

Abstract
Imprinted genes are monoallelically expressed from either the maternal or paternal genome. Because cancer develops through genetic and epigenetic alterations, imprinted genes affect tumorigenesis depending on which parental allele undergoes alteration. We have shown previously in a mouse model of neurofibromatosis type 1 (NF1) that inheriting mutant alleles of Nf1 and Trp53 on chromosome 11 from the mother or father dramatically changes the tumor spectrum of mutant progeny, likely due to alteration in an imprinted gene(s) linked to Nf1 and Trp53. In order to identify imprinted genes on chromosome 11 that are responsible for differences in susceptibility, we tested candidate imprinted genes predicted by a bioinformatics approach and an experimental approach. We have tested 30 candidate genes (Havcr2, Camk2b, Ccdc85a, Cntnap1, Ikzf1, 5730522E02Rik, Gria1, Zfp39, Sgcd, Jup, Nxph3, Spnb2, Asb3, Rasd1, Map2k3, Map2k4, Trp53, Serpinf1, Crk, Rasl10b, Itga3, Hoxb5, Cbx1, Pparbp, Igfbp4, Smarce1, Stat3, Atp6v0a1, Nbr1, and Meox1), two known imprinted genes (Grb10 and Impact) and Nf1, which has not been previously identified as an imprinted gene. Although we confirmed the imprinting of Grb10 and Impact, we found no other genes imprinted in the brain. We did, however, find strain-biased expression of Camk2b, 5730522E02Rik, Havcr2, Map2k3, Serpinf1, Rasl10b, Itga3, Asb3, Trp53, Nf1, Smarce1, Stat3, Cbx1, Pparbp, and Cntnap1. These results suggest that the prediction of imprinted genes is complicated and must be individually validated.
Keywords: Imprinting, Taqman PCR, Neurofibromatosis type 1, p53, Allelic Imbalance
Introduction
Genes that are imprinted show monoallelic expression from either the maternal or the paternal genome. Imprinted marks are established during spermatogenesis and oogenesis to label the maternal and paternal genomes differently and control expression of particular genes. Although much has been learned about how individual genes are marked for imprinting,1 the general rules for establishing imprinting marks are not completely known making it difficult to predict which genes are imprinted from the genomic DNA sequence.
Recently two studies have taken different approaches to genome-wide prediction of imprinted genes. Nikaido and colleagues2, 3 generated embryos derived entirely from the maternal genome (parthengenetic embryos) or from the paternal genome (androgenetic embryos) and compared the expression of 27,663 genes using a high-throughput microarray approach. They identified 2114 transcripts differentially regulated between the two types of experimentally derived embryos. In this analysis they found 11 known imprinted genes and further validated two novel transcripts by sequencing single nucleotide polymorphisms (SNPs) in cDNA derived from F1 reciprocal cross embryos. This study has provided a wealth of candidate imprinted genes, but also likely includes candidates that are not themselves imprinted, but whose expression are dependent directly or indirectly on the expression of an imprinted gene.
A more recent study by Luedi and colleagues4 used a bioinformatics rather than an experimental approach to identify candidate imprinted genes. Based on 44 known imprinted genes, a machine-learning algorithm was used to predict the imprinting status and whether the genes were maternally or paternally imprinted for 23,788 annotated mouse genes. Six hundred candidate genes were identified by the computer program and ranked by the P value for statistical significance. Statistical methods were used to validate the approach and the resulting data was used to predict the features important for imprinting. As with the study by Nikaido and colleagues, this computer modeling approach provided many candidates that can be further validated for their imprinting status.
We are interested in the role of imprinted genes in cancer. Cancer is fundamentally a disease of gene alteration, both at the genetic and at the epigenetic level.5, 6 Imprinted genes can play a role in cancer through epigenetic changes resulting in gene silencing or ectopic expression, or through genetic changes that cause loss or amplification of specific parental alleles of imprinted genes. For example, rhabdomyosarcomas have been shown to specifically lose the maternal copy of human chromosome 11,7 the paternal copy of human chromosome 19 is lost in oligodendrogliomas,8 and the maternal copy of human chromosome 11 is lost in SDHD-linked familial paraganglioma.9
Neurofibromatosis type 1 (NF1) is a familial cancer syndrome in which patients carry mutations in the NF1 gene encoding the neurofibromin protein. Neurofibromin functions as a rasGAP protein to downregulate ras signaling and when neurofibromin is mutated ras becomes hyperactivated, contributing to tumors as well as many other disease manifestations.10 NF1 patients develop benign neurofibromas and optic nerve gliomas, as well as malignant astrocytomas and malignant peripheral nerve sheath tumors. A study of monozygotic twins and more distantly related family members has demonstrated that genes unlinked to NF1 affect the severity of the disease.11 Because of this, we have been studying NF1 to better understand the mechanisms underlying cancer susceptibility. Using a mouse model of the malignancies associated with NF112–14 we have identified several genetic loci that alter susceptibility to astrocytoma and malignant peripheral nerve sheath tumors.15, 16
In our mouse model of NF1 malignancies, the mouse Nf1 gene and the Trp53 gene encoding the p53 protein are mutated on the same chromosome 11 in cis (Nf1−/+;Trp53−/+cis mice) such that they are inherited as a single mutation in genetic crosses. Interestingly, one of the strongest effects on astrocytoma and malignant peripheral nerve sheath tumor susceptibility is inheritance of the Nf1;Trp53 mutant chromosome from the mother or the father. Mice that inherit the Nf1;Trp53 mutation from their father are at an increased risk for developing malignant peripheral nerve sheath tumor15 and mice that inherit the Nf1;Trp53 mutation from their mother are at an increased risk for developing astrocytoma.16 Because these effects can be seen on a highly inbred C57BL/6J (B6) strain background, we hypothesize that an imprinted gene (or genes) on chromosome 11, as opposed to maternal effects, is responsible for the differences in susceptibility. When the wild-type copies of Nf1 and Trp53 are lost at the initiation of tumorigenesis, imprinted genes on chromosome 11 may be lost, amplified, or dysregulated leading to changes in susceptibility depending on whether the lost allele is the maternal or paternal copy. Four imprinted genes have been confirmed on chromosome 11 (Grb10, Commd1, U2af1-rs1, and the Ddc-exon1a transcript17) (http://www.geneimprint.com/site/genes-by-species.Mus+musculus, http://www.mgu.har.mrc.ac.uk/research/imprinting/maps/new/imprin-viewmaps11.html), however, many more are predicted to be imprinted. We have developed a sensitive method to test candidate imprinted genes using Taqman real-time PCR and have tested many of the candidate imprinted genes on mouse chromosome 11. This method allows one to determine whether a gene of interest shows an imprinted pattern of expression in tissue that can then be followed up by confirmation using bisulfite analysis of genomic methylation. We have tested the imprinting status of the candidates in normal adult brain of wild-type intra-species F1 hybrids to avoid any potential changes in imprinting due to inter-species hybrids.18
Results
We examined 28 genes on chromosome 11 that were predicted to be imprinted using a bioinformatic approach.4 Of these 28 genes, Aipl1, Zfp750, and Lyzl6 were predicted to have little to no expression in the brain based on microarray expression data available online from GNF SymAtlas v1.2.4 (http://symatlas.gnf.org/SymAtlas/). Of the remaining 25 genes, 11 (Stra13, Wscd1, 0610010K14Rik, Tmem98, Osbp2, Lrrc45, Mtmr3, Plekhh3, 1110067D22Rik, Tbcd, and 1110031I02Rik) did not have any available single nucleotide polymorphisms (SNPs) between C57BL/6J (B6) and either A/J or 129S1 or 129X1 in the mRNA sequence or the available SNPs were too close to exon-intron boundaries to design assays that amplify both mRNA and genomic DNA sequence equally (http://phenome.jax.org/pub-cgi/phenome/mpdcgi?rtn=snps/door). Of the remaining 18 genes, one was predicted to be a processed pseudogene (ENSMUSG00000050692) and another was an olfactory receptor (Olfr43). We therefore excluded these two genes from our analysis as being unlikely to affect cancer susceptibility in our model. The remaining 12 genes that we designed quantitative PCR assays for were Havcr2, Camk2b, Ccdc85a, Cntnap1, Ikzf1, 5730522E02Rik, Gria1, Zfp39, Sgcd, Jup, Nxph3, and 0610025P10Rik. The assay for 0610025P10Rik failed in the first round of design and was not pursued further, giving us 11 genes to test from the study by Leudi et al.
We also examined 122 Riken clones that had been reported to be differentially expressed in parthenogenetic embryos and androgenotic embryos.2, 3 Because parthenogenetic embryos are generated entirely from the maternal genome and androgenetic embryos are generated entirely from the paternal genome, genes that are differentially expressed between these two types of embryos are predicted to be imprinted or dependent on imprinted genes for their expression. Of the 121 Riken genes on chromosome 11 (http://fantom2.gsc.riken.jp/EICODB/imprinting/cgi-bin/him.cgi?chr=11&sp=Mm), 29 were discontinued records or not found in the NCBI database and were excluded from our analysis. Thirty-four genes did not have polymorphic SNPs between B6 and A/J or 129S1 or 129X1 in the mRNA sequence (Drg1, Selm, Plek, 0610010F05Rik, Kcnip1, Dock2, Adra1b, 9530066K23Rik, Rasgef1c, Canx, Zfp354c, Zfp354a, Cdkl3, Tcf7, 9530068E07Rik, Flcn, Mpdu1, Alox12, Pfn1, Ksr1, Evi2a, Rab11fip4, Zfp207, Ccl11, Fkbp10, Tmem101, Mettl2, Pitpnc1, Sumo2, Cbx8, Cbx4, Aatk, Mtmr3, and 2810410L24Rik). Of the remaining 59 genes, five were predicted to have little or no expression in the brain (Itk, Krt20, Klhl10, Birc5, and Tex19). From the remaining 54 genes we chose 20 to design quantitative PCR assays. These genes are Ikzf1, Spnb2, Asb3, Rasd1, Map2k3, Map2k4, Trp53, Serpinf1, Crk, Rasl10b, Itga3, Hoxb5, Cbx1, Pparbp, Igfbp4, Smarce1, Stat3, Atp6v0a1, Nbr1, and Meox1. Ikzf1 was identified in both the bioinformatics gene list4 and the experimental gene list2 giving us a total of 30 experimental genes to test.
In addition to the 30 candidate genes, we designed assays for two known imprinted genes (Grb10 and Impact)19–22 and Nf1 because we wanted to test whether Nf1 or Trp53 are monoallelically expressed in our Nf1−/+;Trp53−/+cis mouse model (Trp53 was identified as a candidate in parthenogenetic/androgenetic embryos). The 32 genes on chromosome 11 are diagramed in Figure 1. The positive control gene Impact is located on mouse chromosome 18 and is not shown.
Figure 1.
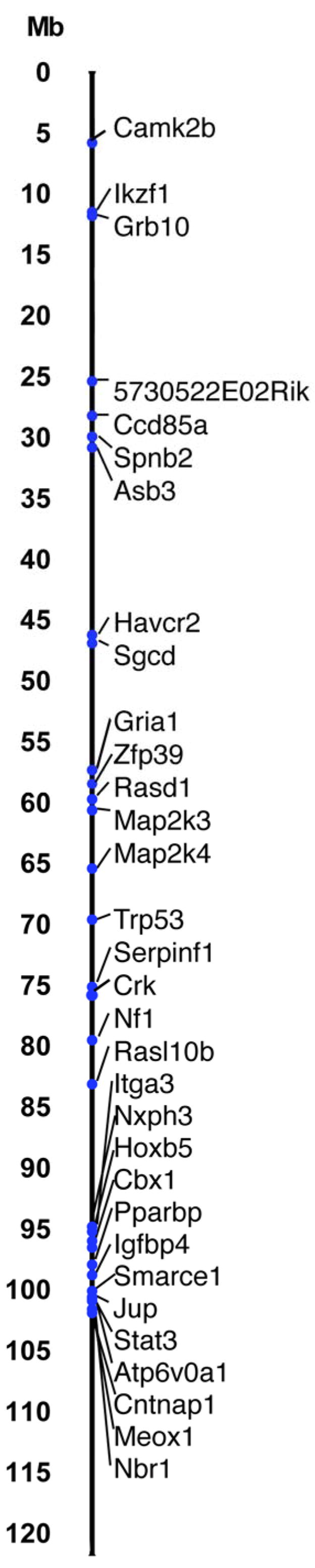
Diagram of tested genes on mouse chromosome 11. Candidate genes were chosen that covered the length of chromosome 11. The centromere is at the top of the diagram and the telomere is at the bottom, with the Mb distance from the centromere indicated along the left side of the diagram.
We designed Taqman quantitative PCR assays for the 33 tested genes as described previously15 (Tsang et al, in preparation). We ran the assays on different ratios of B6:A/J genomic DNA or B6:129S4Sv/Jae (129S4) in which inbred B6 DNA and inbred A/J or 129S4 DNA were mixed such that constant amounts of DNA ranged from 0% B6:100% A/J (or 129S4) to 100% B6:0% A/J (or 129S4). We determined the cubic regression for each assay from this dilution curve and used it to calculate the %B6 in each experimental samples from the difference in Ct between the B6 allele probe and the A/J or 129S4 allele probe. The absolute Ct values were examined to determine whether a gene was expressed at a very low level in adult brain.
To determine whether the candidate genes were imprinted we isolated RNA from adult brains of polymorphic reciprocal F1 progeny. Shi et al have demonstrated that hybrids between Mus musculus and Mus spretus show loss of normal imprinting patterns,18 so we used only Mus musculus derived inbred strains to make reciprocal crosses, although this limited the available polymorphisms to distinguish parental gene alleles. Half the tested samples were from mice that inherited the B6 allele from their father and half inherited the B6 allele from their mother. The opposite parent was either A/J or 129S4 depending on the SNP being tested. We isolated genomic DNA from the tails of the same mice and used this F1 genomic DNA in which the alleles are balanced to control for biased amplification of one allele or the other. We compared the ratio of alleles in cDNA samples made from brain RNA to the ratio of alleles in the matched genomic DNA samples. Supplemental Table 2 shows the level of amplification of the different samples (given as Ct, where Ct is the cycle number at which the signal exceeds the threshold of detection and the lower the Ct value the greater the amount of the SNP allele present in the sample). We averaged the Ct values from three or four separate brains run each in duplicate for the B6 SNP and the A/J or 129S4 SNP for cDNA and gDNA separately. In addition to determining the average Ct, we tested for outliers using Grubbs’ test23 and removed them from the analysis. All outliers had higher Ct values, suggesting that they resulted from a failure of the Taqman assay likely due to pipetting errors in the high-throughput experiment. Two genes were expressed at very low levels (Sgcd and Meox1) with the Ct values for both probes being greater than 34 cycles, making it more difficult to determine the allelic ratios with certainty. The standard deviation of the calculated value of %B6 was very large (Table 1), such that it was not possible to determine the statistical significance of any differences seen in these samples. In addition, three assays showed low amplification of the genomic DNA (Gria1, Meox1, and Impact), although in most cases the standard deviations were still small.
Table 1.
Calculated percentage B6 allele in cDNA and gDNA
| Maternal B6 | Paternal B6 | ANOVA | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cDNA | gDNA | CDNA | gDNA | Tukey post-hoc P | |||||||||||
| Gene | Mb | Avg | SD | Avg | SD | Avg | SD | Avg | SD | imprint | strain (mat) | strain (pat) | Rank | Ref 1 | Ref 2 |
| Camk2b* | 5.9 | 44.4 | 1.5 | 50.4 | 2.3 | 42.6 | 0.8 | 49.7 | 1.5 | < 0.001 | < 0.001 | 4 | P | ||
| Ikzf1* | 11.6 | 45.5 | 3.3 | 48.2 | 1.9 | 45.6 | 3.8 | 46.1 | 3.0 | 10 | M | P | |||
| Grb10* | 11.9 | 5.7 | 3.1 | 57.6 | 2.4 | 98.3 | 1.4 | 56.8 | 4.7 | < 0.001 | < 0.001 | < 0.001 | P | ||
| 5730522E02Rik | 25.5 | 30.7 | 6.0 | 49.4 | 5.8 | 36.7 | 3.0 | 49.6 | 2.4 | < 0.001 | < 0.001 | 16 | M | ||
| Ccdc85a (1133) | 28.3 | 54.5 | 2.9 | 54.5 | 1.5 | 52.7 | 1.7 | 51.1 | 1.9 | 6 | P | ||||
| Ccdc85a (1134) | 28.3 | 54.5 | 3.5 | 51.4 | 3.1 | 56.2 | 1.5 | 52.7 | 1.8 | 6 | P | ||||
| Spnb2 | 30.0 | 53.7 | 1.2 | 54.1 | 1.5 | 53.6 | 0.7 | 54.6 | 1.5 | M | |||||
| Asb3 | 30.9 | 56.6 | 1.4 | 51.9 | 1.1 | 57.8 | 1.5 | 53.7 | 1.2 | < 0.001 | < 0.001 | M | |||
| Havcr2 | 46.3 | 48.6 | 3.0 | 54.8 | 1.0 | 48.6 | 2.0 | 54.1 | 1.2 | < 0.01 | < 0.01 | 3 | M | ||
| Sgcd*† | 47.0 | 0.7 | 0.9 | 52.4 | 0.7 | 47.8 | 7.8 | 52.2 | 1.4 | 23 | M | ||||
| Gria1†† | 57.4 | 58.2 | 1.9 | 54.1 | 2.1 | 48.1 | 14.8 | 55.3 | 1.9 | 17 | M | ||||
| Zfp39 | 58.7 | 54.7 | 1.3 | 54.8 | 1.2 | 53.5 | 1.1 | 54.7 | 1.8 | 22 | P | ||||
| Rasd1 | 59.8 | 50.1 | 1.9 | 51.7 | 1.4 | 49.5 | 1.3 | 50.4 | 1.1 | M | |||||
| Map2k3 | 60.7 | 49.7 | 0.7 | 58.7 | 3.6 | 49.3 | 0.6 | 55.5 | 2.0 | < 0.001 | < 0.001 | P | |||
| Map2k4 | 65.5 | 53.0 | 1.3 | 55.6 | 1.3 | 51.6 | 3.7 | 52.4 | 5.5 | P | |||||
| Trp53 | 69.7 | 82.6 | 4.7 | 52.7 | 3.1 | 83.2 | 2.1 | 58.7 | 2.3 | < 0.001 | < 0.001 | P | |||
| Serpinf1 | 75.2 | 49.5 | 1.1 | 51.4 | 1.9 | 49.8 | 1.9 | 50.6 | 1.3 | P | |||||
| Crk* | 75.5 | 54.4 | 0.8 | 54.6 | 1.5 | 54.8 | 0.5 | 53.3 | 1.2 | M | |||||
| Nf1 | 79.6 | 53.5 | 1.8 | 50.3 | 3.3 | 53.8 | 2.2 | 52.3 | 2.6 | ||||||
| Rasl10b* | 83.2 | 37.8 | 2.2 | 53.5 | 3.2 | 37.7 | 1.5 | 54.5 | 0.7 | < 0.001 | < 0.001 | M | |||
| Itga3 | 94.9 | 48.0 | 2.0 | 53.3 | 2.1 | 49.8 | 1.0 | 54.9 | 1.3 | < 0.001 | < 0.001 | P | |||
| Nxph3 | 95.3 | 53.4 | 1.2 | 53.0 | 1.5 | 53.5 | 1.1 | 54.5 | 2.1 | 25 | M | ||||
| Hoxb5* | 96.1 | 50.2 | 9.7 | 52.6 | 1.2 | 47.6 | 7.0 | 52.4 | 2.0 | P | |||||
| Cbx1 | 96.6 | 51.0 | 3.2 | 52.9 | 0.7 | 49.5 | 3.0 | 53.8 | 1.6 | P | |||||
| Pparbp (1151)* | 98.0 | 52.5 | 2.0 | 50.6 | 1.8 | 51.7 | 0.8 | 52.0 | 0.8 | P | |||||
| Pparbp (1152)* | 98.0 | 56.3 | 1.0 | 56.7 | 0.9 | 54.4 | 1.6 | 57.0 | 1.5 | < 0.01 | P | ||||
| Igfbp4 (1149) | 98.9 | 51.7 | 1.7 | 55.2 | 2.8 | 53.2 | 1.6 | 55.2 | 3.1 | P | |||||
| Igfbp4 (1150) | 98.9 | 51.7 | 1.1 | 50.7 | 0.8 | 51.8 | 1.6 | 50.5 | 0.5 | P | |||||
| Smarce1 | 99.0 | 51.3 | 1.4 | 54.0 | 0.8 | 51.5 | 1.4 | 53.6 | 1.5 | < 0.05 | M | ||||
| Jup | 100.2 | 51.5 | 1.1 | 51.4 | 0.8 | 52.7 | 1.8 | 50.8 | 1.5 | 24 | M | ||||
| Stat3 | 100.7 | 53.6 | 4.5 | 49.3 | 1.8 | 52.9 | 1.1 | 49.7 | 1.2 | M | |||||
| Atp6v0a1 | 100.8 | 52.8 | 0.8 | 53.3 | 1.1 | 53.3 | 1.8 | 53.7 | 1.2 | ||||||
| Cntnap1* | 101.0 | 37.7 | 3.1 | 53.8 | 7.5 | 40.4 | 4.4 | 61.4 | 6.1 | < 0.001 | < 0.001 | 7 | M | ||
| Meox1*† | 101.7 | 32.9 | 12.2 | 40.5 | 3.8 | 42.3 | 13.0 | 41.8 | 5.1 | P | |||||
| Nbr1 | 102.0 | 48.7 | 3.2 | 48.7 | 2.3 | 48.5 | 0.8 | 48.6 | 3.0 | P | |||||
| IMPACT*†† | chr 18 | 6.3 | 4.7 | 43.8 | 2.5 | 100.0 | 0.0 | 45.8 | 2.5 | < 0.001 | < 0.001 | < 0.001 | |||
outlier removed (Grubbs test)
expression below level of reliable detection
low amplification of genomic DNA
P (imprint) is post hoc comparison of %B6 in maternal cDNA to %B6 in paternal cDNA
P (strain (mat)) is post hoc comparison of %B6 in maternal cDNA to %B6 in maternal gDNA
P (strain (pat)) is post hoc comparison of %B6 in paternal cDNA to %B6 in paternal gDNA
Rank indicates the P value ranking for genes on chromosome 11 predicted to be imprinted by Luedi et al, 2005, with 1 being the lowest P value and 28 being the highest P value.
Ref 1: Luedi et al 2005
Ref 2: Nikaido et al 2003
We calculated the percentage of B6 allele in the cDNA and gDNA samples from the Ct values and the results are shown in Table 1. Genes with very low expression levels show high variability as evidenced by the high standard deviations. Only two of the genes tested showed evidence of monoallelic expression, Grb10 (Figure 2A) and Impact (Figure 2B), the positive controls included in the experiment. Both were expressed from the paternal chromosome as expected for brain tissue.19–22 Overall, our data does not support the predictions of imprinting from previous bioinformatic4 or experimental2 approaches.
Figure 2.

Percentage B6 allele in maternal B6 cDNA, maternal B6 gDNA, paternal B6cDNA, and paternal B6 gDNA samples. Graphs show the average percentage of the B6 allele in samples with the standard deviation indicated as error bars. Grb10 (panel A) and Impact (panel B) are known imprinted genes and show very low expression of the B6 allele when it is inherited from the mother (red bar) and very high expression of the B6 allele when it is inherited from the father (blue bar), indicating that these genes are expressed from the paternal allele. The B6 allele makes up approximately 50% of the F1 genomic DNA when the B6 allele is inherited from the mother (red checked bar) or from the father (blue checked bar) as expected. Camk2b (panel C) is expressed less from the B6 allele, whereas Asb3 (panel D), Trp53 (panel E), and Nf1 (panel F) are expressed more from the B6 allele.
We found several genes in the set we tested that show greater expression in one strain or the other. We compared the percentage B6 allele for cDNA samples to the percentage B6 allele for genomic DNA samples. Eleven genes showed less expression from the B6 allele than from the other allele (Camk2b, 5730522E02Rik, Havcr2, Map2k3, Serpinf1, Rasl10b, Cbx1, Itga3, Pparbp, Smarce1 and Cntnap1). A graph of Camk2b allelic balance is shown in Figure 2C as an example. Six genes showed slightly higher expression from the B6 allele than from the other allele (Asb3, Trp53, Nf1, and Stat3). Graphs of Asb3 (Figure 2D), Trp53 (Figure 2E) and Nf1 (Figure 2F) are shown for example. We had previously shown that Nf1 was expressed higher in the brains of inbred B6 mice than inbred 129S4 mice.24 As shown in Figure 2F, we do see the B6 allele expressed slightly higher than the 129 allele in B6X129S4 F1 hybrids brains, although this difference was only statistically significant when the paternal and maternal samples are combined (P=0.02, t-test). For six genes (Serpinf1, Nf1, Stat3, Cbx1, Pparbp, and Smarce1), we combined the cDNA and gDNA samples because there was no statistically significant difference between maternal B6 and paternal B6 cDNA or gDNA and we determined the statistical significance of the strain expression bias in the combined samples.
Discussion
Our data demonstrates that genome-wide prediction of imprinted genes is not yet reliable, at least for expression in the adult brain, and that validation of candidate imprinted genes is very important. Of the genes we tested, only the previously known imprinted genes, Grb10 and Impact, were imprinted in the brain and none of the reported candidates from the literature2, 4 on chromosome 11 showed evidence of imprinting. Our data support previous findings that only a small percentage of the candidate imprinted genes identified by Nikaido et al are confirmed as imprinted in normal hybrid embryos.25 Grb10 has been shown to have complex imprinting patterns in different tissues, being paternally, maternally, or biallelically expressed depending on the tissue studied19–21 so it certainly remains possible that these candidate genes show imprinted expression in tissues other than the brain. It is important to note that it is not possible to prove that a gene is never imprinted, since imprinted expression could be specific to a different tissue, time of development, or possibly to the strain background. However, our results provide evidence that these genes are not universally imprinted and can show biallelic expression in the brain. This presents the interesting possibility that as more candidate imprinted genes are validated in different tissues it may be possible to develop machine learning programs to identify the tissue-specific imprinting signatures surrounding these genes. Studies of parthegenotes and androgenotes will not distinguish these tissue-specific imprinted genes, and so it is important to develop methods such as the one we present here to look at imprinted expression in specific tissues.
It is interesting to note that for the genes on chromosome 11 identified as imprinted by the studies of Nikaido et al and Luedi et al, there were only two genes found in both datasets, suggesting very little overlap of these two methods. We were unable to test Mtmr3 due to a lack of exon SNPs polymorphic for the strains we studied. We tested Ikzf1 and found no imprinting or strain bias. The computer model of Luedi et al predicted that Ikzf1 should be maternally imprinted, whereas the experimental data from Nikaido et al demonstrated expression from the paternal genome. In addition, a recent study26 examined the differential expression of genes in mice with maternal or paternal duplications of chromosome 11 and also found paternal expression of Ikzf1 in the brain. The Nikaido study uses androgenotes and parthegenotes that may not undergo normal maintenance of imprinted genes through the process of manipulating embryos,27 whereas the Schulz study uses translocated chromosomes to generate uniparental disomies. In the case of uniparental disomies it is not clear whether the translocated chromosome pieces are packaged properly in the nucleus with the homologous chromosome or whether they are located with the centromeric chromosome in the nucleus, and whether this could potentially lead to subtle changes in gene expression.28 It is therefore formally possible that the expression of Ikzf1 is more sensitive to changes in chromatin packaging. There is precedence for this in the work of Ling et al who showed that normal expression of the paternal Nf1 allele depends on the stable interaction of the paternal mouse chromosome 11 and the maternal mouse chromosome 7 through the CTCF insulator protein, whereas expression of the maternal Nf1 allele is not affected by this interaction29 It is also unclear how a mixed strain background could affect the interpretation of imprinted candidates in these different experiments, given we see very clear effects of strain polymorphisms on expression on many genes.
The parental expression bias we see in our data ranges from very subtle (3% difference in allelic expression) to quite substantial (54% difference). Although these differences are statistically significant, it is unlikely that the smaller differences affect tumor susceptibility or other biological function. We report these subtle differences here because they may reflect subtle differences in the epigenetic marks surrounding these genes that could explain why they were identified in the studies of Nikaido et al and Luedi et al. For most of the genes that show a strain-specific bias, there are polymorphic SNPs between B6 and A/J or 129S1/129X1 in the 5′UTR and intron 1 (Jackson Laboratory Mouse Phenome Database SNP website, http://phenome.jax.org/pub-cgi/phenome/mpdcgi?rtn=snps/wiz1), suggesting that the difference in expression levels could be due in part to cis-acting polymorphisms affecting transcription. Two exceptions are Havcr2 that has SNPs between B6 and A/J in intron 2, but not intron 1 or the 5′UTR, and Serpinf1 that has a synonymous coding SNP polymorphic between B6 and A/J in exon 8. These two genes could be differentially expressed due to polymorphisms in trans-acting factors, or other more indirect effects.
We have previously shown using quantitative PCR and SYBR green detection methods that the level of expression of Nf1 in wild-type B6 brains is 30% higher than wild-type 129 brains.24 In these experiments using Taqman quantitative PCR on B6X129S4 F1 brains we find higher expression from the B6 allele than from the 129S4 allele ranging from 5–16% depending on the experimental run. In both experiments the B6 allele is expressed at a higher level than the 129S4 allele, however the size of the effect is smaller in the F1 hybrid than in the inbred strains. This may be due to trans-acting factors that are matched to the strain of the Nf1 gene, such that there is a mixture of trans-acting alleles in the F1 hybrid and a dampening of the expression difference.
It is interesting to note that we see a similar strain effect on the Trp53 gene, such that the B6 allele is expressed 24–54% higher on average than the 129S4 allele depending on the experimental run. It has been shown that the phenotype of Trp53 null mice differs on a 129 inbred strain background compared to a B6,129 mixed strain background30 and this data suggests that there may be additional strain specific effects in the presence of p53. In the case of both the Nf1 and Trp53 tumor suppressors, expression is higher in the brains of mice that are more susceptible to brain cancer16 suggesting that the higher level of the tumor suppressor gene is not protective against cancer. We hypothesize that the B6 strain may have evolved higher levels of oncogenic factors in balance and opposing Nf1 and Trp53, such that when the tumor suppressor genes become mutated or lost the oncogenic effects are more pronounced in the B6 strain, increasing susceptibility to cancer. Additional experiments are needed to test whether this accounts for the difference in strain susceptibility to cancer.
Although we have not identified any novel fully imprinted genes in this study, the validation of candidates and demonstration of their biallelic expression in the adult brain will be useful for refinement of future computational predictors of imprinted genes. In particular, this method of expression analysis distinguishes imprinting from other variations in expression, and provides support to pursue further proof of imprinting by bisulfite analysis of genomic DNA. These data also emphasize the importance of assaying imprinted genes on well-defined genetic backgrounds to separate out strain-specific effects from parental origin effects. There remain 120 candidate genes on chromosome 11 from the studies of Nikaido et al and Luedi et al that may be imprinted; however, because 30 out of the 30 we tested were biallelically expressed, it may be worth refining the methods of predicting imprinted candidates before analyzing these additional genes. Finally, our data suggest that Grb10 remains a top candidate for the imprinted effect on chromosome 11 responsible for variation in tumor susceptibility in Nf1−/+;Trp53−/+cis mice.
Methods
Generation of F1 hybrid mice
To generate B6 paternal F1 hybrids on a B6XA background, we crossed B6-Nf1−/+;Trp53−/+cis males to wild-type A/J females. We used wild-type F1 progeny from this cross for RNA and genomic DNA. To generate B6 maternal F1 hybrids, we crossed Nf1−/+;Trp53−/+cis females to wild-type A/J males, and used wild-type F1 progeny. We chose progeny matched for age and sex as much as possible given the available mice. The B6XA progeny were all males, the three B6 paternal cohort mice were euthanized at 2.1, 2.1, and 6.0 months of age and the three B6 maternal cohort mice were all euthanized at 3.3 months of age. To generate B6X129S4 F1 hybrids, 129S4-Nf1−/+;Trp53−/+cis mice were crossed to wild-type B6. The B6X129S4 progeny were half males and half females. The four B6 paternal cohort mice were two females and two males all euthanized at 3.4 months of age. The four B6 maternal cohort mice were two females and two males all euthanized at 3.3 months of age. The 129S4-Nf1−/+;Trp53−/+cis we used to generate the F1 hybrids have been maintained on inbred 129S4 since the generation of the knockout Nf1 and Trp53 alleles.31, 32 The B6-Nf1−/+;Trp53−/+cis we used to generate the B6XA/J F1 hybrids have been backcrossed approximately 30 generations onto C57BL/6J. We purchased wild-type A/J from Jackson Laboratory (Bar Harbor, ME)
Isolation of brain RNA and tail genomic DNA
We euthanized mice by CO2 asphyxiation and removed whole brains. The tissue was weighed and 100 mg was homogenized in Trizol Reagent (Invitrogen, Carlsbad, CA) using an Omni power homogenizer (Omni International, Marietta, GA). We isolated total RNA from the homogenized tissue using Trizol Reagent, following the manufacturer’s protocol. The isolated total RNA was quantified using a NanoDrop (NanoDrop Technologies, Wilmington, DE). Prior to cDNA synthesis the total RNA was DNased-treated with DNAfree (Ambion, Austin, TX) following the manufacturer’s protocol. The cDNA was synthesized using Superscript II First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA).
Genomic DNA was isolated from tails by digestion with proteinase K (100 mM Tris, pH 8.5, 5 mM EDTA, 0.2% SDS, 200 mM NaCl, 400 ug/ml proteinase K). After digestion, the genomic DNA was precipitated, resuspended in dH2O and treated with two rounds of phenol/chloroform purification.
Taqman assay design
SNPs predicted to be polymorphic between B6 and A/J or B6 and 129S1 or 129X1 for our genes of interest were chosen from the Jackson Laboratory Mouse Phenome Database SNP website (http://phenome.jax.org/pub-cgi/phenome/mpdcgi?rtn=snps/wiz1). We designed Taqman assays for these SNPs using Assay-by-Design or Primer-Express software as described previously.15 The SNP identification numbers from the NCBI dbSNP database and the primer sequences are given in Supplemental Table 2. We used a standard curve of mixtures of B6 DNA with either A/J DNA or 129S4 DNA to determine the percentage of B6 DNA in each sample. Briefly, we determined the third order polynomial function for each assay using the LINEST function in Excel or regression analysis in GraphPad Prism 4.0a with the percentage of B6 DNA in the standard curve and the Ct difference between the FAM and VIC probes. The Ct difference between the FAM and VIC probes from the experimental samples was then plugged into the polynomial equation to determine the percentage B6 DNA in the sample. All sequence and SNP data were from the NCBI Mouse Genome Build 36.1. Gene names were updated from NCBI Mouse Genome Build 37.1.
Statistical analysis
All samples were run in duplicate, and three to four independent samples were run for each group. For the Ct values, outliers were identified using the Grubbs’ test.23 For each Ct value, the Z ratio was calculated according to the formula Z = |mean − Ct value|/SD where the mean was the average of all measurements for a single probe within a single group and SD was the standard deviation. Ct values for which the Z ratio exceeded a critical cut-off value were excluded from the analysis. For samples run on three brains in duplicate, the critical Z value was taken as 1.89 and for samples run on four brains in duplicate, the critical Z value was taken as 2.13. After outliers were removed, we calculated the average and standard deviation for the Ct values (Supplemental Table 1) and the calculated %B6 (Table 1). The cDNA samples with Ct values for both the FAM and VIC probes greater than 34 were considered to be low expressing for the gene under consideration. We determined the 34 Ct cut-off as being roughly one standard deviation away from the mean of all Ct values for all probes examined.
To determine whether genes were differentially expressed based on parental inheritance or strain, we analyzed the average %B6 for maternal B6 cDNA, maternal B6 gDNA, paternal B6 cDNA, and paternal B6 gDNA samples using ANOVA with a Tukey post-hoc test to make pairwise comparisons. To determine whether a gene showed an imprinted bias, we compared maternal B6 cDNA to paternal B6 cDNA. To determine whether a gene showed strain-specific expression, we compared maternal B6 cDNA to maternal B6 gDNA and paternal B6 cDNA to paternal B6 gDNA. We also looked at whether maternal B6 gDNA and paternal B6 gDNA were significantly different, as this indicates a lack of reproducibility in the assay. For genes that showed no significant difference between maternal and paternal cDNA the average %B6 was recalculated combining all cDNA samples and all gDNA samples and tested for significant differences to determine whether a subtle strain specific bias was present in the larger sample size. The average %B6 values were compared using an unpaired t-test with Welch’s correction for unequal variances in GraphPad Prism 4.0a. The difference in allelic expression was calculated as [(%B6cDNA − %B6gDNA) × 2].
Supplementary Material
Acknowledgments
We would like to thank Erika Truffer for technical assistance and Bev Mock for helpful comments on the manuscript. This project has been funded by the Intramural Research Program of the NIH, National Cancer Institute and with federal funds from the National Cancer Institute under contract NO1-CO-12400 to SAIC Frederick. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsements by the U.S. Government.
Footnotes
This manuscript includes supplementary data listing primer sequences for Taqman assays and Ct values for Taqman PCR.
References
- 1.Wood AJ, Oakey RJ. Genomic imprinting in mammals: emerging themes and established theories. PLoS Genet. 2006;2:e147. doi: 10.1371/journal.pgen.0020147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nikaido I, Saito C, Mizuno Y, Meguro M, Bono H, Kadomura M, Kono T, Morris GA, Lyons PA, Oshimura M, Hayashizaki Y, Okazaki Y. Discovery of imprinted transcripts in the mouse transcriptome using large-scale expression profiling. Genome Res. 2003;13:1402–9. doi: 10.1101/gr.1055303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nikaido I, Saito C, Wakamoto A, Tomaru Y, Arakawa T, Hayashizaki Y, Okazaki Y. EICO (Expression-based Imprint Candidate Organizer): finding disease-related imprinted genes. Nucleic Acids Res. 2004;32:D548–51. doi: 10.1093/nar/gkh093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luedi PP, Hartemink AJ, Jirtle RL. Genome-wide prediction of imprinted murine genes. Genome Res. 2005;15:875–84. doi: 10.1101/gr.3303505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–53. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 7.Scrable H, Cavenee W, Ghavimi F, Lovell M, Morgan K, Sapienza C. A model for embryonal rhabdomyosarcoma tumorigenesis that involves genome imprinting. Proc Natl Acad Sci U S A. 1989;86:7480–4. doi: 10.1073/pnas.86.19.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanson M, Leuraud P, Marie Y, Delattre JY, Hoang-Xuan K. Preferential loss of paternal 19q, but not 1p, alleles in oligodendrogliomas. Ann Neurol. 2002;52:105–7. doi: 10.1002/ana.10217. [DOI] [PubMed] [Google Scholar]
- 9.Hensen EF, Jordanova ES, van Minderhout IJ, Hogendoorn PC, Taschner PE, van der Mey AG, Devilee P, Cornelisse CJ. Somatic loss of maternal chromosome 11 causes parent-of-origin-dependent inheritance in SDHD-linked paraganglioma and phaeochromocytoma families. Oncogene. 2004;23:4076–83. doi: 10.1038/sj.onc.1207591. [DOI] [PubMed] [Google Scholar]
- 10.Huson S, Hughes R. The Neurofibromatoses: A pathogenetic and clinical overview. London: Chapman & Hall Medical; 1994. [Google Scholar]
- 11.Easton D, Ponder M, Huson S, Ponder B. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes. Am J Hum Genet. 1993;53:305–13. [PMC free article] [PubMed] [Google Scholar]
- 12.Cichowski K, Shih T, Schmitt E, Santiago S, Reilly K, McLaughlin M, Bronson R, Jacks T. Mouse models of tumor development in neurofibromatosis type I. Science. 1999;286:2172–6. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 13.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109–13. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- 14.Vogel K, Klesse L, Velasco-Miguel S, Meyers K, Rushing E, Parada L. Mouse tumor model for neurofibromatosis type 1. Science. 1999;286:2176–9. doi: 10.1126/science.286.5447.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reilly KM, Broman KW, Bronson RT, Tsang S, Loisel DA, Christy ES, Sun Z, Diehl J, Munroe DJ, Tuskan RG. An imprinted locus epistatically influences Nstr1 and Nstr2 to control resistance to nerve sheath tumors in a neurofibromatosis type 1 mouse model. Cancer Res. 2006;66:62–8. doi: 10.1158/0008-5472.CAN-05-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reilly KM, Tuskan RG, Christy E, Loisel DA, Ledger J, Bronson RT, Smith CD, Tsang S, Munroe DJ, Jacks T. Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc Natl Acad Sci U S A. 2004;101:13008–13. doi: 10.1073/pnas.0401236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menheniott TR, Woodfine K, Schulz R, Wood AJ, Monk D, Giraud AS, Baldwin HS, Moore GE, Oakey RJ. Genomic imprinting of Dopa decarboxylase in heart and reciprocal allelic expression with neighbouring Grb10. Mol Cell Biol. 2007 doi: 10.1128/MCB.00862-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi W, Krella A, Orth A, Yu Y, Fundele R. Widespread disruption of genomic imprinting in adult interspecies mouse (Mus) hybrids. Genesis. 2005;43:100–8. doi: 10.1002/gene.20161. [DOI] [PubMed] [Google Scholar]
- 19.Arnaud P, Monk D, Hitchins M, Gordon E, Dean W, Beechey CV, Peters J, Craigen W, Preece M, Stanier P, Moore GE, Kelsey G. Conserved methylation imprints in the human and mouse GRB10 genes with divergent allelic expression suggests differential reading of the same mark. Hum Mol Genet. 2003;12:1005–19. doi: 10.1093/hmg/ddg110. [DOI] [PubMed] [Google Scholar]
- 20.Hikichi T, Kohda T, Kaneko-Ishino T, Ishino F. Imprinting regulation of the murine Meg1/Grb10 and human GRB10 genes; roles of brain-specific promoters and mouse-specific CTCF-binding sites. Nucleic Acids Res. 2003;31:1398–406. doi: 10.1093/nar/gkg232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blagitko N, Mergenthaler S, Schulz U, Wollmann HA, Craigen W, Eggermann T, Ropers HH, Kalscheuer VM. Human GRB10 is imprinted and expressed from the paternal and maternal allele in a highly tissue- and isoform-specific fashion. Hum Mol Genet. 2000;9:1587–95. doi: 10.1093/hmg/9.11.1587. [DOI] [PubMed] [Google Scholar]
- 22.Hagiwara Y, Hirai M, Nishiyama K, Kanazawa I, Ueda T, Sakaki Y, Ito T. Screening for imprinted genes by allelic message display: identification of a paternally expressed gene impact on mouse chromosome 18. Proc Natl Acad Sci U S A. 1997;94:9249–54. doi: 10.1073/pnas.94.17.9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Motulsky HJ. Prism 4 Statistics Guide - Statistical Analysis for Laboratory and Clinical Researchers. San Diego, CA: GraphPad Software, Inc; 2003. [Google Scholar]
- 24.Hawes JJ, Tuskan RG, Reilly KM. Nf1 expression is dependent on strain background: implications for tumor suppressor haploinsufficiency studies. Neurogenetics. 2007;8:121–30. doi: 10.1007/s10048-006-0078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruf N, Dunzinger U, Brinckmann A, Haaf T, Nurnberg P, Zechner U. Expression profiling of uniparental mouse embryos is inefficient in identifying novel imprinted genes. Genomics. 2006;87:509–19. doi: 10.1016/j.ygeno.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Schulz R, Menheniott TR, Woodfine K, Wood AJ, Choi JD, Oakey RJ. Chromosome-wide identification of novel imprinted genes using microarrays and uniparental disomies. Nucleic Acids Res. 2006;34:e88. doi: 10.1093/nar/gkl461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fauque P, Jouannet P, Lesaffre C, Ripoche MA, Dandolo L, Vaiman D, Jammes H. Assisted Reproductive Technology affects developmental kinetics, H19 Imprinting Control Region methylation and H19 gene expression in individual mouse embryos. BMC Dev Biol. 2007;7:116. doi: 10.1186/1471-213X-7-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kosak ST, Scalzo D, Alworth SV, Li F, Palmer S, Enver T, Lee JS, Groudine M. Coordinate gene regulation during hematopoiesis is related to genomic organization. PLoS Biol. 2007;5:e309. doi: 10.1371/journal.pbio.0050309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ling JQ, Li T, Hu JF, Vu TH, Chen HL, Qiu XW, Cherry AM, Hoffman AR. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science. 2006;312:269–72. doi: 10.1126/science.1123191. [DOI] [PubMed] [Google Scholar]
- 30.Harvey M, McArthur M, Montgomery C, Jr, Bradley A, Donehower L. Genetic background alters the spectrum of tumor that develop in p53-deficient mice. FASEB J. 1993;7:938–42. doi: 10.1096/fasebj.7.10.8344491. [DOI] [PubMed] [Google Scholar]
- 31.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Current Biology. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 32.Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nature Genetics. 1994;7:353–61. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.