential therapeutic intervention targets of basal ganglia in the prevention and treatment of Parkinson’s disease. Curr. Drug Targets. 2004;5:197–206. doi: 10.2174/1389450043490596. [DOI] [PubMed] [Google Scholar]
- 36.Ribeiro-da-Silva A, Cuello AC, Henry JL. Neuroanatomical localisation of Substance P in the CNS and sensory neurons. Neuropeptides. 2000;34:256–271. doi: 10.1054/npep.2000.0834. [DOI] [PubMed] [Google Scholar]
- 37.Kennedy PG, Rodgers J, Bradley B, Hunt SP, Gettinby G, Leeman SE, de Felipe C, Murray M. Clinical and neuroinflammatory responses to meningoencephalitis in substance P receptor knockout mice. Brain. 2003;126:1683–1690. doi: 10.1093/brain/awg160. [DOI] [PubMed] [Google Scholar]
- 38.Hartung HP, Heininger K, Schafer B, Toyka KV. Substance P and astrocytes: stimulation of the cyclooxygenase pathway of arachidonic acid metabolism. FASEB J. 1988;2:48–51. doi: 10.1096/fasebj.2.1.2446942. [DOI] [PubMed] [Google Scholar]
- 39.Martin FC, Charles AC, Sanderson MJ, Merrill JE. Substance P stimulates IL-1 production by astrocytes via intracellular calcium. Brain Res. 1992;599:13–18. doi: 10.1016/0006-8993(92)90846-2. [DOI] [PubMed] [Google Scholar]
- 40.Gitter BD, Regoli D, Howbert JJ, Glasebrook AL, Waters DC. Interleukin-6 secretion from human astrocytoma cells induced by substance P. J. Neuroimmunol. 1994;51:101–108. doi: 10.1016/0165-5728(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 41.Giulian D, Corpuz M, Richmond B, Wendt E, Hall ER. Activated microglia are the principal glial source of thromboxane in the central nervous system. Neurochem. Int. 1996;29:65–76. doi: 10.1016/0197-0186(95)00140-9. [DOI] [PubMed] [Google Scholar]
- 42.Lieb K, Fiebich BL, Berger M, Bauer J, Schulze-Osthoff K. The neuropeptide substance P activates transcription factor NF-κB and κB-dependent gene expression in human astrocytoma cells. J. Immunol. 1997;159:4952–4958. [PubMed] [Google Scholar]
- 43.Maeda K, Nakai M, Maeda S, Kawamata T, Yamaguchi TC, Tanaka C. Possible different mechanism between amyloid-β (25-35)-and substance P-induced chemotaxis of murine microglia. Gerontology. 1997;43:11–15. doi: 10.1159/000213881. [DOI] [PubMed] [Google Scholar]
- 44.Fiebich BL, Schleicher S, Butcher RD, Craig A, Lieb K. The neuropeptide substance P activates p38 mitogen-activated protein kinase resulting in IL-6 expression independently from NF-κB. J. Immunol. 2000;165:5606–5611. doi: 10.4049/jimmunol.165.10.5606. [DOI] [PubMed] [Google Scholar]
- 45.Laurenzi MA, Arcuri C, Rossi R, Marconi P, Bocchini V. Effects of microenvironment on morphology and function of the microglial cell line BV-2. Neurochem. Res. 2001;26:1209–1216. doi: 10.1023/a:1013911205494. [DOI] [PubMed] [Google Scholar]
- 46.McCluskey LP, Lampson LA. Local immune regulation in the central nervous system by substance P vs. glutamate. J. Neuroimmunol. 2001;116:136–146. doi: 10.1016/s0165-5728(01)00295-8. [DOI] [PubMed] [Google Scholar]
- 47.Palma C, Minghetti L, Astolfi M, Ambrosini E, Silberstein FC, Manzini S, Levi G, Aloisi F. Functional characterization of substance P receptors on cultured human spinal cord astrocytes: synergism of substance P with cytokines in inducing interleukin-6 and prostaglandin E2 production. Glia. 1997;21:183–193. doi: 10.1002/(sici)1098-1136(199710)21:2<183::aid-glia2>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 48.Pachner AR, Gelderblom H, Cadavid D. The rhesus model of Lyme neuroborreliosis. Immunol. Rev. 2001;183:186–204. doi: 10.1034/j.1600-065x.2001.1830115.x. [DOI] [PubMed] [Google Scholar]
- 49.Haddjeri N, Blier P. Sustained blockade of neurokinin-1 receptors enhances serotonin neurotransmission. Biol. Psychiatry. 2001;50:191–199. doi: 10.1016/s0006-3223(01)01162-3. [DOI] [PubMed] [Google Scholar]
- 50.Holmes A, Heilig M, Rupniak NM, Steckler T, Griebel G. Neuropeptide systems as novel therapeutic targets for depression and anxiety disorders. Trends Pharmacol. Sci. 2003;24:580–588. doi: 10.1016/j.tips.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 51.Adell A. Antidepressant properties of substance P antagonists: relationship to monoaminergic mechanisms? Curr. Drug Targets CNS Neurol. Disord. 2004;3:113–121. doi: 10.2174/1568007043482516. [DOI] [PubMed] [Google Scholar]