

Abstract
This study examined the role of oxidative stress in neurotoxic effects of cadmium chloride (Cd) in rat primary mid-brain neuron-glia cultures. Cd accumulated in neuron-glia cultures and produced cytotoxicity in a dose-dependent manner, with IC50 of 2.5μM 24 h after exposure. 3H-dopamine uptake into neuron-glia cultures was decreased 7 days after Cd exposure, with IC50 of 0.9μM, indicative of the sensitivity of dopaminergic neurons to Cd toxicity. To investigate the role of microglia in Cd-induced toxicity to neurons, microglia-enriched cultures were prepared. Cd significantly increased intracellular reactive oxygen species production in microglia-enriched cultures, as evidenced by threefold increases in 2′,7′-dichlorofluorescein signals. Using 5,5-dimethyl-1-pyrroline N-oxide as a spin-trapping agent, Cd increased electron spin resonance signals by 3.5-fold in microglia-enriched cultures. Cd-induced oxidative stress to microglia-enriched cultures was further evidenced by activation of redox-sensitive transcription factor nuclear factor kappa B and activator protein-1 (AP-1), and the increased expression of oxidative stress-related genes, such as metallothionein, heme oxygenase-1, glutathione S-transferase pi, and metal transport protein-1, as determined by gel-shift assays and real-time reverse transcription–PCR, respectively, in microglia-enriched cultures. In conclusion, Cd is toxic to neuron-glia cultures, and the oxidative stress from microglia may play important roles in Cd-induced damage to dopaminergic neurons.
Keywords: cadmium, neurotoxicity, neuron-glia cultures, oxidative stress, NF-κB and AP-1, gene expression
Cadmium (Cd) is a toxic heavy metal in the environment. Cd is a highly accumulative toxicant with very long biological half-life (Friberg et al., 1986). Cd is not biodegradable and its levels in the environment are increasing due to industrial activities and human exposures to Cd are inevitable (Friberg et al., 1986; Goering et al., 1995). Acute Cd exposure produced toxicities to the lung, liver, testes, and brain, while chronic exposure to Cd often leads to renal dysfunction, anemia, osteoporosis, and bone fractures (Friberg et al., 1986; Goering et al., 1995; Klaassen et al., 1999). Cd is a potent carcinogen in a number of tissues of rodents and classified as a human carcinogen (Waalkes, 2003).
The neurotoxic effects of Cd have been reported in neonatal mouse brain (Webster and Valois, 1981) and young rat brain (Wong and Klaassen, 1982). Cd produces oxidative damage to isolated rat optic nerve (Fern et al., 1996) and cultured rat cortical neurons (Lopez et al., 2003). In humans, occupational exposure to Cd is associated with neuropsychological disorders (Hart et al., 1989), and Cd exposure is reported to be a cause of amyotrophic lateral sclerosis (Bar-Sela et al., 2001). Cd is shown to selectively damage striatum (O'Callaghan and Miller, 1986), and Parkinsonism has been reported in a 64-year-old man exposed to Cd at a high dose (Okuda et al., 1997). Thus, accumulating evidence clearly indicates that Cd is neurotoxic in a number of settings.
The mechanisms involved in neurotoxicity of Cd are poorly understood. Oxidative stress has been proposed as a mechanism for Cd toxicity in a number of tissues such as the kidney (Bagchi et al., 1997), liver (Liu et al., 2002), and brain (Kumar et al., 1996). However, little is known about the neurotoxic effects of Cd to dopaminergic neurons, and little is known about the role of microglia in Cd-induced radical generation and oxidative stress in the brain. To fill these gaps would be helpful in understanding of Cd neurotoxicity and would be useful in setting appropriate measures to prevent and/or reduce Cd toxicity.
Thus, the present study was undertaken to examine Cd-induced neurotoxicity using rat primary neuron-glia cultures, which has been proven to be a good model to elucidate the role of oxidative stress from microglia in dopaminergic neuron damages from environmental neurotoxicants (Block et al., 2007; Liu and Hong 2003). Thus, the effects of Cd on mitochondrial and dopaminergic neuron functions in neuron-glia cultures were evaluated via the MTT assay and 3H-dopamine uptake, respectively. To define Cd-induced free radical generation, 5,5-dimethyl-1-pyrroline N-oxide (DMPO) was used as a free radical trapping agent, followed by electron spin resonance (ESR) detection, and 2′,7′-dichlorofluoresin was used for intracellular ROS levels. The redox-sensitive transcription factors nuclear factor kappa B (NF-κB) and activator protein-1 (AP-1), as well as oxidative stress-related gene expression were also examined to define the role of oxidative stress from microglia in Cd toxicity to dopaminergic cultures. The results demonstrated that Cd is toxic to dopaminergic neurons, probably through oxidative stress produced by activated microglia.
MATERIALS AND METHODS
Chemicals and Reagents
Cadmium chloride (CdCl2, Cd) was purchased from Sigma Chemical Co. (St Louis, MO, USA). 3H-dopamine (30 Ci/mmol) and γ32P-adenosine triphosphate (ATP) (3000 Ci/mmol) were purchased from PerkinElmer Life Sciences (Boston, MA). DMPO was purchased from Alexis Biochemicals (San Diego, CA), purified by double distillation, stored under N2 atmosphere at −80°C before use.
Primary Rat Mesencephalic Neuron-glia Cultures
Primary rat mid-brain neuron-glia cultures were prepared following the published protocol (Liu and Hong, 2003; Liu et al., 2000). Briefly, the ventral mesencephalic tissues of brain from Fisher 344 rat fetus at the gestation day 14 were removed and dissociated by a mechanical triturating. The isolated cells were seeded at 5 × 105/well to 24-well culture plates or 1.0 × 105/well to 96-well culture plates precoated with poly-D-lysine (20 μg/ml), and maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air in minimum essential medium containing 10% fetal bovine serum (FBS) and 10% horse serum, 1 g/l glucose, 2mM L-glutamine, 1mM sodium pyruvate, 100μM nonessential amino acids, 50 U/ml penicillin, and 50 μg/ml streptomycin. Fresh medium was added to each well 3 days later. Seven-day-old cultures were used for treatment. Immunocytochemical (ICC) analysis indicated that the cultures were made up of ∼11% microglia, 48% astroglia, and 40% neurons, of which 1−2% were tyrosine hydroxylase-immunoreactive (dopaminergic neurons) (Liu et al., 2000; Yang et al., 2006).
Primary Microglia-Enriched Cultures
Microglia-enriched cultures were prepared from the whole brains of 1- or 2-day-old Fisher 344 rats, as described previously (Liu and Hong, 2003). Briefly, brain tissues, devoid of meninges and blood vessels, were dissociated by a mechanical trituration. The isolated cells (5 × 107) were seeded in 175-cm2 culture flasks in Dulbecco's modified Eagle medium/F12 containing 10% FBS, 2mM L-glutamine, 1mM sodium pyruvate, 100μM nonessential amino acids, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Fresh medium was changed 3 days later. When reaching confluence (10−14 days), microglia were separated from astroglia by shaking the flasks for 1 h. Purity of the microglia-enriched cultures was > 98%, as determined by ICC staining (Yang et al., 2006).
Assessment of Cd Toxicity to Neuron-glia Cultures
MTT assay
The rat primary neuron-glia cultures were seeded in 96-well plates at 1 × 105 cells/per well, and treated with various concentrations of Cd for 24 h. Cell viability was assessed by colorimetric measurement of the reduction product of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT). Cells were incubated for 4 h at 37°C in the presence of MTT (0.5 mg/ml in phosphate buffered saline [PBS]), and culture medium was removed, 100 μl of DMSO was added to dissolve the formazan. Reduced MTT was measured on a microplate reader (Molecular Devices, Palo Alto, CA) at 570 nm. Values were expressed as percentage of controls.
3H-dopamine uptake
Degeneration of dopaminergic neurons was assessed by measuring the ability of cultures to take up 3H-dopamine (Liu et al., 2003b). Briefly, The rat primary neuron-glia cultures were incubated continuously with Cd for 7 days, and then washed twice with warm Krebs–Ringer buffer (KRB, 16mM sodium phosphate, 119mM NaCl, 4.7mM KCl, 1.8mM CaCl2, 1.2mM MgSO4, 1.3mM ethylenediaminetetraacetic acid [EDTA], and 5.6mM glucose; pH 7.4). Cultures were then incubated for 20 min at 37°C with 1μM 3H-dopamine in KRB. After washing cultures three times with ice-cold KRB, cells were then dissolved in 1N NaOH. Radioactivity was determined by liquid scintillation. Nonspecific DA uptake in the presence of mazindol (10μM) was subtracted. In our previous publications, the 3H-dopamine uptake is paralleled well with the number of dopaminergic neurons (TH-immunoreactive cells) (Liu et al., 2000; Yang et al., 2006)
Measurement of Cd Concentration in the Cells
Cd-treated rat primary neuron-glia cultures were washed three times with PBS and harvested in 1 ml of PBS. An aliquot of sonicated cell suspension was used for cellular protein assay, and the remaining cell suspension was digested in nitric acid at 70°C overnight, and diluted in distilled water. Cd concentration was determined by atomic absorption spectrometer (Pekin–Elmer, AAnalst 100, Norwalk, CT), and expressed as ng Cd/mg cellular protein.
Assay of Intracellular ROS
Intracellular ROS was assayed in Cd-treated rat primary microglia-enriched cultures by using 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2-DCFDA) (Molecular Probes, Eugene, OR). During reactions with cellular oxidizing species CM-H2-DCFDA is hydrolyzed to 2′,7′-dichlorofluorescein (DCF) yielding fluorescence. After washing cells twice with warm Hank's balanced salt solution, CM-H2-DCFDA was added to cultures in a final concentration of 1μM, and incubated for 30 min at 37°C (Liu et al., 2000). The cells were then treated with Cd for 1 h at 37°C, and fluorescence intensity was measured at 485 nm for excitation and 530 nm for emission using a SpectraMax Gemini XS fluorescence microplate reader (Molecular Devices, Sunnyvale, CA).
Electron Spin Resonance
Enriched microglia cells were washed and suspended in PBS (5 × 105/ml). Microglia cells were then treated with Cd and the spin-trapping agent DMPO (80mM), and immediately transferred into ESR flat cell inside cavity to measure radical formation up to 40 min, and ESR signals were analyzed on a Bruker ElexSys spectrometer at 9.76 GHz and room temperature. ESR settings were center filed 3484 G; sweep width 100 G; modulation frequency 100 kHz; modulation amplitude 1.0 G; microwave power 20 mW; receiver gain 1.6 × 105; time constant 0.6 s; and sweep time 671 s.
Gel-shift Assay
The gel-shift assay systems from Promega (Madison, WI) were used for NF-κB and AP-1 analysis. Briefly, enriched microglia cells were homogenized in HEGD buffer (25mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 1.5mM EDTA, 10% glycerol, 1mM dithiotreitol, 0.5mM phenylmethylsulfonyl fluoride, and 1× protease inhibitor cocktail (CalBiochem, La Jolla, CA), centrifuged at 12,000 × g for 10 min at 4°C, and washed two times. Nuclear proteins were extracted by incubation with 50 μl of HEGD buffer containing 0.5M KCl on ice for 1 h, followed by centrifugation at 12,000 × g for 15 min and supernatant was stored at −80°C prior to use. The AP-1 and NF-κB oligonucleotide probe were labeled with γ32P-ATP and T4 polynucleotide kinase. Nuclear protein (10 μg) was incubated with gel-shift binding buffer and labeled probe for 20 min and electrophoresed on Novex 6% DNA retardation gel in 0.5× Tris-Borate-EDTA buffer at 300 V. Gels were then dried for autoradiography. The negative controls were used to verify the specificity as indicated in our previous publications (Liu et al., 2002; Qu et al., 2005).
Real-Time RT-PCR
Total RNA was isolated from Cd-treated enriched microglia cells with Trizol and purified with RNeasy kit (Qiagen, Valencia, CA). RNA was reverse transcribed with MuLV reverse transcriptase and oligo-dT primers. The primers for selected genes were designed using Primer Express software (Applied Biosystems, Foster City, CA). The SYBR green Master Mix (Applied Bio-systems) was used for real-time PCR analysis. The Ct values for interested genes were first normalized with that of β-actin in the same sample, and expressed as percentage of controls.
Data Analysis
Means ± SEM of three determinations were calculated. The data were analyzed by analysis of variance, followed by Duncan's test for multiple comparisons. The significance level was set at p < 0.05.
RESULTS
Cd is Toxic to Rat Primary Neuron-Glia Cultures
The rat primary neuron-glia cultures contain both neurons and glial cells to mimic neuron-glia interactions in vivo (Liu et al., 2003). As illustrated in Figure 1A, exposure of the primary neuron-glia cultures to Cd produced a concentration-dependent toxicity, as evidenced by mitochondrial integrity (MTT assay) 24 h after Cd exposure, with IC50 of Cd 2.5μM. Cd-induced toxicity to cells paralleled the cellular Cd accumulation in a concentration-dependent manner (Fig. 1B).
FIG. 1.

Cytotoxicity by MTT assay (top) and cellular accumulation of Cd (bottom) in rat primary neuron-glia cultures 24-h posttreatment. The data are mean ± SEM from three independent experiments. *p < 0.05, compared to control.
To identify the microglia-mediated toxicity of Cd to dopaminergic neurons, rat primary neuron-glia cultures were continuously exposed to low concentrations of Cd for 7 days (Liu and Hong, 2003), and the ability of dopaminergic cells to take 3H-dopamine was measured. Cd exposure produced a concentration-dependent reduction of 3H-dopamine uptake into neuron-glia cultures at concentrations as low as 0.625μM, with IC50 of 0.9μM, indicative of dopaminergic neurons degeneration (Fig. 2). At concentrations higher than 1.25μM, 3H-dopamine uptake capacity decreased by more than 80%.
FIG. 2.

Toxicity of Cd to dopaminergic neurons in rat primary neuron-glia cultures 7-day posttreatment using 3H-dopamine uptake assay. The data are mean ± SEM from three independent experiments. *p < 0.05, compared to control.
Cd Induces Intracellular ROS Production
To define the role of oxidative stress from microglia in Cd-induced toxicity to dopaminergic neurons, intracellular reactive oxygen species (iROS) were measured by oxidizing CM-H2-DCFDA to DCF, and the intensity of fluorescence in the microglia-enriched cultures was measured. As shown in Figure 3, Cd significantly increased DCF signals by threefold 1 h after exposure to Cd at 0.625 and 1.25μM. However, at higher concentration of 2.5μM, the DCF signals were only increased 1.5-fold, probably due to cytotoxicity of Cd to rat primary microglia-enriched cultures at this high concentration. However, the oxidation of DCF to ROS is subject to artifact under certain conditions (Bonini et al., 2006). Thus, the more direct measure of Cd-induced free radical generation is needed to support this conclusion. Microglia-enriched cultures were treated with Cd (0.625μM) and the spin-trapping agent DMPO was used for ESR analysis. As shown in Figure 4, Cd treatment increased DMPO-trapped radical signals 3.5-fold over DMPO alone controls at the 40-min time point (Chang et al., 2000). The four-line ESR signals were characterized by hyperfine coupling constants of aN = 15.06 G, = 14.76, which correspond to DMPO/.OH. However, it is hard to distinguish whether the observed DMPO/.OH spectrum is derived from the trapping of superoxide anion or hydroxyl radical by DMPO (Chang et al., 2000). It should also be noted that the free radical species are mainly from microglia, rather than from astroglia or neurons (Block et al., 2007; Chang et al., 2000; Liu et al., 2000)
FIG. 3.

Effect of Cd on intracellular ROS generation in rat primary microglia-enriched cultures 2-h posttreatment. The data are mean ± SEM from three independent experiments. *p < 0.05, compared to control.
FIG. 4.

Effect of Cd on DMPO–radical adduct formation in rat primary microglia-enriched cultures 1-h posttreatment by ESR analysis. (Top) Representative ESR spectra; (bottom) ESR signal intensity quantification from data of three independent experiments as mean ± SEM. *p < 0.05, compared to control.
Cd Activates Redox-Sensitive Transcription Factors
The transcription factors NF-κB and AP-1 are known to be sensitive to oxidative stress and acute Cd exposure has been shown to activate these transcription factors in various settings (Hart et al., 1999; Liu et al., 2002; Qu et al., 2005). Thus, the ability of Cd to activate these redox-sensitive transcription factors was examined 6 h in rat primary microglia-enriched cultures after Cd exposure. Clearly, Cd treatment dramatically increased NF-κB binding to the DNA (Fig. 5). Similarly, the activation of AP-1 by Cd was also dramatically increased (Fig. 5). The AP-1 activation at higher Cd concentration was less than at the lower concentration, probably due to the death of microglia cells at the higher Cd concentration. Thus, Cd treatment activated redox-sensitive transcription factor NF-κB and AP-1.
FIG. 5.
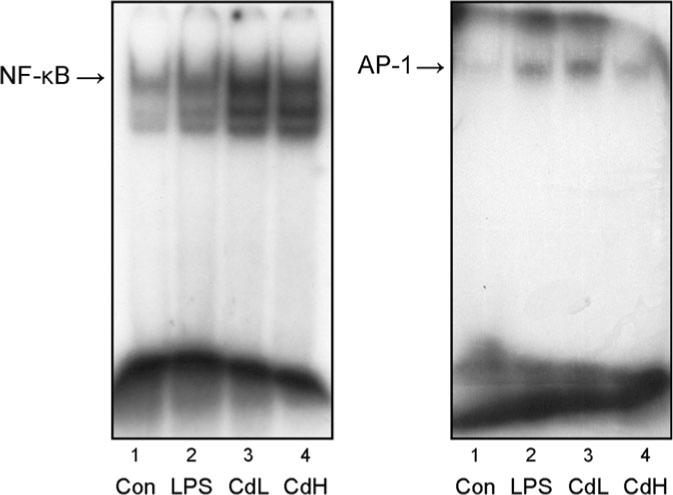
Effect of Cd on NF-κB (left) and AP-1 (right) activation in rat primary microglia-enriched cultures 6-h posttreatment by gel-shift assay. Lane 1, control, lane 2, LPS 1 ng/ml, lane 3, Cd 0.625μM, lane 4, Cd 2.5μM. The data represent one of three independent experiments.
Cd Induces Oxidative Stress-Related Gene Expression
Cd-induced oxidative stress-related gene expression in rat primary microglia-enriched cultures was further examined by real-time RT-PCR (Fig. 6), which usually peaked at 12−24 h. Heme oxygenase-1 (HO-1) is a hallmark of cellular oxidative stress (Applegate et al., 1991), and was increased dose-dependently by Cd up to sixfold; metallothionein-1 (MT-1), a protein inducible by oxidative stress (Bauman et al., 1991) and Cd (Klaassen et al., 1999), was increased in a concentration-dependent manner up to 60-fold; the expression of glutathione S-transferase pi (GST-pi) was increased concentration dependently up to 2.7-fold, which acts as antioxidants and cellular transporters. The expression of the metal transport protein-1 (MTP-1) was increased up to 4.5-fold. The enhanced expression of these genes could be envisioned as important cellular mechanisms in response to Cd-induced oxidative stress.
FIG. 6.

Effect of Cd on the expression of HO-1, MT-1, GST-pi, and MTP-1 in rat primary microglia-enriched cultures 24-h posttreatment using real-time RT-PCR. The data are expressed as mean ± SEM of three independent experiments. *p < 0.05, compared to control.
DISCUSSION
The present study clearly demonstrated that Cd is toxic to rat primary neuron-glia cultures, with IC50 of 2.5μM at 24 h by the MTT assay. Cd is also more toxic to dopaminergic neurons with IC50 of 0.9μM 7 days after Cd exposure, as evidenced by decreases in 3H-dopamine uptake. Neurotoxic effects of Cd have been observed in various brain cell cultures, including rat cortical neurons in culture (Lopez et al., 2003), isolated rat optic nerve preparation (Fern et al., 1996), anterior pituitary cells (Poliandri et al., 2004), the dissociated mesencephalic trigeminal neurons from adult rats (Yoshida, 2001), and glioma and neuron blastoma cells (Huang et al., 1993). The present study adds to our understanding of Cd neurotoxicity in cultured brain cells by demonstrating the involvement of oxidative stress from activated microglia in Cd toxicity to neurons, at least to dopaminergic neurons. Oxidative stress from activated microglia is evidenced by increased DMPO-trapped ESR signals and intracellular ROS levels, as well as by the activation of redox-sensitive NF-κB and AP-1, and the increased expression of oxidative stress-related gene expression. Thus, oxidative stress from activated microglia could play important roles in Cd toxicity to dopaminergic neurons.
Parkinson's disease is a neurodegenerative disorder that progresses over years affecting prominently the dopaminergic neurons of the substantia nigra pars compacta. It is characterized by the loss of the nigrostriatal ascending dopaminergic pathway, which results in symptoms of motor dysfunction such as rigidity tremors, difficult in movements, and loss of balance. Brain dopamine levels are decreased following Cd exposure in rodents (Pillai et al., 2003), and rat brain dopamine secretion pattern can be altered by Cd (Lafuente et al., 2005). The uptake of dopamine into rat brain synaptosomes can also be inhibited by Cd (Rajanna et al., 1990), and in the present study a dramatic decrease in 3H-dopamine uptake into neuron-glia cultures is evident 7 days after continuous exposure to low doses of Cd. Taken together, Cd exposure clearly represents a potential risk for Parkinson's disease (O'Callaghan and Miller, 1986; Okuda et al., 1997).
Activated microglia is present in the vicinity of degenerating neurons in the substantia nigra, and is likely a major contribution factor in Parkinsonism (Block et al., 2007; Langston et al., 1999; Liu et al., 2003). Dopaminergic neurons in the substantia nigra are relatively deficient in oxidative defenses (Yoo et al., 2003), rendering it more susceptible to ROS-induced damage. Several studies have proposed the overproduction of ROS as a mechanism for Cd neurotoxicity (Kumar et al., 1996), similar to Cd-induced ROS damage in the liver (Liu et al., 2002). Excessive production of ROS may overwhelm normal protective mechanisms, leading to selective toxicity to dopamine neurons (Yoo et al., 2003). In the present study, Cd-induced oxidative stress from microglia-enriched cultures was initially demonstrated by marked increases in DCF fluorescence, which has been proven to be a sensitive measure on endotoxin-induced intracellular ROS production (Liu and Hong, 2003). Cd-induced radicals were further evidenced by 3.5-fold increases in DMPO-trapped radical adduct ESR signals in microglia-enriched cultures. DMPO is a sensitive spin-trapping agents to detect radical generation in microglia-enriched cultures (Chang et al., 2000), and the free radicals generated by activated macroglia are likely superoxide anion (Chang et al., 2000; Liu et al., 2003). Superoxide anion radical is also observed from POBN- or PBN-trapped radicals following Cd administrated in vivo (Liu et al., 2002) and in Cd-treated J774A macrophages (Hassoun and Stohs, 1996). Nonetheless, the spin-trapping experiment provides clear evidence that Cd is able to produce free radicals in rat primary microglia-enriched cultures, which in turn could produce oxidative damage to neurons, especially to dopaminergic neurons.
To further support the microglia activation and the ROS-mediated mechanism of Cd toxicity in neuron-glia cultures, the activation of redox-sensitive transcription factors such as NF-κB and AP-1 was examined. Consistent with the literature (Hart et al., 1999; Liu et al., 2002; Qu et al., 2005), both NF-κB and AP-1 were clearly activated by Cd in rat primary microglia-enriched cultures, indicating that Cd-induced radical production in microglia-enriched cultures can further activate the redox-sensitive transcription factors such as NF-κB and AP-1, which in turn leads to overexpression of oxidative stress-related genes.
Induction of HO-1 has been proposed as a general response to oxidative stress in mammalian cells (Applegate et al., 1991), and Cd-induction of HO-1 is used as a biomarker for oxidative stress (Ossola and Tomaro, 1995). In this study HO-1 was increased sixfold, fortifying the oxidative mechanism of Cd toxicity. Similar to HO-1, induction of MT-1 is also an important cellular response to oxidative stress (Bauman et al., 1991; Klaassen et al., 1999), and dramatic induction of MT-1 observed in this study could imply that oxidative stress occurred after Cd treatment. MT-1 and HO-1 induction are important cellular defense mechanisms against Cd-induced oxidative stress in the brain. Induction of GST-pi has also been implicated in Cd-induced oxidative stress in the lung cells (Hart et al., 1999; Shukla et al., 2000), and increases in GSTs not only play a role in cellular antioxidant machinery, but also play a role in cellular transport system for Cd efflux. In support of this role, the MTP-1 was also dramatically induced in rat primary microglia-enriched cultures. The induction of these oxidative stress-related genes could play an integrated role in protecting against Cd-induced oxidative damage to neurons.
Cd-induced oxidative stress in neuron-glia cultures could be mediated by several cellular mechanisms. The activated micro-glia can release proinflammatory mediators to trigger oxidative stress (Block et al., 2007; Liu et al., 2003). Indeed, Cd-induced radical induction in the liver could be suppressed by inactivation of Kupffer cells by gadolinium chloride (Liu et al., unpublished data). The dramatic increase in MTP-1 observed in this study could facilitate iron transport into cells. Iron has been implicated in Cd-induced radical production in the liver, and the iron chelator deferral abolished Cd-induced radical production (Liu et al., submitted). The dramatic induction of MT observed in this study could sequester Cd and render it toxicologically inert (Klaassen et al., 1999), but interaction of MT with Cd may also produce hydroxyl radicals (O'Brien and Salacinski, 1998). Cd is shown to inhibit the electron transfer chain in the brain mitochondria, resulting in accumulation of semiquinones, which are unstable and prone to transfer one electron to molecular oxygen to form superoxide (Wang et al., 2004). Thus, multiple mechanisms are associated with Cd-induced oxidative stress in the brain, and they are not mutually exclusive.
In summary, this study clearly demonstrated that oxidative stress produced from microglia is involved in Cd toxicity to dopaminergic neurons, as evidenced by DMPO–radical adducts, the production of intracellular ROS, the activation of redox-sensitive transcription factor NF-κB and AP-1, and by increased expression of oxidative stress responsive genes. Thus, microglia-mediated oxidative stress could play an important role in Cd-induced dopaminergic neuron damage and Parkinson's disease in humans.
ACKNOWLEDGMENTS
This work is supported in part by the intramural research program of National Institute Environmental Sciences, Center for Cancer Research, National Cancer Institute, and National Center for Toxicogenomics. The authors thank Drs Michelle Block, Chikara Kojima, and Larry. K. Keefer for their critical comments during the preparation of this manuscript.
REFERENCES
- Applegate LA, Luscher P, Tyrrell RM. Induction of heme oxygenase: A general response to oxidant stress in cultured mammalian cells. Cancer Res. 1991;51:974–978. [PubMed] [Google Scholar]
- Bagchi D, Vuchetich PJ, Bagchi M, Hassoun EA, Tran MX, Tang L, Stohs SJ. Induction of oxidative stress by chronic administration of sodium dichromate and cadmium chloride to rats. Free Radic. Biol. Med. 1997;22:471–478. doi: 10.1016/s0891-5849(96)00352-8. [DOI] [PubMed] [Google Scholar]
- Bar-Sela S, Reingold S, Richter ED. Amyotrophic lateral sclerosis in a battery-factory worker exposed to cadmium. Int. J. Occup. Environ. Health. 2001;7:109–112. doi: 10.1179/107735201800339470. [DOI] [PubMed] [Google Scholar]
- Bauman JW, Liu J, Liu YP, Klaassen CD. Increase in metallothionein produced by chemicals that induce oxidative stress. Toxicol. Appl. Pharmacol. 1991;110:347–354. doi: 10.1016/s0041-008x(05)80017-1. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bonini MG, Rota C, Tomasi A, Mason RP. The oxidation of 2’,7’-dichlorofluorescin to reactive oxygen species: A self-fulfilling prophesy? Free Radic. Biol. Med. 2006;40:968–975. doi: 10.1016/j.freeradbiomed.2005.10.042. [DOI] [PubMed] [Google Scholar]
- Chang RC, Rota C, Glover RE, Mason RP, Hong JS. A novel effect of an opioid receptor antagonist, naloxone, on the production of reactive oxygen species by microglia: A study by electron paramagnetic resonance spectroscopy. Brain Res. 2000;854:224–229. doi: 10.1016/s0006-8993(99)02267-2. [DOI] [PubMed] [Google Scholar]
- Fern R, Black JA, Ransom BR, Waxman SG. Cd(2+ )-induced injury in CNS white matter. J. Neurophysiol. 1996;76:3264–3273. doi: 10.1152/jn.1996.76.5.3264. [DOI] [PubMed] [Google Scholar]
- Friberg L, Elinder CG, Kjellstrom T, Nordberg GF. Cadmium and Health: A Toxicological and Epidemiological Appraisal. CRC Press; Boca Raton, FL: 1986. [Google Scholar]
- Goering PL, Waalkes MP, Klaassen CD. Toxicology of cadmium. In: Goyer RA, Cherian MG, editors. Toxicology of Metals: Biological Aspects. Handbook of Experimental Pharmacology. Vol. 115. Springer-Verlag; New York: 1995. pp. 189–213. [Google Scholar]
- Hart BA, Lee CH, Shukla GS, Shukla A, Osier M, Eneman JD, Chiu JF. Characterization of cadmium-induced apoptosis in rat lung epithelial cells: Evidence for the participation of oxidant stress. Toxicology. 1999;133:43–58. doi: 10.1016/s0300-483x(99)00013-x. [DOI] [PubMed] [Google Scholar]
- Hart RP, Rose CS, Hamer RM. Neuropsychological effects of occupational exposure to cadmium. J. Clin. Exp. Neuropsychol. 1989;11:933–943. doi: 10.1080/01688638908400946. [DOI] [PubMed] [Google Scholar]
- Hassoun EA, Stohs SJ. Cadmium-induced production of superoxide anion and nitric oxide, DNA single strand breaks and lactate dehydrogenase in J774A.1 cell cultures. Toxicology. 1996;112:219–226. doi: 10.1016/0300-483x(96)03404-x. [DOI] [PubMed] [Google Scholar]
- Huang J, Tanii H, Kato K, Hashimoto K. Neuron and glial cell marker proteins as indicators of heavy metal-induced neurotoxicity in neuroblastoma and glioma cell lines. Arch. Toxicol. 1993;67:491–496. doi: 10.1007/BF01969920. [DOI] [PubMed] [Google Scholar]
- Klaassen CD, Liu J, Choudhuri S. Metallothionein: An intracellular protein to protect against cadmium toxicity. Annu. Rev. Pharmacol. Toxicol. 1999;39:267–294. doi: 10.1146/annurev.pharmtox.39.1.267. [DOI] [PubMed] [Google Scholar]
- Kumar R, Asic K, Agarwal K, Seth PK. Oxidative stress-mediated neurotoxicity of cadmium. Toxicol. Lett. 1996;89:65–69. doi: 10.1016/s0378-4274(96)03780-0. [DOI] [PubMed] [Google Scholar]
- Lafuente A, Gonzalex-Carracedo A, Romero A, Cabaleiro T, Esquifino AI. Toxic effects of cadmium on the regulatory mechanism of dopamine and serotonin on prolactin secretion in adult male rats. Toxicol. Lett. 2005;155:87–96. doi: 10.1016/j.toxlet.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J. Pharmacol. Exp. Ther. 2000;293:607–617. [PubMed] [Google Scholar]
- Liu B, Hong JS. Primary rat mesencephalic neuron-glia, neuron-enriched, microglia-enriched, and astroglia-enriched cultures. Methods Mol. Med. 2003;79:387–395. doi: 10.1385/1-59259-358-5:387. [DOI] [PubMed] [Google Scholar]
- Liu J, Kadiiska MB, Corton JC, Qu W, Waalkes MP, Mason RP, Liu Y, Klaassen CD. Acute cadmium exposure induces stress-related gene expression in wild-type and metallothionein-I/II null mice. Free Radic. Biol. Med. 2002;32:525–535. doi: 10.1016/s0891-5849(01)00826-7. [DOI] [PubMed] [Google Scholar]
- Lopez E, Figueroa S, Oset-Gasque MJ, Gonzalez MP. Apoptosis and necrosis: Two distinct events induced by cadmium in cortical neurons in culture. Br. J. Pharmacol. 2003;138:901–911. doi: 10.1038/sj.bjp.0705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien P, Salacinski HJ. Evidence that the reactions of cadmium in the presence of metallothionein can produce hydroxyl radicals. Arch. Toxicol. 1998;72:690–700. doi: 10.1007/s002040050562. [DOI] [PubMed] [Google Scholar]
- O'Callaghan JP, Miller D. Diethyldithiocarbamate increases distribution of cadmium to brain but prevents cadmium-induced neurotoxicity. Brain Res. 1986;370:354–358. doi: 10.1016/0006-8993(86)90493-2. [DOI] [PubMed] [Google Scholar]
- Okuda B, Iwamoto Y, Tachibana H, Sugita M. Parkinsonism after acute cadmium poisoning. Clin. Neurol. Neurosurg. 1997;99:263–265. doi: 10.1016/s0303-8467(97)00090-5. [DOI] [PubMed] [Google Scholar]
- Ossola JO, Tomaro ML. Heme oxygenase induction by cadmium chloride: Evidence for oxidative stress involvement. Toxicology. 1995;104:141–147. doi: 10.1016/0300-483x(95)03157-b. [DOI] [PubMed] [Google Scholar]
- Pillai A, Priya L, Gupta S. Effects of combined exposure to lead and cadmium on the hypothalamic-pituitary axis function in proestrous rats. Food Chem. Toxicol. 2003;41:379–384. doi: 10.1016/s0278-6915(02)00247-8. [DOI] [PubMed] [Google Scholar]
- Poliandri AH, Velardez MO, Cabilla JP, Bodo CC, Machiavelli LI, Quinteros AF, Duvilanski BH. Nitric oxide protects anterior pituitary cells from cadmium-induced apoptosis. Free Radic. Biol. Med. 2004;37:1463–1471. doi: 10.1016/j.freeradbiomed.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Qu W, Diwan BA, Reece JM, Bortner CD, Pi J, Liu J, Waalkes MP. Cadmium-induced malignant transformation in rat liver cells: Role of aberrant oncogene expression and minimal role of oxidative stress. Int. J. Cancer. 2005;114:346–355. doi: 10.1002/ijc.20736. [DOI] [PubMed] [Google Scholar]
- Rajanna B, Hobson M, Harris L, Ware L, Chetty CS. Effects of cadmium and mercury on Na(+)-K+, ATPase and uptake of 3H-dopamine in rat brain synaptosomes. Arch. Int. Physiol. Biochim. 1990;98:291–296. doi: 10.3109/13813459009113989. [DOI] [PubMed] [Google Scholar]
- Shukla GS, Shukla A, Potts RJ, Osier M, Hart BA, Chiu JF. Cadmium-mediated oxidative stress in alveolar epithelial cells induces the expression of gamma-glutamylcysteine synthetase catalytic subunit and glutathione S-transferase alpha and pi isoforms: Potential role of activator protein-1. Cell Biol. Toxicol. 2000;16:347–362. doi: 10.1023/a:1007696610186. [DOI] [PubMed] [Google Scholar]
- Waalkes MP. Cadmium carcinogenesis. Mutat. Res. 2003;533:107–120. doi: 10.1016/j.mrfmmm.2003.07.011. [DOI] [PubMed] [Google Scholar]
- Wang Y, Fang J, Leonard SS, Rao KM. Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic. Biol. Med. 2004;36:1434–1443. doi: 10.1016/j.freeradbiomed.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Webster WS, Valois AA. The toxic effects of cadmium on the neonatal mouse CNS. J. Neuropathol. Exp. Neurol. 1981;40:247–257. doi: 10.1097/00005072-198105000-00003. [DOI] [PubMed] [Google Scholar]
- Wong KL, Klaassen CD. Neurotoxic effects of cadmium in young rats. Toxicol. Appl. Pharmacol. 1982;63:330–337. doi: 10.1016/0041-008x(82)90261-7. [DOI] [PubMed] [Google Scholar]
- Yang S, Yang J, Yang Z, Chen P, Fraser A, Zhang W, Pang H, Gao X, Wilson B, Hong JS, et al. Pituitary adenylate cyclase-activating polypeptide (PACAP) 38 and PACAP4−6 are neuroprotective through inhibition of NADPH oxidase: Potent regulators of microglia-mediated oxidative stress. J. Pharmacol. Exp. Ther. 2006;319:595–603. doi: 10.1124/jpet.106.102236. [DOI] [PubMed] [Google Scholar]
- Yoo MS, Chun HS, Son JJ, DeGiorgio LA, Kim DJ, Peng C, Son JH. Oxidative stress regulated genes in nigral dopaminergic neuronal cells: Correlation with the known pathology in Parkinson's disease. Brain Res. Mol. Brain Res. 2003;110:76–84. doi: 10.1016/s0169-328x(02)00586-7. [DOI] [PubMed] [Google Scholar]
- Yoshida S. Re-evaluation of acute neurotoxic effects of Cd2+ on mesencephalic trigeminal neurons of the adult rat. Brain Res. 2001;892:102–110. doi: 10.1016/s0006-8993(00)03240-6. [DOI] [PubMed] [Google Scholar]