

Abstract
Multipotent cardiac progenitor cells are found in the fetal and adult heart of many mammalian species including humans and form as intermediates during the differentiation of embryonic stem cells. Despite similar biological properties, the molecular identities of these different cardiac progenitor cell populations appear to be distinct. Elucidating the origins and lineage relationships of these cell populations will accelerate clinical applications such as drug screening and cell therapy as well as shedding light on the pathogenic mechanisms underlying cardiac diseases.
Introduction
Modern translational medicine rests upon the progressive study of pathways and principles from model organisms such as yeast, fly, fish, and mouse to clinical study in humans. However, as noted by the physician William Osler, much of our knowledge of human disease is based on studies in patients themselves. More recently, this approach has extended into models of human disease based on cultured human cells, where the discovery of the low-density lipoprotein (LDL) receptor pathway in skin fibroblasts from patients with familial hypercholesterolemia ultimately led to one of the biggest advances in clinical cardiovascular medicine. However, many of the most important and puzzling human cardiovascular diseases cannot be adequately studied because specific human cardiovascular cell types—such as cardiomyocytes, endothelial cells (ECs), and vascular smooth muscle cells (VSMCs)—cannot be obtained. Although animal models will continue to be invaluable, there is a huge advantage to studying specific cardiovascular cell types from patients with specific forms of heart disease. The discovery of multipotent cardiovascular progenitor cells not only in mammalian embryos and postnatal (adult) heart but also as an intermediate stage during differentiation of embryonic stem (ES) cells is an important step toward reaching this goal.
“He who studies medicine without books sails an uncharted sea, but he who studies medicine without patients does not go to sea at all.”
—William Osler (1849–1919)
Sophisticated genetic approaches in model organisms provide unique opportunities for determining the embryonic origins and fates of cardiac progenitor cells. This has taught us much about their developmental potency and ability to differentiate into the major functional cell lineages of the heart: cardiomyocytes, ECs, VSMCs, and cardiac fibroblasts. The existence of cardiac progenitor cells in adult heart is of particular interest because the heart was long considered to be without a resident stem cell population. Here, we discuss cardiac progenitor cells from fetal and adult heart and from in vitro differentiated pluripotent stem cells because (1) alterations in the pool of cardiac progenitors during development may be causally related to congenital heart defects; (2) expansion of cardiac progenitors in culture is potentially the most efficient way of producing large numbers of cardiovascular cells for future cell therapy and drug screens; (3) gene targeting in human ES cells is a promising approach for generating cardiac progenitors and their derivatives with specific, clinically relevant gene mutations for elucidating disease mechanisms. In this context, the recent reports on direct reprogramming of human skin fibroblasts to induced pluripotent stem (iPS) cells with an ES cell-like phenotype are particularly exciting because if derived from patients carrying gene mutations affecting the cardiovascular system, it should be possible to obtain cardiac progenitors with the same mutations (see Review by C.E. Murry and G. Keller, and Review by R. Jaenisch and R. Young, in this issue). This may allow pathogenesis to be followed at the cellular level “in a dish” and should enable molecular and genetic screens to find drugs to halt or reverse the disease phenotype.
Cardiac Progenitors in Mouse Fetal and Adult Heart
The origin of heart-forming cells and their roles in organ development have fascinated biologists for over a century. Pioneering work in lower vertebrate species such as frog and chick have laid the blueprint for modern cardiac developmental biology by identifying the mesoderm as the germ layer responsible for mammalian cardiogenesis (Rawles, 1943). Precursors for heart-forming cells in the vertebrate mesoderm transition from expressing Brachyury T, a T-box transcription factor, to expressing mesoderm posterior 1 (Mesp1) when they enter the precardiac mesoderm stage of development (Solloway and Harvey, 2003) (Figure 1). Mesp1+ cells encompass all cardiac progenitor cells and their expression of Mesp1 is turned off as they migrate away from the primitive streak. During their migration, cardiac precursor cells expand rapidly to form the anterior and lateral plate mesoderm where they eventually generate a crescent-shaped structure called the cardiac crescent (Figure 1). Mesp1+ cells have not yet committed to the cardiogenic fate as some also give rise to derivatives of the paraxial mesoderm and skeletal muscle of the head and neck (Saga et al., 1999). It is at the cardiac crescent stage that heart precursor cells commit irreversibly to the cardiac lineage and become cardiac progenitor cells expressing key developmental transcription factors such as Nkx2.5 and Isl-1. Progressive restriction of developmental potency is believed to take place during this time given that cells expressing Mesp1 contribute broadly to all four main cell types in the heart. Cardiogenic Isl-1+ or Nkx2.5+ cells contribute primarily to cardiomyocytes and VSMCs with limited contribution to ECs (Saga et al., 2000; Sun et al., 2007; Moses et al., 2001; Stanley et al., 2002). This stepwise commitment of cardiac progenitor cells toward their differentiated progeny has allowed the use of genetic marking techniques, such as Cre-Lox, to assign a precursor/descendant relationship to cells within the developing heart. With this approach it has been shown that cells in the first heart field (marked by the expression of Tbx5 or the first wave of Nkx2.5) give rise to the left ventricle and portions of the right and left atrium, whereas cells in the second heart field (marked by the expression of Isl-1 or the second wave of Nkx2.5) contribute to the right ventricle, the outflow tract, and portions of both atria (Cai et al., 2003). Interestingly, using retrospective clonal analysis, a powerful genetic approach to tag single cells during early development, Meilhac et al. (2004) have demonstrated a common embryonic origin for some cells in the first and second heart fields (reviewed in Buckingham et al., 2005). Indeed, given that Cre-Lox-based approaches are dependent on the expression domain of key cardiac transcription factors, which may extend beyond a single heart field, it is essential that clonal analysis be performed when multipotentiality is being ascribed to new progenitor cell populations that are identified based on the expression of a specific gene.
Figure 1. Origins of Cardiac Progenitor Cells in the Developing Heart.
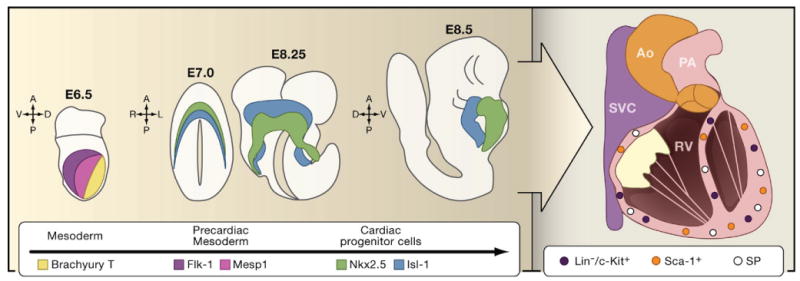
Cells from the developing mesoderm of the mouse embryo are marked by the expression of Brachyury T at E6.5 of embryonic development (yellow). As they transition into precardiac mesoderm, they start to express Mesp1 (pink) and Flk-1 (blue). As these cardiac precursor cells reach the anterior and lateral plate mesoderm, they commit irreversibly to become cardiac progenitor cells by expressing Nkx2.5 (green) or Isl-1 (red) at E7.5. Midline fusion of lateral plate mesoderm and differentiation of these two cell populations results in the formation of Nkx2.5+ embryonic heart tube (green; E8.25 and E8.75) and Isl+ pharyngeal mesoderm (red; E8.25 and E8.75). Some of the pharyngeal mesoderm cells also express Nkx2.5 (not shown). As the heart progresses through embryonic and postnatal development, it acquires a four-chambered identity and is functionally integrated with the systemic vasculature. Within the adult heart reside several different populations of cardiac stem cells including side population (SP) cells and cells that express c-Kit or Sca-1. (Ao, aorta; SVC, superior vena cava; PA, pulmonary artery; RV, right ventricle).
By combining transgenic expression of fluorescent proteins and fluorescence-activated cell sorting (FACS), the biological phenotype and molecular behavior of these embryonic cardiac progenitor cells have recently been elucidated (Wu et al., 2006; Moretti et al., 2006; Kattman et al., 2006). Depending on the markers used and the developmental stage examined, embryonic cardiac progenitor cells may be bi or tri-potent and may differentiate spontaneously into one or more of the three cell lineages: cardiomyocytes, VSMCs, and ECs. Collectively, these studies provide a shift in our understanding of mammalian cardiogenesis, that is, multipotent progenitor cells contribute to the formation of a functional heart by making lineage choice decisions at a single-cell level. This is similar to the development of other stem cell-mediated systems such as blood, skin, and intestine.
The existence of these embryonic multipotent cardiac progenitor cells has prompted a search for their presence in the postnatal heart. Laugwitz et al. (2005) have identified a neonatal Isl-1+ cardiac progenitor cell population with the capacity for differentiation into fully mature cardiomyocytes. It remains to be seen whether these multipotent embryonic cardiac progenitor cells possess the capability for self-renewal and asymmetric division to meet the strict definition of cardiac stem cells. If so, these embryonic cardiac stem cells may be the developmental precursor to adult cardiac stem cells that have been recently described (see below), although they may not necessarily have the same molecular identity.
The notion that the adult mammalian heart may harbor stem cells with replicative and regenerative capacity was suggested initially by a study in patients with myocardial infarction (Beltrami et al., 2001). This study showed an increased number of immature cardiomyocytes with the capacity for mitotic division in the infarct border zone that may have originated from a circulating or endogenous stem cell pool. The same investigators subsequently isolated a lineage-negative, c-Kit-positive (Lin−, Kit +) cell population from adult mice that was reportedly clonogenic, self-renewing, and capable of differentiating into cardiomyocytes, VSMCs, and ECs (Beltrami et al., 2003). Shortly thereafter, two other groups reported the isolation of adult cardiac stem cells based on the expression of Sca1 or the ATP-binding cassette transporter (ABCG2; these cells are also known as side population or SP cells because of their ability to pump out Hoechst and rhodamine dyes) (Oh et al., 2003; Martin et al., 2004). These three populations of adult cardiac progenitor cells (c-Kit +, Sca1+, or SP) are phenotypically different and show differential expression of surface markers (Murry et al., 2006; Evans et al., 2007; Laugwitz et al., 2005; Parmacek and Epstein, 2005; Table S1). These three cell populations represent 1%–2% of the total cell number in the heart (10% of the total side population), enter the cell cycle when growth of the heart is attenuated, proliferate in culture, and form cells expressing cardiomyogenic markers (Hierlihy et al., 2002; Pfister et al., 2005). The exact lineage relationships between these adult cardiac progenitor cell populations and embryonic cardiac progenitor cells is unknown. Analysis of cells expressing c-Kit following transplantation of GFP-labeled bone marrow-derived mononuclear cells into sublethally irradiated wild-type adult mice showed that c-Kit+ cells in the adult heart are derived mostly from the transplanted marrow cells (Fazel et al., 2006); those not derived from transplanted cells but present in the host could be the recently discovered c-Kit+ population found in multiple organs including heart (Massberg et al., 2007). These c-Kit+ cells exit the bone marrow in minute numbers and reside in peripheral tissues where they scout for pathogenic molecules and promote a local innate immune response. Nevertheless, injection of adult cardiac stem cells directly into infarcted mouse myocardium has been reported to provide short-term improvement in heart function (Beltrami et al., 2003; Oh et al., 2003; Messina et al., 2004) although evidence of cardiomyocyte differentiation is limited. Paracrine factors or angiogenesis may have been partly responsible for the reported benefit. Indeed, clinical trials using bone marrow-derived stem cell infusions into patients with myocardial infarction also suggest that paracrine factors may be responsible for the transient improvement in cardiac function in humans (Meyer et al., 2006).
Cardiac Progenitors in Human Fetal and Adult Heart
The presence of endogenous cardiac progenitors in the mouse fetal and adult heart prompted studies into whether similar populations exist in the human heart. An initial study described the isolation of a heterogeneous population of cells from human atrial and ventricular biopsies that form clonal multicellular clusters in suspension culture called “cardiospheres” (Messina et al., 2004). These cardiospheres consist of c-Kit+ cells at the core and cells expressing cardiac and EC markers at the periphery. Cardiomyocytes derived from cardiospheres will only contract if cocultured with neonatal rat cardiomyocytes. More recently, cardiosphere-derived cells were isolated with improved efficiencies from endomyocardial right ventricular biopsies from adult patients (Smith et al., 2007). Concurrently, several studies identified an endogenous cardiac c-Kit+ cell population from patients with aortic stenosis or post-heart transplantation (Quaini et al., 2002; Urbanek et al., 2003) (Table S1). These cells were MDR1+ (multidrug resistance gene 1) but did not express hematopoietic or endothelial progenitor cell markers. When isolated by FACS, human cardiac c-Kit+ cells were reported to give rise to cardiomyocytes, VSMCs, and ECs in vitro and following transplantation into immunodeficient mice (Bearzi et al., 2007). Interestingly, some of the c-Kit+ cells identified as a minute population in human heart were recently shown to be mast cells, based on the presence of the enzyme tryptase and absence of Nkx2.5 or Isl-1 expression, independent of whether they were present in atrium or ventricle or whether they were sorted by FACS and then cultured (J. Pouly, personal communication). Although not all c-Kit+ cells may have been accounted for, these results raise a cautionary note with respect to speculation on their future clinical applications.
Different cell populations with the capacity to proliferate and form cardiomyocytes in adherent cell culture have been isolated independently from both human adult heart (biopsies) and fetal heart on the basis of their ability to bind to an anti-mouse Sca-1 antibody. Their authenticity as cardiac progenitors, however, has been debated because Sca-1 is not a determinant on human cells and 5-azacytidine is required to induce differentiation. 5-azacytidine induces genomewide demethylation and its effects are nonspecific. Nevertheless, the differentiated progeny of these progenitors do have properties of bona fide cardiomyocytes (such as sarcomeric proteins organized into sarcomeres and spontaneous action potentials). The differentiation of these cells may depend on another important population of cells in the heart termed cardiac fibroblasts, which generate extracellular matrix and provide the heart with elasticity and mechanical strength. Cardiac fibroblasts lack markers for cardiomyocytes, ECs, and nerve cells but express discoidin domain receptor 2 and Thy-1 (Hudon-David et al., 2007). They are derivatives of the portion of the epicardium that underwent epithelial-mesenchymal transformation during cardiogenesis. These cells also give rise to coronary VSMCs and ECs. Epicardial cells regulate the formation of the compact myocardium, coronary vasculature, and Purkinje fiber network as well as the fibrous structures of the heart (Winter and Gittenberger-de Groot, 2007). If adult cardiac “fibroblasts” recapitulate the essential properties of epicardium-derived cells in a diseased myocardium, it would support the hypothesis that the epicardium may also be the origin of progenitors in the adult heart (Lepilina et al., 2006). This would be compatible with the fact that Isl-1+ cells are detectable in human neonates (as a “carry over” of second heart field progenitors) but are absent in adult hearts (Laugwitz et al., 2005; Qyang et al., 2007), suggesting that cardiac progenitors identified during later stages of life may arise de novo from the epicardium. Studies aimed at defining the precise lineage relationship between Isl-1+ second heart field cells and progenitor cells for the proepicardial organ will help to clarify the intrinsic differences between these two cell populations.
Cardiac Progenitors from ES Cells and iPS Cells
Pluripotent ES cells have been instrumental in identifying and characterizing cell populations of the early stages of development and lineage commitment, which are difficult to study in the embryo. They represent a renewable source of multiple progenitors including those of the cardiac, cardiovascular, and hemangioblast lineages. In humans, ES cells are derived from early embryos discarded after in vitro fertilization. They can be induced by a variety of methods to undergo stepwise differentiation to mesoderm (expressing Brachyury T) and then to cardiac progenitors (expressing GATA-4, Nkx2.5, and Isl-1). Finally, they differentiate into cardiomyocytes (expressing MHC, cTNI, α-actinin, and other proteins of the contractile machinery; Beqqali et al., 2006; Kehat et al., 2001; Passier et al., 2005), ECs (Levenberg et al., 2002; Ferreira et al., 2007), and VSMCs. Whether the cardiomyocytes or ECs derived from mouse or human ES cells represent derivatives of the first or second heart fields or the proepicardial organ remains to be established, but approaches using genetic markers or physical selection of subpopulations using cell-surface antibodies as in mouse ES cells (Kouskoff et al., 2005; Kattman et al., 2006) should provide an answer. ECs can also develop independently of cardiac progenitors via the hemangioblast (Kennedy et al., 2007; Lu et al., 2007). It will be of interest to compare these cells directly with those derived from cardiac progenitors.
Cardiovascular cells derived from ES cell sources are highly desirable for several reasons: they can carry natural or induced gene mutations for functional analysis and drug screens, they can in principle be scaled up reproducibly for cell-based therapies, and they can be genetically marked for cell selection (Figure 2). However, the best targets for expansion in culture are probably not the undifferentiated stem cells but rather committed progenitors derived from them as they are most likely to yield homogeneous populations of differentiated cells without contaminating (potentially tumorigenic) ES cells. Being able to identify, select, and expand these progenitors is then of immediate importance. Ectopic reporter gene expression or, even better, gene targeting has already been successful in selecting cells at different stages and with different identities in the cardiovascular differentiation pathway of mouse ES cells (Meyer et al., 2000; Kouskoff et al., 2005; Wu et al., 2006; Moretti et al., 2006). However, gene transfer has been challenging in human ES cells so that only now are the first reporter lines for human cardiovascular lineages emerging (Anderson et al., 2007; Huber et al., 2007). Alternatively, an option circumventing the need for genetic modification would be selection on the basis of known cell-surface antigens, which has been used successfully with mouse ES cells to enrich for cell populations with cardiovascular differentiation potential (Kattman et al., 2006; Kouskoff et al., 2005). Even better would be sets of antibodies able to select cardiac progenitors at different stages of development. However, with the exception of ECs, cell type-specific surface antibodies do not yet exist for most lineages.
Figure 2. Therapeutic Implications of Cardiac Progenitor Cells.
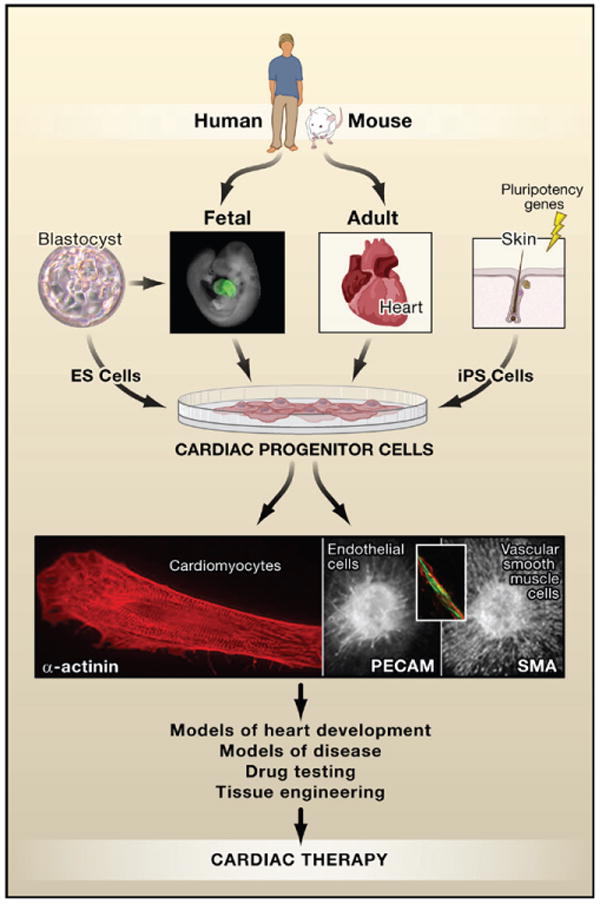
Progenitor cells have been described in fetal and adult heart in multiple species including humans. They also form as an intermediate in the differentiation of embryonic stem (ES) cells giving rise to cardiomyocytes, smooth muscle cells, and endothelial cells. Cardiac progenitors may be uni-, bi-, or tri- potent, depending on their molecular signatures although this relationship is still under investigation. Cardiomyocytes are morphologically readily identifiable by α-actinin staining (red, left panel) of sarcomeric structures, endothelial cells by expression of surface markers such as PE-CAM (green, middle panel and inset), and vascular smooth muscle cells that surround blood vessels by smooth muscle actin (sma; red, right panel). Cells expressing Nkx2.5 in the fetal mouse heart are indicated by green fluorescent protein (GFP) in a transgenic Nkx2.5-GFP mouse. Photos courtesy of C. Mummery (blastocyst; cardiomyocyte); F. Lebrin, D. Ward, and L. Tertoolen (endothelial cell and vascular smooth muscle cell); Sean Wu (fetal heart).
Most human ES cell derivatives acquire only a fetal phenotype and do not fully mature unless placed in an environment resembling normal tissue. Cardiomyocytes derived from human ES cells are also generally immature with radially organized sarcomeres and low action potentials (Mummery et al., 2003). Although this may be beneficial for future cell-based therapies as only fetal, not adult, cardiomyocytes survive transplantation in the heart (Koh et al., 1995; Klug et al., 1996), many other challenges presently preclude therapeutic application. Aside from perceived tumorigenic risk, such challenges include stable integration of transplanted cells into the host myocardium, control of fibrosis and abrogation of immune attack of the cell graft, and a sustained contribution to contractile activity that is lost after myocardial infarction. This is independent of the particular source of cardiac progenitors from which the fully differentiated cardiomyocytes are derived. Several studies have described successful transplantation and long-term survival of grafts from contracting (beating) human ES cell cultures containing cardiomyocytes (Caspi et al., 2007; Laflamme et al., 2007; van Laake et al., 2007); however, none have reported long-term functional improvements and the grafts may form isolated syncytia rather than becoming truly integrated into existing myocardium. In contrast to cell therapies, drug screens or physiological analysis of disease properties in vitro will most likely require an adult cardiomyocyte phenotype. This will require ways to drive fetal and adult cardiac progenitors and human ES cells toward cardiomyocyte maturation, perhaps using cardiac tissue constructs that allow cells to contract in unison and exert positive mechanical forces (Zimmerman et al., 2006; Feinberg et al., 2007).
Although human ES cells represent an exciting platform for technology development, gene targeting of these cells and creation of disease models in culture are challenging. The alternative strategy—somatic cell nuclear transfer (using adult somatic cells from patients with specific diseases) and then deriving the ES cell lines from cloned embryos—seems feasible given recent successes in nonhuman primates (Byrne et al., 2007); however, low efficiency and the scarcity of human eggs are severely rate limiting. Both of these approaches have associated ethical issues. Extremely exciting, however, is the recent demonstration that human skin fibroblasts can be converted to human ES cell-like cells (Takahashi et al., 2007; Yu et al., 2007). These iPS cells express markers of human ES cells and are capable of multilineage differentiation in vitro and form teratomas in vivo, characteristic of pluripotent stem cells. The requirement for virally inserting three or four genes into the adult human somatic cells will likely preclude cell therapy applications in the immediate future. Also, epigenetic differences between iPS cells and human ES cells suggest that iPS cells may not behave identically to human ES cells. However, iPS cells may be particularly useful for bedside-to-bench research (that is, reverse translational medicine) by allowing the creation of accurate models of genetic disease in the culture dish. For example, patients with either Long QT Syndrome (where the electrical action potential duration during heart muscle contraction is prolonged leading to sudden cardiac death) or hypertrophic cardiomyopathy (where excess growth of heart muscle cells induces impaired heart function and increases risks for lethal arrythmia) could donate their skin cells to be converted into iPS cells. The resulting iPS cells could then be differentiated into cardiac progenitors and their derivatives that would bear the same genetic mutations as the patients. Using the techniques developed for human ES cells, basic electrophysiology, genome and proteome comparisons, and rescue experiments could then be carried out to understand the mechanisms of disease pathogenesis and to develop strategies for ameliorating its manifestation or for developing cures. Clearly, iPS cells will not preclude the need to continue research on human ES cells, and undoubtedly a decade of information derived from human ES cells will further the significance and applicability of iPS cells.
Cardiac Progenitors and Implications for Therapy
Stem cell and progenitor cell-based therapies hold tremendous promise for restoring cardiac functions in a variety of degenerative diseases including ischemic cardiomyopathy and conduction system diseases (such as sinus node dysfunction and atrial-ventricular block) as well as congenital heart diseases (such as atrial or ventricular septal defect where the wall between atrial or ventricular chambers is incompletely formed). The prospect for achieving a cure for ischemic cardiomyopathy with stem cells was felt to be so great that clinical trials were undertaken using autologous adult stem cells from a variety of noncardiac sources (Janssens et al., 2006; Assmus et al., 2006; Lunde et al., 2006; Schächinger et al., 2006). These trials set out to demonstrate improved cardiac function via neocardiomyogenesis with developmental plasticity as the scientific principle underpinning the strategy. It is clear now that noncardiac stem cells are unlikely to result in the formation of sufficient numbers of new cardiomyocytes to affect heart function. Nevertheless, regardless of the cell type injected, there appears to be a small but statistically significant improvement in heart function. The clinical significance of this improvement is being debated and there is no evidence thus far of reduction in important end-points such as improved survival or reduced heart failure-related hospitalization. The new hypothesis that paracrine action of the transplanted cells or induced vasculogenesis may be responsible for the observed functional improvement now awaits further confirmation (reviewed in Laflamme et al., 2007). Will the experiences from this initial foray into stem cell-based therapy predict the outcome of future trials using cardiac stem or progenitor cells from adult heart or derived from ES cells or iPS cells? Regardless of whether endogenous or ES/iPS cell-derived cardiac progenitor cells ever make their way into transplantable cell sources for therapy, it is clear that these cells will continue to shed light on fundamental mechanisms important for heart formation and regeneration. By defining the molecular identity of multipotent cardiac progenitor cells and the means by which they can make lineage choice decisions to become either cardiomyocytes, VSMCs, ECs, or mesenchyme fibroblasts, we place ourselves at the helm in determining the most promising means for cardiac therapy. This may require the creation of an engineered tissue graft (containing cardiomyocytes, with ECs and VSMCs organized into vasculature), the identification of new pathways to generate drug targets, or even the generation of a bioartificial heart. In any case, accessibility of cardiac progenitors represents a significant advantage over differentiated cells or undifferentiated pluripotent stem cells to achieve large-scale production of tumor-free cardiac cells for clinical and translational applications.
Perspectives
Recent advances in our understanding of developmental and stem cell biology of the cardiovascular system are bringing us closer to making mammalian cardiac regeneration a clinical reality. This calls for a new experimental paradigm of bedside-to-bench research where the initial fundamental observations are made in human patients and then are followed up by more rigorous mechanistic analyses in model organisms. This strategy may be applicable to a number of diseases for which there are few valid animal model systems, such as complex chromosomal disorders, polygenic disorders where common genetic variants confer susceptibility or resistance to disease, and well-characterized genetic disorders that are not adequately recapitulated in animal models. In addition, drug discovery, cardiotoxicity screening of new drugs, and identification and validation of therapeutic targets should soon be conducted directly on human cardiovascular cells derived directly from patients. Cardiovascular stem cell biology may well herald a new era of reverse translational medicine.
Supplementary Material
Supplemental Data include one table and can be found with this article online at http://www.cell.com/cgi/content/full/132/4/XXX/DC1/.
Acknowledgments
C.L.M. is supported by the Harvard Stem Cell Institute, the Radcliffe Fellowship Programme, and the EU 6th Framework program (Heart Repair) LSHM-CT-2005-018630. S.M.W. is supported by the National Institutes of Health and the GlaxoSmithKline Education and Research Foundation. K.R.C. is supported by the LeDucq Foundation and the Harvard Stem Cell Institute. We thank A. Gittenberger-de Groot for helpful comments.
References
- Anderson D, Self T, Mellor IR, Goh G, Hill SJ, Denning C. Mol Ther. 2007;15:2027–2036. doi: 10.1038/sj.mt.6300303. [DOI] [PubMed] [Google Scholar]
- Assmus B, Honold J, Schachinger V, Britten MB, Fischer-Rasokat U, Lehmann R, Teupe C, Pistorius K, Martin H, Abolmaali ND, et al. N Engl J Med. 2006;355:1222–1232. doi: 10.1056/NEJMoa051779. [DOI] [PubMed] [Google Scholar]
- Bearzi C, Rota M, Hosoda T, Tillmanns J, Nascimbene A, De Angelis A, Yasuzawa-Amano S, Trofimova I, Siggins RW, Lecapitaine N, et al. Proc Natl Acad Sci USA. 2007;104:14068–14073. doi: 10.1073/pnas.0706760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beqqali A, Kloots J, Ward-van Oostwaard D, Mummery C, Passier R. Stem Cells. 2006;24:1956–1967. doi: 10.1634/stemcells.2006-0054. [DOI] [PubMed] [Google Scholar]
- Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, et al. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P. N Engl J Med. 2001;344:1750–1757. doi: 10.1056/NEJM200106073442303. [DOI] [PubMed] [Google Scholar]
- Buckingham M, Meilhac S, Zaffran S. Nat Rev Genet. 2005;6:826–835. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- Byrne JA, Pedersen DA, Clepper LL, Nelson M, Sanger WG, Gokhale S, Wolf DP, Mitalipov SM. Nature. 2007;450:497–502. doi: 10.1038/nature06357. [DOI] [PubMed] [Google Scholar]
- Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S. Dev Cell. 2003;5:877–889. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi O, Huber I, Kehat I, Habib M, Arbel G, Gepstein A, Yankelson L, Aronson D, Beyar R, Gepstein L. J Am Coll Cardiol. 2007;50:1884–1893. doi: 10.1016/j.jacc.2007.07.054. [DOI] [PubMed] [Google Scholar]
- Evans SJ, Mummery C, Doevendans PA. Semin Cell Dev Biol. 2007;18:153–160. doi: 10.1016/j.semcdb.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P, Verma S, Weisel RD, Keating A, Li RK. J Clin Invest. 2006;116:1865–1877. doi: 10.1172/JCI27019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AW, Feigel A, Shevkoplyas SS, Sheehy S, Whitesides GM, Parker KK. Science. 2007;317:1366–1370. doi: 10.1126/science.1146885. [DOI] [PubMed] [Google Scholar]
- Ferreira LS, Gerecht S, Shieh HF, Watson N, Rupnick MA, Dallabrida SM, Vunjak-Novakovic G, Langer R. Circ Res. 2007;101:286–294. doi: 10.1161/CIRCRESAHA.107.150201. [DOI] [PubMed] [Google Scholar]
- Hierlihy AM, Seale P, Lobe CG, Rudnicki MA, Megeney LA. FEBS Lett. 2002;530:239–243. doi: 10.1016/s0014-5793(02)03477-4. [DOI] [PubMed] [Google Scholar]
- Huber I, Itzhaki I, Caspi O, Arbel G, Tzukerman M, Gepstein A, Habib M, Yankelson L, Kehat I, Gepstein L. FASEB J. 2007;21:2551–2563. doi: 10.1096/fj.05-5711com. [DOI] [PubMed] [Google Scholar]
- Hudon-David F, Bouzeghrane F, Couture P, Thibault G. J Mol Cell Cardiol. 2007;42:991–1000. doi: 10.1016/j.yjmcc.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Janssens S, Dubois C, Bogaert J, Theunissen K, Deroose C, Desmet W, Kalantzi M, Herbots L, Sinnaeve P, Dens J, et al. Lancet. 2006;367:113–121. doi: 10.1016/S0140-6736(05)67861-0. [DOI] [PubMed] [Google Scholar]
- Kattman SJ, Huber TL, Keller GM. Dev Cell. 2006;11:723–732. doi: 10.1016/j.devcel.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, Livne E, Binah O, Itskovitz-Eldor J, Gepstein L. J Clin Invest. 2001;108:407–414. doi: 10.1172/JCI12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M, D'Souza SL, Lynch-Kattman M, Schwantz S, Keller G. Blood. 2007;109:2679–2687. doi: 10.1182/blood-2006-09-047704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug MG, Soonpaa MH, Koh GY, Field LJ. J Clin Invest. 1996;98:216–224. doi: 10.1172/JCI118769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh GY, Soonpaa MH, Klug MG, Pride HP, Cooper BJ, Zipes DP, Field LJ. J Clin Invest. 1995;96:2034–2042. doi: 10.1172/JCI118251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouskoff V, Lacaud G, Schwantz S, Fehling HJ, Keller G. Proc Natl Acad Sci USA. 2005;102:13170–13175. doi: 10.1073/pnas.0501672102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, Police S, et al. Nat Biotechnol. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- Laugwitz KL, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, Lin LZ, Cai CL, Lu MM, Reth M, et al. Nature. 2005;433:647–653. doi: 10.1038/nature03215. Erratum: (2007). Nature 446, 934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepilina A, Coon AN, Kikuchi K, Holdway JE, Roberts RW, Burns CG, Poss KD. Cell. 2006;127:607–619. doi: 10.1016/j.cell.2006.08.052. [DOI] [PubMed] [Google Scholar]
- Levenberg S, Golub JS, Amit M, Itskovitz-Eldor J, Langer R. Proc Natl Acad Sci USA. 2002;99:4391–4396. doi: 10.1073/pnas.032074999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SJ, Feng Q, Caballero S, Chen Y, Moore MA, Grant MB, Lanza R. Nat Methods. 2007;4:501–509. doi: 10.1038/nmeth1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde K, Solheim S, Aakhus S, Arnesen H, Abdelnoor M, Egeland T, Endresen K, Ilebekk A, Mangschau A, Fjeld JG, et al. N Engl J Med. 2006;355:1199–1209. doi: 10.1056/NEJMoa055706. [DOI] [PubMed] [Google Scholar]
- Martin CM, Meeson AP, Robertson SM, Hawke TJ, Richardson JA, Bates S, Goetsch SC, Gallardo TD, Garry DJ. Dev Biol. 2004;265:262–275. doi: 10.1016/j.ydbio.2003.09.028. [DOI] [PubMed] [Google Scholar]
- Massberg S, Schaerli P, Knezevic-Maramica I, Köllnberger M, Tubo N, Moseman EA, Huff IV, Junt T, Wagers AJ, Mazo IB, von Andrian UH. Cell. 2007;131:994–1008. doi: 10.1016/j.cell.2007.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meilhac SM, Esner M, Kelly RG, Nicolas JF, Buckingham ME. Dev Cell. 2004;6:685–698. doi: 10.1016/s1534-5807(04)00133-9. [DOI] [PubMed] [Google Scholar]
- Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F, Salio M, Battaglia M, Latronico MV, Coletta M, et al. Circ Res. 2004;95:911–921. doi: 10.1161/01.RES.0000147315.71699.51. [DOI] [PubMed] [Google Scholar]
- Meyer N, Jaconi M, Landopoulou A, Fort P, Pucéat M. FEBS Lett. 2000;478:151–158. doi: 10.1016/s0014-5793(00)01839-1. [DOI] [PubMed] [Google Scholar]
- Meyer GP, Wollert KC, Lotz J, Steffens J, Lippolt P, Fichtner S, Hecker H, Schaefer A, Arseniev L, Hertenstein B, et al. Circulation. 2006;113:1287–1294. doi: 10.1161/CIRCULATIONAHA.105.575118. [DOI] [PubMed] [Google Scholar]
- Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. Genesis. 2001;31:176–180. doi: 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, Qyang Y, Bu L, Sasaki M, Martin-Puig S, et al. Cell. 2006;127:1151–1165. doi: 10.1016/j.cell.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Mummery C, Ward-van Oostwaard D, Doevendans P, Spijker R, van den Brink S, Hassink R, van der Heyden M, Opthof T, Pera M, de la Riviere AB, et al. Circulation. 2003;107:2733–2740. doi: 10.1161/01.CIR.0000068356.38592.68. [DOI] [PubMed] [Google Scholar]
- Murry CE, Reinecke H, Pabon LM. J Am Coll Cardiol. 2006;47:1777–1785. doi: 10.1016/j.jacc.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, et al. Proc Natl Acad Sci USA. 2003;100:12313–12318. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmacek MS, Epstein JA. Cell. 2005;120:295–298. doi: 10.1016/j.cell.2005.01.025. [DOI] [PubMed] [Google Scholar]
- Passier R, Oostwaard DW, Snapper J, Kloots J, Hassink RJ, Kuijk E, Roelen B, de la Riviere AB, Mummery C. Stem Cells. 2005;23:772–780. doi: 10.1634/stemcells.2004-0184. [DOI] [PubMed] [Google Scholar]
- Pfister O, Mouquet F, Jain M, Summer R, Helmes M, Fine A, Colucci WS, Liao R. Circ Res. 2005;97:52–61. doi: 10.1161/01.RES.0000173297.53793.fa. [DOI] [PubMed] [Google Scholar]
- Pouly J, Bruneval P, Mandet C, Proksch S, Peyrard S, Amrein C, Bousseaux C, Guillemain R, Deloche P, Fabiani JN, Menasche PJ. Thorac Cardiovasc Surg. 2008 doi: 10.1016/j.jtcvs.2007.10.024. in press. [DOI] [PubMed] [Google Scholar]
- Quaini F, Urbanek K, Beltrami AP, Finato N, Beltrami CA, Nadal-Ginard B, Kajstura J, Leri A, Anversa PN. N Engl J Med. 2002;346:5–15. doi: 10.1056/NEJMoa012081. [DOI] [PubMed] [Google Scholar]
- Qyang Y, Martin-Puig S, Chiravuri M, Chen S, Xu H, Bu L, Jiang X, Lin L, Granger A, Moretti A, et al. Cell Stem Cell. 2007;1:165–179. doi: 10.1016/j.stem.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Rawles ME. Physiol Zool. 1943;16:22–42. [Google Scholar]
- Saga Y, Miyagawa-Tomita S, Takagi A, Kitajima S, Miyazaki J, Inoue T. Development. 1999;126:3437–3447. doi: 10.1242/dev.126.15.3437. [DOI] [PubMed] [Google Scholar]
- Saga Y, Kitajima S, Miyagawa-Tomita S. Trends Cardiovasc Med. 2000;10:345–352. doi: 10.1016/s1050-1738(01)00069-x. [DOI] [PubMed] [Google Scholar]
- Schächinger V, Erbs S, Elsässer A, Haberbosch W, Hambrecht R, Hölschermann H, Yu J, Corti R, Mathey DG, Hamm CW, et al. N Engl J Med. 2006;355:1210–1221. doi: 10.1056/NEJMoa060186. [DOI] [PubMed] [Google Scholar]
- Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marbán E. Circulation. 2007;115:896–908. doi: 10.1161/CIRCULATIONAHA.106.655209. [DOI] [PubMed] [Google Scholar]
- Solloway MJ, Harvey RP. Cardiovasc Res. 2003;58:264–277. doi: 10.1016/s0008-6363(03)00286-4. [DOI] [PubMed] [Google Scholar]
- Stanley EG, Biben C, Elefanty A, Barnett L, Koentgen F, Robb L, Harvey RP. Int J Dev Biol. 2002;46:431–439. [PubMed] [Google Scholar]
- Sun Y, Liang X, Najafi N, Cass M, Lin L, Cai CL, Chen J, Evans SM. Dev Biol. 2007;304:286–296. doi: 10.1016/j.ydbio.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Urbanek K, Quaini F, Tasca G, Torella D, Castaldo C, Nadal-Ginard B, Leri A, Kajstura J, Quaini E, Anversa P. Proc Natl Acad Sci USA. 2003;100:10440–10445. doi: 10.1073/pnas.1832855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Laake LW, Passier R, Monshouwer-Kloots J, Verkelij A, Lips D, Freund C, den Ouden K, Ward-van Oostwaard D, Korving J, Tertoolen L, et al. Stem Cell Res. 2007;1:9–24. doi: 10.1016/j.scr.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Winter EM, Gittenberger-de Groot AC. Cell Mol Life Sci. 2007;64:692–703. doi: 10.1007/s00018-007-6522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SM, Fujiwara Y, Cibulsky SM, Clapham DE, Lien CL, Orkin SH. Cell. 2006;127:1137–1150. doi: 10.1016/j.cell.2006.10.028. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. Published online November 20, 2007. [DOI] [PubMed] [Google Scholar]
- Zimmermann WH, Melnychenko I, Wasmeier G, Didié M, Naito H, Nixdorff U, Hess A, Budinsky L, Brune K, Michaelis B, et al. Nat Med. 2006;12:452–458. doi: 10.1038/nm1394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data include one table and can be found with this article online at http://www.cell.com/cgi/content/full/132/4/XXX/DC1/.