

Abstract
Activated natural killer (NK) cells mediate potent cytolytic and secretory effector functions, and are vital components of the early antiviral immune response. NK cell activities are regulated by the assortment of inhibitory receptors that recognize major histocompatibility class I ligands expressed on healthy cells and activating receptors that recognize inducible host ligands or ligands that are not well characterized. The activating Ly49H receptor of mouse NK cells is unique in that it specifically recognizes a virally encoded ligand, the m157 glycoprotein of murine cytomegalovirus (MCMV). The Ly49H-m157 interaction underlies a potent resistance mechanism (Cmv1) in C57BL/6 mice, and serves as an excellent model in which to understand how NK cells are specifically activated in vivo, as similar receptor systems are operative for human NK cells. For transduced cells expressing m157 in isolation and for MCMV-infected cells, we show that m157 is expressed in multiple isoforms with marked differences in abundance between infected fibroblasts (high) and macrophages (low). At the cell surface m157 is exclusively a glycosylphosphatidylinositol-associated protein in MCMV-infected cells. Through random and site-directed mutagenesis of m157 we identify unique residues that provide for efficient cell surface expression of m157, but fail to activate Ly49H-expressing reporter cells. These m157 mutations are predicted to alter the conformation of a putative m157 interface with Ly49H, one that relies on the position of a critical α0-helix of m157. These findings support an emerging model for a novel interaction between this important NK cell receptor and its viral ligand.
Introduction
The vital role for NK cells in defense against certain viral infections is well established for human infections and in mouse models, and requires both cytotoxic and cytokine/chemokine effector functions of activated NK cells (1-5). NK cell activation by appropriate targets involves the integration of both inhibitory and stimulatory input. Inhibitory NK cell receptors such as those of the human killer immunoglobulin-like receptor (KIR) and mouse Ly49 receptor families recognize specific major histocompatibility complex (MHC) class I ligands. As MHC I molecules are ubiquitously expressed, KIR and Ly49 receptors constitute a major mechanism for self-tolerance among NK cells (6-9). Peripheral NK cells may be activated en masse by a variety of stimuli, including inducible host ligands for the NKG2D receptor and IgG-coated targets recognized by the low-affinity FcγR-IIIA/CD16 receptor (reviewed in (10-12)). In addition, subset-specific NK cell activation has been demonstrated in mice for NK cells bearing the Ly49H activation receptor in the context of MCMV infection (13-15). Ly49H-mediated recognition of the MCMV-encoded protein, m157, underlies the dominant MCMV resistance trait, Cmv1, expressed by the C57BL/6 and related inbred mouse strains (16, 17). In part, allelic differences in the ly49 gene cluster explains the strain-to-strain differences in resistance to MCMV. BALB/c and other strains lacking ly49h are much more susceptible to MCMV infection, relying primarily upon NKG2D-mediated activation (18-20). More recently, additional MCMV resistance loci have been described (Cmv2-4) (21-24), although the precise receptor-ligand interactions are not completely understood. Thus, among these NK cell-mediated responses the Ly49H-m157 interaction represents a unique model in which physiologically relevant parameters regulating NK cell activation may be dissected.
Curiously, although m157 is not recognized by any other Ly49 receptor in the C57BL/6 strain, it is a specific ligand for the inhibitory Ly49I receptor in the 129 strain, prompting speculation that m157 has been maintained in the MCMV genome for its ability to function as a MHC I decoy ligand (14, 25). In support of this, the putative m157 polypeptide was predicted to fold with MHC I-like topology (15), and a recently reported crystal structure for m157 indicates strong homology to mouse T22, a beta-2-microglobulin (β2m)-associated MHC class Ib protein (26). MCMV m157 does not require β2m for surface expression or Ly49H activation (14, 15, 27); indeed the absence of a β2m association for the α3 domain of m157 is predicted to impart an interaction with Ly49H that is significantly different from that observed between inhibitory Ly49 receptors and their MHC I ligands (26). This interaction is also unique in that m157 is predicted to be a glycosylphosphatidylinositol-associated protein (GPI-AP), a rare cell surface linkage for virally encoded glycoproteins. While GPI-association has been reported for cells transduced with m157 alone (27), GPI-linkage for m157 in MCMV-infected cells has not been demonstrated.
Deletion of ly49h locus in BxD8 mice (inbred recombinant congenic strain containing the Nkc region derived from the C57BL/6 parental strain), or in vivo blockade of the m157-Ly49H interaction abrogates MCMV resistance (28). Conversely, transgenic expression of Ly49 in BALB/c or FVB mice converts these animals from a susceptible to a resistant phenotype with regard to MCMV infection (29). The importance of this NK mediated immune mechanism is highlighted further by the observation that serial passage of MCMV in C57BL/6 mice lacking adaptive immunity quickly results in the selection of MCMV escape viruses that have acquired novel mutations in m157 (30-32). Infection of wild-type C57BL/6 mice with plaque-purified MCMV harboring m157 mutations results in loss of NK cell control and exacerbated disease (32). However, the majority of these m157 variants are null mutants predicted to result in no surface expression of m157; the expression of m157 on the surface of variant MCMV-infected cells was not examined. Thus the specific role for Ly49H-mediated recognition of MCMV-infected cells expressing variant m157 could not be addressed.
In this study we demonstrate that m157 is a GPI-AP on MCMV-infected fibroblasts and macrophages, and that infected cells express multiple m157 isoforms ranging from ∼42-50 kDa. Random and site-directed mutagenesis of m157 and selection for novel mutants stably expressed at the cell surface revealed the identity of critical m157 residues required for functional activation of Ly49H. The m157 mutants that fail to activate Ly49H impart a reduced stability of the Ly49H-m157 interaction over time, suggesting that duration of binding may be a significant determining factor for delivery of an activating signal. These findings advance the current understanding of a successful herpesvirus immune evasion mechanism and how NK cells recognize these viral infections.
Materials and Methods
Antibodies
Monoclonal antibodies (mAb) specific for native m157 were generated (with the assistance of the University of Iowa Hybridoma Core) by immunizing BALB/c mice with m157-transduced BaF3 cells. Supernatants from pooled hybridoma clones were initially screened for the ability to block activation mediated by Ly49H-expressing reporter cells stimulated by BaF3-m157 cells (CPRG assay for β-galactosidase activity). Pooled hybridomas showing >80% inhibition of Ly49H activation were then cloned by limiting dilution, expanded and re-screened for blocking activity. Supernatants from clones showing blocking activity were then tested for staining of both BaF3-m157 and C1498-m157 cells (and the parental controls) prior to subcloning and tertiary screening. We verified specific cell surface staining for multiple m157-transduced cell types by flow cytometry and identified two clones that were purified for further analyses: 6D5 (mouse IgG2a) and 1F2 (mouse IgG1). Another m157-specific mAb, clone 6H121, (27) was a generous gift from W. Yokoyama (Washington University, St. Louis, MO). All monoclonal antibodies were affinity-purified on Protein-G columns (Amersham Biosciences, Piscataway, NJ). Allophycocyanin-conjugated streptavidin was obtained from BD Pharmingen and FITC-conjugated goat anti mouse was obtained from Southern Biotechnology Associates, Inc. Anti-HSA (clone M1/69) and anti-Thy1 (clone H013.4) were kindly provided T. Waldschmidt (University of Iowa). Control mAb against β-actin and caspase 3 were obtained from Sigma-Aldrich (Saint Louis, MO) and Upstate Biologicals (Lake Placid, NY) respectively.
Cell Lines, Transfections, and Flow Cytometry
The pMX-based retroviral vectors and retroviral packaging cell line, Platinum-E (PLAT-E; kindly provided by T. Kitamura, University of Tokyo, Tokyo, Japan), BWZ.36 cells expressing an inducible lacZ reporter cassette (kindly provided by Nilabh Shastri, University of California, Berkeley, CA), and the derivative reporter line expressing Ly49H + DAP12 (HD12 cells) have been previously described (15). BWZ.36 and HD12 cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT) and 2 mM L-glutamine (“R10”). NIH-3T3 fibroblasts and transduced derivative lines were maintained in complete Dulbeco's Modified Eagle's Medium (DMEM/high glucose, GIBCO/Invitrogen, Carlsbad, CA) supplemented with 10% bovine calf serum (BCS) and 2 mM L-glutamine (“D10”). C1498 (C57BL/6J myeloid leukemia) cells and transduced derivatives were maintained in D10 plus 1 mM sodium pyruvate and 50 μM 2-mercaptoethanol. BaF3 (BALB/c pre-B) cells and derivative lines were maintained in HEPES buffered R10 supplemented with 10% X63-Ag8-653 cell culture supernatant as a source of IL-3. IC-21 (C57BL/6J peritoneal macrophage line) and DC2.4 (C57BL/6 dendritic cells, kindly provided by Kenneth Rock, University of Massachusetts Medical School, Worcester, Massachusetts and the Dana-Farber Cancer Institute, Boston, MA) were cultured in HEPES buffered R10 plus 1 mM sodium pyruvate.
Wild-type (wt) and mutant m157 cDNA constructs were directionally subcloned into the Bam HI and Not I sites of the pMX-Puro retroviral vector. Transduced cells were selected in puromycin (2.0-3.0 μg/mL) for at least one week prior to use. Transduced cells were surface stained with either unconjugated or biotinylated anti-m157 mAbs 6D5, 1F2, and/or 6H121 followed by the appropriate secondary detection reagent. Fluorescently labeled cells were analyzed on a FACSCalibur flow cytometer (BD, Mountain View, CA) and the data were processed using FlowJo software (version 8.4.6, Treestar, Ashland, OR).
MCMV infection
NIH-3T3 fibroblasts, IC-21 macrophages and DC2.4 dendritic cells were infected with wild-type Smith strain or the K181-derived green fluorescent protein (GFP)-expressing RM4503.3 strain (33) (kindly provided by L. Geist, University of Iowa, Iowa City, IA) of MCMV at a multiplicity-of-infection (MOI) of five. Mock-infected controls were treated with the cell lysates from uninfected fibroblast equivalents prepared in parallel to MCMV viral stocks. Infected cells were rocked gently every 10 minutes for one hour at 37°C (adsorption time) and subsequently harvested between 12 and 24 hours post-infection.
Immunoprecipitation, Western blot analysis, and PI-PLC treatment
For immunoprecipitation, cells were lysed in IP buffer (30 mM Tris-HCl (pH.7.5), 150 mM NaCl, 1% Triton X-100, 10% (v/v) glycerol, 1 mM PMSF, and protease inhibitors 1:100 (Sigma-Aldrich, St. Louis, MO) on ice for 30 minutes followed by 20 minutes of centrifugation at 10,000×g. Supernatants (1 mg of protein) were incubated with 2 μg of specific mAb overnight at 4°C. 20 μl of Protein G-Sepharose (Amersham Biosciences, Piscataway, NJ) were then added to cell lysates and incubated for two hours at room temperature. Beads were washed seven times with IP buffer and proteins were eluted by the addition of 50 μl 2x Laemmli reducing sample buffer and analyzed by Western blotting using anti-m157 mAb 6H121. For Co-IP experiments, transduced C1498 cells expressing different m157 mutants were coincubated with HD12 cells (2:1) for 10, 40 or 180 minutes (37°C, 5% CO2). Cell lysates were then prepared as described above. 3D10 antibodies were used for Ly49H immunoprecipitation and co-immunoprecipitated m157 was developed with 6H121 antibodies by Western blot. Initial amount of m157 and total protein (β-actin) in each IP sample were controlled by Western blotting. Since available anti-Ly49H reagents (including mAb 3D10) do not recognize denatured Ly49H in Western analyses, we verified comparable cell surface Ly49H expression on HD12 cells for each sample by FACS analysis for each analysis.
For Western blot analysis of cell lysates, cells were washed in PBS and lysed in 1% Triton X-100 buffer (20mM Tris-HCl pH 7.5, 1% Triton X-100 (v/v) in Versene). 20 μg of proteins were separated on 10% or 12% SDS–PAGE, and blotted onto a nitrocellulose membrane (BioRad, Hercules, CA). Equal loading was controlled by reversible staining of the membrane with Ponceau S solution and with mAb to β-actin (Sigma, St Louis, MO). Membranes were blocked with 5% nonfat dry milk in PBS containing 0.1% Tween-20 and then incubated overnight with specific mAb at 4°C. The blots were counterstained with anti-mouse IgG conjugated with HRP (Pierce, Rockford, IL) or with anti-mouse IgG TrueBlot™ conjugated with HRP (eBioscience, San Diego, CA) for analysis of IP samples. Mouse IgG TrueBlot™ preferentially detects the non-reduced form of mouse IgG (IgG1, IgG2a, IgG2b, IgG3) over the reduced, SDS-denatured form of IgG thereby eliminating interference by the heavy and light chains of the immunoprecipitating antibody in IP/immunoblotting applications.
For Triton X-114 phase separation 1×107 cells were lysed in buffer A (2% Triton X-114, 20 mM Tris pH 7.4, 2mM EDTA, protease inhibitors 1:100) for 15 min on ice. Lysates were incubated at 37°C for 2 min and subjected to centrifugation (600×g). Aqueous and detergent phases were separated and analyzed by Western blotting. For phosphatidylinositol-specific phospholipase C (PI-PLC) treatment, detergent phases were prepared from C1498-m157 cells or MCMV-infected IC-21 and NIH-3T3 cells and divided in half. Both halves were diluted with buffer A without Triton X-114 to a final detergent concentration of 2%. One aliquot was incubated in buffer alone while 1 unit of PI-PLC (Invitrogen; Carlsbad, CA) was added to the other aliquot. The lysates were incubated in enzymes at 30°C for 1 hour and analyzed by Western blotting as above.
CPRG Assay for β-galactosidase activity
BWZ.36 cells (34) transduced with Ly49H and the signaling adapter protein, DAP12 (HD12 cells) (15), were co-incubated with either m157-transfectants or MCMV-infected cells (or their appropriate parental and mock-infected controls) for 6-16 hours (37°C, 5% CO2) in a 96-well plate to induce β-galactosidase production within the HD12 reporter cells. The amount of β-galactosidase activity on chlorophenol red β-D-galactopyranoside (CPRG, 0.15M; Calbiochem, San Diego, CA) substrate was quantitatively determined at several points during the linear phase of enzymatic activity by measuring the absorbance at 575 nm (635 nm reference) using the μ–Quant plate reader and K.C.-Jr. software package (Bio-tek Instruments, Inc., Winooski, VT). Results are shown as a percentage of maximal stimulation obtained upon culturing HD12 reporter cells with 5 ng/mL phorbol-myristate acetate (PMA) and 1 μM ionomycin.
Construction, screening and sequencing of m157 mutant library
Random mutations in m157 were generated using the Genemorph II Random Mutagenesis kit (Stratagene; La Jolla, CA). Optimizing the PCR cycle number and amount of input template DNA yielded a library of m157 molecules averaging 1-2 amino acid substitutions per cDNA copy (verified by sequencing individual clones from 6 independent libraries). This library was directionally subcloned into the Bam HI and Not I sites of the pMX-Puro retroviral vector and then transduced into 5×106 C1498 cells. Four days after transduction, puromycin-resistant C1498 cells were dually stained with 6D5 + goat anti-mouse FITC and biotinylated 1F2 + streptavidin-allophycocyanin to detect cell surface expression of m157. Transduced C1498 cells were sorted into two distinct m157-expressing populations—6D5/1F2 double-positives and a smaller 6D5lo/1F2+ subset in order to maximize the number of surface-expressed m157 variants that could be assessed for Ly49H activation. Sorted m157-expressing cells were cloned by limiting dilution in 96 well plates at a density of 0.5 cells per well. Six hundred and fifty clones were initially screened using the HD12 reporter cells and the CPRG assay described above. Clones were tested in duplicate and those that failed to activate Ly49H (low or no induction of β-galactosidase) in both samples were subsequently expanded and re-stained for the surface expression of m157 as detected by 6D5 and/or 1F2. Those demonstrating a stable surface m157 phenotype were then retested for HD12 activation. Total RNA was isolated using TRIzol reagent (Invitrogen; Carlsbad, CA) from all non-HD12 activating clones expressing detectable surface m157. Copy DNA was then reverse-transcribed using random hexamers and Superscript II polymerase (Invitrogen; Carlsbad, CA) and amplified with high-fidelity DNA polymerase (Phusion; Finnzymes distributed by New England Biolabs, Ipswich, MA) prior to sequencing. All clones showing reduced or absent HD12-stimulating capacity (defined as <20% maximal stimulation by C1498 cells expressing wt-m157) were shown to harbor at least one m157 mutation. All mutations and their corresponding Ala-substitution variants were regenerated using PCR-mediated site-directed mutagenesis, and then transduced independently into C1498 cells as above. The resulting puromycin-resistant clones and regenerated single-mutant m157-expressing cell lines were FACS-sorted for high-level m157 expression using 6H121 mAb in order to normalize m157 expression levels compared to C1498-m157 (wt) cells.
Computer-assisted analysis and modeling of m157
Numbering of m157 residues follows that reported for the native protein (26), beginning at Ile22 of the full-length predicted amino acid sequence. Protein analysis and predictions for m157 sequences were carried out using the following algorithms available at the ExPASy Proteomics Tools server (http://ca.expasy.org/tools/) (35) and the Biological Information Resource server at the University of Washington (http://courses.washington.edu/bioinfo/BIR/): including ScanProsite (http://au.expasy.org/tools/scanprosite/), NetNGly glycosylation predictor (http://www.cbs.dtu.dk/services/NetNGlyc/), DGPI online GPI predictor (http://129.194.185.165/dgpi/DGPI_demo_en.html), and big-PI Predictor (http://mendel.imp.ac.at/sat/gpi/gpi_server.html). Structural analyses were performed using the 3D-PSSM algorithm (36) (http://www.sbg.bio.ic.ac.uk/∼3dpssm/index2.html). Mapping of m157 mutations was carried out using the deposited m157 structural data file (PDB ID code: 2NYK) and the PyMOL modeling program (http://www.pymol.org) (37).
Results
Generation of functionally distinct anti-m157-specific monoclonal antibodies
In order to more fully characterize the MCMV-encoded ligand for the activating NK cell receptor Ly49H, we generated mAb specific for m157 by immunizing BALB/c mice with m157-transduced BaF3 cells. We purified two clones, 6D5 and 1F2, which specifically stained BaF3-m157 and C1498-m157 cells but not their GFP-expressing parental counterparts (Figure 1A). In addition, dual staining of C1498-m157 cells using both purified mAb indicated that 6D5 and 1F2 bind distinct epitopes of m157 (Figure 1B). Isotyping analysis identified 6D5 as an IgG2a-kappa and 1F2 as an IgG1-kappa (not shown).
FIGURE 1.

Specificity and differential functional activity of m157-specific monoclonal antibodies. A, Flow cytometric analysis of BaF3 and C1498 cells transduced with control GFP (top row of histograms) or m157 (bottom row) retroviral expression vectors after staining with biotinylated anti-m157 monoclonal antibody 6D5 (left set of four histograms) or 1F2 (right set of four histograms). Histograms stained with secondary streptavidin-APC reagent alone are depicted in thin dashed lines. B, Flow cytometric analysis of parental and m157-transduced C1498 cells (left and right panels, respectively) following dual staining with mAbs 6D5 (secondary = goat anti-mouse FITC, x-axis) and biotinylated 1F2 (secondary = streptavidin APC, y-axis). C, β-galactosidase activity (CPRG assay) of Ly49H-expressing HD12 reporter cells following 16 hour co-culture with BaF3-m157 cells (4×104 each) in the absence (open inverted triangles) or presence of increasing concentrations of anti-m157 mAbs 6D5 (black squares), 1F2 (black triangles), or isotype control (open diamonds). D, Lysates of C1498-m157 cells were immunoprecipitated with 6D5 or 1F2 conjugated to protein G-agarose beads and detected by Western blot analysis with 6H121 antibody. Top panels (I) show blots counterstained with HRP-conjugated IgG (H: IgG heavy chain; L: IgG light chain). Bottom panels (II) show blots counterstained with HRP-conjugated IgG TrueBlot™, which detects native antibody, but not denatured IgG heavy (H) and light (L) chains. The residual signal is specific for m157.
Next, we tested the inhibitory activity of each m157-specific mAb in our in vitro reporter assay for Ly49H-mediated activation (15). When pre-incubated with BaF3-m157 cells mAb 6D5 and 1F2 show strikingly different blocking capacity on induction of HD12 reporter activity—6D5 potently blocked HD12 activation by BaF3-m157 stimulators (>90% inhibition at 0.5 μg/mL), whereas 1F2 showed only ∼50% inhibition even at very high concentrations (e.g., 50 μg/mL, Figure 1C). Similar results were obtained with C1498-m157 stimulators and MCMV-infected targets (not shown). Taken together, these data indicate that the m157-specific mAbs 6D5 and 1F2 bind distinct epitopes and that the 6D5-specific epitope may be proximal to a critical Ly49H recognition site on m157.
Using a similar strategy, Tripathy et al. generated anti-m157 mAb clone 6H121 and demonstrated specific staining of surface-expressed m157 by flow cytometry on transduced BaF3-m157 cells and MCMV-infected primary bone marrow macrophages by immunofluorescence microscopy (27). Using this reagent, we were able to further characterize m157 by immunoprecipitation with 6D5 or 1F2 followed by Western blotting with 6H121. As shown in Figure 1D, m157 is detected as an amorphous band at 42-48 kDa, expressed in transduced C1498 cells. Note that 6D5 and 1F2 immunoprecipitate m157 proteins of similar size, but that 6D5 is more efficient than 1F2, consistent with its higher mean fluorescence intensity surface staining capacity in flow cytometry (Fig. 1A).
Distinct m157 isoforms among m157-transduced and MCMV-infected cells
To characterize m157 expressed in MCMV-infected cells, we performed Western blot analysis on lysates from NIH-3T3 fibroblasts and IC-21 macrophages infected with either wt-Smith strain or the GFP-expressing strain RM4503.3 of MCMV (derived from strain K181, (33)), comparing them to lysates obtained from C1498 or 3T3 cells transduced with m157-only (Figure 2). Interestingly, the pattern of detected m157 isoforms differs between cell types for transduced cells (Fig. 2A) and MCMV-infected cells (Fig. 2B). For example, in MCMV-infected cells there is a shared m157 isoform at ∼42 kDa (low abundance, but present in MCMVSmith-infected IC-21 cells), whereas a larger species of ∼50 kDa present in macrophages is not detected in fibroblasts, and the most abundant m157 species in MCMV-infected fibroblasts (∼46 kDa) is absent in infected macrophages. There is also a striking difference in relative abundance of m157 present in MCMV-infected fibroblasts (high) compared to macrophages (low). Densitometric analysis of a summation of all m157 species for each infected cell line indicated that NIH-3T3 fibroblasts contain 10-fold more m157 than IC-21 macrophages. There is little difference in the isoforms or abundance of m157 between the different strains of MCMV; this is not surprising given the high degree of conservation of m157 and flanking sequences in the Smith vs. K181 strains (Fig. 2B). We have also observed a consistently low level of m157 expression in MCMV-infected DC2.4 dendritic cells, similar to IC-21 macrophages (data not shown), suggesting that m157 expression may be differentially regulated in these infected cell types.
FIGURE 2.

Western blot analysis of m157 reveals multiple isoforms differentially expressed in m157-transduced and MCMV-infected cells. A, Triton X-100 lysates prepared from parental and m157-transduced C1498 or NIH-3T3 cells were analyzed by Western blotting with anti-m157 mAb 6H121; multiple m157 isoforms ranging between 42-48 kDa are detected (β-actin loading control shown below). B, NIH-3T3 fibroblasts and IC-21 macrophages were infected with MCMV (Smith strain or the K181-derived GFP-expressing RM4503.3 strain, as indicated) at a MOI of 5 for 24 hours. Triton X-100 lysates were subjected to Western blotting as in A, including the actin loading control. Note that 10-fold less m157 is produced in MCMV-infected IC-21 macrophages (digital image analysis), and that the m157 isoforms above 42 kDa migrate with a different relative molecular mass compared to either m157-transduced cells or to MCMV-infected fibroblasts.
MCMV m157 is expressed as a GPI-associated protein on the surface of transduced and MCMV-infected cells
Protein analysis software predicts m157 to be a glycosylphosphatidylinositol-associated protein (GPI-AP) with potential GPI-linkages at Ser 269 (native m157 numbering (26); DGPI online GPI predictor: http://129.194.185.165/dgpi/DGPI_demo_en.html) or Ser 286 (DGPI online GPI predictor and I.M.P. big-PI Predictor: http://mendel.imp.ac.at/sat/gpi/gpi_server.html). Tripathy et al. recently demonstrated that m157 is expressed as a GPI-AP on the surface of transduced RMA-m157 cells (27). However, we wanted to verify the cell surface linkage in other cell types and, more importantly, in MCMV-infected cells. First, we examined C1498 (H2b myeloid leukemia) cells transduced with m157. Triton X-114 lysates of C1498 and C1498-m157 cells were separated into a soluble fraction containing cytoplasmic proteins and a detergent fraction containing membrane-associated proteins, and then analyzed by Western blotting using anti-m157 mAb 6H121. As shown in Fig. 3A, m157 is found almost exclusively in the membrane fraction (fraction II, along with the HSA internal control). However, following treatment with PI-PLC, a majority of m157 moved into the soluble (aqueous) fraction, consistent with GPI-cleavage (Fig. 3B). Similar results were observed in Triton X-114 lysates prepared from MCMV-infected fibroblasts and macrophages, indicating that m157 is expressed as a GPI-AP following infection of these cells (Fig. 3C). At least 3 PI-PLC-sensitive m157 isoforms were detected in MCMV-infected NIH-3T3 fibroblasts, but only a single predominant isoform of ∼45 kDa was detected in MCMV-infected IC-21 macrophages. Note that the slower migration of m157 in fraction II is artifactual, due to the high concentration of detergent.
FIGURE 3.

The m157 protein is expressed exclusively as a glycosylphosphatidylinositol-associated protein (GPI-AP) in m157-transduced and MCMV-infected cells. A, Triton X-114 lysates from parental and m157-transduced C1498 cells were separated into aqueous (I) and membrane (II) fractions and subjected to Western blotting as described above. Antibodies against heat-stable antigen (HSA) and caspase-3 serve as membrane-associated and cytoplasmic controls, respectively. B, Aqueous and membrane fractions of Triton X-114 lysates prepared from C1498-m157 cells were treated with PI-PLC and subsequently analyzed by Western blot as above. Prior to PI-PLC treatment, the majority of m157 is associated with the membrane fraction (as in A), but following treatment with PI-PLC, m157 is released from the membrane fraction (II) into the aqueous fraction (I). C, NIH-3T3 and IC-21 cells were infected with MCMVSmith (MOI=5; 20 hours) after which Triton X-114 lysates were separated into membrane and aqueous fraction, subjected to PI-PLC treatment and analyzed by Western blotting as in A and B above. Note that m157 is expressed exclusively as a GPI-AP in infected fibroblasts and macrophages. The slightly slower migration of m157 in the membrane fraction (II) is artifactual due to the presence of detergent.
These results were corroborated by flow cytometric analysis of intact C1498-m157 cells following PI-PLC treatment; a >95% reduction in cell surface m157 is observed as compared to mock-treated controls (Fig. 4A). Importantly, PI-PLC- treatment of C1498-m157 cells abrogated Ly49H-mediated activation of HD12 reporter cells (Fig. 4B). This reduction in β-galactosidase induction was not due to blocking activity of cleaved m157, since PI-PLC-treated cells were washed prior to co-incubation with HD12 reporters. De novo regeneration of cell surface m157 occurring during the 6 hour co-incubation period accounts for the minimal residual β-galactosidase activity as determined by flow cytometry of cells immediately following PI-PLC and at the end of the co-incubation period (data not shown). In addition, treatment of MCMV-infected IC-21 or DC2.4 cells with PI-PLC prior to co-incubation with HD12 reporter cells also abrogates their Ly49H stimulation capacity (Fig. 4C); in these experiments, residual β-galactosidase activity is not apparent, reflecting a lower level of m157 expression (Fig. 2) and perhaps a concomitantly diminished capacity for regeneration of cell surface m157 in MCMV-infected (vs. transduced; Fig. 4B) host cells. These data are consistent with essentially exclusive expression of m157 as a GPI-AP in both retrovirally-transduced and MCMV-infected cells.
FIGURE 4.
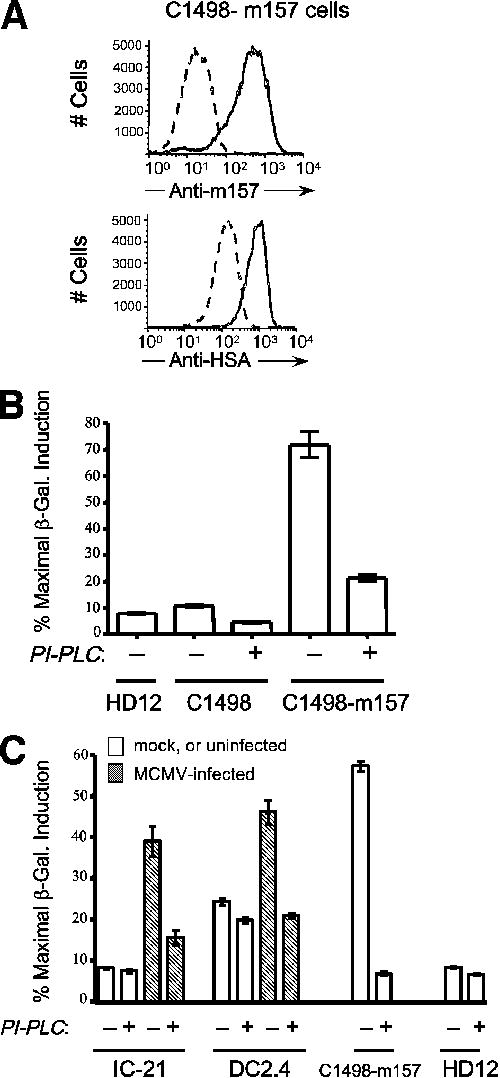
PI-PLC treatment abrogates activation of Ly49H by m157-transduced and MCMV-infected cells. A, C1498-m157 cells (1×106) were treated with 1 unit of PI-PLC for one hour at 37°C and then stained for surface expression of m157 (mAb 6D5) or heat-stable antigen (HSA; clone M1/69). Dashed and solid histograms represent PI-PLC-treated and mock-treated samples, respectively. Note that >95% of surface m157 is lost following PI-PLC treatment. B, Mock- or PI-PLC treated parental and m157-expressing C1498 cells were co-incubated with HD12 reporter cells at a 1:1 (4×104 each) for 16 hours after which β-galactosidase activity was determined by CPRG assay. Data are expressed as a percent of the maximal β-galactosidase induction obtained with PMA and ionomycin. Note that a majority of Ly49H activation is lost following PI-PLC treatment. C, IC-21 macrophages and DC2.4 dendritic cells were mock- (white bars) or MCMVSmith -infected (MOI = 5; hatched bars) and plated in 96 well plates at 1×105 cells/well for 12 hours after which they were treated with 0.1 units (IC-21) or 0.2 units (DC2.4) of PI-PLC for 1 hour at 37°C. Following PI-PLC treatment HD12 reporter cells (5×104) were added for an additional 6 hours co-incubation. Brefeldin A (5 ng/ml) was included during the PI-PLC treatment and the length of the co-incubation for the DC2.4 cells to inhibit regeneration of cell surface m157. Induction of β-galactosidase was quantitatively measured as in B above.
Random mutagenesis of m157 and selection of expressed m157 variants
Recent reports describe the rapid emergence of MCMV ‘escape’ viruses in vivo in C57BL/6 mice lacking adaptive immunity, but retaining NK cell-mediated selective pressure exerted through Ly49H recognition of m157. The majority of the escape viruses harbor null mutations in m157 (deletions or frameshifts); other mutations predicting expression of a mutant cell surface m157 glycoprotein were detected as FLAG-tagged surface proteins (30) or were not evaluated for the capacity to produce detectable surface m157 (31, 32). We sought to determine key residues in m157 required for activation of Ly49H; that is, mutations that result in detectable expression of cell surface m157, but fail to activate Ly49H. We optimized the parameters for PCR-mediated random mutagenesis in order to generate m157 cDNA containing 1-2 nonconservative mutations per copy, as verified by sequence analysis of at least 10 clones per library (data not shown). This m157 cDNA library was subcloned into the pMX-Puro retroviral vector and transduced into C1498 cells. We chose C1498 as the host cell line because m157 is expressed as a GPI-AP at a high level in C1498 cells, resulting in a consistently potent stimulation of HD12 reporters compared to other m157-transduced cell lines (AHD and JWH, unpublished observations). This strategy allowed us to have a higher degree of confidence in identifying key m157 mutations expressed in C1498 cells that fail to activate Ly49H. Flow cytometric analysis of C1498 cells transduced with the mutant m157 library revealed that at least 30% of the clones were expressing m157 to a similar level as control C1498-m157 cells previously transduced with wt-m157 (Fig. 5A). In order to maximize the number of surface-expressed m157 mutants in our screen, we sorted approximately one-half of the total mutant library-transduced C1498 cells for m157 expression based on dual staining with both 6D5 and 1F2. Two sorted groups of dual-positive cells (6D5+/1F2+ and 6D5lo/1F2+) were then cloned by limiting dilution and 630 surviving clones were tested in duplicate for activation of HD12 reporter cells. All clones showing <20% maximal induction of β-galactosidase activity (CPRG assay) were expanded and retested for surface m157 expression. Clones that stained with either 6D5 or 1F2 were then tested again for Ly49H activation (CPRG assay, secondary screen) and those failing to achieve 20% maximal β-galactosidase induction were analyzed further as candidate clones harboring m157 mutations critical for activation of Ly49H.
FIGURE 5.

Generation and characterization of m157 mutants in C1498 cells. A, Flow cytometric analysis of C1498 cells transduced with the m157 cDNA library generated with the Genemorph II Random Mutagenesis kit (Genemorph) as compared to control C1498-m157 cells stained with mAb 6D5 (dashed histogram). Approximately 30% of the transduced C1498 cells are expressing m157 at the surface. Mutant m157-transduced C1498 cells were then dually stained with 6D5 and 1F2, sorted, and sub-cloned as described in materials and methods. B, Flow cytometric analysis of C1498 cells transduced with selected m157 mutants generated by site-directed mutagenesis. Expression of m157 as detected by three anti-m157 mAb (6D5, 6H121, and 1F2) is shown for the parental C1498, C1498-m157 wild-type control, and clones expressing m157 with mutations at positions Ile153 or Lys161 (I153T, K161N). Note that both m157 mutants are detected by mAb 6H121, but are differentially recognized by mAbs 6D5 and 1F2. Grey histograms with dashed lines represent staining with secondary reagent alone. C, Activation of Ly49H reporter cells by C1498 cells expressing single mutations in m157. C1498 cells expressing m157 variants were co-incubated with HD12 reporter cells (4×104 each) for 16 hours at 37°C. β-galactosidase induction was measured 2 hours after the addition of CPRG and expressed as a percentage of maximal induction obtained by using PMA and ionomycin.
Total RNA from clones expressing m157 but failing to activate Ly49H was harvested, reverse-transcribed, and sequenced to determine the identity of underlying m157 mutations. In some cases a clone had more than one mutation, and several clones contained the same mutation in combination with a second unique mutation. In addition, the density of m157 expression among the clones was not consistent (data not shown). For these m157-low (mean fluorescence intensity) clones, it was possible that suboptimal Ly49H activation was simply a reflection of low ligand density. Thus, in order to resolve informative mutations from confounding mutations resulting in low-level cell surface m157 expression, we reconstructed each identified random mutation as a single, isolated mutant by site-directed PCR mutagenesis. In parallel, we created alanine substitutions for each novel, non-alanine mutation (Table I).
Table I. Summary of cell surface expressed m157 mutations in C1498 cells.
| Founder clonea | m157 mutation(s)b | m157 mutant MFIc | Ly49H activationd | Position in tertiary m157 structuree | ||
|---|---|---|---|---|---|---|
| 6D5 | 1F2 | 6H121 | ||||
| wt-m157 control | none | 361 | 112 | 619 | 100% | not applicable |
| CF5 | T9S | 301 | 84 | 471 | 135% | exposed; amino-terminal α0-helix |
| T9A | 233 | 62 | 344 | 117% | ||
| 1E8 | N43K | 169 | 58 | 310 | 79% | exposed; loop between 5th and 6th β-strand in platform |
| N43A | 119 | 42 | 286 | 62% | ||
| I153T | (02) | (02) | 218 | 0% | buried; long α2-helix | |
| I153A | (0) | (02) | 140 | 12% | ||
| DA10 | S67A | 235 | (0) | 612 | 80% | exposed; loop connecting α1-helix to β-strand platform |
| T261A | 226 | 64 | 335 | 114% | exposed; loop within β-stranded α3 domain | |
| AC9 | K161N | 84 | 42 | 248 | 2% | buried; long α2-helix |
| K161A | 297 | 114 | 589 | 21% | ||
| 5B3 | K170R | 181 | (04) | 369 | 76% | exposed; long α2-helix |
| K170A | 160 | (05) | 310 | 95% | ||
only five (of 630) clones survived tertiary screening; clones1E8 and DA10 contained multiple mutations.
only those mutations resulting in a nonconservative, in-frame amino acid substitution are listed. Each unique m157 mutation (shown in bold) and its corresponding alanine substitution variant was regenerated by site-directed PCR mutagenesis.
each unique m157 mutation was transduced into C1498 cells and subsequently sorted by flow cytometry for high-level m157 expression; mean fluorescence intensity (MFI) values are listed after subtracting background MFI from parental C1498 cells. MFI for control C1498-m157 cells (wt-m157) is also indicated as a reference.
represents percentage of wt-m157 activity for each mutant after subtracting the background from parental C1498 cells.
positions mapped using PyMOL analysis software and the published PDB structure file for m157 (ID code: 2NYK); see also Figure 8.
C1498 cells were then independently transduced with the single-mutation m157 cDNA, selected and sorted by flow cytometry for high-level m157 expression. Subsequent flow cytometric analysis of each mutant m157 cell line using three different anti-m157 reagents indicated a consistent level of m157 expression among the mutant lines, although not all of the m157 mutants are recognized by all anti-m157 mAbs. In particular, the mutant line harboring a mutation at Ile153 retains binding by 6H121 and, to a lesser extent 1F2, but shows no binding to 6D5, suggesting that these mAbs discriminate an epitope containing or affected by Ile153 (Fig. 5B and Table I). We also tested each variant m157-expressing cell line for sensitivity to PI-PLC treatment, and observed that, like wt-m157, all m157 mutants were expressed exclusively as GPI-AP in C1498 cells (data not shown). Finally, we mapped each mutated m157 residue onto the published structure for m157 (26) and found that residues at mutated positions 9 (Thr), 43 (Asn), 67 (Ser), 170 (Lys) and 261 (Thr) are solvent exposed and scattered across the entire m157 structure, whereas mutant residues at positions 153 (Ile) and 161 (Lys) are buried on either side of the bend within the long α2-helix (Table I).
Identification of m157 residues mediating a functional interaction with Ly49H
Next, with comparable surface expression of individual m157 mutants, we were able to assess the capacity of each for Ly49H activation using our HD12 reporter assay for β-galactosidase induction. Fig. 5C shows a CPRG assay for β-galactosidase activity in HD12 cells following co-incubation with each of the cell lines expressing site-directed m157 mutations at positions identified above. In multiple CPRG assays, the individual activating capacity of each m157-expressing line varies modestly, but the hierarchy of activating potency is remarkably consistent from experiment-to-experiment (n = 4). Importantly, a mutation at either position 153 (Ile→Thr), or 161 (Lys→Asn) completely abrogates activation mediated by Ly49H. The alanine-substitution mutants at these positions are also significantly deficient, but retain partial activity (12% and 21% of wt-m157, respectively). Further, the HD12-mediated activation by all m157 variants was completely blocked by preincubation of the stimulator cells with anti-m157 mAb 6D5 or 6H121 (not shown). Thus, despite robust cell surface expression of m157 as a GPI-AP and in a conformation recognized by at least two anti-m157 mAbs, the m157 I153T/A and K161N/A variants fail to activate Ly49H, consistent with their proximity to a critical Ly49H recognition site (or sites). Mutations at other positions, including Tyr261, resulted in robust or only moderately reduced Ly49H activation, suggesting that individually these residues contribute little or no influence upon the functional interaction with Ly49H. It is noteworthy that the majority of these ‘irrelevant’ mutations were initially identified in combination with at least one other m157 mutation (e.g., N43K, T67A, T261A), most likely reflecting a combined effect of partially deleterious mutations. The conservative mutation at Lys170 (K170R) has a modest, but reproducibly negative impact on Ly49H activation, one that is partially rescued by replacement with an even less conserved Ala at this position (K170A, Fig. 5C). It is likely that the reduced density of cell surface m157 expression (not shown) combined with the modulating effect of the K170R mutation in the original clone (5B3) resulted in an overall Ly49H activating capacity that fell below our screening threshold (meeting our criteria for a clone of interest).
One striking feature of the m157 variants that fail to activate Ly49H is that both contain mutations at buried residues within the m157 structure. The absence of m157 mutations at solvent-exposed residues identified from our random mutagenesis and screening strategy led us to generate additional site-directed mutations at exposed residues predicted to interact with Ly49H. These surface-exposed sites form two clusters: Asp109, Ile111 and Arg158 are positioned at the amino terminus of the α1 helix, along the opposite face to that lying just above the buried Lys161 (analogous to ‘Site 1’ for Ly49 receptor interactions with MHC I ligands), while Thr117 and Asp119 form a second site along the outer edge of the β-stranded platform lying between the α1 and α2 helicies (26). Using our retroviral transduction system, we were able to generate C1498 cell lines expressing m157 at the surface with mutations at D109N or R158Q, mutations predicted to affect Ly49H binding based upon the m157 structure (26) and originally described for strains of MCMV that fail to activate Ly49H (30). As shown in Fig. 6A, both D109N and R158Q single-mutation m157 variants show robust cell surface expression as detected by all three anti-m157 mAbs, similar to that of the wt-m157-expressing control. When co-incubated with HD12 reporter cells, both the D109N- and R158Q-expressing cells stimulated Ly49H-mediated induction of β-galactosidase with comparable potency to wt-m157 (85% and 102% of wt-m157 activity, respectively) that was completely abrogated by addition of blocking mAb 6D5 (Fig. 6B). These results are in stark contrast for what was found for the I153T and K161N mutations above (Fig. 5C), and indicate that either the D109 and R158 residues are not critical for Ly49H recognition of m157, or that as isolated mutations these positions are tolerant to significant substitutions (polar uncharged residues replacing an acidic Asp or basic Arg residue).
FIGURE 6.

Characterization of m157 variants with putative Ly49H contact site mutations expressed in C1498 cells. A, Flow cytometric analysis of C1498 cells transduced with m157 mutations Asp109 or Arg158 generated by site-directed mutagenesis. Cell surface expression of m157 as detected by three anti-m157 mAbs (6D5, 6H121, and 1F2) is shown for the parental C1498, C1498-m157 (wt) control, C1498-m157 D109N and C1498-m157 R158Q. Note that expression of m157 for both mutants is essentially equivalent to that for wt-m157. B, Activation of Ly49H reporter cells by C1498 cells expressing single mutations in m157. C1498 cells expressing m157 variants were co-incubated with HD12 reporter cells (4×104 each) for 16 hours at 37°C. β-galactosidase induction was measured 5 hours after the addition of CPRG and expressed as a percentage of maximal induction obtained by using PMA and ionomycin. Duplicate aliquots of stimulator cells were preincubated without (black bars) or with (white bars) blocking anti-m157 mAb 6D5 (20 μg/mL) prior to coincubation with HD12 reporters. Note that the variant D109N and R158Q m157-expressing cells activate Ly49H with essentially equal potency as wt-m157.
m157 variants that fail to activate Ly49H show reduced stability of the Ly49H-m157 complex
The failure of I153T/A and K161N/A to activate Ly49H reporters could be due to reduced binding to Ly49H, or a failure to induce a conformational change in Ly49H required for ITAM-dependent signaling. To resolve these possibilities, we incubated C1498 cells expressing wt-m157 or each of the four single-mutant variants with Ly49H-expressing HD12 cells at 37°C /5% CO2 for 10-to-180 minutes and then performed immunoprecipitation of Ly49 using the anti-Ly49H mAb 3D10. The immunoprecipitates were visualized by Western blot using anti-m157 mAb 6H121. As shown in Figure 7, wt-m157 forms a stable interaction with Ly49H that remains detectable even after 180 minutes of co-incubation, whereas the I153T and K161N variants show significantly reduced interaction with Ly49H after only 10 minutes. At 40 minutes there is no detectable m157 in the Ly49H immunoprecipitates, despite comparable expression of m157 in these cell lines (Fig. 5B and Fig. 7 immunoblot inset showing actin and m157 lysate input controls). Since available Ly49H-specific reagents do not recognize denatured Ly49H in Western blotting, we verified essentially equivalent Ly49H content in each mixture by flow cytometry (data not shown). Furthermore, while the activating m157 variants D109N and R158Q show a stable interaction with Ly49H at 10 minutes, there is a marked reduction (R158Q) or complete absence (D109N) of m157 after 40 minutes, indicating that these residues may be important in maintaining a durable m157-Ly49H complex. These results indicate that isolated mutations in m157 have a variable effect on the overall affinity of Ly49H for m157, and that Ly49H-dependent signaling in HD12 cells requires a stable interaction with m157 lasting at least 10 minutes.
FIGURE 7.

Assessment of Ly49H-m157 interactions by co-immunoprecipitation reveals a hierarchy of stability for m157 variants containing single mutations compared to wt-m157. C1498 cells expressing different m157 mutants were incubated with HD12 cells expressing Ly49H (ratio = 2:1) for the indicated time periods. Cell mixtures were lysed in IP buffer and immunoprecipitated with anti-Ly49H mAb 3D10 and resolved on 10% SDS-PAGE. To detect co-immunoprecipitated m157, Western blot analysis was performed with mAb 6H121. “Lysate input” inset shows m157 and total protein (actin) loading controls for each sample.
Discussion
MCMV infection in mice is a robust animal model for understanding the complex biology of this unique family of highly successful and pathogenic DNA viruses, as well as the integrated anti-viral immune response of the host. The observation that MCMV encodes a potent activating ligand for NK cells was striking, and serves as an opportunity to dissect the molecular parameters regulating NK cell activation. Our findings advance the understanding of how NK cells recognize and respond to MCMV-infected cells at the molecular interface between Ly49H and m157. Although this unique interaction has vital consequences for the host (28) and is capable of exerting potent selective force upon MCMV (30-32), it is clear that m157 is evolving in response to additional selective pressures, including those exerted by other Ly49 receptors (38). Thus, our findings are important for understanding the activation of a critical lymphocyte subset in antiviral defense, but also have implications for understanding the mechanism of m157-mediated immune evasion.
We utilized three distinct anti-m157 mAbs to characterize m157 expression. These reagents recognize unique epitopes and manifest dramatically different blocking activity in our Ly49H reporter assay: mAbs 6D5 and 6H121 potently block Ly49H activation, whereas 1F2 does not (Figures 1 and 6). Although we have not mapped the m157 epitopes recognized by each mAb in detail, these results suggest that 6D5 and 6H121 recognize closely approximated regions overlapping a single Ly49H recognition site, or identify distinct sites required for Ly49H binding and/or activation. This conclusion is supported by the observation that mutation at Ile153 abrogates binding by 6D5, but not 6H121 (Figure 5B). Although immunoprecipitation with either 6D5 or 1F2 followed by Western blotting with 6H121 identifies a single m157 species (Figure 1D), more detailed analyses are underway to determine whether m157 interacts with other host cell or virally-encoded factors in transduced or MCMV-infected cells. The markedly lower level of m157 protein detected in macrophages compared to fibroblasts (Figure 2B) may reflect reduced transcription, processing, or ‘accessibility’ of m157 in these cells. In addition, the relative sizes of m157 isoforms vary between infected cell types, sharing only the ∼42 kDa band (Figure 2 and data not shown). There are at least five predicted N-linked glycosylation sites in the full-length m157 polypeptide, four of which are retained in fully processed, GPI-linked m157 at the cell surface (Asn178, Asn187, Asn213, and Asn267). Analysis of site-directed m157 mutants at each site indicates that differences in Mw for the m157 isoforms expressed in transduced and MCMV-infected cells are due to differential glycosylation (N.V. Guseva, C. Fullenkamp, P. Naumann, and J. W. Heusel, manuscript in preparation).
We show that MCMV-infected cells express m157 exclusively as a GPI-AP (Figs. 3C and 4C). In addition, we and others have successfully transduced m157 into a variety of murine cell lines including BaF3 (15), C1498, NIH-3T3, R1.1 and RMA (27); m157 is expressed as a GPI-AP in all of these cells. The utilization of a GPI linkage for virally encoded proteins is relatively rare; the Dengue virus non-structural protein 1 (NS1) was reported as the first such example (39). Since Dengue NS1 and other GPI-AP are capable of signaling, a similar signaling capacity for m157 potentially exists (40-42). The GPI-linkage of m157 may also provide a clue as to its origin. In this regard, it is noteworthy that other GPI-AP in the extended MHC I family are ligands for inhibitory receptors expressed on NK cells, including human HLA-G (43, 44) and murine Qa-2 (45). We are currently investigating the biologic significance of GPI-linkage for m157 in the context of Ly49 receptor interactions (both inhibitory and activating) and overall host resistance to MCMV.
Under the direct selection of Ly49H, MCMV rapidly evolves escape mutants in m157 when serially passaged in C57BL/6 and derivative strains, the majority of which are deletions, frameshift or nonsense mutations precluding expression of m157 protein (30-32). However, selected m157 escape mutants and variant sequences from wild MCMV strains have been reported that are predicted to result in m157 protein at the cell surface, including delThr111, D119Y, a small deletion near the predicted GPI-addition site (Thr261-Arg263), D109N, I111L, and R158Q (30, 31). Cell surface expression was demonstrated only for a subset of m157 variants (as FLAG-tagged versions), and although the MCMV strains from which these mutations were derived failed to activate Ly49H reporters, these strains also harbored numerous other nonconservative m157 substitutions (30), suggesting that a combination of m157 polymorphisms is required to escape detection by Ly49H. This idea is also supported by our identification of C1498-m157 clones harboring multiple m157 mutations that together fail to activate Ly49H, but as isolated mutations retain full activating capacity (Table I, clones CF5 and DA10). In our analyses, the m157 mutations D109N, R158Q and T261A result in abundant cell surface expression detected by all three anti-m157 mAbs (Figure 6A and Table I) and all three of these m157 variants retain the capacity to activate Ly49H reporters (Figs. 5B and 6B). While one or more of these sites may contribute to Ly49H binding affinity (Fig. 7), none is absolutely required for the activating Ly49H-m157 interaction.
The co-immunoprecipitation of m157 variants with Ly49H revealed at clear hierarchy of stability in the Ly49H-m157 interactions between HD12 cells and the m157 variants that has important functional consequences (Fig. 7). Mutations that show reduced Ly49H-m157 interaction as early as 10 minutes fail to activate Ly49H (I153T and K161N), while other mutations that show reduced Ly49H-m157 interactions only after 40 minutes provide for full Ly49H activation (D109N and R158Q). This is the first demonstration of a minimal interaction requirement for signaling between an isolated NK cell receptor and its ligand. These data may reflect both a minimum affinity and a minimum duration of interaction between Ly49H and m157 in order to achieve an activation threshold in the BWZ.36 reporter cell line, and are reminiscent of the well documented effect of signal sustainability for TCR-MHC/peptide interactions on T cell responses (reviewed in (46, 47). More detailed studies are needed to determine the magnitude of individual affinities of these m157 variants for Ly49H, and whether lower affinity interactions may be compensated by accessory molecular interactions in Ly49H+ NK cells. Similarly, the effect of affinity and duration of receptor engagement between m157 variants and relevant inhibitory Ly49 receptors (e.g., Ly49I129) should be addressed, since the rules for Ly49 receptor activation and inhibition may differ (48-50).
The recent solution of a crystal structure for m157 allows for predictions about how it may engage not only Ly49H, but other Ly49 receptors as well. When mapped onto this m157 structure, the activation-deficient mutations at Ile153 and Lys161 were striking in that both are buried residues (Fig. 8A). In particular, the buried Lys161 occupies a unique position near the bend in the long α2 helix: through hydrogen bonding this residue anchors adjacent residues in the amino-terminal α0 helix (Asp16, Phe17 and Phe19) and in the β-stranded floor of the α1/α2 platform (Gly107; Fig. 8B). Replacement of lysine with the bulkier asparagine would likely disrupt these hydrogen bonds and result in the destabilization the amino-terminal α0 helix in particular. Replacement of Lys161 with alanine would be expected to have a less deleterious effect, since the smaller alanine residue would permit access of water molecules to provide some stabilization of the structure. Indeed, the K161A mutation retains some partial Ly49H activating capacity (Fig. 5C). In this regard, the T9S mutation is notable for its exposed position in the α0-helix. Interestingly, this mutation does not inhibit Ly49H activation, and may even enhance it (Fig. 5C). Interpretation of the Ile153 mutation is not straightforward. The failure of the alanine substitution at Ile153 suggests that there are strict size constraints at this buried position that impact Ly49H recognition. Finally, the observation that mutations in the exposed Asp109 or Arg158 residues of m157 did not affect Ly49H activation was unexpected (Fig. 6), and suggests that additional polymorphisms must also contribute to the loss of Ly49H recognition of the m157 variants from which these isolated mutations were derived (30). Regardless, the Lys161 mutations are informative, supporting a critical role for this residue in securing the amino-terminal α0 helix to the larger m157 superstructure. A more rigidly fixed α0 helix may be contribute directly (e.g., key Ly49H binding site residues) or indirectly (allowing access to a site that is blocked by an untethered α0 helix) toward Ly49H-mediated activation.
FIGURE 8.
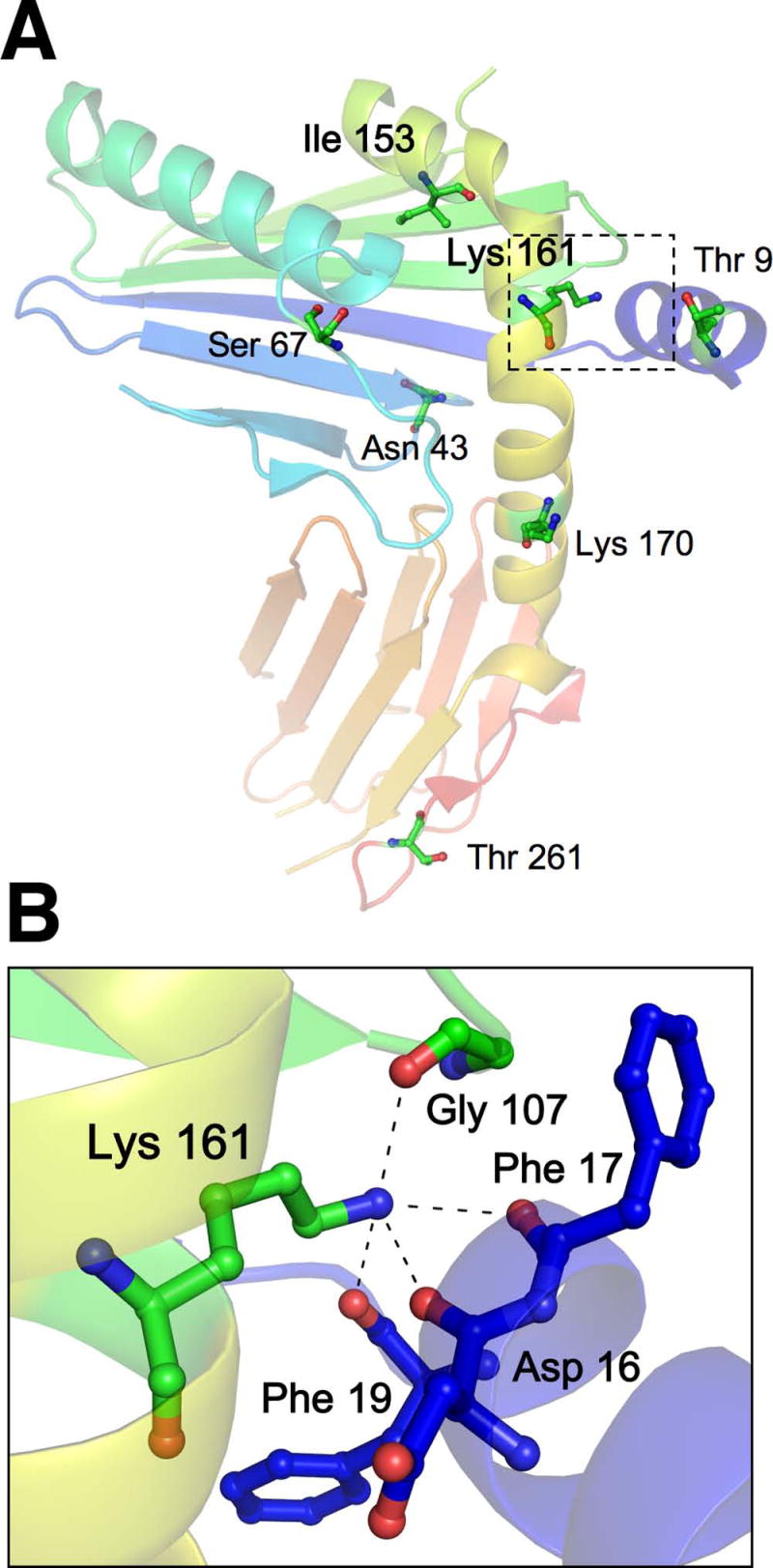
Mapping of m157 mutations onto the existing m157 structure reveals an unanticipated role for buried residues Ile153 and Lys161 in mediating Ly49H recognition. A, The positions of selected m157 mutations (described in Table I) are depicted according to the published structure for m157, which is rendered in color from amino (blue) to carboxyl (red) termini (PDB ID code 2NYK (26)). Only residues Ile153 and Lys161 (in bold) are buried and lie on either side of the bend in the long α2 helix (yellow). The dashed box indicates the enlarged area of interest surrounding Lys161 in B. B, Lys161 forms hydrogen bonds with four other residues: Gly107 located in a small loop of the β-stranded region (green) forming the floor of the α1/α2 platform, and Asp16, Phe17, and Phe19 of the amino-terminal α0 helix (blue). Thus, Lys161 anchors the α0 helix in position relative to the m157 superstructure. Figure generated using PyMOL software (37).
Our findings advance the understanding of how NK cells recognize MCMV-infected cells at the molecular interface between Ly49H and m157. While the overall theme is similar, in that NK cell receptors recognize ligands with MHC I-like structure, the individual receptor-ligand interactions are much more diverse than previously appreciated. The dual nature of m157 as both decoy (for inhibitory Ly49 receptors) and danger signal (for activating Ly49 receptors) will allow for the comparison of molecular features that govern both immune evasion and activation. Considering the recent m157 atomic structure and Ly49H mutagenesis data, a novel structural interaction between Ly49H and its MCMV-encoded ligand is emerging that will help to explain how competing mechanisms of immune evasion and NK cell activation are operating during the earliest phases of viral infection.
Acknowledgments
The authors thank Colleen Fullenkamp for preparation of key reagents and other experimental assistance, Subramanian Ramaswamy for assistance in mapping and interpretation of m157 mutations, and Jon Houtman, Thomas Waldschmidt and Kevin Legge for helpful discussions and critical review of the manuscript.
This work was supported in part by NIH R56 AI53226-01.
Abbreviations used in this paper
- β2m
beta-2-microglobulin
- CPRG
chlorophenolred-β-D-galactopyranoside
- DC
dendritic cell(s)
- GPI-AP
glycosylphosphatidylinositol-associated protein
- MCMV
murine cytomegalovirus
- MOI
multiplicity of infection
- PI-PLC
phosphatidylinositol-specific phospholipase C
Footnotes
Disclosures: The authors have no conflicting interests to report.
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
References
- 1.Orange JS. Human natural killer cell deficiencies and susceptibility to infection. Microbes & Infection. 2002;4:1545–1558. doi: 10.1016/s1286-4579(02)00038-2. [DOI] [PubMed] [Google Scholar]
- 2.Biron CA, Brossay L. NK cells and NKT cells in innate defense against viral infections. Current Opinion in Immunology. 2001;13:458–464. doi: 10.1016/s0952-7915(00)00241-7. [DOI] [PubMed] [Google Scholar]
- 3.Biron C, Byron K, Sullivan J. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. 1989;320:1731–1735. doi: 10.1056/NEJM198906293202605. [DOI] [PubMed] [Google Scholar]
- 4.Loh J, Chu DT, O'Guin AK, Yokoyama WM, Virgin HWIV. Natural Killer Cells Utilize both Perforin and Gamma Interferon To Regulate Murine Cytomegalovirus Infection in the Spleen and Liver. J Virol. 2005;79:661–667. doi: 10.1128/JVI.79.1.661-667.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lodoen MB, Lanier LL. Natural killer cells as an initial defense against pathogens. Current Opinion in Immunology. 2006;18:391–398. doi: 10.1016/j.coi.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yokoyama WM, Kim S. Licensing of natural killer cells by self-major histocompatibility complex class I. Immunological Reviews. 2006;214:143–154. doi: 10.1111/j.1600-065X.2006.00458.x. [DOI] [PubMed] [Google Scholar]
- 7.Gasser S, Raulet DH. Activation and self-tolerance of natural killer cells. Immunological Reviews. 2006;214:130–142. doi: 10.1111/j.1600-065X.2006.00460.x. [DOI] [PubMed] [Google Scholar]
- 8.Bashirova AA, Martin MP, McVicar DW, Carrington M. The Killer Immunoglobulin-Like Receptor Gene Cluster: Tuning the Genome for Defense. Annual Review of Genomics and Human Genetics. 2006;7:277–300. doi: 10.1146/annurev.genom.7.080505.115726. [DOI] [PubMed] [Google Scholar]
- 9.Parham P. MHC class I molecules and KIRs in human history, health and survival. Nature Reviews Immunology. 2005;5:201–214. doi: 10.1038/nri1570. [DOI] [PubMed] [Google Scholar]
- 10.Perussia B. Fc receptors on natural killer cells. Curr Top Microbiol Immunol. 1998;230:63–88. doi: 10.1007/978-3-642-46859-9_6. [DOI] [PubMed] [Google Scholar]
- 11.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003 Oct;3(10):781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 12.Tassi I, Klesney-Tait J, Colonna M. Dissecting natural killer cell activation pathways through analysis of genetic mutations in human and mouse. Immunological Reviews. 2006;214:92–105. doi: 10.1111/j.1600-065X.2006.00463.x. [DOI] [PubMed] [Google Scholar]
- 13.Daniels KA, Devora G, Lai WC, O'Donnell CL, Bennett M, Welsh RM. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. Journal of Experimental Medicine. 2001;194:29–44. doi: 10.1084/jem.194.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [comment] [DOI] [PubMed] [Google Scholar]
- 15.Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, Scalzo AA, Fremont DH, Yokoyama WM. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci U S A. 2002;99:8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scalzo AA, Fitzgerald NA, Simmons A, La Vista AB, Shellam GR. Cmv-1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J Exp Med. 1990;171:1469–1483. doi: 10.1084/jem.171.5.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shellam GR, A J, Papadimitriou JM, Bancroft GJ. Increased susceptibility to cytomegalovirus infection in beige mutant mice. Proc Natl Acad Sci U S A. 1981;78:5104–5108. doi: 10.1073/pnas.78.8.5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown MG, Scalzo AA, Stone LR, Clark PY, Du Y, Palanca B, Yokoyama WM. Natural killer gene complex (Nkc) allelic variability in inbred mice: evidence for Nkc haplotypes. Immunogenetics. 2001;53:584–591. doi: 10.1007/s002510100365. [DOI] [PubMed] [Google Scholar]
- 19.Krmpotic A, Busch DH, Bubic I, Gebhardt F, Hengel H, Hasan M, Scalzo AA, Koszinowski UH, JonjicStipan MCMV glycoprotein gp40 confers virus resistance to CD8+ γ/δ−T cells and NK cells in vivo. Nat Immunol. 2002;3:529–535. doi: 10.1038/ni799. [DOI] [PubMed] [Google Scholar]
- 20.Scalzo AA, Manzur M, Forbes CA, Brown MG, Shellam GR. NK gene complex haplotype variability and host resistance alleles to murine cytomegalovirus in wild mouse populations. Immunol Cell Biol. 2005;83:144–149. doi: 10.1111/j.1440-1711.2005.01311.x. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez M, Sabastian P, Clark P, Brown MG. Cmv1-Independent Antiviral Role of NK Cells Revealed in Murine Cytomegalovirus-Infected New Zealand White Mice. J Immunol. 2004;173:6312–6318. doi: 10.4049/jimmunol.173.10.6312. [DOI] [PubMed] [Google Scholar]
- 22.Desrosiers MP, Kielczewska A, Loredo-Osti JC, Adam SG, Makrigiannis AP, Lemieux S, Pham T, Lodoen MB, Morgan K, Lanier LL, Vidal SM. Epistasis between mouse Klra and major histocompatibility complex class I loci is associated with a new mechanism of natural killer cell-mediated innate resistance to cytomegalovirus infection. Nat Genet. 2005;37:593–599. doi: 10.1038/ng1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dighe A, Rodriguez M, Sabastian P, Xie X, McVoy M, Brown MG. Requisite H2k Role in NK Cell-Mediated Resistance in Acute Murine Cytomegalovirus-Infected MA/My Mice. J Immunol. 2005;175:6820–6828. doi: 10.4049/jimmunol.175.10.6820. [DOI] [PubMed] [Google Scholar]
- 24.Adam SG, Caraux A, Fodil-Cornu N, Loredo-Osti JC, Lesjean-Pottier S, Jaubert J, Bubic I, Jonjic S, Guenet JL, Vidal SM, Colucci F. Cmv4, a New Locus Linked to the NK Cell Gene Complex, Controls Innate Resistance to Cytomegalovirus in Wild-Derived Mice. J Immunol. 2006;176:5478–5485. doi: 10.4049/jimmunol.176.9.5478. [DOI] [PubMed] [Google Scholar]
- 25.Arase H, Lanier LL. Virus-driven evolution of natural killer cell receptors. Microbes & Infection. 2002;4:1505–1512. doi: 10.1016/s1286-4579(02)00033-3. [DOI] [PubMed] [Google Scholar]
- 26.Adams EJ, Juo ZS, Venook RT, Boulanger MJ, Arase H, Lanier LL, Garcia KC. Structural elucidation of the m157 mouse cytomegalovirus ligand for Ly49 natural killer cell receptors. PNAS. 2007;104:10128–10133. doi: 10.1073/pnas.0703735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tripathy SK, Smith HRC, Holroyd EA, Pingel JT, Yokoyama WM. Expression of m157, a Murine Cytomegalovirus-Encoded Putative Major Histocompatibility Class I (MHC-I)-Like Protein, Is Independent of Viral Regulation of Host MHC-I. J Virol. 2006;80:545–550. doi: 10.1128/JVI.80.1.545-550.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown MG, Dokun AO, Heusel JW, Smith HR, Beckman DL, Blattenberger EA, Dubbelde CE, Stone LR, Scalzo AA, Yokoyama WM. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science. 2001;292:934–937. doi: 10.1126/science.1060042. [DOI] [PubMed] [Google Scholar]
- 29.Lee SH, Zafer A, de Repentigny Y, Kothary R, Tremblay ML, Gros P, Duplay P, Webb JR, Vidal SM. Transgenic expression of the activating natural killer receptor Ly49H confers resistance to cytomegalovirus in genetically susceptible mice. Journal of Experimental Medicine. 2003;197:515–526. doi: 10.1084/jem.20021713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voigt V, Forbes CA, Tonkin JN, Degli-Esposti MA, Smith HRC, Yokoyama WM, Scalzo AA. Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. PNAS. 2003;100:13483–13488. doi: 10.1073/pnas.2233572100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.French AR, Pingel JT, Wagner M, Bubic I, Yang L, Kim S, Koszinowski U, Jonjic S, Yokoyama WM. Escape of Mutant Double-Stranded DNA Virus from Innate Immune Control. Immunity. 2004;20:747–756. doi: 10.1016/j.immuni.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 32.French AR, Pingel JT, Kim S, Yang L, Yokoyama WM. Rapid emergence of escape mutants following infection with murine cytomegalovirus in immunodeficient mice. Clinical Immunology. 2005;115:61–69. doi: 10.1016/j.clim.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 33.van Den Pol AN, Mocarski E, Saederup N, Vieira J, Meier TJ. Cytomegalovirus cell tropism, replication, and gene transfer in brain. Journal of Neuroscience. 1999;19:10948–10965. doi: 10.1523/JNEUROSCI.19-24-10948.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karttunen J, Shastri N. Measurement of Ligand-Induced Activation in Single Viable T Cells Using the lacZ Reporter Gene. Proceedings of the National Academy of Sciences. 1991;88:3972–3976. doi: 10.1073/pnas.88.9.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gasteiger E, G A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelley LA, M R, S MJE. Recognition of Remote Protein Homologies Using Three-Dimensional Information to Generate a Position Specific Scoring Matrix in the program 3D-PSSM. In: Sorin Istrail PP, Waterman Michael, editors. RECOMB 99, Proceedings of the Third Annual Conference on Computational Molecular Biology; New York, New York 10036: The Association for Computing Machinery; 1999. pp. 218–225. [Google Scholar]
- 37.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]
- 38.Scalzo AA, Corbett AJ, Rawlinson WD, Scott GM, Degli-Esposti MA. The interplay between host and viral factors in shaping the outcome of cytomegalovirus infection. Immunol Cell Biol. 2007;85:46–54. doi: 10.1038/sj.icb.7100013. [DOI] [PubMed] [Google Scholar]
- 39.Jacobs MG, Robinson PJ, Bletchly C, Mackenzie JM, Young PR. Dengue virus nonstructural protein 1 is expressed in a glycosyl-phosphatidylinositol-linked form that is capable of signal transduction. FASEB J. 2000;14:1603–1610. doi: 10.1096/fj.14.11.1603. [DOI] [PubMed] [Google Scholar]
- 40.Gunter KC, M T, Shevach EM. T cell-activating properties of an anti-Thy-1 monoclonal antibody. Possible analogy to OKT3/Leu-4. J Exp Med. 1984;159:716–730. doi: 10.1084/jem.159.3.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shenoy-Scaria AM, Kwong J, Fujita T, Olszowy MW, Shaw AS, Lublin DM. Signal transduction through decay-accelerating factor. Interaction of glycosyl-phosphatidylinositol anchor and protein tyrosine kinases p56lck and p59fyn 1. J Immunol. 1992;149:3535–3541. [PubMed] [Google Scholar]
- 42.Stefanova I, Horejsi V, Ansotegui IJ, Knapp W, Stockinger H. GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science. 1991;254:1016–1019. doi: 10.1126/science.1719635. [DOI] [PubMed] [Google Scholar]
- 43.Rajagopalan S, Long EO. A Human Histocompatibility Leukocyte Antigen (HLA)-G-specific Receptor Expressed on All Natural Killer Cells. J Exp Med. 1999;189:1093–1100. doi: 10.1084/jem.189.7.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khalil-Daher I, Riteau B, Menier C, Sedlik C, Paul P, Dausset J, Carosella ED, Rouas-Freiss N. Role of HLA-G versus HLA-E on NK function: HLA-G is able to inhibit NK cytolysis by itself. Journal of Reproductive Immunology. 1999;43:175–182. doi: 10.1016/s0165-0378(99)00032-7. [DOI] [PubMed] [Google Scholar]
- 45.Chiang EY, Henson M, Stroynowski I. The nonclassical major histocompatibility complex molecule Qa-2 protects tumor cells from NK cell- and lymphokine-activated killer cell-mediated cytolysis. Journal of Immunology. 2002;168:2200–2211. doi: 10.4049/jimmunol.168.5.2200. [DOI] [PubMed] [Google Scholar]
- 46.Iwashima M. Kinetic perspectives of T cell antigen receptor signaling. A two-tier model for T cell full activation. Immunological Reviews. 2003;191:196–210. doi: 10.1034/j.1600-065x.2003.00024.x. [DOI] [PubMed] [Google Scholar]
- 47.Lanzavecchia A, Lezzi G, Viola A. From TCR engagement to T cell activation: a kinetic view of T cell behavior. Cell. 1999;96:1–4. doi: 10.1016/s0092-8674(00)80952-6. [DOI] [PubMed] [Google Scholar]
- 48.Kielczewska A, Kim HS, Lanier LL, Dimasi N, Vidal SM. Critical Residues at the Ly49 Natural Killer Receptor's Homodimer Interface Determine Functional Recognition of m157, a Mouse Cytomegalovirus MHC Class I-Like Protein. J Immunol. 2007;178:369–377. doi: 10.4049/jimmunol.178.1.369. [DOI] [PubMed] [Google Scholar]
- 49.Dam J, Guan R, Natarajan K, Dimasi N, Chlewicki LK, Kranz DM, Schuck P, Margulies DH, Mariuzza RA. Variable MHC class I engagement by Ly49 natural killer cell receptors demonstrated by the crystal structure of Ly49C bound to H-2K(b) Nat Immunol. 2003;4:1213–1222. doi: 10.1038/ni1006. [DOI] [PubMed] [Google Scholar]
- 50.Tormo J, Natarajan K, Margulies DH, Mariuzza RA. Crystal structure of a lectin-like natural killer cell receptor bound to its MHC class I ligand. Nature. 1999;402:623–631. doi: 10.1038/45170. [DOI] [PubMed] [Google Scholar]