

Abstract
Mycobacterium tuberculosis α–isopropylmalate synthase (MtIPMS) catalyzes the condensation of AcCoA with α–ketoisovalerate (α–KIV) and the subsequent hydrolysis of α–isopropylmalyl-CoA to generate the products CoA and α–isopropylmalate (α–IPM). This is the first committed step in L–leucine biosynthesis. We have purified recombinant MtIPMS and characterized it using a combination of steady-state kinetics, isotope effects, isotopic labeling, and 1H-NMR spectroscopy. The α–keto acid specificity of the enzyme is narrow and the acyl-CoA specificity is absolute for AcCoA. In the absence of α–KIV MtIPMS does not enolize the α–protons of AcCoA, but slowly hydrolyzes acyl-CoA analogs. Initial velocity studies, product inhibition, and dead-end inhibition studies indicate that MtIPMS follows a nonrapid equilibrium random Bi Bi kinetic mechanism, with a preferred pathway to the ternary complex. MtIPMS requires two catalytic bases for maximal activity (both with pKa values of ca. 6.7), and we suggest that one catalyzes deprotonation and enolization of AcCoA and the other activates the water molecule involved in the hydrolysis of α–isopropylmalyl-CoA. Primary deuterium and solvent kinetic isotope effects indicate that there is a step after chemistry that is rate limiting, although with poor substrates such as pyruvate, hydrolysis becomes partially rate-limiting. Our data is inconsistent with the suggestion that a metal-bound water is involved in hydrolysis. Finally, our results indicate that the hydrolysis of α–isopropylmalyl-CoA is direct, without the formation of a cyclic anhydride intermediate. Based on these results, a chemical mechanism for the MtIPMS-catalyzed reaction is proposed.
Keywords: α-Isopropylmalate Synthase, Steady-State Kinetics, Chemical Mechanism
Mycobacterium tuberculosis, the causative agent of Tuberculosis, is intrinsically resistant to many first line antibiotics used to treat other common bacterial infections and resistance to effective drugs is increasing. The characterization of new targets and the development of new drugs are urgently needed. Mycobacteria, like many other bacteria, synthesize the branched-chain amino acids L-valine and L –leucine, and pantothenic acid, from α-ketoisovalerate1 (α–KIV). Studies using transposon mutagenesis in mycobacteria have demonstrated that the L-leucine biosynthetic pathway is essential for Mycobacterium bovis both in vitro and in vivo (1-3). These results suggest that mycobacteria, engulfed within the macrophage phagolysosome-like compartment, cannot obtain sufficient L-leucine from the surrounding environment. These findings also validate the L-leucine biosynthetic pathway as a target for inhibitor design and drug development to treat Tuberculosis. In spite of its importance, the L-leucine biosynthetic pathway is among the least studied amino acid biosynthetic pathways.
The L-leucine branch of the branched-chain amino acid pathway (Scheme 1) starts with the AcCoA-dependent carboxymethylation of α–KIV catalyzed by α–isopropylmalate synthase (EC 2.3.3.13). This enzyme is subject to feedback inhibition by L-leucine in many organisms and the M. tuberculosis IPMS has recently been shown to exhibit slow-onset, feedback inhibition by L-leucine (4). α–Isopropylmalate is subsequently converted to β-isopropylmalate by α–isopropylmalate isomerase (EC 4.2.1.33) and then oxidatively decarboxylated to α–ketoisocaproate by β-isopropylmalate dehydrogenase (EC 1.1.1.85). α–Ketoisocaproate is converted directly to L-leucine by the action of a branched-chain amino acid transaminase (5). All of the genes involved in L-leucine biosynthesis have defined orthologues in the M. tuberculosis genome (6, 7).
Scheme 1.
α–Isopropylmalate synthase (IPMS) catalyzes a Claisen-type condensation between α–KIV and AcCoA. The most well-characterized members of this family are malate synthase (MS), a component of the glyoxylate pathway, citrate synthase (CS), a component of the Krebs cycle, and most recently homocitrate synthase (HCS), a component of the yeast α–aminoadipate pathway (8). Despite the similarities in their respective substrates and the chemical reactions catalyzed, MS and CS perform the condensation reaction using quite different catalytic mechanisms. MS from both yeast and M. tuberculosis use a Mg2+ ion (9, 10) to bind and polarize the carbonyl group of glyoxylate, while CS does not use a divalent metal, but rather a pair of histidine residues to polarize the carbonyl group of oxaloacetate. In addition, CS from different organisms can display quite different pH dependences, with the Thermoplasma acidophilum CS reaction being independent of pH and the pig heart CS reaction being dependent on two ionizable enzyme groups (11).
IPMS from Salmonella enterica serovar typhimurium (12-15), Alcaligenes eutrophus H16 (16-18), Neurospora (19), and Saccharomyces (20) have been partially characterized. The common features of most α–IPM synthases are a requirement for monovalent cations for maximal activity, feedback inhibition by L-leucine, and narrow substrate specificity for analogs of α–KIV. In contrast, there are many differences revealed by these studies including the effects of divalent metals, inhibition by some α-keto acids, feedback regulation, and allosteric properties. Chanchaem and Palittapongarnpim have cloned, expressed and purified active MtIPMS, but no rigorous kinetic studies were performed (21). Recently, Koon et al. solved the three-dimensional crystal structure of IPMS from M. tuberculosis with Zn2+ and α–KIV bound at the active site (22). The insertion of two residues of one monomer into the active site of the other in the dimeric enzyme suggests how regulation by L-leucine may be achieved (22). Our own studies have revealed an absolute dependence for a monovalent cation for activity and potassium appears to be the physiologically relevant monovalent ion. Additionally, the monovalent cation is required for the binding of, and activation of catalysis by, a divalent metal that is used to coordinate the carbonyl and carboxyl oxygens of α-KIV (25).
This paper describes the detailed biochemical characterization of the recombinant α-isopropylmalate synthase from M. tuberculosis H37Rv. Using a combination of steady-state kinetics, primary deuterium and solvent kinetic isotope effects, isotopic labeling, and 1H-NMR spectroscopy, the following questions were addressed. (i) What is the substrate specificity for α–keto acids and acyl-CoA analogs? (ii) What is the kinetic mechanism of MtIPMS? (iii) Does acid/base chemistry play an important role in catalysis? (iv) Does the enzyme hydrolyze acyl-CoA analogs? (v) What is the rate-limiting step in the reaction? (vi) What is the chemical mechanism used for intermediate cleavage?
Materials and Methods
Materials
(R)-Hydroxyisovalerate was obtained from Fluka. α-Keto acids, acyl-CoAs, buffers and all other chemicals were obtained from Sigma, Aldrich or Fisher. Nickel-NTA resin was from Novagen. Chromatographic columns and resins were obtained from Pharmacia. Chelex® 100 was purchased from Bio-Rad.
General Methods
Protein concentration was measured using the bicinchoninic acid method (Pierce), using bovine serum albumin as standard. Protein electrophoresis was performed using the Phast System (Pharmacia) and 10−15% SDS-PAGE Gradient PhastGels. All spectrophotometric assays were performed using a UVIKON XL UV-Vis spectrophotometer equipped with a circulating water bath and thermospacers. All experiments analyzed using Lineweaver-Burke plots were performed in duplicate, or triplicate when the measured parameters were greater than 10% different in duplicate experiments. NMR spectroscopy experiments were performed on a Bruker DRX300 NMR Spectrometer at 25°C (AECOM Structural NMR Resource). Cloning of the leuA gene (Rv3710) and expression and purification of MtIPMS are described elsewhere (4).
Characterization of IPMS
The monomer molecular mass was estimated by SDS-PAGE. Protein samples were analyzed by electrospray ionization/mass spectrometry (Laboratory of Mass Spectrometry and Proteomics - A.E.C.OM.) for accurate determination of the monomer mass.
Oligomeric State
Analytical gel filtration experiments were performed with a Superose 12 Column (Pharmacia) in 20 mM TEA, pH 7.8 containing 100 mM KCl at a flow rate of 0.2 mL/min. Dynamic light scattering was measured with a DynaPro MS/X dynamic light-scattering instrument (Protein Solutions) with samples of MtIPMS at 10 mg/mL in 20 mM TEA, pH 7.8. Sedimentation velocity experiments were performed in a Beckman Optima XL-1 analytical centrifuge at 25°C and a rotor speed of 35,000 rpm using an An-Ti 60 rotor. Sedimentation boundaries were followed at 280 nm using the absorption optics. The samples were loaded into double sector centrifuge cells and equilibrated at 25°C for 15−30 min prior to initiating centrifugation. The sedimentation and diffusion coefficients were determined using the program SVEDBERG v6.39 (23) using a v̄ = 0.7173 calculated from the amino acid composition. These parameters were normalized to the standard conditions of 20°C using SEDNTERP v1.08.
Measurement of Enzymatic Activity
Initial velocities for the forward reaction of α-isopropylmalate synthase were determined using 4,4’-dithiodipyridine (DTP) to detect the formation of CoA at 324 nm (ε = 19,800 M−1 cm−1) at 25°C (24). A typical reaction mix contained 50 mM HEPES, pH 7.5, 20 mM KCl, 20 mM MgCl2, 100 μM DTP, 1 mM AcCoA, and 0.5 mM α-KIV. Reactions were initiated by the addition of enzyme, typically 20 nM final concentration. Potassium and magnesium were included because they are required for maximal activity (25). An alternative assay monitored the decrease in absorbance at 232 nm due to the hydrolysis of the thioester of AcCoA (Δε = 4,500 M−1 cm−1), using either phosphate or Tris buffers (26).
Product Analysis by 1H-NMR
The reaction of MtIPMS with AcCoA and α-KIV was followed by 1H-NMR. Reaction mixtures consisted of 50 mM potassium phosphate buffer pH 7.0, 12 mM MgCl2, 1.1 mM α-KIV, 1 mM AcCoA, and 100 μg of MtIPMS. Reactions containing propionyl-CoA and α-KIV were performed in a similar manner.
Preparation of [2H3-methyl]-AcCoA
[2H3-methyl]-AcCoA was prepared by S-acetylation of CoA using deuterated acetic anhydride (Aldrich). A typical acetylation reaction contained 100 mM TEA, pH 7.8, 40 mM [2H3-methyl]-acetic anhydride and 20 mM CoA in 1 mL of 50% ethanol. The reaction was monitored using DTP, and the reaction was complete in 10 min at 37°C. Reaction mixtures were stored at −20°C and the exact concentration of [2H3-methyl]-AcCoA was determined spectrophotometrically (26), using the adenine ring absorbance at 260 nm (ε = 16,400 M−1 cm−1) and the thioester bond absorbance at 232 nm (ε = 8,700 M−1 cm−1). The values obtained agreed to ± 1%, indicating stability of AcCoA under these conditions. The extent of deuteration of [2H3-methyl]-AcCoA was determined by 1H-NMR.
pH studies
The pH dependence of V/Ka-KIV (kcat/Km) was determined by varying the concentration of α-KIV at a fixed, saturating concentration of AcCoA. The pH dependence of V/KAcCoA (kcat/Km) and V (kcat) were determined by varying the concentration of AcCoA at a fixed, saturating concentration of α-KIV (27). The following buffers were used at the indicated pH ranges: MES, pH 6.50; PIPES, pH 6.38 − 7.15; HEPES, pH 7.11 − 7.87; TAPS, pH 8.05 − 8.79; CHES, pH 8.97. Experiments were performed with Good buffers, which have low metal-binding capabilities, using overlapping pH values (27, 28).
Primary Deuterium and Solvent Isotope Effects
Isotope effects were determined in 100 mM TAPS, pH 8.3 at saturating concentrations of Mg2+ and K+. Primary deuterium kinetic isotope effects on V and V/KAcCoA were determined using [1H3-methyl]- and [2H3-methyl]-AcCoA in the presence of saturating concentrations of either α–KIV or pyruvate. Solvent kinetic isotope effects on V and V/KAcCoA were determined in water or 80% deuterium oxide, in the presence of saturating concentrations of either α–KIV or pyruvate at pH 8.3. The proton inventory on the Vmax was performed by varying the atom fraction of deuterium oxide from 0 to 0.8 in increments of 0.2, in quadruplicate in the presence of saturating concentrations of substrates.
Enolization and hydrolysis of Ac-CoA
Enzyme-catalyzed enolization of AcCoA was assessed by 1H-NMR in D2O. The intensity of the peak corresponding to the α–methyl protons from AcCoA was measured as a function of time. Enzyme-catalyzed hydrolysis of AcCoA was also determined by 1H-NMR since the methyl proton resonances of AcCoA and acetic acid appear at different frequencies. Typical incubation mixtures contained 100 mM NaH2PO4, pH 7.0, 10 mM KCl, and 2 mM AcCoA, and 11.6 μM IPMS in 600 μL of D2O. The effect of Mg2+ was probed by adding 12 mM of MgCl2 to the reaction mixture. Enolization in the presence of either (R)- or (S)-α-hydroxyisovalerate were tested at concentrations of the alcohols of 3.5 mM. The intensity of the peaks were normalized relative to the peaks from the gem dimethyl groups of the pantetheine moiety of CoA.
Mechanism of hydrolysis by 18O isotope-induced shift on 13C-NMR signal
In order to probe the possible existence of an anhydride intermediate during the hydrolysis of the α-isopropylmalate-CoA thioester intermediate, we measured the 18O-induced isotope shifts of the 13C carbon resonances of the product, α-isopropylmalate (29, 30). The reaction mixture contained 100 mM NaH2PO4, pH 7.0, 8 mM KCl, 12 mM MgCl2, 20 mM AcCoA, 15 mM α-KIV, and 5 μM IPMS in 1 mL (70 % v/v, H218O). The reaction was allowed to proceed at 37°C for 120 min. The enzyme was removed by ultrafiltration and the ultrafiltrate was lyophilized. The lyophilized powder was suspended in 300 μL of D2O and the 13C spectrum was determined using 1H-decoupled 13C-NMR, averaging 60,000 scans.
Data analysis
Kinetic data were fitted using the nonlinear, least-square, curve-fitting programs of SigmaPlot 2000 for Windows, version 6.00. Individual saturation curves were fitted to
| (1) |
where V is the maximal velocity, A is the substrate concentration and K is the Michaelis constant for the substrate (Km). Individual saturating curves showing linear substrate inhibition were fitted to
| (2) |
where Ki is the apparent inhibition constant for substrate A. Data showing an intersecting initial velocity pattern on double reciprocal plots were fitted to
| (3) |
where A and B are the concentrations of substrates and KA and KB the Michaelis constants. Inhibition data showing linear, competitive or noncompetitive patterns in double reciprocal plots were fitted to eqs. 4 and 5, respectively,
| (4) |
| (5) |
where I is the inhibitor concentration and Kis and Kii the slope and intercept inhibition constants, respectively. Inhibition data, performed under saturating concentrations of substrates and variable concentrations of inhibitors were fitted to
| (6) |
where v is the rate in the presence of inhibitor at concentration I, v0 is the rate without inhibitor, IC50 is the concentration of inhibitor which gives 50% inhibition, and nH is the Hill coefficient. pH profile data were fitted to eq 7 for two nonresolvable acidic ionizable groups, eq. 8 for two acidic nonresolvable and one basic ionizable group, and eq. 9 for two acidic nonresolvable and two basic nonresolvable ionizable groups,
| (7) |
| (8) |
| (9) |
where C is the pH-independent plateau value, H is the hydrogen ion concentration, Ka and Kb are the respective acid and basic pKa constants for the ionizable groups. Primary deuterium and solvent isotope effects were fitted to eqs. 10, 11, and 12, for isotope effects on V only, V/K only, or both V and V/K, respectively
| (10) |
| (11) |
| (12) |
where Fi is the fraction of the isotopic label, and EV and EV/K are the isotope effects minus one on V and V/K, respectively. The linear proton inventory on Vmax was fitted to
| (13) |
where n is the atom fraction of deuterium in the mixed isotopic solvent, Vn is the velocity in the solvent with atom fraction of deuterium n, Vo is the velocity in H2O and kD/kH is the isotope effect. Errors were propagated as described in Skoog and West for indeterminate errors (31).
Results
General Properties of MtIPMS
Gel filtration of the homogeneous protein over a Superose 12 column generated a single peak that eluted at the position of the dimer in the presence of 100 mM KCl (data not shown). Dynamic light scattering revealed that in the absence, or presence, of 100 mM KCl, the protein is a dimer (data not shown). The sedimentation and diffusion coefficients were determined for MtIPMS at protein concentrations ranging from 2.4 to 9.3 μM in 40 mM TEA, pH 7.8 containing 8 mM KCl and 12 mM MgCl2, in the absence or presence of L-leucine. The sedimentation boundaries are well described by a single component; S, and D linearly decreased with increasing protein concentration, consistent with the protein behaving as a non-interacting monodisperse particle (data not shown). Extrapolation to infinite dilution yields values of S°20,w of 8.6 ± 0.3 and 8.7 ± 0.3 S, respectively and D°20,w of 7.8 ± 0.6 and 9.2 ± 0.8 F, respectively. The values of S and D calculated (32) from the crystal structure of the MtIPMS dimer (22) are 8.97 and 5.08 F, respectively.
Initial Velocity Studies
In order to characterize the kinetic mechanism, we performed initial velocity studies. The intersecting initial velocity pattern obtained by varying the concentration of α–KIV at fixed variable concentrations of AcCoA (Supplementary Information) is consistent with a sequential Bi Bi kinetic mechanism (33).
Substrate Specificity
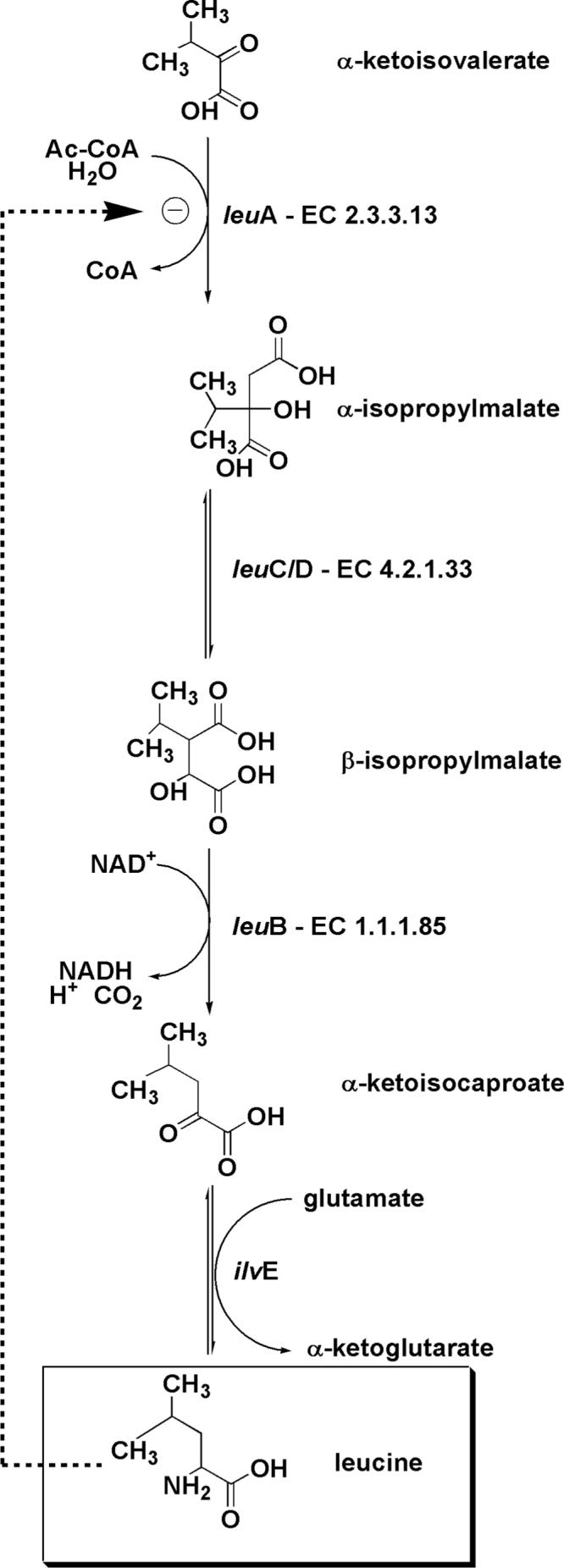
Kinetic parameters for α–keto acids and AcCoA are summarized in Table 1. No activity was detected up to 5 mM concentration of the following analogs: glyoxalate, α-ketoisocaproate, α-keto-β-methyl-n-valerate, α-ketoglutarate, phenylpyruvate and phenylglyoxalate. The apparent substrate inhibition observed with some α-keto acids (Supplementary Information) will be discussed below. The α–keto acids that are substrates for MtIPMS are α–ketoisovalerate, pyruvate, α–ketovalerate and α–ketobutyrate. The data presented in Figure 1A reveals that there is no “uncoupled” hydrolysis of AcCoA in the presence of α–KIV, and that the reaction rate measured in steady-state experiments is due entirely to hydrolysis of the initially formed condensation product, α-isopropylmalyl-CoA. Using propionyl-CoA, a very small quantity of the condensation product of α–KIV and propionyl-CoA is observed by 1H-NMR (Fig. 1B). However, greater than 95% of the steady-state rate observed with propionyl-CoA is due to hydrolysis. The rate of hydrolysis of propionyl-CoA is much slower than the rate of condensation and hydrolysis observed using AcCoA and α-KIV, which is complete in a few minutes under these conditions. Measurements of the rates of hydrolysis of AcCoA, propiony-CoA and crotonyl-CoA in the absence of α–KIV, under steady-state conditions, indicate that MtIPMS does very slowly hydrolyze acyl-CoA analogs (Table 2, and data not shown). These results confirm that AcCoA is the only acyl-CoA analog accepted in the condensation reaction by MtIPMS.
Table 1.
Steady-State Kinetic Parameters for M. tuberculosis IPMSa
| Fixed Substrate | Variable Substrate | kcat (s−1) | Km (μM) | kcat/Km (s−1 M−1) (×10−2) |
|---|---|---|---|---|
| AcCoA | α—KIV | 3.5 ± 0.1 | 12 ± 1 | 34000 ± 260 |
| AcCoA | α—KV | 7.0 ± 1.3 | 410 ± 180 | 5900 ± 80 |
| AcCoA | α—KB | 10.7 ± 0.4 | 860 ± 160 | 8000 ± 25 |
| AcCoA | PY | 6.1 ± 0.5 | 9500 ± 1680 | 16 ± 1 |
| α—KIV | AcCoA | 2.1 ± 0.1 | 136 ± 5 | 150 ± 9 |
| α—KV | AcCoA | 5.0 ± 0.2 | 279 ± 31 | 180 ± 20 |
| α—KB | AcCoA | 7.6 ± 0.1 | 57 ± 2 | 1300 ± 49 |
| PY | AcCoAb | 4.5 ± 0.3 | 74 ± 13 | 610 ± 120 |
At pH 7.4 and 25°C. The following concentrations of fixed co-substrate were used: 1 mM AcCoA, 250 μM α—KIV, 4 mM α—KV, 10 mM α—KB, and 100 mM PY.
Saturation curves displaying apparent linear substrate inhibition were fitted to eq. 2. The Ki for α—KV at a fixed saturating concentration of AcCoA was 10.1 ± 4.4 mM, and the Ki for AcCoA in the presence of a fixed saturating concentration of pyruvate was 4.0 ± 1.3 mM.
Figure 1. 1H-NMR analysis of the reaction of MtIPMS.
(A) Region of spectral interest of the reaction of MtIPMS with AcCoA and α-KIV. Reaction mixture without enzyme (bottom spectrum) and reaction after reaching completion (top spectrum). (B) Region of interest of the spectra of reaction of MtIPMS with propionyl-CoA and α–KIV. Reaction mixture without enzyme (bottom spectrum) and reaction after reaching completion (top spectra). Although data was recorded throughout the experiments only the last spectrum is shown in each case.
Table 2.
Steady-State Kinetic Parameters for the α—Keto Acid-Independent Hydrolysis of Acyl-CoA Analogs Catalyzed by M. tuberculosis IPMSa.
| Without added MgCl2 |
With added MgCl2 |
|||||
|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km (s−1 M−1) | Km (μM) | kcat (s−1) | kcat/Km (s−1 M−1) | |
| Acetyl-CoA | 370 ± 40 | (6 ± 0.3) × 10−2 | 150 ± 19 | 160 ± 29 | (3 ± 0.2)× 10−2 | 170 ± 36 |
| Propionyl-CoA | 220 ± 40 | (4 ± 0.3) × 10−2 | 190 ± 36 | NDb | ND | - |
| Crotonyl-CoA | 50 ± 3 | (5 ± 0.2) × 10−2 | 940 ± 72 | 95 ± 15 | (4 ± 0.2)× 10−2 | 400 ± 70 |
. Performed at 25°C and pH 7.4.
. (ND) Not determined.
Product and Dead-End Inhibition Studies
Due to the nature of our assay that precludes the use of any compound with free thiol (e.g., CoA), and the high Km for AcCoA, we did not attempt to use CoA as a product inhibitor to study the order of substrate binding. Product inhibition studies with α–IPM and dead-end inhibition studies with (S)-α-hydroxyisovalerate (α-IPM analog) and dethio-CoA (CoA analog) were performed and the results are summarized in Table 3. The competitive patterns obtained with α–IPM versus α–KIV and dethio-CoA versus AcCoA indicate a random Bi Bi kinetic mechanism. Further support for the random nature of substrate binding come from the apparent linear substrate inhibition observed under some experimental conditions (Table 1 and Supplementary Information). These patterns are a hallmark of nonrapid equilibrium random Bi Bi kinetic mechanisms where there is a preferred route to the ternary complex (34, 35).
Table 3.
Product and Dead-End Inhibition Patterns for M. tuberculosis IPMSa
| varied substrate | inhibitor | conc. of the co-substrate | patternb | Kis (mM) | Kii (mM) |
|---|---|---|---|---|---|
| AcCoA | α—IPM | sat α—KIV | NIc | - | - |
| AcCoA | α—IPM | 5 X Km α—KIV | NC | 0.02 ± 0.03 | 0.07 ± 0.04 |
| α—KIV | α—IPM | sat AcCoA | C | 0.20 ± 0.15 | - |
| α—KIV | (S)-α-HIV | sat AcCoA | NC | 1.7 ± 0.6 | 20.7 ± 5.3 |
| α—KIV | Dethio-CoA | sat AcCoA | NC | 2.4 ± 0.6 | 1.8 ± 0.2 |
| AcCoA | Dethio-CoA | sat α—KIV | C | 1.06 ± 0.04 | - |
At pH 7.4, 25°C, 20 mM KCl, and 20 mM MgCl2. The concentration of co-substrates were held fixed and saturating (α—KIV, 500 μM and AcCoA, 1 mM), unless otherwise stated.
C = competitive and NC = noncompetitive.
NI = no inhibition.
Enolization and Hydrolysis of AcCoA
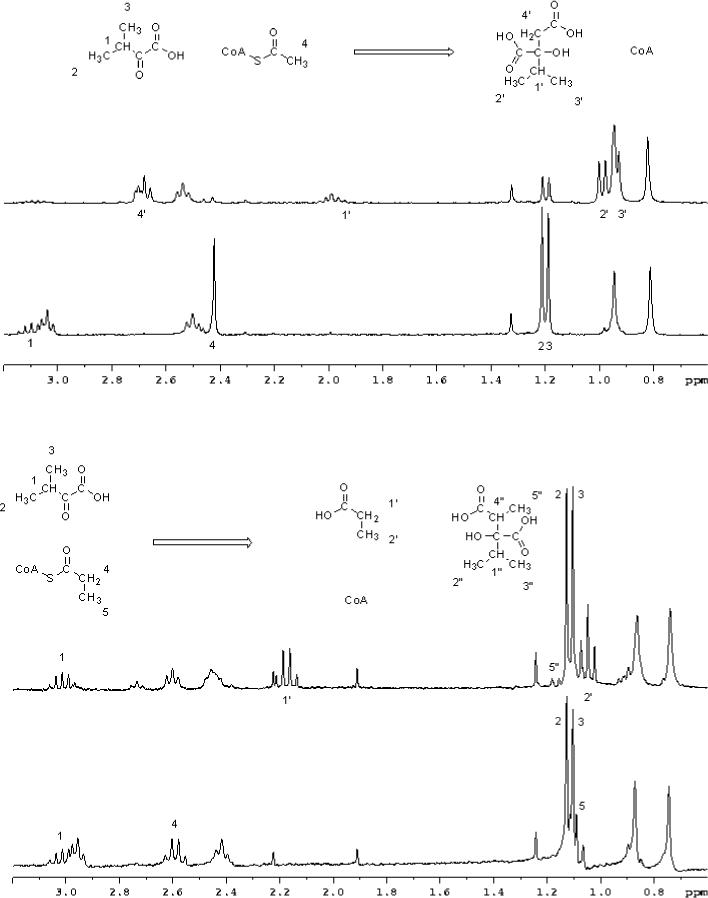
To gain information about the chemical mechanism of MtIPMS, we examined the enzyme-catalyzed exchange of the α–methyl protons of AcCoA in D2O in the absence of α-KIV using 1H-NMR spectroscopy. Figure 2A shows a representative stack plot of 1H-NMR spectra over the course of the reaction. The singlet at 2.45 ppm corresponds to the three methyl hydrogens from the acetyl group of AcCoA, the singlet at 1.95 ppm arises from the methyl hydrogens of acetate and the singlet at 0.95 ppm corresponds to the pantetheine gem dimethyl hydrogens used as an internal calibration. Figure 2B is a plot of the corrected intensities of the peaks shown in Figure 2A versus time. The excellent correspondence between the disappearance of the AcCoA peak and the appearance of an acetate peak indicates that MtIPMS does not catalyze the exchange of the α–methyl protons of AcCoA with solvent at any significant rate, but does slowly hydrolyze AcCoA. This rate is (3.0 ± 0.2) × 10−2 s−1 in the presence of saturating mono- and divalent metals (Table 2). The presence of MgCl2, or product analogs, like (R)- or (S)-α-hydroxyisovalerate, did not have any effect on the rates of hydrolysis or enolization of AcCoA (data not shown).
Figure 2. MtIPMS-catalyzed enolization and hydrolysis of AcCoA probed by 1H-NMR.

(A) Stack plot of the region of interest from the 1H-NMR spectra of reaction of MtIPMS with AcCoA. The spectra were recorded at 0, 15, 30, 45, 60, 75, and 90 min. The peak at 2.3 ppm corresponds to the protons of the acetyl group of the AcCoA, the peak at 1.95 corresponds to the protons of the free acetate and the peak at 0.94 ppm corresponds to the protons of the gem dimethyl groups of the pantetheine moiety of AcCoA, used as internal control. (B) Plot of the corrected intensity of each peak versus time. Control peak is represented by open circles, AcCoA protons by open squares, acetate peak by open triangles, and the difference between acetyl group and acetate group, which correspond to the exchange or enolization are represented by open diamonds.
pH Studies
To probe the role of general acid/base chemistry in the mechanism of MtIPMS, we determined the pH dependence of kcat and kcat/Km for both substrates, in the region of 6.00 ≤ pH ≤ 9.00 (Fig. 3). The kcat profile displays a sharp decrease at low pH, with a slope of two. A fit of the data to eq. 7, for two nonresolvable2 ionizable basic groups, provided estimates of the pKa values of 6.7 ± 0.1, suggesting that there are two active site bases that are essential for chemistry. The kcat/Kα–KIV profile is bell-shaped, with a slope of two on the acidic side and a slope of one on the basic side. A fit of the data to eq. 8 provided estimates of the pKa values of the two ionizable groups on the acidic side of 7.0 ± 0.1 and the basic ionizable group of 8.6 ± 0.3. This group may correspond to Arg 80 that is observed in the crystal structure to be within hydrogen bonding distance of the carbonyl group of the metal-bound α–KIV. The kcat/KAcCoA profile is also bell-shaped, with slopes of two on both the acidic and basic limbs. The basic residues exhibit pKa values of 6.5 ± 0.1, and the acidic residues exhibit pKa values of 8.5 ± 0.2 (eq. 9). The bases seen in both kcat/Km profiles are most likely the same seen in the kcat profile and their slight shift from the values observed in the kcat profiles is probably due to stickiness of the substrates3. The pKa of the enzymic acidic residues are most likely shifted outward due to the same reason. The groups seen at high pH values in the kcat/Km profiles are involved in substrate binding (27) and not catalysis, but we have no structural information with which to assign these groups to the corresponding amino acids.
Figure 3. pH profiles of MtIPMS reaction.

. The kcat and Km values were determined at each pH by varying the concentration of one substrate at a fixed saturating concentration of the other substrate, and saturating concentrations of K+ and Mg2+. The AcCoA data is represented by closed circles and the best fit of the data by the continuous line. The α–KIV data is represented by the open circles and the best fit of the data by the dashed line.
Primary Deuterium Isotope Effects
In order to define the rate-limiting step and characterize the chemical mechanism, we performed isotope effect experiments using [1H3-methyl]- and [2H3-methyl]-AcCoA4. No kinetic isotope effect was observed at fixed saturating concentrations of α-KIV and variable concentrations of [1H3-methyl]- or [2H3-methyl]-AcCoA at pH values of 6.3, 7.2 and 9.0. Substitution of α-KIV with α-KB, or pyruvate, which have Km values 70 and 800-fold higher than α-KIV, respectively, yielded similar results at pH 7.2. Similar results were obtained using saturating concentrations of [1H3-methyl]- or [2H3-methyl]-AcCoA and varying α-KIV (data not shown). The absence of any measurable primary deuterium kinetic isotope effect using deuterated AcCoA, at various pH values and with different substrates, strongly suggests that deprotonation and enolization of AcCoA are not the rate-determining steps in the MtIPMS reaction.
Solvent Isotope Effects
Solvent isotope effects were performed at pH 8.3 using 100 mM TAPS buffer. Using α-KIV as the α-keto acid substrate, small solvent isotope effects (0.9 − 1.2) were observed that are unlikely to be statistically significant. Using pyruvate as substrate, we were able to measure larger, normal solvent isotope effects5 (Table 4). The proton inventory on Vmax, using saturating concentrations of AcCoA and pyruvate, was linear. The linear proton inventory indicates that there is only one proton being transferred in the transition state for hydrolysis. These results support the idea that although with the natural substrate, hydrolysis is not rate-limiting, using less sticky substrates the hydrolytic step becomes partially rate-limiting.
Table 4.
Solvent Isotope Effects for M. tuberculosis IPMSa
| Solvent Isotope Effects | |||
|---|---|---|---|
| Fixed Substrate | Variable Substrate | D2OV | D2OV/K |
| Pyruvate | AcCoA | 2.0 ± 0.2 | 1 |
| AcCoA | Pyruvate | 1.8 ± 0.2 | 2.2 ± 0.5 |
Solvent isotope effects were determined in 100% H2O or 80 % D2O, at pH 8.3 and 25°C.
Hydrolysis of α–isopropylmalyl-CoA
To determine the mechanism used by MtIPMS to hydrolyze the α-isopropylmalyl-CoA intermediate (Fig. 4, inset), we used an 18O-labeling strategy and measuring the 18O induced shift on the 13C-NMR resonance for the product. Figure 4 shows the downfield carboxylate region of the 13C-NMR spectra of the α–isopropylmalate product generated in 72% H218O. The peak corresponding to the C1 carboxylate of α–IPM (182.6 ppm) is a singlet, while the C4 carboxylate resonance (180.3 ppm) is composed of two singlets, one corresponding to the carboxylate containing 18O (ca. 70%) and the other corresponding to the 16O containing carboxylate. The isotope-induced shift on the 13C resonance expected for a carboxylate containing 18O is about 0.035 ppm (36, 37), and we observed a 0.029 ppm isotope-induced shift. This result strongly suggests that hydrolysis of the α–isopropylmalyl-CoA thioester is unlikely to occur via intramolecular attack by the distal carboxylate on the thioester to generate the anhydride and CoA in solution. However, the formation of an intramolecular anhydride and its regiospecific hydrolysis on the enzyme cannot be formally ruled out by this result.
Figure 4. 18O labeling of α–isopropylmalyl-CoA hydrolysis.
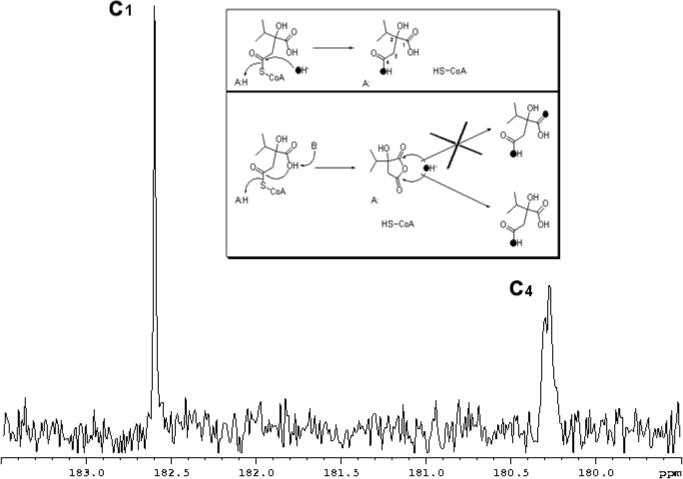
Region of interest of the 1H-decoupled 13C spectra showing the resonances of the C1 and C4 atoms of α–isopropylmalate and the unique 18O isotope-induce shift on the C4 carboxylate of α–isopropylmalate. Inset shows the scheme for the possible mechanisms for hydrolysis of α–isopropylmalyl-CoA, and the labeling patterns generated. The top panel represents the direct hydrolysis where 18O is represented by filled circles. The bottom panel represents the two mechanisms that could promote hydrolysis of the intramolecular anhydride formed by attack of C1 on the C4 thioester. The upper pathway represents a non-regioselective hydrolysis (in solution) and the lower pathway represents a regioselective hydrolysis (at the active site).
Discussion
General Properties
Gel filtration, dynamic light scattering, and sedimentation velocity experiments demonstrate that MtIPMS is a dimer in solution and this oligomeric state is not influenced by the K+ concentration, as is observed for bacterial citrate synthases (38). In addition, the sedimentation velocity studies show that L-leucine has no effect, within experimental error, on the oligomerization state of MtIPMS at the protein concentrations analyzed. The correspondence of the measured coefficients suggests that MtIPMS is a stable dimer in solution. The difference between the measured and calculated diffusion coefficient is intriguing in light of the observation from the MtIPMS structure that MtIPMS forms an extended dimer (22). The sedimentation data suggests a more compact arrangement of the regulatory and catalytic domains in solution than is observed in the crystal.
Substrate Specificity
The α–keto acid specificity of MtIPMS is very similar to what was reported for the IPMS of yeast, Salmonella, Alcaligenes, and Neurospora (14, 16, 19, 20). Only small α–keto acids can be accommodated in the active site, in agreement with the small size and hydrophobicity of the α–KIV binding site observed in the crystal structure (22).
The broad acyl-CoA specificity reported for some IPMS enzymes from other organisms should be viewed cautiously. The lack of a rigorous analysis of the products of the reaction, and the nature of the spectrophotometric assay used in these studies suggests that previous studies were incapable of distinguishing between bona fide condensation of the acyl-CoA substrate and simple hydrolysis uncoupled from condensation (14, 16, 19, 20). In addition, the rates of hydrolysis of these acyl-CoA analogs were not performed in the absence of α–KIV. Our data, specifically the 1H-NMR analysis of the reaction of AcCoA or propionyl-CoA with α–KIV indicates that propionyl-CoA is a very poor substrate and that the rate observed in the presence of α–KIV is due to hydrolysis of propionyl-CoA rather than its condensation and hydrolysis (Fig. 2B). Kinetic evidence comparing the rates of hydrolysis of propionyl-CoA and crotonyl-CoA in the absence or presence of α–KIV argues that MtIPMS cannot use CoA-linked acyl chains longer than two carbons. Inspection of the structure confirms that there is not enough room to accommodate a longer acyl group in the active site (22). In summary, MtIPMS has very narrow acyl donor substrate specificity, using only AcCoA as substrate.
Kinetic Mechanism
The intersecting initial velocity pattern obtained indicates that MtIPMS requires formation of a ternary complex before chemistry can take place. The patterns obtained using product and dead-end inhibitors are most consistent with a random Bi Bi kinetic mechanism (Scheme 2), with α–IPM being a competitive inhibitor versus α–KIV and dethio-CoA being a competitive inhibitor versus AcCoA. The two noncompetitive patterns observed (Table 3) are also indicative of a random mechanism with the formation of the abortive E:α–KIV:CoA and E:α–IPM:AcCoA complexes (Scheme 2).
Scheme 2.

Further indications of random substrate binding comes from the non-hyperbolic kinetics observed using variable amounts of α–KV at saturating concentrations of AcCoA, showing concave downward curvature on a double reciprocal plot (Supplementary Information), and using variable concentrations of AcCoA in the presence of saturating concentrations of pyruvate, displaying concave up curvature on the double reciprocal plot (Supplementary Information). In random, but not ordered mechanisms, upward curvature in a double reciprocal plot indicates that the rate of the chemical steps is greater than the rate of substrate release (39, 40). These substrate inhibition patterns are commonly found in nonrapid equilibrium random systems, where there is a preferred pathway to the ternary complex (34, 35, 39-41). The rapid equilibrium assumption is not valid for MtIPMS since the kcat/Km values for the all the α–keto acid substrates are much lower than one would expect for a diffusion controlled reaction, around 109 s−1 M−1 (42).
A non-unitary V/K isotope effect is observed when pyruvate is varied, at a fixed saturating concentration of AcCoA. The unitary value of D2OV/KAcCoA at a saturating pyruvate concentration indicates either that AcCoA is sticky under these conditions (dissociates more slowly than chemistry occurs), which is also in agreement with the nonrapid equilibrium kinetic mechanism (Scheme 2), or that with pyruvate the kinetic mechanism becomes ordered, with AcCoA binding before pyruvate (43).
Enolization and Condensation
Enolization of AcCoA, the first chemical step in the reaction of MtIPMS, is catalyzed by a general base with a pKa value of 6.7 (Fig. 3). The assignment of the active site base is not an easy task in the case of MtIPMS due to the presence of several strictly conserved ionizable residues in the active site (e.g., Arg80, Glu218, Glu317, Arg318, His379’, and Tyr410’) and the lack of a structure with AcCoA bound. Therefore, we will not speculate on the identity of the active site base without further mutagenesis data.
We were unable to detect enolization of AcCoA under a variety of conditions, including in the presence of α–hydroxy acid analogs of the substrate. This indicates a strong degree of coupling between the enolization and condensation reactions. The fact that enolization only takes place when the α–keto acid substrate is present indicates that MtIPMS might use a concerted mechanism for enolization and condensation. Concerted mechanisms are a common strategy used by enzymes to avoid formation of very unstable species, like the carbanionic enolate of AcCoA, and for rate-acceleration of enzymatic enolization reactions (e.g. mandelate racemase uses concerted general acid/base catalysis to promote rate acceleration of the proton abstraction from mandelate) (44-46). Figure 5 depicts the mechanism we propose for MtIPMS with a concerted enolization and condensation step. An alternative stepwise mechanism can be easily accommodated by assuming that enolization only occurs when α–KIV is bound and that the enzyme base does not exchange with solvent from the enolate complex. Malate synthase follows a stepwise mechanism (47), but the absence of primary deuterium isotope effects precludes the use of multiple isotope effects in our case. In the case of pig heart CS, the enolization of AcCoA is easily demonstrated, however enolization was not observed for the T. acidophilum CS (11), which could indicate that the latter follows a concerted pathway.
Figure 5. Proposed chemical mechanism for MtIPMS-catalyzed reaction.

B1 and B2 represent the two bases whose ionization are observed in the kcat pH profile. H:A is likely to be Arg 80 based on the structure of the E-Zn2+-α-KIV structure (22) and may be the additional ionizable group observed in the kcat/Km α-KIV pH profile. The acidic ionizable groups observed in the kcat/Km AcCoA pH profile cannot be assigned since no structural data on an AcCoA complex exist.
In summary, after deprotonation of AcCoA (1), condensation takes place more rapidly than reprotonation, and the α–hydroxyl group of the intermediate α–isopropylmalyl-CoA (2) is either rapidly protonated or stabilized as the alkoxide by Mg2+ coordination before release and protonation (Fig. 5).
Hydrolysis of α–isopropylmalyl-CoA
After condensation, a second enzymic base also exhibiting a pKa value of 6.7 abstracts a proton from a water molecule, which attacks the thioester carbon of the intermediate (2) forming a short-lived tetrahedral intermediate (3). The linear proton inventory obtained indicates that there is only a single proton in flight during the transition state for hydrolysis (Fig. 5). In addition, the normal solvent isotope effect observed indicates that the water molecule that is hydrolyzing the thioester intermediate is not bound to the divalent metal, which would be expected to yield an inverse solvent isotope effects (48). The intermediate 3 then collapses releasing the two products, CoA (4) and α–isopropylmalate. With the physiological substrates, this step is not rate-limiting. We do not believe that there is an α–isopropylmalate anhydride formed in the reaction cycle of MtIPMS. This conclusion is derived from three major observations. First, there is no incorporation of 18O label in the carboxylate derived from α–KIV during the reaction (Fig. 4), which excludes the possibility of release of an anhydride in solution. Second, MtIPMS is able to hydrolyze a variety of acyl-CoA analogs that cannot undergo cyclization in the absence of α–keto acids (Table 2). Third, inspection of the crystal structure of MtIPMS does not show any space in the active site pocket for a five-membered ring, without major structural rearrangements (22).
Finally, our inability to detect isotope effects with the physiological substrates under a variety of conditions suggests that a step other than chemistry is rate-limiting. This could be caused by slow substrate binding or product dissociation, a conformational change that allows for substrate binding or product release, or by a combination of both. Although there are other explanations for the lack of observable isotope effects in carbanion formation, such as very asymmetrical or nonlinear transition states, and very fast internal return (49), the slow dissociation of the substrates/products seems to fit with our kinetic and mechanistic data.
Conclusions
MtIPMS is a stable dimer in solution and this is not affected by the addition of K+, Mg2+, or the feedback inhibitor L-leucine. The α–keto acid specificity of MtIPMS is narrow and the acyl-CoA specificity is essentially absolute for AcCoA. MtIPMS is the first Claisen-condensing enzyme characterized to date that does not follow an ordered kinetic mechanism. It follows a nonrapid equilibrium random Bi Bi kinetic mechanism, with a preferred pathway to the ternary complex. MtIPMS uses general base chemistry for both deprotonation-condensation and hydrolytic reactions. With the physiological substrates a step other than chemistry is rate-limiting for the overall reaction. Using pyruvate, hydrolysis of α-methylmalyl-CoA is partially rate-limiting. Our solvent kinetic isotope effect data argues that the metal is not involved in the activation of the water molecule for hydrolysis. Finally, the α-isopropylmalyl-CoA thioester intermediate is directly hydrolyzed by water.
Supplementary Material
Acknowledgments
We thank Drs. Jordan Kriakov and William R. Jacobs Jr. for the generous donation of M. tuberculosis H37Rv genomic DNA. We also wish to thank Dr. Sean Cahill for his contributions to the NMR experiments, to Dr. Michael Brenowitz for assistance with the sedimentation velocity experiments and the interpretation of its results, and to Dr. James Errey for his assistance with preparation and purification of dethio-CoA. Finally, the authors wish to thank Mr. Rafael Guimarães da Silva for careful reading of this manuscript and to Dr. Argyrides Argyrou for thoughtful discussions.
This work was supported by NIH Grant A133696.
Footnotes
MtIPMS, M. tuberculosis α-isopropylmalate synthase; TEA, triethanolamine; HEPES, N-[2-hydroxyethyl]piperazine-N’-[2-ethanesulfonic acid]; TAPS, (N-tris[hydroxymethyl]methyl-2-aminopropanesulfonic acid); MES, 2-(N-morpholino)ethanesulfonic acid; CHES, 2-(N-cyclohexylamino) ethanesulfonic acid; PIPES, piperazine-N,N’-bis[2-ethanesulfonic acid]); Tris, tris(hydroxymethyl) aminomethane; α-KIV, α-ketoisovalerate; α-KB, α-ketobutyrate; α-KV, α-ketovalerate; PY, pyruvate; α-IPM, α-isopropylmalate; α-HIV, α-hydroxyisovalerate; CoA, Coenzyme A; AcCoA, acetyl-Coenzyme A; MS, malate synthase; CS, citrate synthase; HCS, homocitrate synthase; LB, Luria Broth; DTP, 4,4’-dithiodipyridine; IC50, concentration of inhibitor necessary to cause 50 % of loss in activity; NMR, nuclear magnetic resonance; KIE, kinetic isotope effects.
pH profile data showing slopes > 1 or < −1 are usually fitted to equations such as v = C /[(1+ H / Ka1) + (1+ H / Ka2 ) + (Kb1 / H ) + (Kb2 / H )] , which describe the pH-dependence of a rate constant on the ionization of two different conjugated acids and two different conjugated bases. During the analysis of our data it became clear that this equation did not fit our data well (e.g. very large standard error were associated with the K values). This might be due to the two pK values being very close one to another (e.g., nonresolvable) or that the addition of the extra variables requires many additional data points at lower and higher pH values, which are difficult to obtain with high accuracy, to increase the precision of the fit.
The pKa values seen in kcat/Km profiles, but not in kcat profiles, of sticky substrates are usually displaced outward by 0.3 to 0.5 pH units, to lower pH when protonation decreases activity and to higher pH when deprotonation decreases activity (26).
Using UV and 1H-NMR spectroscopy we were able to determine that greater than 99 % of CoA was converted to [2H3-methyl]-AcCoA, under the experimental conditions described (data not shown). MALDI-TOF analysis indicates that 9.6 ± 0.4 % of the product was bis-acetylated (data not shown). In the presence of α-KIV, the rates observed using [2H3-methyl]-AcCoA or commercial [1H3-methyl]-AcCoA were identical, under a variety of conditions suggesting that the 9.6 % of the [2H3-methyl]-AcCoA that was bis-acetylated does not have any effect that invalidates our conclusions.
The observation of normal solvent isotope effects for the reaction catalyzed by MtIPMS is inconsistent with the prediction, based on the crystal structure, that a metal bound water molecule is involved in the hydrolytic step (22). Metal bound water molecules have fractionation factors smaller than one and therefore will give rise to inverse solvent isotope effects (42).
Supporting Information Available
Initial velocity patterns for MtIPMS, plots showing apparent substrate inhibition by AcCoA (in the presence of saturating concentrations of pyruvate) and α–ketovalerate (in the presence of saturating concentrations of AcCoA) are shown. This material is available free of charge on the Internet at http://pubs.acs.org.
Bibliography
- 1.McAdam RA, Weisbrod TR, Martin J, Scuderi JD, Brown AM, Cirillo JD, Bloom BR, Jacobs WR., Jr. In vivo growth characteristics of leucine and methionine auxotrophic mutants of Mycobacterium bovis BCG generated by transposon mutagenesis. Infect Immun. 1995;63:1004–12. doi: 10.1128/iai.63.3.1004-1012.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bange FC, Brown AM, Jacobs WR., Jr. Leucine auxotrophy restricts growth of Mycobacterium bovis BCG in macrophages. Infect Immun. 1996;64:1794–9. doi: 10.1128/iai.64.5.1794-1799.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hondalus MK, Bardarov S, Russell R, Chan J, Jacobs WR, Jr., Bloom BR. Attenuation of and protection induced by a leucine auxotroph of Mycobacterium tuberculosis. Infect Immun. 2000;68:2888–98. doi: 10.1128/iai.68.5.2888-2898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Carvalho LP, Argyrou A, Blanchard JS. Slow-onset feedback inhibition: inhibition of Mycobacterium tuberculosis alpha-isopropylmalate synthase by L-leucine. J Am Chem Soc. 2005;127:10004–5. doi: 10.1021/ja052513h. [DOI] [PubMed] [Google Scholar]
- 5.Umbarger HE. In: Escherichia coli and Salmonella. Cellular and Molecular Biology. Neidhardt FC, editor. ASM Press; Washington, D.C.: 1996. pp. 442–449. [Google Scholar]
- 6.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Barrell BG, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–44. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 7.Wheeler PR, Blanchard JS. In: Tuberculosis and the Tubercle Bacillus. Cole ST, Eisenach KD, McMurray DN, Jacobs WR Jr., editors. ASM Press; Washington, D.C.: 2005. pp. 312–314. [Google Scholar]
- 8.Andi B, West AH, Cook PF. Kinetic mechanism of histidine-tagged homocitrate synthase from Saccharomyces cerevisiae. Biochemistry. 2004;43:11790–5. doi: 10.1021/bi048766p. [DOI] [PubMed] [Google Scholar]
- 9.Smith CV, Huang CC, Miczak A, Russell DG, Sacchettini JC, Honer zu Bentrup K. Biochemical and structural studies of malate synthase from Mycobacterium tuberculosis. J Biol Chem. 2003;278:1735–43. doi: 10.1074/jbc.M209248200. [DOI] [PubMed] [Google Scholar]
- 10.Anstrom DM, Kallio K, Remington SJ. Structure of the Escherichia coli malate synthase G:pyruvate:acetyl-coenzyme A abortive ternary complex at 1.95 A resolution. Protein Sci. 2003;12:1822–32. doi: 10.1110/ps.03174303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurz LC, Drysdale G, Riley M, Tomar MA, Chen J, Russell RJ, Danson MJ. Kinetics and mechanism of the citrate synthase from the thermophilic archaeon Thermoplasma acidophilum. Biochemistry. 2000;39:2283–96. doi: 10.1021/bi991982r. [DOI] [PubMed] [Google Scholar]
- 12.Teng-Leary E, Kohlhaw GB. Mechanism of feedback inhibition by leucine. Binding of leucine to wild-type and feedback-resistant alpha-isopropylmalate synthases and its structural consequences. Biochemistry. 1973;12:2980–6. doi: 10.1021/bi00740a006. [DOI] [PubMed] [Google Scholar]
- 13.Teng-Leary E, Kohlhaw GB. Binding of alpha-ketoisovalerate to alpha-isopropylmalate synthase. Half-of-the-sites and all-of-the-sites availability. Biochim Biophys Acta. 1975;410:210–9. doi: 10.1016/0005-2744(75)90221-1. [DOI] [PubMed] [Google Scholar]
- 14.Kohlhaw G, Leary TR, Umbarger HE. Alpha-isopropylmalate synthase from Salmonella typhimurium. Purification and properties. J Biol Chem. 1969;244:2218–25. [PubMed] [Google Scholar]
- 15.Leary TR, Kohlhaw G. Dissociation of alpha-isopropylmalate synthase from Salmonella typhimurium by its feedback inhibitor leucine. Biochem Biophys Res Commun. 1970;39:494–501. doi: 10.1016/0006-291x(70)90605-4. [DOI] [PubMed] [Google Scholar]
- 16.Wiegel J, Schlegel HG. alpha-Isopropylmalate synthase from Alcaligenes eutrophus H 16. II. Substrate specificity and kinetics. Arch Microbiol. 1977;112:247–54. doi: 10.1007/BF00413088. [DOI] [PubMed] [Google Scholar]
- 17.Wiegel J, Schlegel HG. Alpha-Isopropylmalate synthase from Alcaligenes eutrophus H 16 I. Purification and general properties. Arch Microbiol. 1977;112:239–46. doi: 10.1007/BF00413087. [DOI] [PubMed] [Google Scholar]
- 18.Wiegel J, Schlegel HG. Alpha-isopropylmalate synthase from Alcaligenes eutrophus H 16. III. Endproduct inhibition and its relief by valine and isoleucine. Arch Microbiol. 1977;114:203–10. doi: 10.1007/BF00446863. [DOI] [PubMed] [Google Scholar]
- 19.Webster RE, Gross SR. The α-isopropylmalate synthetase of Neurospora. I. The Kinetics and End Product Control of a-Isopropylmalate Synthetase Function. Biochemistry. 1965;4:2309–2318. [Google Scholar]
- 20.Kohlhaw GB. Alpha-isopropylmalate synthase from yeast. Methods Enzymol. 1988;166:414–23. doi: 10.1016/s0076-6879(88)66054-x. [DOI] [PubMed] [Google Scholar]
- 21.Chanchaem W, Palittapongarnpim P. A variable number of tandem repeats result in polymorphic alpha-isopropylmalate synthase in Mycobacterium tuberculosis. Tuberculosis (Edinb) 2002;82:1–6. doi: 10.1054/tube.2001.0314. [DOI] [PubMed] [Google Scholar]
- 22.Koon N, Squire CJ, Baker EN. Crystal structure of LeuA from Mycobacterium tuberculosis, a key enzyme in leucine biosynthesis. Proc Natl Acad Sci U S A. 2004;101:8295–300. doi: 10.1073/pnas.0400820101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Philo JS. An improved function for fitting sedimentation velocity data for low-molecular-weight solutes. Biophys J. 1997;72:435–44. doi: 10.1016/S0006-3495(97)78684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jocelyn PC. In: Sulfur and Sulfur Amino Acids. Jakoby WB, Griffith OW, editors. Academic Press, Inc.; Orlando: 1987. pp. 44–50. [Google Scholar]
- 25.de Carvalho LP, Blanchard JS. Kinetic Analysis of the Effects of Monovalent Cations and Divalent Metals on the Activity of Mycobacterium tuberculosis α-Isopropylmalate Synthase. Arch Biochem Biophys In Press. 2006 doi: 10.1016/j.abb.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 26.Dawson RMC, Elliot DC, Elliot WH, Jones KM. Oxford University Press; New York: 1991. [Google Scholar]
- 27.Cleland WW. The use of pH studies to determine chemical mechanisms of enzyme-catalyzed reactions. Methods Enzymol. 1982;87:390–405. doi: 10.1016/s0076-6879(82)87024-9. [DOI] [PubMed] [Google Scholar]
- 28.Stoll VS, Blanchard JS. Buffers: principles and practice. Methods Enzymol. 1990;182:24–38. doi: 10.1016/0076-6879(90)82006-n. [DOI] [PubMed] [Google Scholar]
- 29.Risley J, Van Etten R. An 18O Isotope Shift upon 13C NMR Spectra and Its Application to the Study of Oxygen Exchange Kinetics. Journal of the American Chemical Society. 1979;101:252–253. [Google Scholar]
- 30.Risley J, Van Etten R. 18O-Isotope Effect in 13C Nuclear Magnetic Resonance Spectroscopy. 2. The Effect of Structure. Journal of the American Chemical Society. 1980;102:4609–4614. [Google Scholar]
- 31.Skoog DA, West DM. Fundamentals of Analytical Chemistry. Saunders College Publishing; Philadelphia, PA: 1982. [Google Scholar]
- 32.Garcia De La Torre J, Huertas ML, Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys J. 2000;78:719–30. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cleland WW. Determining the chemical mechanisms of enzyme-catalyzed reactions by kinetic studies. Adv Enzymol Relat Areas Mol Biol. 1977;45:273–387. doi: 10.1002/9780470122907.ch4. [DOI] [PubMed] [Google Scholar]
- 34.Segel IH. Enzyme Kinetics, Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. John Wiley & Sons, Inc.; New York: 1993. [Google Scholar]
- 35.Ferdinand W. The interpretation of non-hyperbolic rate curves for two-substrate enzymes. A possible mechanism for phosphofructokinase. Biochem J. 1966;98:278–83. doi: 10.1042/bj0980278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Risley JM, Etten RLV. Oxygen-18 isotope effect in carbon-13 nuclear magnetic resonance spectroscopy. 2. The effect of structure. J Am Chem Soc. 1980;102:4609–4614. [Google Scholar]
- 37.Risley JM, Etten RLV. An oxygen-18 isotope shift upon carbon-13 NMR spectra and its application to the study of oxygen exchange kinetics. J Am Chem Soc. 1979;101:252–253. [Google Scholar]
- 38.Tong EK, Duckworth HW. The quaternary structure of citrate synthase from Escherichia coli K12. Biochemistry. 1975;14:235–41. doi: 10.1021/bi00673a007. [DOI] [PubMed] [Google Scholar]
- 39.Bar-Tana J, Cleland WW. Rabbit muscle phosphofructokinase. II. Product and dead end inhibition. J Biol Chem. 1974;249:1271–6. [PubMed] [Google Scholar]
- 40.Williams JW, Northrop DB. Kinetic mechanisms of gentamicin acetyltransferase I. Antibiotic-dependent shift from rapid to nonrapid equilibrium random mechanisms. J Biol Chem. 1978;253:5902–7. [PubMed] [Google Scholar]
- 41.Williams KR, Schofield P. J Biol Chem. Vol. 252. 1977. Kinetic mechanism of tRNA nucleotidyltransferase from Escherichia coli; pp. 5589–97. [PubMed] [Google Scholar]
- 42.Fersht A. Structure and Mechanism in Protein Science. W.H Freeman and Company.; New York: 1999. [Google Scholar]
- 43.Cook PF. In: Enzyme Mechanism from Isotope Effects. Cook PF, editor. CRC Press, Inc.; Boca Raton: 1991. pp. 203–207. [Google Scholar]
- 44.Gerlt JA, Kozarich JW, Kenyon GL, Gassman PG. Electrophilic catalysis can explain the unexpected acidity of carbon acids in enzyme-catalyzed reactions. J Am Chem Soc. 1991;113:9667–9669. [Google Scholar]
- 45.Gerlt JA, Gassman PG. Understanding the rates of certain enzyme-catalyzed reactions: proton abstraction from carbon acids, acyl-transfer reactions, and displacement reactions of phosphodiesters. Biochemistry. 1993;32:11943–52. doi: 10.1021/bi00096a001. [DOI] [PubMed] [Google Scholar]
- 46.Mitra B, Kallarakal AT, Kozarich JW, Gerlt JA, Clifton JG, Petsko GA, Kenyon GL. Mechanism of the reaction catalyzed by mandelate racemase: importance of electrophilic catalysis by glutamic acid 317. Biochemistry. 1995;34:2777–87. doi: 10.1021/bi00009a006. [DOI] [PubMed] [Google Scholar]
- 47.Clark JD, O'Keefe SJ, Knowles JR. Malate synthase: proof of a stepwise Claisen condensation using the double-isotope fractionation test. Biochemistry. 1988;27:5961–71. doi: 10.1021/bi00416a020. [DOI] [PubMed] [Google Scholar]
- 48.Quinn DM, Sutton LD. In: Enzyme Mechanism from Isotope Effects. Cook PF, editor. CRC Press, Inc.; Boca Raton: 1991. pp. 73–126. [Google Scholar]
- 49.Buncel E, Dust JM. Carbanion Chemistry. Structure and Mechanisms. Oxford University Press; New York, NY: 2003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.