

Abstract
Overexpression of zinc finger E-box binding homeobox transcription factor 1 (ZEB1) in cancer leads to epithelial-to-mesenchymal transition (EMT) and increased metastasis. As opposed to overexpression, we show that mutation of the ZEB1 gene in mice causes a mesenchymal-epithelial transition in gene expression characterized by ectopic expression of epithelial genes such as E-cadherin and loss of expression of mesenchymal genes such as vimentin. And in contrast to rapid proliferation in cancer cells where ZEB1 is overexpressed, this mesenchymal-epithelial transition in mutant mice is associated with diminished proliferation of progenitor cells at sites of development defects including the forming palate, skeleton and CNS. ZEB1 gene dosage-dependent deregulation of epithelial and mesenchymal genes extends to mouse embryonic fibroblasts (MEFs), and mutant MEFs also display diminished replicative capacity in culture, leading to premature senescence. Replicative senescence in MEFs is classically triggered by products of the INK4a gene. However, this INK4a pathway is not activated during senescence of ZEB1 gene mutant MEFs. Instead, there is ectopic expression of two other cell cycle inhibitory cyclin dependent kinase inhibitors, p15INK4b and p21CDKN1a. And, we demonstrated that this ectopic expression of p15INK4b extends in vivo to sites of diminished progenitor cell proliferation and developmental defects in ZEB1 gene null mice.
Introduction
Epithelial-to-mesenchymal transition (EMT) is an important step toward acquisition of a metastatic phenotype in cancer (reviewed in Hay and Zuk, 1995; Thiery, 2003). This transition involves repression of key epithelial genes, highlighted by the cell-cell contact modulator, E-cadherin. Downregulation of E-cadherin, and the resulting release of cell-cell adhesion in tumors, is thought to contribute a motile phenotype that facilitates metastasis. Conversely, mesenchymal genes including vimentin and smooth muscle actin as well as various matrix and matrix degrading enzymes are induced during EMT. The resulting alterations in cell shape, matrix expression and ability to digest and move through different tissues are thought to work in conjunction with loss of cell-cell contact to generate the metastatic phenotype. However, EMT is not simply a cancer-driven process, it is crucial early in embryogenesis for delamination of neural crest from the neural tube, and defining an ectodermal-mesodermal boundary (Hay and Zuk, 1995; Thiery, 2003; Cheung et al., 2005). Later in gestation, EMT is important for proper kidney and heart formation. Further, EMT is also a crucial feature of the pathologic fibrotic response to injury (e.g., wound healing and fibrotic organ diseases) (Hay and Zuk, 1995; Thiery, 2003; Zavadil and Bottinger, 2005). EMT during embryogenesis, in cancer metastasis and in fibrotic responses is thought to be driven by TGF–β family members (reviewed in Zavadil and Bottinger, 2005).
Zinc finger E-box binding homeobox 1 (ZEB1) (also known as ZFHX1A, δEF1, TCF8, and Zfhep) binds a set of E-box-like elements which overlap with those bound by ZEB2/SIP1 and the Snail family (Genetta et al., 1994; Sekido et al., 1994; Postigo and Dean, 2000). Each of these E-box-binding proteins can act as a transcriptional repressor through recruitment of the co-repressor, CtBP (Postigo and Dean, 1999; Grooteclase and Frisch, 2000; Chinnadurai, 2002; Hemavathy et al., 2005). CtBP is a part of a larger complex including polycomb proteins and CoREST that causes epigenetic modification of DNA and histones leading to heterochromatin assembly and thus transcriptional silencing (Ringrose et al., 2004 and refs. therein). A CtBP-CoREST repressor complex is targeted, via interaction with ZEB1, to genes crucial for late-stage pituitary organogenesis (Wang et al 2007).
Overexpression of ZEB1 in cancer is associated with repression of E-cadherin and EMT (Guaita 2002; Eger et al., 2005; Pena et al., Spoelstra et al., 2006; Witta et al., 2006; Peinado et al 2007). Additionally, ZEB1 can also serve as a transcriptional activator, and this seems to be directed at least in part toward mesenchymal genes such as collagens, smooth muscle actin and myosin, vimentin, and genes in the vitamin D signaling pathway which is important in mesenchymal differentiation (Chamberlain and Sanders, 1999; Lazarova et al., 2001; Dillner and Sanders, 2002; Postigo 2003; Postigo et al., 2003; van Grunsven et al., 2006; Nishimura et al., 2006). Heterozygous mutation of the ZEB1 gene leads to impaired smooth muscle actin and myosin expression and TGF-β-dependent smooth muscle cell differentiation following vascular injury (Nishimura et al., 2006). Thus, ZEB1 can contribute to repression of epithelial genes as well as activation of mesenchymal genes.
TGF–β expression by tumors and surrounding stroma drives EMT in cancer, and TGF–β family members are also crucial for EMT during development (Zavadil and Bottinger, 2005). Accordingly, TGF–β represses epithelial genes such as E-cadherin and induces mesenchymal genes such as vimentin. In vivo ZEB1 is important for TGF–β-dependent smooth muscle cell differentiation, and TGF–β-dependent expression of smooth muscle actin and myosin genes (Nishimura et al., 2006). ZEB1 binds activated Smads as well as the histone acetyl transferase p300, which is an essential Smad co-activator, and this binding facilitates assembly of a Smad-p300 complex while leading to dissociation of ZEB1 from its co-repressor, CtBP (Postigo et al., 2003; Postigo, 2003; van Grunsven et al., 2006). Thus, in response to TGF-β and in the presence of activated Smads and p300, ZEB1 is switched from a repressor to a co-activator.
ZEB1 is expressed in proliferating mesenchymal and neural progenitors, and mutation of the ZEB1 gene leads to cleft secondary palate and other craniofacial abnormalities (Takagi et al., 1998). Forming cartilage at these sites appears to be hypoplastic histologically. In addition to craniofacial defects, the mice have skeletal abnormalities including shortened limbs and digits as well as fusion and curvatures in the skeleton and tail. A subset of the embryos exhibited severe CNS defects including failure of neural tube closure at both cranial and caudal ends, and exencephaly (Takagi et al., 1998). A molecular basis for these various defects is unknown.
As opposed to EMT seen when ZEB1 is overexpressed in rapidly proliferating cancer cells, we present evidence here that mutation of the ZEB1 gene causes mesenchymal-epithelial transition in gene expression and diminished proliferation in progenitor cells at sites of developmental defects in mouse embryos. This phenotype extends to mouse embryo fibroblasts (MEFs) derived from mutant mice. These cells ectopically express E-cadherin, which is associated with an abnormal epithelial-like morphology. Additionally, they undergo premature replicative senescence in culture, which is associated with ectopic expression of cell cycle inhibitory cyclin dependent kinase inhibitors (CDKIs), p15INK4b and p21CDKN1a. This ectopic expression of p15INK4b is also seen in vivo at sites of diminished proliferation of progenitor cells (and developmental defects) in ZEB1 gene mutant embryos.
Materials and Methods
Mouse embryonic fibroblast isolation and cell culture
Embryo fibroblasts were isolated from crosses of mice heterozygous for ZEB1 and genotyped as described (Takagi et al.,. 1998). Cells of different genotypes were cultured in DMEM medium with 10% FBS at 10% CO2, and split at 1:3 ratio as soon as confluent. For experiments with TGF-β, one day after splitting, TGF-β1 (BioSource, Camarillo, CA) was added into the cultures at final concentrations of 5 pm, 25 pm, and 75 pm, and RNA was isolated 24 hrs later.
RNA extraction and real-time PCR
RNA was extracted using TRIzol solution (Invitrogen, Carlsbad, CA). cDNA was synthesized using the Invitrogen RT kit according to the manufacturer's protocol (Invitrogen, Carlsbad, CA). SYBR Green real-time quantitative PCR was performed in Mx3000P Real-Time PCR System (Stratagene, Cedar Creek, TX) according to the manufacturer's instructions. The RT-PCR primers and annealing temperature are shown in Supplementary Table 1. Three independent samples were analyzed for each condition and/or cell type, and each sample was compared in at least 3 independent RT-PCR amplifications.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were based on the UpState protocol (http://www.upstate.com/misc/protocol) using formaldehyde to crosslink genomic DNA. Polyclonal antiserum for ZEB1 (Darling et al., 2003), histone 3 and histone 4 (Santa Cruz Biotechnology, Inc. Santa Cruz, CA) were used for immunoprecipitation. Equal amounts of anti-IgG or pre-immune serum were used as controls. ChIP PCR reactions were similar to those described above for real-time PCR using primer sets (Supplementary Table 2) to amplify promoter sequences of E-cadherin and GAPDH genes.
Immunohistochemistry
Mouse embryos at E15.5 were fixed, embedded, and sectioned at 5 μm. The primary antibody dilution for ZEB1, vimentin (Santa Cruz Biotechnology, Inc. Santa Cruz, CA), GFAP (Chemicon, Temecula, CA), E-cadherin (BD-Pharmingen, San Jose, CA) was 1:100, 1:10, 1:10, and 1:50, respectively, whereas the secondary antibody dilution was 1:300 for both anti-rabbit IgG conjugated with Alex Flor®-488 (Molecular Probes, Eugene, Oregon) and anti-mouse IgG conjugated with Cy3 (Sigma, St. Louis, MO). The slides were mounted with coverslips using anti-fade medium Permount (Fisher) and viewed using an Olympus confocal microscope.
Analysis of cell proliferation in vivo
Two hours before collection of E15.5 embryos, mothers received an intraperitioneal injection of 40 mg/kg of 5′-bromodeoxyuridine (BrdU) in PBS. Embryos were fixed in 10% buffered formalin, embedded in paraffin, and sectioned at 5 μm. Sections were incubated with 0.1% Tween 20, 4% goat (for ZEB1 antibody; Darling et al., 2003) or sheep serum (for BrdU antibody), and 2% bovine serum albumin (BSA) (Sigma, St. Louis, MO) for 1 hr. Polyclonal primary antibodies against ZEB1 and BrdU (raised in rabbit and mouse, respectively) were applied to the sections at 1:50, and incubated at 4°C overnight. Slides were then incubated at 1:300 dilution either with anti-rabbit IgG conjugated with Alex Flor®-488 (Molecular Probes, Eugene, Oregon) or Cy3 (Sigma, St. Louis, MO) for 1 hr. The slides were viewed with an Olympus confocal microscope.
Cell proliferation in the brain was analyzed as described (Molofsky et al., 2006). For these experiments, five micron sections from corresponding regions of wild-type and null liter mate embryos were immunostained for BrdU and observed using a Zeis LSM 510 confocal laser scanning microscope. One micron optical sections were incorporated into a Z-stack using a 40× objective and LSM510 software was used to create a 3-D image. Three adjacent sections were analyzed from each embryo. The percentage of BrdU positive cells within similar fields was determined. Similar results were also evident by simply counting the number of BrdU-positive cells in the field. Averages from two wild-type and three null embryos (and three adjacent sections from each embryo) are presented.
Cellular senescence assays
Senescent β-galactosidase activity was analyzed using the x-gal-based Sigma Cell Senescence Staining Kit using the manufacturer's protocol (Sigma, St. Louis, MO). Stained cells were photographed, and randomly selected fields were counted for percentage of positive cells.
Results
Mutation of ZEB1 leads to ectopic expression of E-cadherin and loss of vimentin in mesenchymal progenitors
Overexpresion of ZEB1 in cancer is associated with EMT. Thus, we wondered whether ZEB1 has a role normally in regulation of epithelial and mesenchymal genes during development, and whether such regulation might be linked to the developmental defects seen in ZEB1 gene null mice. A variety of developmental defects appear in ZEB1 gene null embryos late in gestation. Therefore, we began by comparing the pattern of ZEB1 to that of the classic epithelial and mesenchymal markers E-cadherin and vimentin, respectively, in embryonic mice late in gestation. We found little or no overlap of ZEB1 and E-cadherin. Specifically, ZEB1 was found in nasal mesenchyme and mesenchyme of the forming palate, while E-cadherin was confined to the epithelial borders (Fig. 1A-B). E-cadherin was found in the skin (Fig. 2C), whereas ZEB1 was evident in mesenchyme and forming muscle and in perichrondrial mesenchyme surrounding forming cartilage (Fig. 2A); E-cadherin was absent in the forming muscle and mesenchyme (Fig. 2C). In the tongue, ZEB1 was present in the forming muscles, whereas E-cadherin was confined to the epithelial border (Fig. 1B; Supplemental Fig. 1). ZEB1 was also evident in neuroectodermally-derived progenitor cells such as those in the ventricular zone of the lateral ventricles (Fig. 4A), the olfactory bulb (Supplemental Fig. 3) (as well as other regions of the brain), the neural retina, optic nerve and the retinal pigmented epithelium (Fig. 3A); E-cadherin was absent from all of these sites (Fig. 3B, 4E, and Supplemental Fig. 3).
Fig 1.
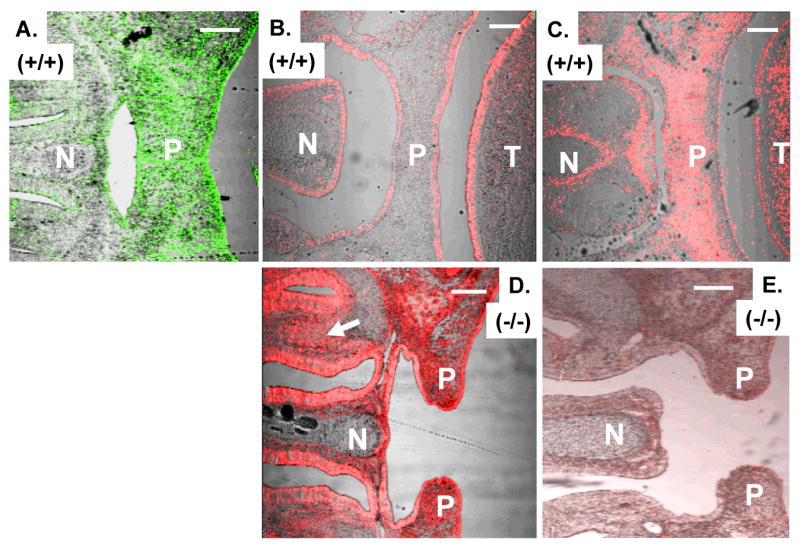
E-cadherin becomes ectopically expressed whereas vimentin is diminished in palate and nasal mesenchyme in ZEB1 gene mutant mice. A. ZEB1 expression in mesenchyme in the palate (P) and surrounding the developing nasal cartilage (N). B. E-cadherin is expressed on the nasal (N), palatal (P) and tongue (T) epithelium. C. Expression of vimentin in palatal and nasal mesenchyme. D. E-cadherin becomes ectopically expressed on nasal and palatal mesenchyme in ZEB1 gene null mice. Note the failure of palate closure. The arrow indicates mesenchyme in the nasal region. E. Loss of vimentin expression in the palatal and nasal mesenchyme of ZEB1 gene null mice. Immunostaining of mice at E16.5 is shown. Bars are 100 μm.
Fig. 2.
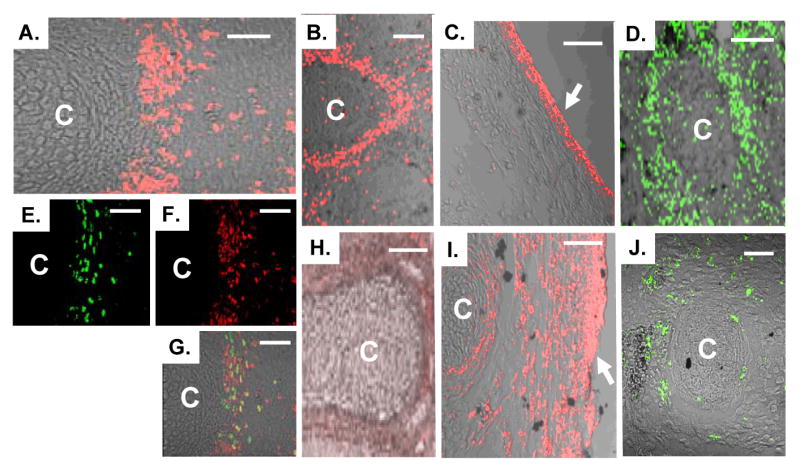
Mutation of the ZEB1 gene leads to ectopic expression of E-cadherin, loss of vimentin, and defective proliferation in the perichondrial region of forming cartilage (C). A. Immunostaining showing expression of ZEB1 in the perichondrium. B. Expression of vimentin in the perichondrium. C. E-cadherin is expressed in the skin but not in underlying mesenchymal cells. D. BrdU incorporation into the perichondrium. E and F. Double immunolabeling for ZEB1 and BrdU in the perichondrium. G. Overlay of panels E and F. H. Diminished immunostaining for vimentin in the perichondrium of ZEB1 gene null mice. I. Ectopic expression of E-cadherin in the perichondrium of ZEB1 gene null mice. J. Loss of BrdU incorporation into the perichondrium of ZEB1 gene null mice. Immunostaining of mice at E15.5 is shown. Bars are 50 μm.
Fig. 4.
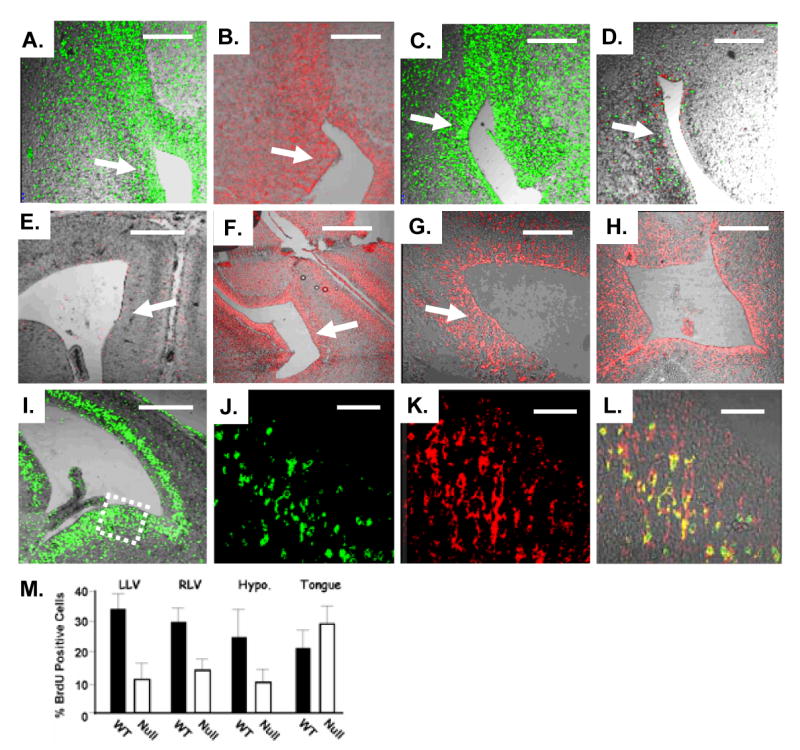
Mutation of the ZEB1 gene leads to ectopic expression of E-cadherin, loss of vimentin and GFAP expression, and decreased proliferation in the ventricular zone of the brain. A. ZEB1 is expressed in the ventricular zone (arrow) of the lateral ventricle. B and C. Expression of vimentin and GFAP, respectively, in the ventricular zone. D. Expression of both vimentin (red) and GFAP (green) is lost in the ventricular zone of ZEB1 gene mutant mice. E. E-cadherin is not expressed in the lateral ventricle in wild-type mice. F-G. E-cadherin becomes ectopically expressed in the ventricular zone of the lateral ventricle in ZEB1 gene null mice. H. E-cadherin is expressed in the third ventricle in ZEB1 gene null mice. I. BrdU incorporation in the ventricular zone of the lateral ventricle. J and K. Double immunolabeling of the boxed region in panel I for BrdU and ZEB1, respectively. L. Overlay of panels J and K. Immunostaining at E15.5 is shown. M. Quantitation of BrdU incorporation into the ventricular zone of the left lateral ventricle (LLV), right lateral ventricle (RLV), hypothalamus (Hypo.), and as a control the tongue. A representative view of areas counted is shown in Supplemental Fig. 3. Bars are 100 μm in panels E, F and I; 50 μm in panels A-D, G and H; 25 μm in J-K.
Fig. 3.
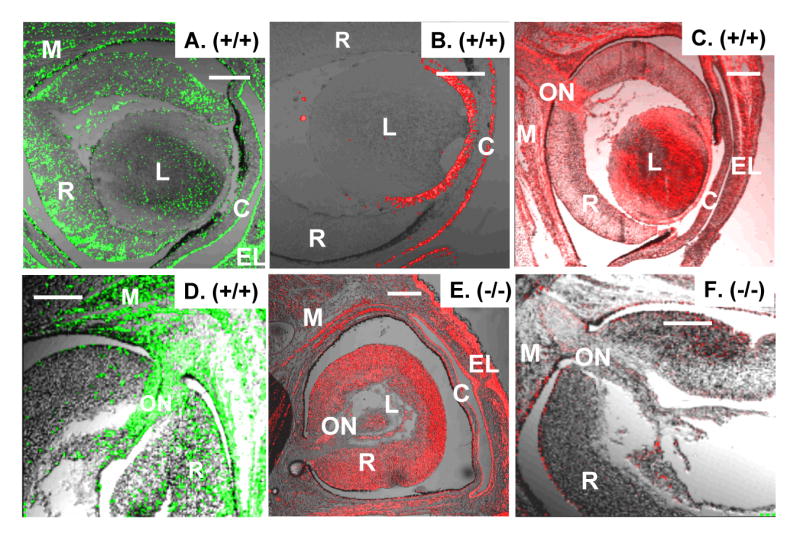
Ectopic E-cadherin expression and diminished vimentin and GFAP expression in the embryonic eye of ZEB1 gene null mice. A. Expression of ZEB1 in the retina (R), forming eye muscles (M). L, lens; C, cornea. B. E-cadherin expression is confined to the epithelium of the lens and cornea. C. Vimentin is expressed in the retina, lens, optic nerve (ON), and forming eyes muscles. D. GFAP is evident in the retina, optic nerve, and forming eye muscles. E. Ectopic expression of E-cadherin in the retina, optic nerve and forming eye muscle in ZEB1 null mice. F. Double immunostaining for vimentin (red) and GFAP (green) show loss of expression of both proteins in the retina, optic nerve and forming eye muscle in ZEB1 gene null mice. Immunostaining of mice at E16.5 is shown. Bars are 100 μm.
By contrast, we found that the patterns of expression of vimentin and ZEB1 were similar; both were expressed in mesenchymal cells of the palate (Fig. 1A and C) and in perichondrium surrounding forming cartilage (Fig. 2A-B), on forming muscles in the tongue (Supplemental Fig. 1) and surrounding the eye (Fig. 3A and C), in neural retina (Fig. 3A and C), optic nerve (Fig. 3C), and the brain (Fig. 4A-B; Supplemental Fig. 3). These results show that ZEB1 is expressed in a variety of mesenchymal and neuroectodermally-derived progenitors along with vimentin, and that it does not overlap with E-cadherin.
Next, we examined ZEB1 gene null mice to determine whether there were changes in E-cadherin or vimentin expression. While expression of E-cadherin was maintained on the epithelial border of the forming palate in ZEB1 gene null mice at E15.5, it became ectopically expressed on palatal mesenchyme (Fig. 1D; Supplemental Fig. 2). E-cadherin was also maintained on the epithelium of the forming nasal region in the null mice, but as in the palate, it became ectopically expressed on the surrounding nasal mesenchyme (Fig. 1D). Again, E-cadherin expression was maintained on the skin epithelium, but it became ectopically expressed on perichondrial mesenchyme surrounding developing cartilage (Fig. 2I), which proliferates to add cells to the forming cartilage. Also, E-cadherin became ectopically expressed on the forming muscles surrounding the eye (Fig. 3E). But interestingly, E-cadherin expression remained confined to the epithelial border of the tongue; it did not become ectopically expressed on forming tongue muscles, despite the fact that ZEB1 is normally expressed on these forming muscles (Supplemental Fig. 2).
We reasoned that if this ectopic expression of E-cadherin in ZEB1 gene null mice reflects a mesenchymal (neuroectodermal)-to-epithelial transition in gene expression, then one would also expect to see diminished or contracted expression of a classic mesenchymal/neuroectodermal marker such as vimentin. Indeed, vimentin expression diminished in the palatal mesenchyme (Fig. 1E) and perichondrium in ZEB1 gene null mice (Fig. 2H).
Mutation of ZEB1 leads to a proliferative defect in mesenchymal progenitor cells
As noted above, a previous report suggested histologically that at least some sites of forming cartilage are hyoplastic in ZEB1 gene null mice. And, we noted that ZEB1 was expressed in populations of mesenchymal progenitors which are known to be proliferative. Therefore, we injected pregnant mice with BrdU to assess cell proliferation, and harvested embryos 2 hours later. Liter mate embryos were then compared for BrdU incorporation by immunostaining. As expected, we found significant overlap between ZEB1 and BrdU in the perichodrium (Fig. 2 E-G). However, BrdU incorporation in these cells was dramatically reduced in null mice (Fig. 2D, J), indicating a proliferative defect in mesenchymal progenitors surrounding forming cartilage.
Mutation of the ZEB1 gene leads to ectopic expression of E-cadherin and loss of vimentin and GFAP in the brain and eye
At E16.5, E-cadherin is expressed on the epithelium of the eye lid, the cornea, and the lens (Fig. 3B). In the ZEB1 gene null mice, E-cadherin expression was maintained at these sites, but it became ectopically expressed on the neural retina, optic nerve, and forming eye muscles (Fig. 3E).
E-cadherin was not detected in the embryonic brain late in gestation (E15.5 to E17.5) (Fig. 4E; Supplemental Fig. 3). However in the null mice, it appeared on cells in the ventricular zone of the lateral ventricles and in the olfactory bulb, which is populated by progenitors from the ventricular zone (Curtis et al., 2007 and refs. therein) (Fig. 4F-G; Supplemental Fig. 3). Additionally, E-cadherin also appeared in proliferative regions of the third ventricle, the telencephalic vesicle, the thalamus and the hypothalamus (Fig. 4H; results not shown). This ectopic expression of E-cadherin was confined to sites in the brain which normally express ZEB1, and which are known to be sites of proliferating progenitor cells late in gestation.
In contrast to ectopic expression of E-cadherin, vimentin expression in the ZEB1 gene null mice was diminished in the retina, optic nerve, and forming eye muscles (Fig. 3C and F). Additionally, vimentin expression was also diminished in the ventricular zone of the lateral ventricles (Fig. 4B and D), in the olfactory bulb (Supplemental Fig. 3) and in other regions of the brain where ZEB1 is normally expressed and where there was ectopic expression of E-cadherin. As with E-cadherin, vimentin expression in the tongue did not change with ZEB1 gene mutation (Supplemental Fig. 2).
While vimentin is expressed in neuroectodermally-derived progenitor cells, glial fibrillary acidic protein (GFAP) is expressed and organized into filaments as these progenitor cells differentiate into glia (reviewed in Messing and Brenner, 2003). As with vimentin, we found diminished GFAP immunostaining in the brain, neural retina and optic nerve of ZEB1 gene null mice (Fig. 3D and F; 4C-D).
ZEB1 mutation leads to a proliferative defect in embryonic brain progenitor cells
A subset of ZEB1 gene null embryos show severe CNS defects with failure of neural tube closure at both the cranial and caudal ends and exencephaly. As with proliferating mesenchymal cells, ZEB1 expression overlapped significantly with BrdU incorporation in the ventricular zone of the lateral ventricles and in other proliferative regions of the brain at E15.5 (Fig. 4I-L; results not shown). Low power views of head sections of embryos immunostained for BrdU suggested that proliferation in the ventricular zone of the lateral ventricles and other regions of the brain was diminished in ZEB1 gene null mice (Supplemental Fig. 4). Therefore, we counted the percentage of BrdU-positive cells at various sites in the brain (see Material and Methods). Corresponding regions from three null and two wild-type liter mates were analyzed. Three adjacent sections were counted for each embryo. A significant decrease in BrdU incorporation was seen in each of the sites in the null embryos (Fig. 4M). This diminished proliferation in progenitors in the ventricular zone of the brain was similar to or greater than that seen with mutation of Bmi1 (Molofsky et al 2005). As a control, no significant difference between wild type and null embryos was seen in BrdU incorporation in the tongue (Fig. 4M; Supplemental Fig. 4).
Ectopic expression of E-cadherin in embryonic fibroblasts from ZEB1 gene mutant mice
We isolated mouse embryo fibroblasts (MEFs) from wild-type, heterozygous and null liter mates, and asked whether ZEB1 gene mutation leads to ectopic E-cadherin expression on the cells. While E-cadherin was not detected by immunostaining on the wild-type cells, it was evident on the mutant cells (Fig. 5). Additionally, there was a ZEB1 gene dosage-dependent induction of E-cadherin mRNA in the cells (Fig. 6B and 8B). Interestingly, we also observed MEFs with abnormal epithelial-like morphology in both the null and heterozygous cell populations (Fig. 5; results not shown). These cells constituted more than 25% of the culture. It is of note, however, that E-cadherin immunostaining was evident uniformly throughout the null cell population; it was not confined to cells exhibiting epithelial-like morphology. These results link ectopic expression of E-cadherin in the mutant MEFs to an abnormal epithelial-like morphology.
Fig. 5.
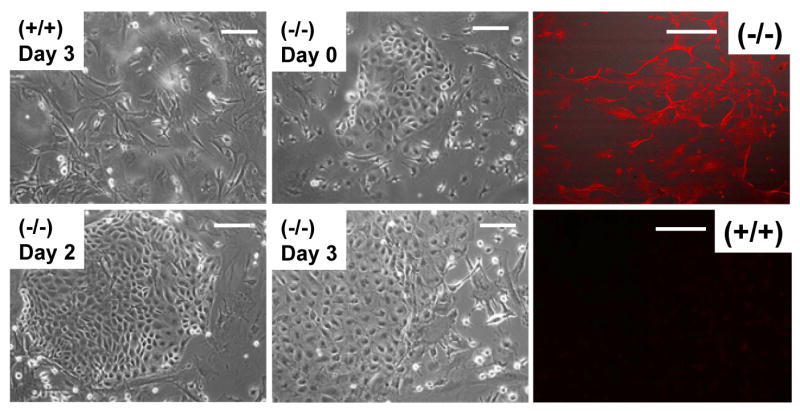
E-cadherin is ectopically expressed on embryo fibroblasts from ZEB1 gene mutant mice, and the cells display an abnormal epithelial-like morphology. Left-hand panels show light micrographs of cells at different time points after isolation. Note the increase in cells with epithelial-like morphology. Such clusters of epithelial-like cells were not evident in the wild-type population. Right-hand panels show immunostaining for E-cadherin. Bars are 25 μm.
Fig. 6.

Effect of ZEB1 gene mutation on expression of ZEB2/ SIP1 and Snail mRNA, and TGF-β-dependent induction of vimentin and repression of E-cadherin mRNA. A. Real time PCR was used to assess the effect of ZEB1 gene mutation on expression of other E-box-binding repressors (ZEB2/SIP1, Snail1 or Snail2) in MEFs. Results were normalized to β-actin and GAPDH mRNA, with similar results. B. TGF–β is unable to repress E-cadherin mRNA levels in ZEB1 gene null MEFs. Concentrations of TGF-β are shown. No E-cadherin mRNA was detected in wild-type cells, see Fig. 8B below. C. Basal vimentin mRNA expression is unaffected by ZEB1 gene mutation, but TGF–β induction is lost in ZEB1 gene null MEFs. D. Basal plasminogen activator inhibitor-1 (PAI-1) mRNA is unaffected by ZEB1 gene mutation, and it remains inducible by TGF–β in ZEB1 gene null MEFs.
Fig. 8.
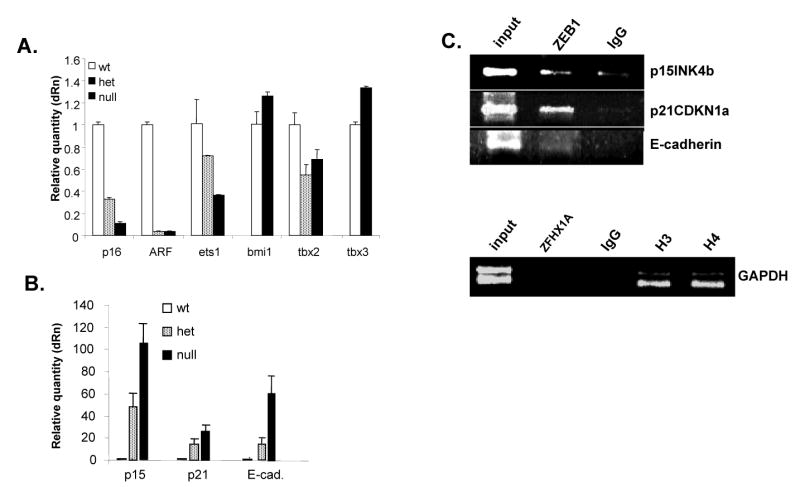
Mutation of the ZEB1 gene does not trigger the classic INK4A replicative senescence pathway in MEFs, instead it is associated with induction of p15INK4b and p21CDKN1a. A. Real-time PCR was used to compare mRNA expression from the INK4a locus (p16INK4a and ARF), and genes know to regulate the INK4a locus, in proliferating wild-type MEFs (P3) and senescent ZEB1 gene heterozygous (P5) and null (P2) cells. B. p15INK4b, p21CDKN1A and E-cadherin mRNAs are induced in a gene dosage-dependent fashion in ZEB1 gene mutant MEFs. Real-time PCR results using the same samples in panel A are shown. C. Chromatin immunoprecipitation assays. “input” indicates starting chromatin used for the immunoprecipitations; “IgG” indicates pre-immune serum. Histone H3 and H4 are positive controls for the GAPDH promoter.
We wondered whether the ectopic expression of E-cadherin seen with ZEB1 gene mutation may result indirectly from the downregulation of one of the other E-box-binding repressors. Therefore, we compared expression of ZEB2/SIP1, Snail1 and Snail2 mRNA levels in wild-type and ZEB1 gene mutant MEFs. No significant change was seen in Snail 1 or 2 mRNA levels in heterozygous or null MEFs, and expression of ZEB2/SIP1 was induced with ZEB1 gene mutation (Fig. 6A). This finding of ZEB2/SIP1 induction is consistent with a recent report showing increased expression of ZEB2/SIP1 in smooth muscle cells of ZEB1 gene mutant mice (Nishimura et al., 2006). Therefore, ectopic expression of E-cadherin in ZEB1 gene mutant MEFs is not an indirect result of downregulation of ZEB2/SIP1, Snail1 or Snail2. Indeed, E-cadherin is expressed in the cells despite the fact that ZEB2/SIP1 mRNA is upregulated (and Snail1/2 mRNA is unchanged).
Next, we performed chromatin immunoprecipitation (ChIP) assays to determine whether ZEB1 is bound to the E-cadherin gene promoter in vivo. We found that ZEB1 was present at the promoter in vivo (Fig. 8C). As a negative control, we found that ZEB1 was not present at the GAPDH promoter (Fig. 8C). Taken together, these results are consistent with the idea that ZEB1 is directly repressing E-cadherin expression by binding to its promoter.
ZEB1 and TGF-β induction of vimentin and repression of E-cadherin
TGF–β drives EMT by inducing mesenchymal genes such as vimentin, and repressing epithelial genes such as E-cadherin. Given the previously documented linkage between ZEB1 and TGF–β signaling discussed above, we wondered whether the effects of ZEB1 on repression of E-cadherin and induction of vimentin might be related to TGF–β signaling. Initially, we asked whether basal vimentin mRNA expression was affected by ZEB1 gene mutation in MEFs. However, we found no significant difference in the level of vimentin mRNA in wild-type vs. null MEFs (Fig. 6C). Therefore, we asked whether ZEB1 might be important for TGF-β-mediated induction of vimentin. Indeed, we found that vimentin mRNA expression was not inducible by TGF–β in the null MEFs (Fig. 6C). However, ZEB1 gene mutation did not lead to a general block in TGF–β signaling because we found that TGF–β-mediated induction of plasminogen activator inhibitor-1 (PAI-1) mRNA was not diminished in null MEFs (Fig. 6D). We then conclude that ZEB1 is required for TGF–β-mediated induction of vimentin in the MEFs.
Our results above show that ZEB1 is required to prevent ectopic E-cadherin expression in MEFs. We then wondered whether TGF–β would be able to repress E-cadherin expression in the null MEFs. Indeed, we found that TGF–β was unable to repress E-cadherin mRNA in the absence of ZEB1 (Fig. 6B). ZEB1 expression is known to be induced by TGF-β (Nishimura et al., 2006). Thus taken together, the results raise the possibility that TGF-β may repress E-cadherin through induction of ZEB1.
Mutation of the ZEB1 gene leads to INK4a-independent premature replicative senescence in MEFs
We then wondered whether the proliferative defects in ZEB1 gene null mice that we observed in ventricular zone and mesenchymal progenitor cells might also be reflected in MEFs from mutant mice. Therefore, ZEB1 gene mutant and wild-type liter mate-matched MEFs were compared for proliferation. We found that ZEB1 gene null cells arrested by P2, whereas heterozygous cells stopped proliferating by P4 (Fig. 7A). Wild-type cells continued proliferating beyond P10 (Fig. 7A; results not shown). We noticed that the arrested cells adopted a flatten morphology and they remained non-proliferative but viable for months in culture (Fig. 7B; results not shown). Because these are properties of senescence, we stained the cells for senescent β-galactosidase activity. We found that essentially all of the arrested heterozygous and null cells expressed senescent β-galactosidase, whereas only ∼30% of wild-type cells were positive at P9 (Fig. 7C). We then concluded that the mutant MEFs undergo premature replicative senescence in a ZEB1 gene dosage-dependent fashion.
Fig. 7.
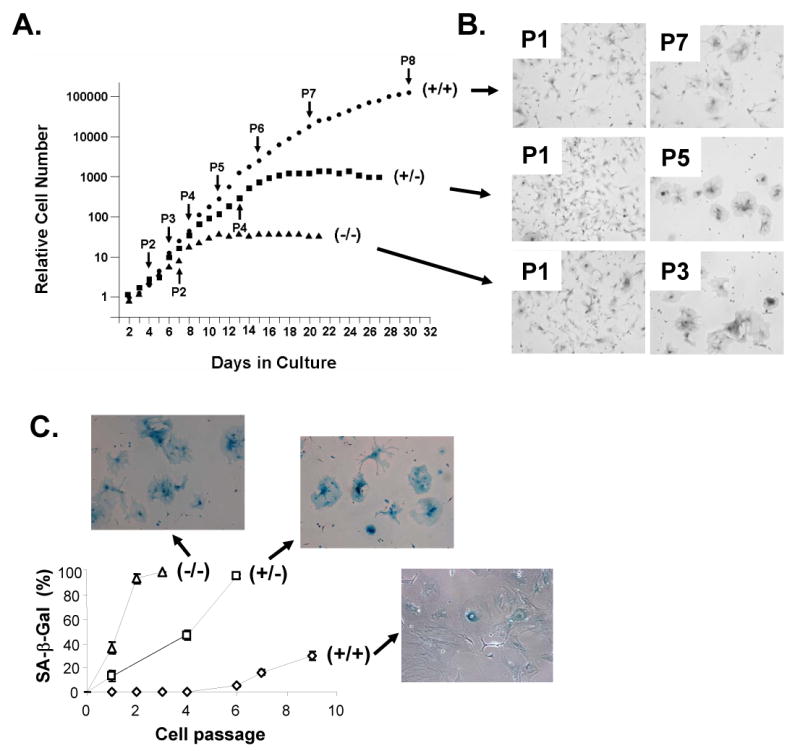
MEFs from ZEB1 gene mutant mice undergo gene dosage-dependent senescence. A. Effect of ZEB1 gene mutation on MEF proliferation. B. Micrographs of cells at early vs. later passage numbers. Note appearance of large flat senescent-like cells associated with growth arrest. C. Arrested ZEB1 gene mutant cells express senescent β-galactosidase (SA-β Gal).
Next, we asked whether the classic INK4a senescence pathway was being activated in ZEB1 gene mutant MEFs. Mutations in the transcriptional repressor, Bmi1, have been shown to trigger proliferative defects and senescence in neural progenitors of the ventricular zone (Valk-Lingbeek et al., 2004). Bmi1 negatively regulates INK4a, and in the absence of Bmi1 this locus expresses the CDKI, p16INK4a, and the p53 regulator, ARF (reviewed in Gil and Peters, 2006). Induction of INK4a in Bmi1 null embryo fibroblasts triggers senescence of MEFs (Valk-Lingbeek et al., 2004), and a cross of the Bmi1 null mice to INK4a null mice prevents senescence of the ventricular zone progenitors in vivo (Jacobs et al., 1999). It is of note that normal replicative senescence of wild-type MEFs results from time-dependent induction of the INK4a locus in culture [usually leading to senescence around passage (P) 10-12] (Lowe and Sherr 2003; Gil and Peters 2006). Mutation of Bmi1 accelerates this INK4a activation, leading to premature replicative senescence.
To investigate whether activation of the INK4a pathway was responsible for premature senescence of the ZEB1 gene mutant MEFs, we used real-time PCR to analyze expression of mRNAs from the INK4a locus, and from genes known to regulate this locus including Bmi1 (repressor), ets-1 (activator), TBX2 (repressor) and TBX3 (repressor) (Jacobs et al., 1999; Lessard and Sauvageau, 2003; Lingbeek et al., 2002; Ohtani et al., 2001). We failed to find upregulation of p16INK4a or ARF (or any of the INK4a regulators) in senescent ZEB1 gene mutant cells (Fig. 8A). Instead, expression of both mRNAs was downregulated in the null and heterozygous cells compared to wild-type cells. It is of note that this downregulation of p16INK4a and ARF mRNA correlates with ZEB1 gene dosage-dependent downregulation of the INK4a inducer, ets-1 (Fig. 8A). As a positive control, we found that p16INK4A expression was induced with passage number in the wild-type MEFs, as reported previously by a number of groups (reviewed in Gil and Peters 2006; Lowe and Sherr 2003; results not shown). We then concluded that the premature replicative senescence seen in the ZEB1 gene mutant MEFs is not the result of activation of the classic INK4a locus.
p15INK4b and p21CDKN1a are ectopically expressed in ZEB1 gene mutant MEFs
TGF-β treatment classically induces growth arrest, but this does not involve induction of INK4a; instead, two other CDKIs, p21CDKN1A and p15INK4b, are induced to trigger the growth arrest (Reynisdottir et al., 1995). Because of the known linkage of ZEB1 to TGF–β signaling, we asked whether these CDKIs might be induced in the ZEB1 gene mutant MEFs. Indeed, we found a gene dosage-dependent induction of both mRNAs, with induction of p15INK4b being the most dramatic (Fig. 8B). These results demonstrate that ZEB1 is required to prevent ectopic expression of these CDKIs in MEFs.
Next, we wondered if ZEB1 acts directly on the p21CDKN1A and p15INK4b gene promoters. Therefore, we performed chromatin immunoprecipitation assays with both promoter regions. Indeed, we found that ZEB1 was bound to both promoters, but no interaction was seen with the control GAPDH promoter (Fig. 8C). These results are consistent with the notion that ZEB1 is repressing these CDKIs directly by binding to their promoters in vivo.
p15INK4b is ectopically expressed in mesenchymal and ventricular zone progenitor cells in ZEB1 gene null embryos
p15INK4b was dramatically induced in ZEB1 gene mutant MEFs (Fig. 8B), and expression of p15INK4b is known to block cell proliferation (Reynisdottir et al., 1995). Therefore, we wondered whether p15INK4b might become ectopically expressed in ZEB1 gene null mice at sites of diminished proliferation. We did not detect p15INK4a expression in mesenchymal cells or in the ventricular zone in wild-type mice (Fig. 9A-B, E). Indeed, the developing skin was the only tissue where we observed significant expression (Supplemental Fig. 5). However as in the MEFs, p15INK4b became ectopically expressed in the perichondrium in ZEB1 gene null mice (Fig. 9C-D; Supplemental Fig. 5), and these cells showed diminished proliferation (Fig. 2D, J).
Fig. 9.

p15INK4b is ectopically expressed in the perichondrium, on forming cartilage, and in the ventricular zone of the lateral ventricle in ZEB1 gene null mice. A and C. Immunostaining for p15INK4b on forming cartilage. B and D. Overlay of the immunostaining in panels A and C with a Nomarski image. E and F. Immunostaining for p15INK4b in the ventricular zone of the lateral ventricle. G and H. Immunostaining for BrdU and p15INK4b, respectively, in the ventricular zone of the lateral ventricle. I. Overlay of panels G and H. Immunostaining of E15.5 embryos is shown. The bar is 50 μm.
p15INK4b expression was also ectopically expressed in the ventricular zone of ZEB1 gene null mice (Fig. 9F). While there was overlap between p15INK4b and BrdU incorporation, it is of note that there were a number of additional p15INK4b-positive cells in this region that were BrdU-negative (Fig. 9G-I), and there was overall diminished proliferation of cells in this region. These results are consistent with the notion that ectopic expression of p15INK4b in null mice in both mesenchymal and ventricular zone progenitors is leading to diminished proliferation. Previous studies have found expression of p15INK4b in the chick embryo hind brain consistent with a role in controlling cell proliferation in this region (Kim et al 2006).
Discussion
ZEB1 is important for regulating epithelial vs. mesenchymal gene expression balance in vivo. Overexpression in cancer leads to EMT, whereas mutation of the gene causes the opposite mesenchymal-epithelial transition. This mesenchymal-epithelial transition in ZEB1 mutant mice is accompanied by diminished proliferation of progenitor cells. The alteration in mesenchymal-epithelial gene expression and proliferative defects are evident in MEFs from ZEB1 gene mutant mice, allowing us to examine these alterations in more detail. The proliferative defects in the MEFs result in cellular senescence. But, this is not mediated through the classic INK4a pathway which can trigger senescence in response to oncogenic mutation and aging. Instead, this senescence is associated with induction of other CDKIs, p15INK4b and p21CDKN1a, and we demonstrate that ZEB1 gene mutation causes ectopic expression of p15INK4b at sites of proliferative defects in vivo.
The epithelial and mesenchymal genes as well as the CDKIs, which are deregulated in response to ZEB1 gene mutation, have in common their regulation by TGF–β to affect EMT and cell cycle arrest, respectively. The linkage of each of these genes to TGF–β together with the fact that ZEB1 participates in TGF–β signaling via binding to activated Smads, suggests that the function of ZEB1 in maintaining mesenchymal vs. epithelial gene expression balance and cell proliferation may be associated with TGF–β superfamily signaling. In this regard, it is of note that ZEB1 was required for TGF–β regulation of key epithelial and mesenchymal genes (e.g., E-cadherin and vimentin). And, the effect of ZEB1 was opposite on the two genes in keeping with the mesenchymal-epithelial gene expression transition seen in ZEB1 mutant mice. Mutation of the ZEB1 gene led to ectopic expression of E-cadherin, and TGF–β was no longer able to repress the gene. Because TGF–β induces ZEB1 (Nishimura et al 2006), we suggest that induction of ZEB1 is a mechanism for TGF–β repression of E-cadherin, and this likely occurs via recruitment of a ZEB1-CtBP repressor complex to the E-cadherin promoter. By contrast to E-cadherin, vimentin expression is induced by TGF–β. Its expression was diminished in ZEB1 gene mutants, and ZEB1 was required for TGF–β-mediated induction of vimentin in MEFs. Gene induction by TGF–β is mediated by activation of Smad transcription factors, and we suggest that ZEB1 dependent, TGF–β-mediated induction of genes such vimentin and smooth muscle actin and myosin may be the result of ZEB1 being required to mediate efficient assembly of a Smad-p300 transcription complex at the promoters (which excludes CtBP; Postigo 2003).
Previously, it was shown that overexpression of ZEB1 facilitated TGF–β induction of the p15INK4b promoter in transfection assays (Postigo 2003). Yet, here we found that p15INK4b is ectopically expressed in ZEB1 gene mutant cells. These findings are not necessarily contradictory. Taken together, they suggest that p15INK4a is under repression by ZEB1. Because the p15INK4b gene is a known target of activated Smads, we propose that recruitment of a Smad-ZEB1-p300 complex to the promoter in response to TGF–β may serve to displace CtBP from ZEB1, leading to derepression of the gene.
We suggest that ZEB1 is important for regulating mesenchymal-epithelial gene expression balance and maintaining proliferation of a subset of progenitor cells late in gestation. But, it is interesting that this phenotype extends to premature replicative senescence in cultured MEFs. Mutations in other genes such as Bmi1 show a similar premature senescence phenotype in MEFs. And, such a phenotype in MEFs is closely linked to diminished proliferation and senescence of progenitor cells in the CNS and bone marrow. While these other mutations involve INK4a regulators and p16INK4a itself, it is of note that we observe diminished CNS progenitor proliferation in ZEB1 gene null mice, and this effect was similar to or even greater than that observed with Bmi1 mutation (Molofsky et al 2005). Interestingly, TGF–β and p15INK4b have key roles in restricting proliferation of T-cell progenitors, and mutation or epigenetic silencing of p15INK4b leads to lymphoproliferative disease (Latres et al., 2000; Wolff et al., 2003; Lessard and Sauvageau, 2003; Mishra et al., 2005), and is common in leukemia (reviewed in Claus and Lubbert, 2003). It is then of note that ZEB1 gene null mice have a diminished number of T-cell progenitors, which fail to populate the thymus (Higashi et al., 1997; Takagi et al., 1998). These results raise the possibility of a proliferative defect in a subset of bone marrow-derived progenitors with ZEB1 gene mutation.
Supplementary Material
Acknowledgments
These studies were supported in part by grants from the NIH to DCD and DSD, and by NIH Center for Biomedical Research Excellence in Molecular Targets Grant RR018733, and core grant EY015636.
References
- Chamberlain EM, Sanders MM. Identification of the novel player deltaEF1 in estrogen transcriptional cascades. Mol Cell Biol. 1999;19:3600–3606. doi: 10.1128/mcb.19.5.3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M, Chaboissier MC, Mynett A, Hisrt E, Shedl A, Briscoe J. The transcriptional control of trunk neural crest induction, survival and delamination. Dev Cell. 2005;8:179–92. doi: 10.1016/j.devcel.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Chinnadurai G. CtBP an unconventional transcriptional corepressor in development and oncogenesis. Mol Cell. 2002;9:213–24. doi: 10.1016/s1097-2765(02)00443-4. [DOI] [PubMed] [Google Scholar]
- Claus R, Lubbert M. Epigenetic targets in hematopoietic malignancies. Oncogene. 2003;22:6489–96. doi: 10.1038/sj.onc.1206814. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Kam M, Nannmark U, Anderson MF, Axell MZ, Wikkelso C, Holtas S, van Roon-Mom WM, Bjork-Eriksson T, Nordborg C, Frisen J, Draqunnow M, Faull RL, Eriksson PS. Human neuroblasts migrate to the olfactory bulb via a lateral ventricular extension. Science. 2007;315:1243–9. doi: 10.1126/science.1136281. [DOI] [PubMed] [Google Scholar]
- Darling DS, Stearman RP, Qi Y, Qiu MS, Feller JP. Expression of Zfhep/deltaEf1 protein in palate, neural progenitors and differentiated neurons. Gene Expr Patterns. 2003;3:709–17. doi: 10.1016/s1567-133x(03)00147-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillner NB, Sanders MM. The zinc finger/homeodomain protein deltaEF1 mediates estrogen-specific induction of the ovalbumin gene. Mol Cell Endocrinol. 2002;192:85–91. doi: 10.1016/s0303-7207(02)00088-6. [DOI] [PubMed] [Google Scholar]
- Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R. DeltaEf1 is a transcriptional repressor of e-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005;24:2375–85. doi: 10.1038/sj.onc.1208429. [DOI] [PubMed] [Google Scholar]
- Genetta T, Ruezinsky D, Kadesch T. Displacement of an E box binding repressor by basic helix-loop-helix proteins, implications for B-cell specificity of the immunoglobulin heavy-chain enhancer. Mol Cell Biol. 1994;14:6153–6163. doi: 10.1128/mcb.14.9.6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–77. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- Grooteclaes ML, Frisch SM. Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene. 2000;19:3823–28. doi: 10.1038/sj.onc.1203721. [DOI] [PubMed] [Google Scholar]
- Guaita S, Puig I, Franci C, Garrido M, Domínguez D, Battle E, Dedhar S, De Herreros AG, Baulida J. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J Biol Chem. 2002;277:39209–16. doi: 10.1074/jbc.M206400200. [DOI] [PubMed] [Google Scholar]
- Hay ED, Zuk A. Transformations between epithelium and mesenchyme, normal, pathological and experimentally induced. Am J Kidney Dis. 1995;26:678–90. doi: 10.1016/0272-6386(95)90610-x. [DOI] [PubMed] [Google Scholar]
- Hemavathy K, Ashraf SL, Ip YT. Snail/slug family of repressors, slowly going into the fast lane of development and cancer. Gene. 2005;257:1–12. doi: 10.1016/s0378-1119(00)00371-1. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Moribe H, Takagi T, Sekido R, Kawakami K, Kikutani H, Kondoh H. Impairment of T cell development in deltaEf1 mutant mice. J Exp Med. 1997;185:1467–79. doi: 10.1084/jem.185.8.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizn M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–8. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- Kim SH, Rowe J, Fujii H, Jones R, Schmierer B, Kong BW, Kuchler K, Foster D, Ish-Horowicz D, Peters G. Upregulation of chicken p15INK4b at senescence and in the developing brain. J Cell Sci. 2006;119:2435–43. doi: 10.1242/jcs.02989. [DOI] [PubMed] [Google Scholar]
- Latres E, Malumbres M, Sotillo R, Martin J, Ortega S, Martin-Caballero J, Flores JM, Cordon-Cardo C, Barbacid M. Limited overlapping roles for p15ink4b and p18ink4c cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 2000;19:3496–506. doi: 10.1093/emboj/19.13.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarova DL, Bordonaro M, Sartorelli AC. Transcriptional regulation of the vitamin D3 receptor gene by ZEB. Cell Growth Differ. 2001;12:319–326. [PubMed] [Google Scholar]
- Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–60. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- Lingbeek ME, Jacobs JJ, van Lohuizen M. The T-box repressors TBX2 and TBX3 specifically regulate the tumor suppressor gene p14ARF via a variant T-site in the initiator. J Biol Chem. 2002;277:1234–46. doi: 10.1074/jbc.M200403200. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Sherr CJ. Tumor suppression by INK4a-Arf, progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- Messing A, Brenner M. GFAP, functional implications gleaned from studies of genetically engineered mice. Glia. 2003;43:87–90. doi: 10.1002/glia.10219. [DOI] [PubMed] [Google Scholar]
- Mishra L, Derynck R, Mishra B. Transforming growth factor-beta signaling in stem cells and cancer. Science. 2005;310:68–71. doi: 10.1126/science.1118389. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, He S, Bydon M, Morrison SJ, Pardal P. Bmi-1 promotes neural stem cell self-renewal and neural development but not growth and survival by repressing the p16INK4a and p19Arf senescence pathways. Genes Dev. 2005;19:1432–7. doi: 10.1101/gad.1299505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph MM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–52. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura G, Manabe I, Tsushima K, Fujiu K, Oishi Y, Maemura K, Miyagishi M, Hagashi Y, Kondoh H, Nagai R. delta EF1 regulates TGF̃ beta signaling in vascular smooth muscle cell differentiation. Dev Cell. 2006;11:93–104. doi: 10.1016/j.devcel.2006.05.011. [DOI] [PubMed] [Google Scholar]
- Ohtani N, Zebedee Z, Huot TJ, Stinson JA, Sugimoto M, Ohashi Y, Sharrocks AD, Peters G, Hara E. Opposing effects of ets and Id proteins on p16INK4A expression during cellular senescence. Nature. 2001;409:1067–70. doi: 10.1038/35059131. [DOI] [PubMed] [Google Scholar]
- Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progresion: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- Pena C, Garcia JM, Silva J, Garcia V, Rodríguez R, Alonso I, Salas C, de Herreros AG, Munoz A, Bonilla F. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer, clinicopathological correlations. Hum Mol Genet. 2005;14:3361–70. doi: 10.1093/hmg/ddi366. [DOI] [PubMed] [Google Scholar]
- Postigo AA. Opposing functions of ZEB proteins in the regulation of the TGFbeta/BMP signaling pathway. EMBO J. 2003;22:2443–2452. doi: 10.1093/emboj/cdg225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo AA, Dean DC. ZEB represses transcription through interaction with the corepressor CtBP. Proc Natl Acad Sci USA. 1999;96:6683–6688. doi: 10.1073/pnas.96.12.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo AA, Dean DC. Differential expression and function of members of the zfh-1 family of zinc finger/homeodomain repressors. Proc Natl Acad Sci USA. 2000;97:6391–6396. doi: 10.1073/pnas.97.12.6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo A, Depp JL, Taylor JT, Kroll KL. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003;22:2453–2462. doi: 10.1093/emboj/cdg226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynisdottir I, Polyak K, Iavarone A, Massague J. Kip/Cip and Ink4a inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev. 1995;9:1831–45. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- Ringrose L, Ehret H, Paro R. Distinct contributions of histone H3 lysine 9 and 27 methylation to locus-specific stability of polycomb complexes. Mol Cell. 2004;16:641–53. doi: 10.1016/j.molcel.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Sekido R, Murai K, Funahashi J, Karachi Y, Fujisawa-Sehara A, Nabeshima Y, Kondoh H. The delta-crystallin enhancer-binding protein delta EF1 is a repressor of E2-box-mediated gene activation. Mol Cell Biol. 1994;14:5692–5700. doi: 10.1128/mcb.14.9.5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoelstra NS, Manning NG, Higashi Y, Darling D, Singh M, Shroyer KR, Broadddus RR, Horwitz KB, Richer JK. The transcription factor ZEB1 is aberrantly expressed in aggressive uterine cancers. Cancer Res. 2006;66:3893–902. doi: 10.1158/0008-5472.CAN-05-2881. [DOI] [PubMed] [Google Scholar]
- Takagi T, Moribe H, Kondoh H, Higashi Y. deltaEF1, a zinc finger and homeodomain transcription factor, is required for skeleton patterning in multiple lineages. Development. 1998;125:21–31. doi: 10.1242/dev.125.1.21. [DOI] [PubMed] [Google Scholar]
- Thiery JP. Epithelia-mesenchymal transitions in development and pathologies. Curr Opion Cell Biol. 2003;15:740–6. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118:409–18. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- van Grunsven LA, Taelman V, Michiels C, Van de Putte T, Nelles L, Wuytens G, Verschueren K, Huylebroeck D. deltaEF1 and SIP1 are differentially expressed and have overlapping activities during Xenopus embryogenesis. Dev Dyn. 2006;235:1491–500. doi: 10.1002/dvdy.20727. [DOI] [PubMed] [Google Scholar]
- Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, Liu F, Taylor H, Jayes FL, Korach KS, Glass CK, Fu XD, Rosenfeld MG. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature. 2007;446:882–7. doi: 10.1038/nature05671. [DOI] [PubMed] [Google Scholar]
- Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita M, Chan Z, Baron A, Franklin W, Drabkin HA, Girad L, Gazdar AF, Minna JD, Bunn PA. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66:944–50. doi: 10.1158/0008-5472.CAN-05-1988. [DOI] [PubMed] [Google Scholar]
- Wolff L, Garin MT, Koller R, Bies J, Liao W, Malumbres M, Tessarollo L, Powell D, Perella C. Hypermethylation of the ink4b locus in murine myeloid leukemia and increased susceptibility to leukemia in p15ink4b-deficient mice. Oncogene. 2003;22:9265–74. doi: 10.1038/sj.onc.1207092. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Bottinger EP. TGF–β and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–74. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.