

Abstract
Arylamine N-acetyltransferase 2 (NAT2) modifies drug efficacy/toxicity and cancer risk due to its role in bioactivation and detoxification of arylamine and hydrazine drugs and carcinogens. Human NAT2 alleles possess a combination of single nucleotide polymorphisms (SNPs) associated with slow acetylation phenotypes. Clinical and molecular epidemiology studies investigating associations of NAT2 genotype with drug efficacy/toxicity and/or cancer risk are compromised by incomplete and sometimes conflicting information regarding genotype/phenotype relationships. Studies in our laboratory and others have characterized the functional effects of SNPs alone, and in combinations present in alleles or haplotypes. We extrapolate this data generated following recombinant expression in yeast and COS-1 cells to assist in the interpretation of NAT2 structure. Whereas previous structural studies used homology models based on templates of N-acetyltransferase enzyme crystal structures from various prokaryotic species, alignment scores between bacterial and mammalian N-acetyltransferase protein sequences are low (~ 30%) with important differences between the bacterial and mammalian protein structures. Recently, the crystal structure of human NAT2 was released from the Protein Data Bank under accession number 2PFR. We utilized the NAT2 crystal structure to evaluate the functional effects of SNPs resulting in the protein substitutions R64Q (G191A), R64W (C190T), I114T (T341C), D122N (G364A), L137F (A411T), Q145P (A434C), E167K (G499A), R197Q (C590A), K268R (A803G), K282T (A845C), and G286E (G857A) of NAT2. This analysis advances understanding of NAT2 structure-function relationships, important for interpreting the role of NAT2 genetic polymorphisms in bioactivation and detoxification of arylamine and hydrazine drugs and carcinogens.
Keywords: Human N-acetyltransferase 2 (NAT2), single nucleotide polymorphism (SNP), arylamine carcinogens, pharmacogenetics, cancer risk, structure/function
INTRODUCTION
Variability in the human arylamine N-acetyltransferase 2 (NAT2; EC 2.3.1.5) phenotype was first identified as a modifier of toxic side-effects in patients prescribed the anti-tubercular drug isoniazid [1]. In addition to the metabolism of many aromatic amine and hydrazine drugs [2], NAT2 modifies cancer predisposition with roles in bioactivation and detoxification of aromatic and heterocyclic amine carcinogens [3]. In the metabolic scheme for these drugs and carcinogens, NAT2 catalyzes not only N-acetylation, but following N-hydroxylation also catalyzes subsequent O-acetylation and N,O-acetylation as depicted in Fig. 1 [4–6].
Fig. 1. Metabolism of arylamines includes both activation and inactivation via N-acetylation, O-acetylation, and N,O-acetylation catalyzed by NAT2.
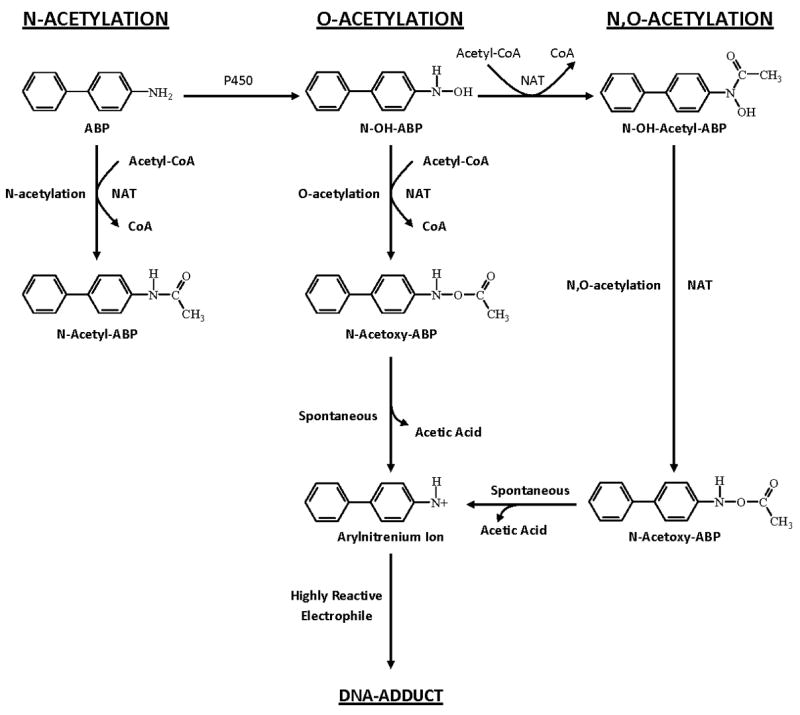
These reactions are depicted for the arylamine carcinogen 4-aminobiphenyl, ultimately leading to the generation of highly reactive electrophiles that bind to DNA potentially leading to mutations and cancer. Adapted from [5].
Like all mammalian arylamine N-acetyltransferase proteins including human N-acetyltransferase 1 (NAT1), NAT2 contains a functional Cys-His-Asp catalytic triad, which resembles that of cysteine proteases [7]. With a ping-pong bi-bi reaction mechanism, the catalytic Cys68 is first acetylated upon binding of acetyl-coenzyme A (AcCoA), followed by CoA release, substrate binding, and acetyl group transfer to the substrate’s exocyclic amine or to the oxygen of substrate’s hydroxylated amine [8]. Human NAT2 substrate selectivity is strikingly different from human NAT1 [9–10] and is strongly influenced by three key serine residues at positions 125, 127, and 129 [11].
Human NAT2 alleles or haplotypes possess a combination of single nucleotide polymorphisms (SNPs), some of which are associated with slow acetylator phenotypes (Table 1). NAT2 polymorphisms modify individual cancer risk and drug response, or susceptibility to adverse drug reactions [2–5, 56–62]. Molecular epidemiologic studies have investigated the relationship between NAT2 allele status and individual risk for various cancers including urinary bladder, colorectal, breast, prostate, pancreas, lung, liver, esophageal, and non-Hodgkin lymphoma. However, except for the smoking-related urinary bladder cancer (25, 63), these studies have been unable to establish a consistent association between acetylator status and human cancers. It needs to be noted, however, that because of the difficulties in determining phenotype, most of these studies used NAT2 phenotypes inferred from genotyping results and the validity of the conclusions derived from these studies may be compromised by the uncertainty of NAT2 genotype-phenotype relationships (64).
Table 1.
Human NAT2 alleles
| NAT2 Allele(Haplotype)a | Nucleotide Change(s) and rs Identifiersb | Amino Acid Change(s)b | Phenotypec | References |
|---|---|---|---|---|
| NAT2*4 | Reference | Reference | Rapid | [12–28] |
| NAT2*5A |
T341C (rs1801280)
C481T (rs1799929) |
I114T
L161L (synonymous) |
Slow | [13,20,21, 29,30] |
| NAT2*5B |
T341C (rs1801280)
C481T (rs1799929) A803G (rs1208) |
I114T
L161L (synonymous) K268R |
Slow | [14,20–28,30,32] |
| NAT2*5C |
T341C (rs1801280)
A803G (rs1208) |
I114T
K268R |
Slow | [20,31,29–31,33] |
| NAT2*5D | T341C (rs1801280) | I114T | Slow | [23,26,28,34–37] |
| NAT2*5E |
T341C (rs1801280)
G590A (rs1799930) |
I114T
R197Q |
Slow | [34] |
| NAT2*5F |
T341C (rs1801280)
C481T (rs1799929) C759T A803G (rs1208) |
I114T
L161L (synonymous) K268R V253V (synonymous) |
Slow | [38] |
| NAT2*5G |
C282T (rs1041983)
T341C (rs1801280) C481T (rs1799929) A803G (rs1208) |
Y94Y (synonymous)
I114T L161L (synonymous) K268R |
Slow | [39] |
| NAT2*5H |
T341C (rs1801280)
C481T (rs1799929) A803G (rs1208) 859Del |
I114T
L161L (synonymous) K268R S287 Frameshift |
Slow | [39] |
| NAT2*5I |
T341C (rs1801280)
A411T (rs4986997) C481T (rs1799929) A803G (rs1208) |
I114T
L137F L161L (synonymous) K268R |
Slow | [27] |
| NAT2*5J |
C282T (rs1041983)
T341C (rs1801280) G590A (rs1799930) |
Y94Y (synonymous)
I114T R197Q |
Slow | [40] |
| NAT2*5K |
C282T (rs1041983)
T341C (rs1801280) |
Y94Y (synonymous) | [41] | |
| NAT2*5L |
T70A
T341C (rs1801280) C481T (rs1799929) A803G (rs1208) |
L24I
I114T L161L (synonymous) K268R |
[41,42] | |
| NAT2*5M |
T341C (rs1801280)
C481T (rs1799929) A803G (rs1208) G838A |
I114T
L161L (synonymous) K268R V289M |
[43] | |
| NAT2*6A |
C282T (rs1041983)
G590A (rs1799930) |
Y94Y (synonymous)
R197Q |
Slow | [12–14,20–28,30,33] |
| NAT2*6B | G590A (rs1799930) | R197Q | Slow | [23,26,28,30,35,36] |
| NAT2*6C |
C282T (rs1041983)
G590A (rs1799930) A803G (rs1208) |
Y94Y (synonymous)
R197Q K268R |
Slow | [35,36,41] |
| NAT2*6D |
T111C
C282T (rs1041983) G590A (rs1799930) |
F37F (synonymous)
Y94Y (synonymous) R197Q |
Slow | [37] |
| NAT2*6E |
C481T (rs1799929)
G590A (rs1799930) |
L161L (synonymous)
R197Q |
Slow | [45] |
| NAT2*6F |
G590A (rs1799930)
A803G (rs1208) |
R197Q
K268R |
[42] | |
| NAT2*6G |
C282T (rs1041983)
A518G G590A (rs1799930) |
Y94Y (synonymous)
K173R R197Q |
[42] | |
| NAT2*6H |
C282T (rs1041983)
G590A (rs1799930) A766G |
Y94Y (synonymous)
R197Q K256E |
[42] | |
| NAT2*6I |
C282T (rs1041983)
G590A (rs1799930) G838A G857A |
Y94Y (synonymous)
R197Q V280M G286E |
[42] | |
| NAT2*6J |
C282T (rs1041983)
G590A (rs1799930) G857A (rs1799931) |
Y94Y (synonymous)
R197Q G286E |
[43] | |
| NAT2*6K |
C282T (rs1041983)
G590A (rs1799930) C638T |
Y94Y (synonymous)
R197Q P213L |
[43] | |
| NAT2*6L |
C282T (rs1041983)
C345T G590A (rs1799930) |
Y94Y (synonymous)
D115D(synonymous) R197Q |
[43] | |
| NAT2*7A | G857A (rs1799931) | G286E | Slow; Substrate dependent? | [13,22,23,26,28,30] |
| NAT2*7B |
C282T (rs1041983)
G857A (rs1799931) |
Y94Y (synonymous)
G286E |
Slow; Substrate dependent? | [12,20–29] |
| NAT2*10 | G499A | E167K | Slow; Substrate dependent? | [22,26–28] |
| NAT2*11A | C481T (rs1799929) | L161L (synonymous) | Rapid | [23,26,28,46] |
| NAT2*11B |
C481T (rs1799929) 8
59Del |
L161L (synonymous)
S287 Frameshift |
Unknown | [39] |
| NAT2*12A | A803G (rs1208) | K268R | Rapid | [23,26,28,30,47,48] |
| NAT2*12B |
C282T (rs1041983)
A803G (rs1208) |
Y94Y (synonymous)
K268R |
Rapid | [20,21,30,36] |
| NAT2*12C |
C481T (rs1799929)
A803G (rs1208) |
L161L (synonymous)
K268R |
Rapid | [20,21,31,35,36,48] |
| NAT2*12D |
G364A (rs4986996)
A803G (rs1208) |
D122N
K268R |
Slow | [27] |
| NAT2*12E |
C282T (rs1041983)
C578T A803G (rs1208) |
Y94Y (synonymous)
T193M K268R |
[41,42] | |
| NAT2*12F |
T622C
A803G (rs1208) |
Y208H
K268R |
[41] | |
| NAT2*12G |
G609T
A803G (rs1208) |
E203D
K268R |
[42] | |
| NAT2*12H |
C403G
A803G (rs1208) |
L135V
K268R |
[43] | |
| NAT2*13A | C282T (rs1041983) | Y94Y (synonymous) | Rapid | [20,21,23–6,28,30,33,34,48,49] |
| NAT2*13B |
C282T (rs1041983)
C578T |
Y94Y (synonymous)
T193M |
[42] | |
| NAT2*14A | G191A (rs1801279) | R64Q | Slow | [20,21,23–26,28,31,50] |
| NAT2*14B |
G191A (rs1801279)
C282T (rs1041983) |
R64Q
Y94Y (synonymous) |
Slow | [50,51] |
| NAT2*14C |
G191A (rs1801279)
T341C (rs1801280) C481T (rs1799929) A803G (rs1208) |
R64Q
I114T L161L (synonymous) K268R |
Slow | [34–36] |
| NAT2*14D |
G191A (rs1801279)
C282T (rs1041983) G590A (rs1799930) |
R64Q
Y94Y (synonymous) R197Q |
Slow | [34,36] |
| NAT2*14E |
G191A (rs1801279)
A803G (rs1208) |
R64Q
K268R |
Slow | [34,42] |
| NAT2*14F |
G191A (rs1801279)
T341C (rs1801280) A803G (rs1208) |
R64Q
I114T K268R |
Slow | [36] |
| NAT2*14G |
G191A (rs1801279)
C282T (rs1041983) A803G (rs1208) |
R64Q
Y94Y (synonymous) K268R |
Slow | [37] |
| NAT2*14H |
G191A (rs1801279)
C282T (rs1041983) C683T |
R64Q
Y94Y (synonymous) P228L |
[42] | |
| NAT2*14I |
G191A (rs1801279
C481T (rs1799929) A803G (rs1208) |
R64Q
L161L (synonymous) K268R |
[44] | |
| NAT2*17 | A434C | Q145P | Slow | [20,21,23,26,52] |
| NAT2*18 | A845C | K282T | Rapid | [20,21,23,26,52] |
| NAT2*19 | C190T (rs1805158) | R64W | Slow | [53–55] |
Adapted from http://N-acetyltransferasenomenclature.louisville.edu [56].
Human NAT2 alleles should be written in upper case and italicized. Protein products of the alleles are also upper case but not italicized and the asterisk is omitted. For example, the allele NAT2*4 encodes the protein NAT2 4. NAT2*4 has historically been designated “wildtype”. Since it is the most common occurring alleles in some but not all ethnic groups the designation of “wildtype” allele is somewhat arbitrary.
Reference gene sequence published in Genbank Assession Number X14672. The frequency of individual SNPs and alleles in various ethnic groups is available online from the National Cancer Institute’s SNP500Cancer database. RS refers to reference SNP. Signature SNP for each cluster is italicized. Although additional SNPs have been identified outside the open reading frame [42,43,57], they will not be named until a functional effect is observed. SNPs should be identified by designating “A” of the ATG translation initiation codon as number 1. SNPs upstream of this site are designated by negative numbers and SNPs downstream of this site are designated by positive numbers [56].
PHENOTYPE of COMMON NAT2 ALLELES OR HAPLOTYPES
Various combinations of SNPs are identified as NAT2 alleles or haplotypes (Table 1). NAT2*4 is considered the “wild-type” or reference allele/haplotype with no SNPs. Variant NAT2 alleles or haplotypes possessing combinations of SNPs are segregated into clusters possessing a signature SNP either alone or in combination with others. A consensus N-acetyltransferase gene nomenclature was first published in 1995 [65]. An international nomenclature committee publishes an internet accessible website for updates at http://N-acetyltransferasenomenclature.louisville.edu [56].
Striking ethnic differences in the frequencies of SNPs (http://snp500cancer.nci.nih.gov) are responsible for the corresponding ethnic differences in frequency of rapid and slow acetylator NAT2 alleles or haplotypes [41,42] and therefore phenotypes [1,2,25,57,58,60]. For example, the G191A (R64Q) SNP common to the NAT2*14 allele cluster is frequent in Africans and African-Americans, but virtually absent in Caucasian, Indian, and Korean populations. Similarly, the NAT2*7 cluster possessing the G857A (K268R) SNP is much more frequent in South India and Korea than other populations while the NAT2*5 cluster containing the T341C (I114T) SNP is much less frequent in Korea than in Europe, North America, India and Africa. Deduction of NAT2 phenotypes is assigned based on co-dominant expression of rapid and slow acetylator NAT2 alleles or haplotypes. Individuals homozygous for rapid NAT2 acetylator alleles are deduced as rapid acetylators, individuals homozygous for slow acetylator NAT2 alleles are deduced as slow acetylators, and individuals possessing one rapid and one slow NAT2 allele are deduced as intermediate acetylators [25]. The functional effects of many of the NAT2 alleles/haplotypes are summarized in Table 1, but as described below, a consensus has not yet been reached on the functional effects of all of them.
MOLECULAR BASIS FOR ALTERED FUNCTION OF NAT2 VARIANTS
Reductions in the amount of NAT2 protein expressed in human liver from individuals with slow acetylator phenotype have been reported [12,66]. Slow acetylator NAT2 alleles recombinantly expressed in COS-1 cells [13,27,28,67], chinese hamster ovary cells [32] and yeast [23,37] show reduced levels of NAT2 protein when compared with NAT2*4. These data suggest that slow acetylator phenotype is conferred, at least for some NAT2 alleles, by reduction(s) in NAT2 protein.
Characterization of NAT2 alleles has been investigated primarily in recombinant expression systems. Recombinant human NAT2 5, NAT2 6, NAT2 7, and NAT2 14 clusters yield variable reductions in catalytic activity associated with slow acetylator phenotype, while recombinant human NAT2 12 and NAT2 13 clusters catalyze N-, O-, and N,O-acetyltransferase activities at levels comparable to the rapid acetylator NAT2 4 [21]. Recently, some controversy has arisen regarding the assignment of NAT2*12 and NAT2*13 as rapid acetylator alleles [48]. As shown in Table 1, NAT2*12 allele clusters possess the signature A803G (K268R) SNP whereas the NAT2*13 allele possesses the synonymous C282T SNP. Previous studies have clearly shown that the A803G (K268R) characteristic of NAT2*12 alleles and C282T characteristic of NAT2*13 alleles do not alter NAT2 catalytic activity [20,21,23,28]. Three studies using caffeine as a phenotype probe suggested that NAT2*12 and NAT2*13 were associated with slow acetylation phenotype [33,48,68], but this was related to an NAT2 genotyping artifact [47,69]. The other studies did not distinguish the NAT2*12 or the NAT2*13 alleles from other NAT2 genotypes. Nevertheless, verification of NAT2*12 and NAT2*13 as rapid acetylator alleles has been shown in vivo with 5 subjects possessing NAT2*13A and 20 subjects possessing NAT2*12A [70].
Recombinant NAT2 proteins associated with some slow acetylator alleles show reduced stability [20,23,28,31,37,58] whereas others appear targeted for proteomic degradation by the proteosome [27,67]. Human NAT2 7B recombinantly expressed in bacteria has altered affinity for some but not other substrates [20,22]. Some human NAT2 SNPs cause reductions in quantity of recombinant NAT2 protein in eukaryotic expression systems [12,13,23,27,28,32,37,67]. Since multiple mechanisms for reductions in NAT2 activity are associated with various combinations of SNPs that make up NAT2 alleles, the ability to distinguish among multiple acetylator phenotypes is complex and a function of the sensitivity and specificity of the phenotyping method. Depending upon the drug probe and analytical method used, acetylation phenotypes often exhibit overlap due to numerous genetic and/or environmental factors, including the large number and diversity of NAT2 genotypes present in human populations [41,42]. The relative specificity of the substrate for NAT2 versus NAT1 at the concentrations obtained in vivo will also affect acetylator phenotype. Caffeine is commonly used as a probe drug for NAT2 phenotype determinations because it is relatively non-invasive. Although excellent NAT2 genotype/phenotype correlations have been reported [33,58], genetic and/or environmental effects on other enzymes (e.g., cytochrome P450s, xanthine oxidases, NAT1) may affect metabolite levels or ratios used to determine NAT2 phenotype. Other potential artifacts in the use of caffeine to determine NAT2 phenotype also have been reported [24,71–73].
Common NAT2 alleles or haplotypes have been investigated following recombinant expression in COS-1 cells. As shown in Fig. 2, NAT2*5B, NAT2*6A, and NAT2*14B each had reduced N-acetyltransferase activities towards sulfamethazine (SMZ) and O-acetyltransferase activities towards N-hydroxy-4-aminobiphenyl (N-OH-ABP) and N-hydroxy-2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (N-OH-PhIP). Catalytic activity for NAT2*7B which possesses the signature SNP G857A differed with respect to substrate as it showed reduced activity towards the N-acetylation of SMZ and the O-acetylation of N-OH-ABP, but not towards the O-acetylation of N-OH-PhIP. As shown in Fig. 3, NAT2*5B, NAT2*6A, NAT2*7B and NAT2*14B reduced maximum N-acetyltransferase activities. NAT2*7B was the only allele/haplotype that changed apparent Km, reducing it towards SMZ and increasing it towards acetyl coenzyme A (AcCoA).
Fig. 2. Comparison of common recombinant NAT2 allozymes towards the N-acetylation of sulfamethazine (SMZ) the O-acetylation of N-hydroxy-4-aminobiphenyl (N-OH-ABP) and N-hydroxy-2-amino-1-methyl-6-phenylimidazo[4,5-b] pyridine (N-OH-PhIP).
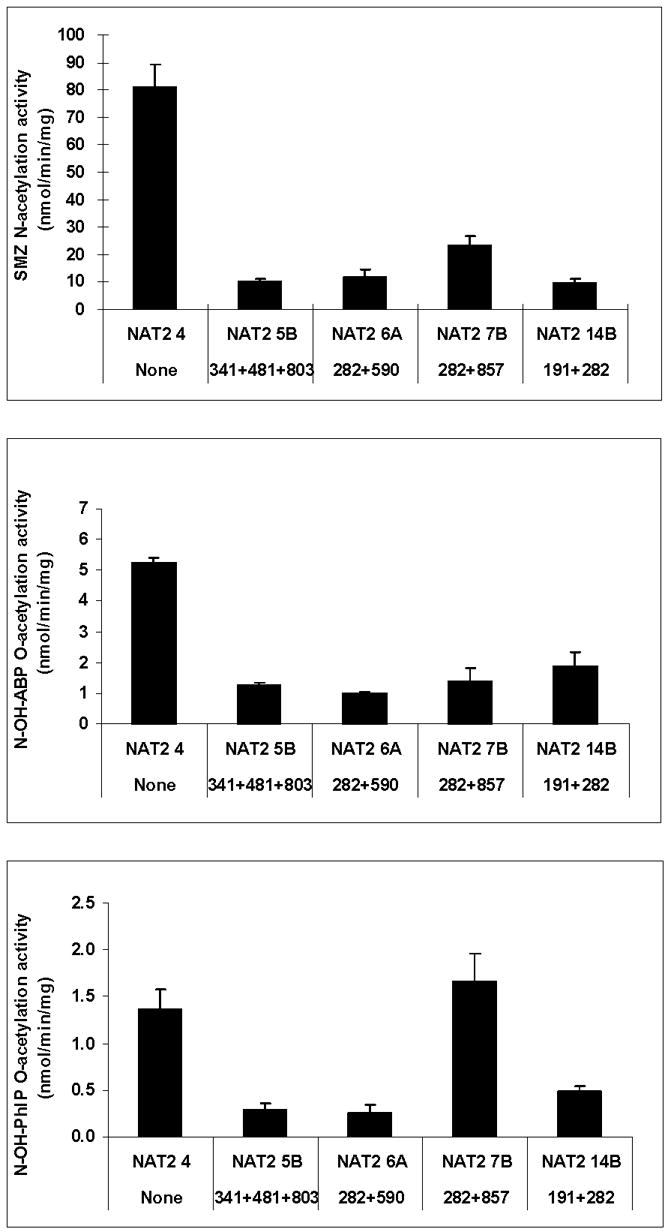
Each bar represents Mean ±SD for three determinations following recombinant expression in COS-1 cells. NAT2 catalytic activity is generally consistent across the substrates with the exception of NAT2 7B, which exhibits reduced activity towards SMZ and N-OH-ABP but not towards N-OH-PhIP. Adapted from [28].
Fig. 3. Michaelis-Menten kinetic constants for sulfamethazine (SMZ) and acetyl coenzyme A cofactor (AcCoA) in common recombinant NAT2 allozymes.
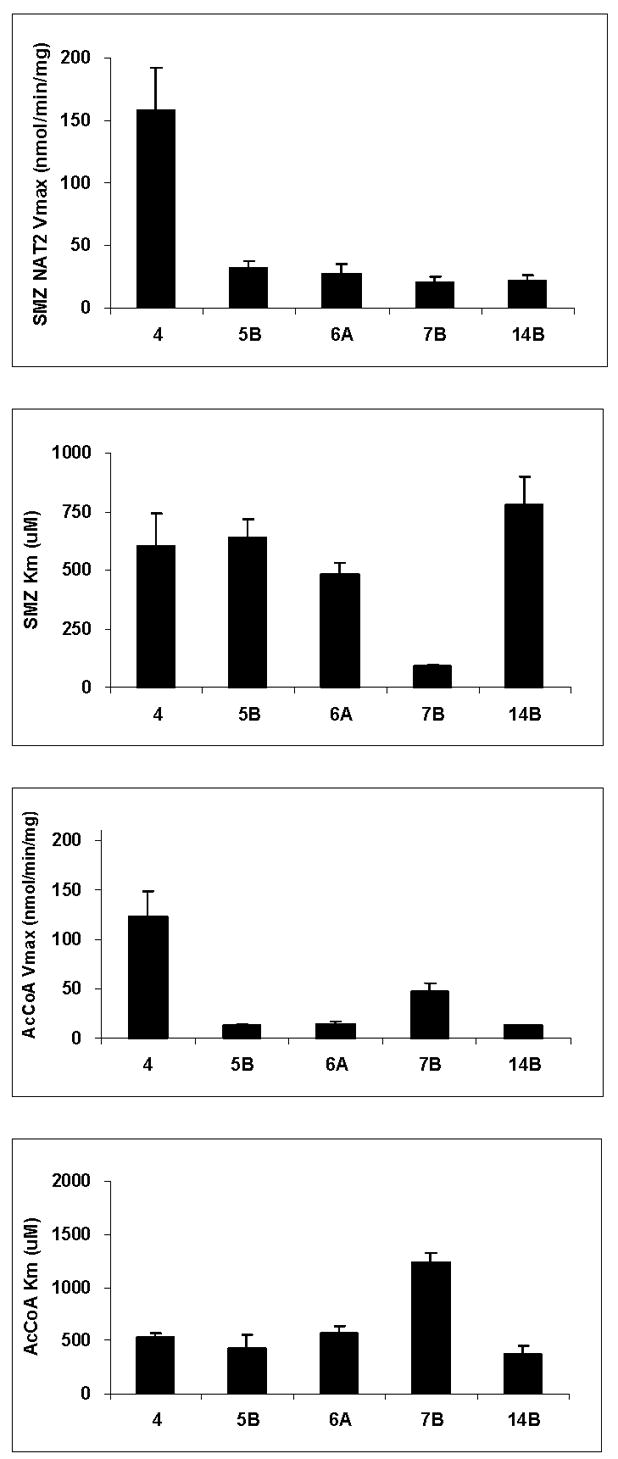
Each bar represents Mean ±SD for three determinations following recombinant expression in COS-1 cells. Adapted from [28].
EFFECTS OF INDIVIDUAL SNPS ON NAT2 FUNCTION
The effects of individual SNPs on NAT2 catalytic activity have been investigated following recombinant expression in yeast. As shown in Fig. 4, C190T (R64W), G191A (R64Q), T341C (I114T), A434C (Q145P) and G590A (R197Q) are associated with reductions in both N- and O-acetyltransferase catalytic activities, whereas T111C (F37F), C282T (Y94Y), C481T (L161L), C759T (V253V), and A803G (K268R) are associated with reductions in neither. The effects of A845C (K282T) and G857A (G286E) differ with substrate, as they reduce N-acetyltransferase activity towards 2-aminofluorene (AF) but not SMZ, and they reduce O-acetyltransferase activity towards N-hydroxy-2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (N-OH-MeIQx) but not N-OH-PhIP. As shown in Fig. 5, similar findings were found for the N-acetylation of SMZ and the O-acetylation of N-OH-PhIP following recombinant expression in COS-1 cells. G857A (G286E) reduced the O-acetylation of N-OH-ABP but not N-OH-PhIP.
Fig. 4. Effects of individual SNPs in NAT2 on the N-acetylation of sulfamethazine (SMZ) and 2-aminofluorene (AF) and the O-acetylation of N-hydroxy-2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline (N-OH-MeIQx) and N-hydroxy-2-amino-1-methyl-6-phenylimidazo[4,5-b] pyridine (N-OH-PhIP).
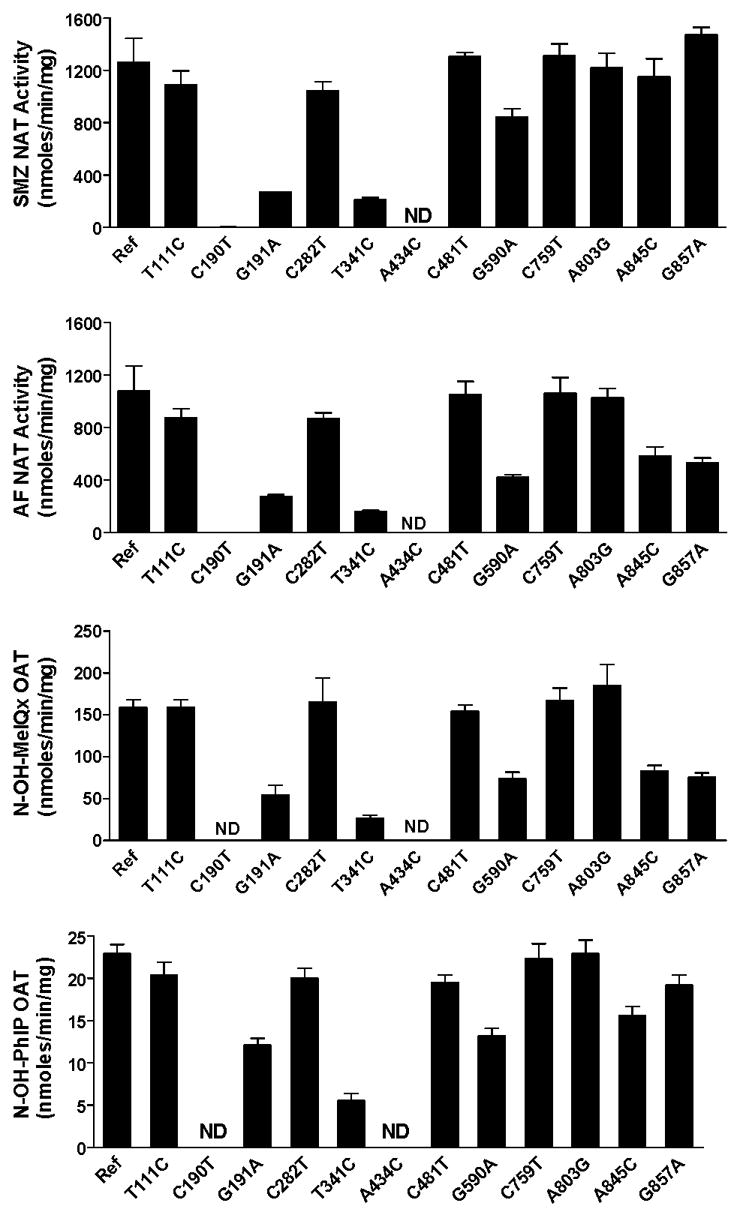
Each bar represents Mean ±SEM for three determinations following recombinant expression in yeast (Schizosaccaromyces pombe). The effect of the SNPs on NAT2 catalytic activity are generally consistent across the substrates with the exception of A845C and G857A, which reduce activity towards AF and N-OH-MeIQx far more than they do towards SMZ and N-OH-PhIP. Ref: Reference allele (NAT2*4, no SNPs); ND; Non-detectable. Adapted from [23,26,55].
Fig. 5. Effects of individual SNPs in NAT2 on the N-acetylation of sulfamethazine (SMZ) the O-acetylation of N-hydroxy-4-aminobiphenyl (N-OH-ABP) and N-hydroxy-2-amino-1-methyl-6-phenylimidazo[4,5-b] pyridine (N-OH-PhIP).
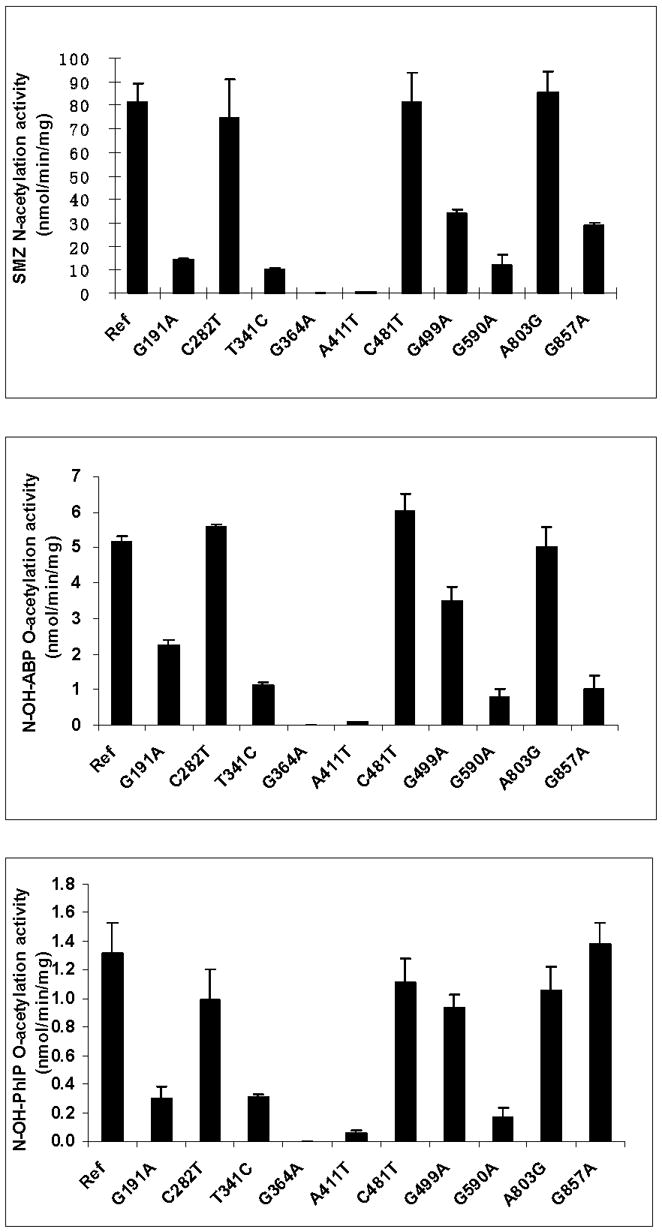
Each bar represents Mean ±SD for three determinations following recombinant expression in COS-1 cells. Ref: Reference allele (NAT2*4, no SNPs). The effect of each SNP on NAT2 catalytic activity is generally consistent across the substrates with the exception of G857A, which reduces activity towards SMZ and N-OH-ABP far more than it does towards N-OH-PhIP. Adapted from [28].
Data from additional studies to investigate mechanisms for the reductions in N- and/or O-acetyltransferase activities are illustrated in Fig. 6. Following recombinant expression in COS-1 cells, the SNPs which reduced N- and O-acetyltransferase activities (Fig. 5) each did so by reductions in expression of recombinant NAT2 protein but not recombinant NAT2 mRNA. The reduction in NAT2 protein appeared to be related to stability of the protein for G191A (R64W), G590A (R197Q) and G857A (G286E), but not for T341C (I114T), A411T (L137F), and G499A (E167K). As shown in Fig. 7, G191A (R64W), T341C (I114T), G499A (E167K), G590A (R197Q), and G857A (G286E) reduced maximum N-acetyltransferase activities whereas C282T (Y94Y), C481T (L161L), and A803G (K268R) did not. G857A (G286E) changed apparent Km, reducing it towards SMZ and increasing it towards the acetyl coenzyme A (AcCoA) cofactor.
Fig. 6. Effects of individual SNPs in NAT2 on relative NAT2 protein expression (top panel), relative mRNA expression (middle panel) and stability of the recombinant NAT2 protein (lower panel).
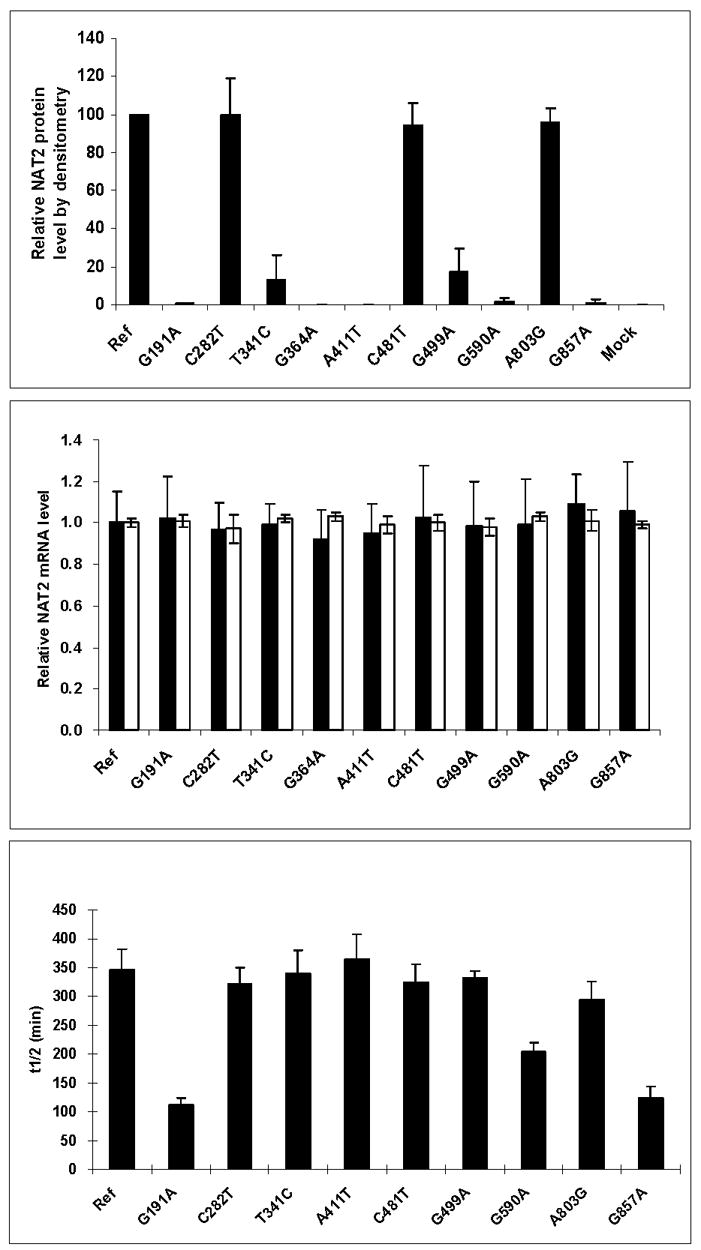
Each bar represents Mean ±SD for three determinations following recombinant expression in COS-1 cells. Ref: Reference allele (NAT2*4, no SNPs). In the top panel, mock refers to transfection with pcDNA5/FRT plasmid without the NAT2 insert. In the middle panel, expression of NAT2 mRNA is shown in solid bars and beta-actin mRNA in open bars. Adapted from [28].
Fig. 7. Effects of individual SNPs in NAT2 on Michaelis-Menten kinetic constants for sulfamethazine (SMZ) and acetyl coenzyme A cofactor (AcCoA).
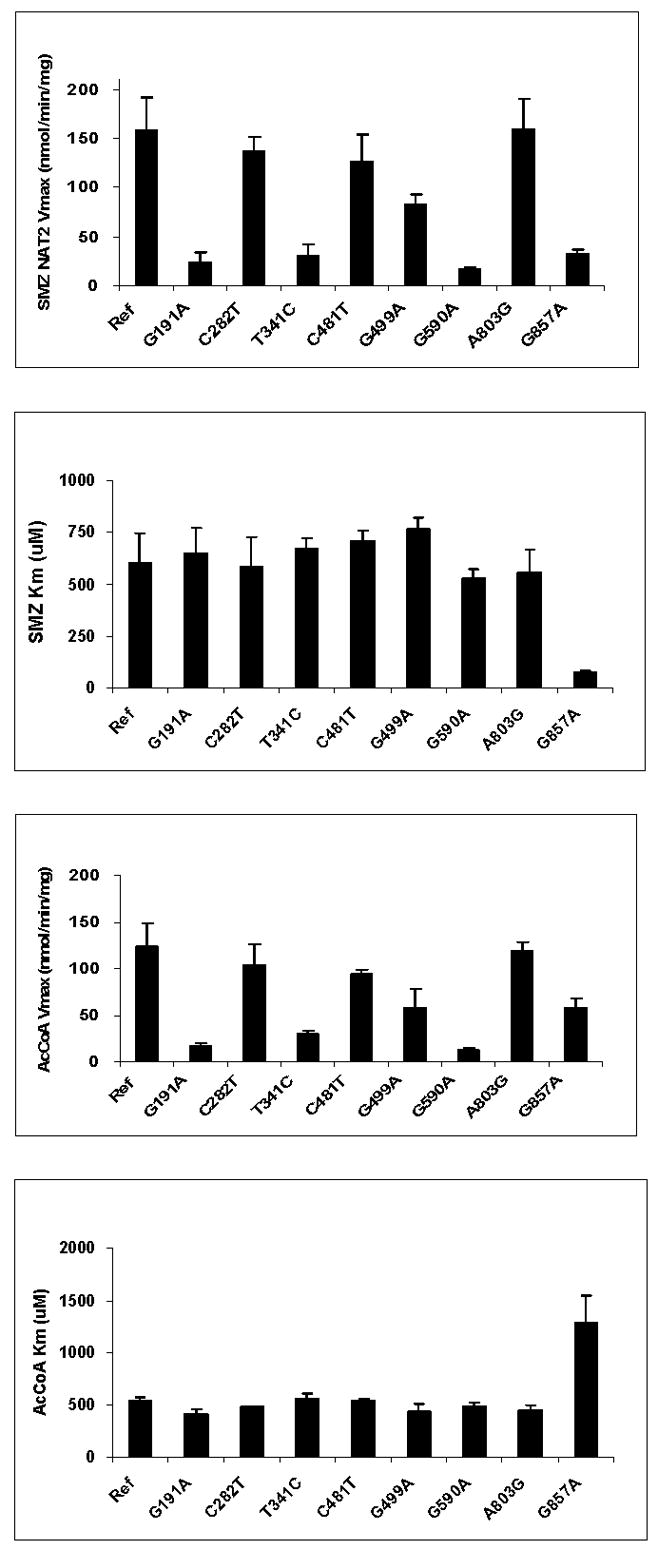
Each bar represents Mean ±SD for three determinations following recombinant expression in COS-1 cells. G191A, T341C, G499A, G590A, and G857A significantly (p< 0.05) reduced apparent Vmax. G857A significantly (p<0.05) decreased apparent Km towards SMZ and increased apparent Km towards AcCoA. Adapted from [28].
MOLECULAR MODELING OF NAT2 NON-SYNONYMOUS SNPs
Previous NAT2 modeling studies sought to better understand structural characteristics, including SNP locations and computational docking of substrates [10,74–77]. These models were generated based on homology to N-acetyltransferase enzyme crystal structures from Salmonella typhimurium, Pseudomonas aeruginosa, Mycobacterium smegmatis, and/or Mesorhizobium loti [7,78–81]. However, the alignment scores between bacterial and mammalian N-acetyltransferase protein sequences are low (~30%), and important differences between the bacterial and mammalian N-acetyltransferase protein structures limit the utility of the mammalian N-acetyltransferase homology models [77].
For the following molecular modeling evaluations, NAT2 crystal structure coordinates were obtained from the Protein Data Bank (PDB) under code 2PFR. The 2PFR PDB file shows that in this case the NAT2 protein crystallizes as a dimer. NAT2 coordinates from chain A (NAT2 protein 1) were used for these analyses because chain B (NAT2 protein 2) is missing coordinates of atoms for residue E167 and have more undetermined atoms than the chain A. None of the missing residues in chain A interact with any of the residues evaluated in this study.
Prior to analysis, the residue name HIS was changed to HIE in the PDB file to define histidine residues as neutral species that are protonated in the epsilon position. Hydrogen atoms were added to the structure using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco [82]. Figures were generated using a ribbon color scheme that distinguishes the previously defined N-acetyltransferase enzyme domains [83]. Solvent accessible surface areas (SASA) were measured for residue side-chains using Naccess V2.1.1 [84] and reported as percent of total side-chain surface area. Hydrogen bonding interactions and donor-acceptor hydrogen bonding distances were calculated using HBPLUS [85] and are reported in angstroms (Å). The location and molecular interactions of NAT2 residues R64, I114, D122, L137, Q145, E167, R197, K268, K282, and G286 were evaluated. Molecular images depicting the nature and location of these interactions were generated using Chimera.
Previous structural evaluations of SNP-induced residue substitutions are compromised by low template-target sequence alignments in homology modeling. In particular, the placement of both the second domain loop and the C-terminal tail constitute major changes to previous homology-based models [77]. The crystal structure for the NAT2 was recently deposited into the RCSB Protein Data Bank under the code 2PFR [86]. Two major structural features distinguish mammalian N-acetyltransferase structure from the bacterial. First, the domain II loop (165–185 based on crystal structure [86]) is associated with the domain III beta sheet, apparently providing structural stability and limiting active site access [77]. Second, the mammalian N-acetyltransferase C-terminal tail is not alpha helical, as is the case for the bacterial N-acetyltransferases, but instead is a coil that reaches around and associates with the active site pocket, thereby playing a key role in defining the size and shape of the active site pocket. Due to the lack of appropriate structural template information for these regions, and the limitations of current homology modeling techniques, these structural features were impossible to model correctly using the bacterial N-acetyltransferase structures as templates. Homology modeling is capable of generating protein structure for sequence that is aligned with a structure template, but is unlikely to accurately predict secondary or tertiary structure in the absence of a template (Fig. 8). The following evaluations of the NAT2 SNP-induced residue changes are based on the mammalian N-acetyltransferase protein crystal structure.
Fig. 8. Human NAT2 crystal structure (2PFR) ribbon diagram.
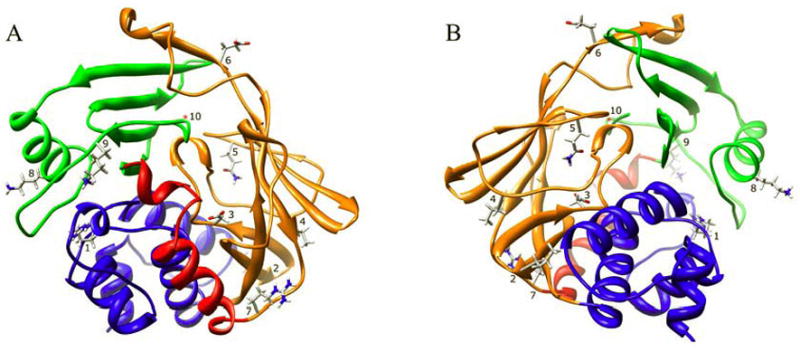
The ribbon is colored to indicate NAT protein domain I (blue), the interdomain region (red), domain II (orange), and domain III (green). The location of residues R64 (1), I114 (2), D122 (3), L137 (4), Q145 (5), E167 (6), R197 (7), K268 (8), K282 (9), and G286 (10) are shown. Two orientations are shown, one at the site of substrate entry into the active site (A) and the other on the reverse side (B) of the structure.
R64Q (G191A) and R64W (C190T)
The side-chain of residue R64 is 2.7% solvent exposed in domain I, and its side-chain hydrogen bonds twice to E38 (R64:NE-E38:OE1 2.74 Å, and R64:NH2-E38:OE2 2.90 Å), and twice to N41 (R64:NH2-N41:OD1 2.82 Å, and R64:NH1-N41:OD1 3.10 Å), both of which are in a stretch of domain I coil (V35-G51) that is mostly absent of secondary structure (Fig. 9A). The hydrogen bonding interactions between positively charged R64 and negatively charged E38 are salt bridges, and are stronger than normal hydrogen bonds. Replacing arginine with either glutamine or tryptophan at residue 64 will result in loss of these interactions, which contribute to the conformation and dynamics of the first domain tertiary structure. The fact that residues R64 and E38 are completely conserved in all known mammalian N-acetyltransferases suggests that these interactions are important for the function and/or stability of the enzyme. Residue N41 is also highly conserved among N-acetyltransferases. Significant structural changes in this region could influence the position of catalytic triad residue C68, thereby altering its interactions with catalytic H107, and/or affecting its acetylation. It is unlikely, however, that residue R64 hydrogen bonding interactions are critical for C68 positioning, since many other hydrogen bonding interactions support the tertiary structure near the domain I helix containing C68. Additionally, residue 64 is adjacent to a pocket that opens to the outside of the protein and is solvent accessible. The exposure of R64 to this pocket is minimal, and is included in the SASA measurement of 2.7% above. It is unknown whether this pocket has any functional purpose. Previous modeling studies have reported interactions between R64 and E39 [75]. However, NAT2 has an asparagine at residue 39, and a glutamate at residue 38.
Fig. 9. Molecular interactions formed by various human NAT2 residues changed as a result of NAT2 SNPs.
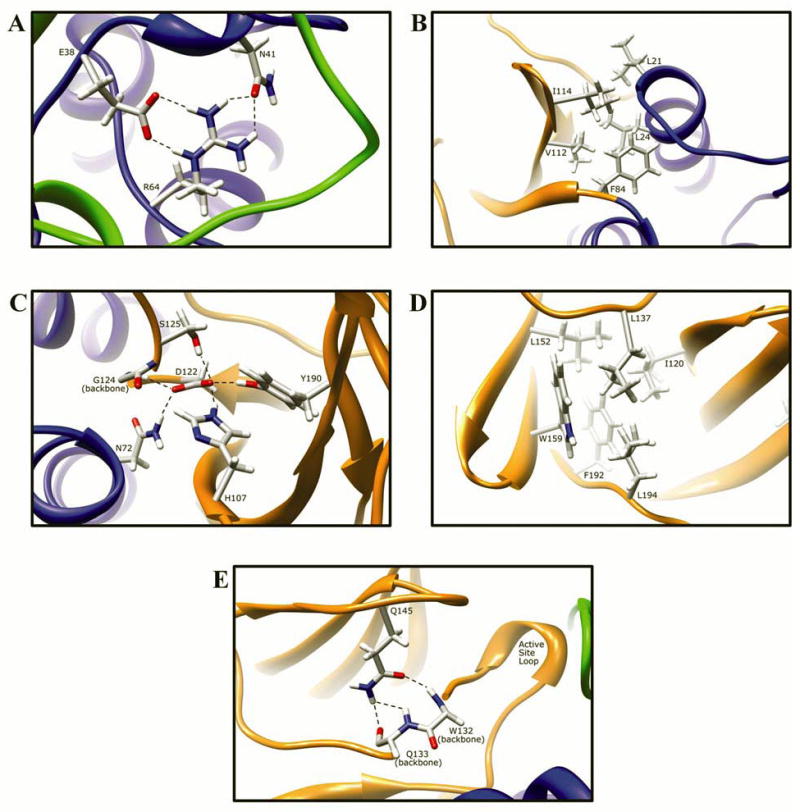
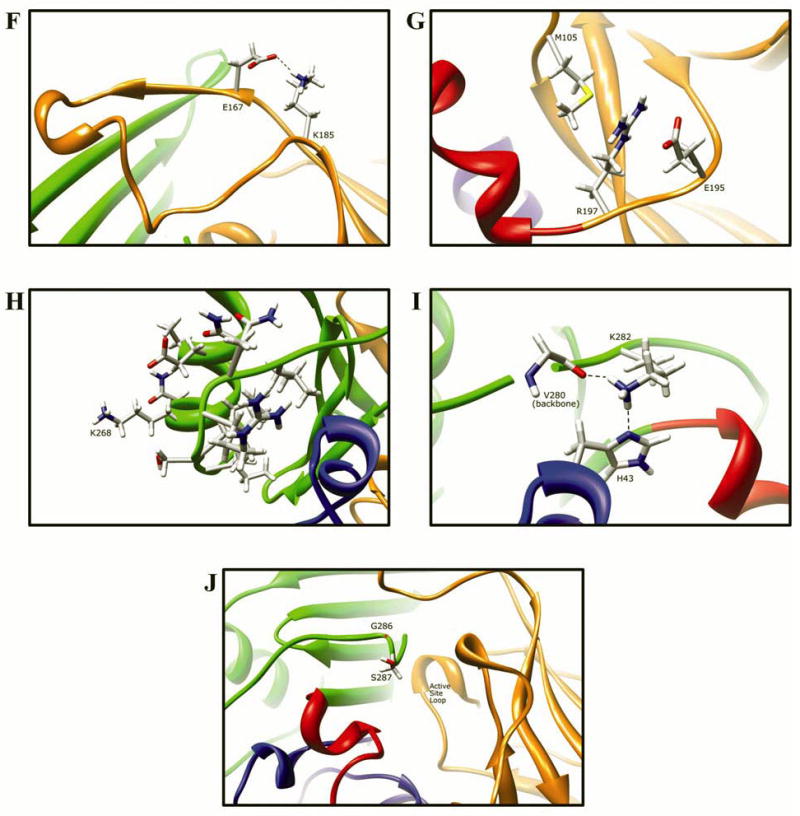
Protein domains are indicated by ribbon color as in Fig. 8, with backbone atoms shown instead of ribbon where necessary. (A) Residue R64 hydrogen bonds to E38 and N41 in domain I. (B) I114 is part of a hydrophobic core at an interface between the domain II beta barrel and a domain I helix. (C) Catalytic core residue D122 hydrogen bonds to N72, H107, S125, Y190, and the backbone of G124. (D) L137 is part of a hydrophobic core in the center of the domain II beta barrel. (E) Q145 hydrogen bonds to the backbone of residues Q133 and W132 in domain II. (F) Domain II loop residue E167 forms a relatively weak hydrogen bond to loop residue K185. (G) Positively charged R197 in domain I forms electrostatic interactions with negatively charged E195 and the M105 sulfur. (H) The side-chain of K268 is surface exposed in domain III, and does not interact with any other residue. (I) Domain III residue K282 hydrogen bonds to H43 in domain I and to the backbone of V280. (J) G286 is located adjacent to the active site pocket in the C-terminal region of domain III and has no interactions with other residues.
Functional studies of the R64Q variant in bacterial [20,21], yeast [23], and mammalian cells [28] demonstrated reduced activity and protein levels due to reduced protein stability. R64Q is a frequent SNP in African and African-American populations [58]. These data suggest that the R64 interactions are necessary for structural stability. Even conservative replacement with a glutamine residue, which is also polar and could maintain some of the R64 hydrogen bonding interactions, was insufficient to maintain stability. Since glutamine is less bulky than arginine, the loss of enzyme stability cannot be attributed to steric clashes.
Likewise, functional studies of the NAT2 R64W variant in yeast demonstrated reduced protein activity and protein levels due to reduced protein stability [55]. A loss of critical hydrogen bond and salt bridge interactions in the R64W variant, as we observed in the NAT2 crystal structure, is consistent with reduced protein thermostability.
I114T (T341C)
The side-chain of residue I114 is 16.2% solvent exposed on the periphery of NAT2 at the end of a beta strand in the domain II beta barrel. The I114 side chain shares hydrophobic interactions with residues L21 and L24 in domain I, and F84 and V112 in domain II (Fig. 9B). Because it is peripheral, the interactions of hydrophobic I114 with the solvent likely also contribute to the structural fold in that region. Changing this hydrophobic residue to a polar hydrophilic threonine residue may reduce hydrophobic interactions at the interface between the domain II beta barrel and the domain I helix, altering the protein folding in that region. However, because the residue is peripheral, and the surrounding protein structure is highly organized into secondary and tertiary structures, it is unlikely that a reduction of hydrophobic forces in this region will result in major structural changes.
I114T is the most common SNP in Caucasian populations [58]. Recombinant expression of the I114T variant in mammalian cells did not result in changes in protein stability or apparent kinetic parameters, but led to a reduction of active enzyme possibly due to enhanced protein degradation [67]. This residue change was also shown to cause slow acetylator phenotype when recombinantly expressed in bacterial [20,21] and yeast [23]. These functional data are consistent with a structural change in the I114T variant, which could increase protein aggregation and/or targeting for degradation without altering the protein’s stability.
D122N (G364A)
The side-chain of catalytic triad residue D122 is completely buried in the protein with hydrogen bonds to the side chains of N72 (D122:OD1-N72:ND2 2.93 Å), H107 (D122:OD2-H107:NE2 2.85 Å), S125 (D122:OD2-S125:OG 2.97 Å), Y190 (D122:OD2-Y190:OH 2.74 Å), and the backbone of G124 (D122:OD1-G124:N 2.83 Å) (Fig. 9C). These multiple hydrogen bonding interactions likely contribute to the stability of the active site loop conformation and the interaction between domains I and II. The location and interactions of residues D122, N72, H107, and Y190 are all highly conserved in mammalian and bacterial N-acetyltransferases (bacterial residues are numbered differently). Likewise, residue 124 is conserved in size, since it is always either a glycine or alanine, and in hydrogen bonding interactions with D122 in mammalian and bacterial N-acetyltransferases. Residue 125, however, is highly variable and plays an important role in substrate selectivity [11]. Although a conservative replacement with an asparagine residue in this location likely also results in hydrogen bonding interactions with surrounding residues, loss of these native and highly conserved interactions is likely to negat ively affect the protein structure. Perhaps more importantly, because D122 is a catalytic triad residue, any change at residue 122 will adversely affect the function of the catalytic triad [87].
Functional studies of the D122N variant demonstrated that the variant is catalytically inactive. Recombinant expression of the NAT2 D122N variant in mammalian cells resulted in reduced protein probably due to protein degradation pathways, though inclusion bodies were not tested for protein aggregation [27]. Disruption of the catalytic triad likely also affects enzyme acetylation, thereby increasing proteasomal degradation [88].
L137F (A411T)
The side-chain of residue L137 is 6.1% solvent exposed and oriented toward the interior of the domain II beta-barrel. The L137 residue shares hydrophobic interactions with residues I120, L152, W159, F192, and L194 (Fig. 9D). The L137F substitution is likely to affect the beta barrel structure due to steric clashes that result from replacing leucine with a larger phenylalanine residue. It is also possible that aromatic interactions between the F137 and W159 residue side chains may alter the native domain II fold. This interaction was observed in an MD optimized homology model of the NAT2 L137F variant, which resulted in repositioning of the 154–158 beta turn.
Functional studies of the L137F variant in mammalian cells demonstrated reduction in protein levels with no change in protein stability, possibly the result of proteasomal degradation [27]. These data are consistent with a change in secondary structure that could trigger mammalian degradation mechanisms.
Q145P (A434C)
The side-chain of Q145 is 11.1% solvent exposed in domain II, and hydrogen bonds to the backbone of residues W132 (Q145:OE1-W132:N 2.94 Å) and Q133 (Q145:NE2-Q133:O 2.86 Å, and Q145:OE1-Q133:N 3.28 Å) on an adjacent strand in domain II (Fig. 9E). Changing this residue to a proline in the Q145P variant is likely to result in disruption of secondary structure due to the loss of stabilizing hydrogen bonding interactions, and due to the introduction of a rigid proline residue [89]. Because residues W132 and Q133 are part of the coil that becomes the active site loop, altering their backbone interactions with residue 145 may affect the conformation of the active site loop and thereby alter enzymatic activity and/or substrate selectivity. The disruption of the beta strand structure was observed in a molecular dynamics (MD) optimized model of the NAT2 Q145P variant (unpublished data).
Functional studies of the Q145P variant in yeast demonstrated reduced or undetectable catalytic activity [23,26] due to reduced immunoreactive protein in the soluble fraction, although the protein stability was not affected [23]. This reduction in protein is probably due to enhanced protein degradation, triggered by structural changes similar to those predicted in our MD optimized model of the NAT2 Q145P variant.
E167K (G499A)
E167 is part of the unstructured “loop” that is problematic when generating homology models of the mammalian N-acetyltransferases using bacterial N-acetyltransferase templates [77]. The side-chain of residue E167 is 71.4% solvent exposed in the domain II loop (165–185), and weakly hydrogen bonds to K185 (acceptor to hydrogen atoms E167:OE2-K185:HZ1 2.76 Å, acceptor to donor atoms E167:OE2-K185:NZ 3.55 Å), which is in an adjacent strand of the domain II loop (Fig. 9F). This loop plays a role in stabilizing the mammalian N-acetyltransferase proteins [77]. Removing this interaction may affect the dynamics or conformation of the loop, which largely lacks defined secondary structure and is therefore more susceptible to dynamic and conformational changes.
Functional studies of the E167K variant in mammalian cells demonstrated a reduction in activity due to reduced protein levels, with no reduction in mRNA levels or protein thermostability [28]. It is possible that the E167K variant has small structural changes that cause protein aggregation and/or increased degradation.
R197Q (G590A)
The R197 side-chain is 19.9% solvent exposed near the surface of the protein in domain II near the inter-domain helix (Fig. 9G). Residue R197 has no hydrogen bonding interactions with other residues, although the side chain of E195 partially blocks solvent access and is close enough (R197:NH1-E195:OE2 3.30 Å, and R197:NH2-E195:OE1 3.57 Å), though not at an optimal angle for hydrogen bonding. Close interactions were also observed between R197 and M105. Electrostatic interactions are likely to exist between the positively charged R197 side-chain and the lone pairs of electrons on the M105 side-chain sulfur. Additionally, the negative side chain of residue E195 could form electrostatic interactions with the positive R197 side-chain. This set of electrostatic interactions results in what appears to be an electrostatic interaction “sandwich”, with positively charged R197 between negatively charged E195 and the methionine sulfur’s electron pairs. Replacing the positively charged arginine with a neutral glutamine residue results in loss of these electrostatic interactions. Although hydrogen bonding between R197 and E195 would still be possible, it would not be expected to contribute to stability of the tertiary structure like the interaction with M105, since E195 and R197 are on the same chain. It is worthwhile to note that the same interactions are observed in NAT1 (PDB ID# 2PQT) between residues M105, E195, and R197. Residue 195 is a positively charged lysine, though only the carbon chain of the lysine residue side-chain interacts with the arginine side-chain, suggesting that steric forces or van der Waals forces are also involved in the close interactions of E195 with R197.
R197Q is a relatively frequent SNP in most populations [58]. Functional studies of the R197Q variant in bacteria, yeast, and mammalian cells demonstrated reduced NAT2 activity and protein levels due to reduced protein thermostability [20,21,23,28] Interestingly, all of these studies have demonstrated only slightly reduced thermostability for the R197Q variant. These results are consistent with loss of the relatively weak electrostatic interactions of R197 with E195 and M105.
K268R (A803G)
The side-chain of residue 268 is 75.6% solvent exposed on the surface of the protein in the domain III alpha helix and has no interactions with surrounding residues (Fig. 9H) or symmetry related crystal neighbors (unpublished data). Replacing K268 with arginine is not expected to affect the alpha helical structure. Functional studies of the K268R variant recombinantly expressed in bacterial [20,21], yeast [23,26] and mammalian cells [28] demonstrated no changes in protein expression, protein stability, or catalytic activity. Evaluation of the NAT2 structure suggest that the conserved activity in the K268R variant is due to the surface location and lack of contacts with surrounding residues, and not because K268R is a conservative residue change.
K282T (A845C)
The side-chain of residue K282 is 37.1% solvent exposed near the C-terminal end in domain III. The K282 side-chain shares hydrogen bonding interactions with the side-chain of H43 (K282:NZ-H43:ND1 2.86 Å) in domain I, and the backbone of residue V280 (K282:NZ-V280:O 2.90 Å) in domain III (Fig. 9I). Since the C-terminal tail from residue I270 to I290 lacks secondary structure, the interaction with domain I residue H43 likely contributes to the stability of the C-terminal tail conformation. Replacing K282 with a threonine residue may result in loss of these interactions and structural destabilization. It is also possible that introduction of a more rigid threonine backbone could influence the conformation of the C-terminal coil. Conformational freedom of the K282 residue in this crystal structure may be limited by steric clashes due to its location in the protein dimer interface (K282[A]:CE-I218[B]:CG2 4.31 Å).
Functional studies of the K282T variant in yeast demonstrated reduced thermostability at 50°C, but no change in the catalytic activity of the enzyme or the expression of the enzyme [23]. The apparent paradox of reduced stability without reduced protein suggests that the stability of the protein is only slightly affected by this residue change, and therefore reduced thermostability is readily observed at 50°C but not at 37°C. Only slight reductions in activity were observed at 37°C in recombinant yeast systems for N-acetylation of 2-aminofluorene, and O-acetylation of two different N-hydroxylated heterocyclic amine substrates [23,26]. Loss of hydrogen bonding interactions when threonine replaces lysine at residue 282 could contribute to this slight reduction in NAT2 stability. The structural impact of losing these H-bonding interactions may be greater in this case due to the lack of secondary structure from I270 to I290.
G286E (G857A)
Residue G286 is 6.8% solvent exposed on the C-terminal tail in domain III, directly adjacent to the active site (Fig. 9J). The backbone of residue G286 does not directly interact with any other residue. Replacing glycine with a much larger glutamate residue at 286 could significantly alter the conformation of the C-terminal tail adjacent to the active site opening due to steric clashes with nearby residues and loss of the highly flexible glycine residue. Since the C-terminal tail residues play a significant role in defining the size and shape of the active site opening, the G286E variant protein is likely to have altered active site access and altered substrate selectivity. Such a significant change to a C-terminal residue adjacent to the active site is also likely to affect AcCoA binding and C68 acetylation. The NAT2 crystal structure (PDB ID# 2PFR) has CoA bound in its active site, with a hydrogen bonding interaction between CoA and residue S287. The C-terminal tail conformational change that may accompany the G286E variant would likely influence this interaction between S287 and CoA.
Consideration of the NAT1 crystal structure (PDB ID# 2PQT) at residue 286 adds to our understanding of the potential effects of the G286E substitution in NAT2. NAT1 has the larger arginine residue, which appears to influence the conformation of the C-terminal tail, since the conformational twist in the NAT2 C-terminal tail coil at residue G286 is missing from the NAT1 C-terminal tail at R286. When considering the influence of NAT2 residue 286 on CoA interactions with residue 287, it is unlikely that the NAT2 S287 residue is required for AcCoA binding; this interaction is not conserved in NAT1, which has a phenylalanine at 287. NAT1/NAT2 differences in substrate selectivities and/or catalytic activities are likely influenced by the difference in bulkiness of the smaller NAT2 G286 residue compared to the larger NAT1 R286 residue sidechain, which is oriented toward the active site opening. In addition to the well-documented differences in substrate selectivity, NAT1 is catalytically more efficient than NAT2 [58]. Differences at residues like 286 that alter enzyme acetylation and/or active site size/shape may play important roles in these differences. Thus, the addition of a bulky glutamate side-chain at residue 286 in the NAT2 G286E variant might be expected to alter substrate selectivity and/or catalytic activity.
G286E is a common SNP in Asian populations [58]. Functional studies of the G286E variant in mammalian cells demonstrated reduced activity for some substrates but not for others, and reduced protein due to reduced thermostability [28]. The same study reported increased apparent Km for AcCoA, a finding that was not observed in a recombinant bacterial system [20] using a different aromatic amine substrate. It is possible that reduced protein acetylation contributes to the overall reduction of protein levels through proteasomal degradation [88]. Our observation that the G286E residue change could alter the size and shape of the active site pocket is consistent with the substrate-dependent activity changes illustrated in figures 4 and 5. In addition, the finding that the G286E residue change alters the Km for AcCoA is consistent with a conformational change in the C-terminal tail which may alter interactions with AcCoA.
CONCLUSIONS
The relationship between NAT2 genotype and acetylator phenotype has been investigated through recombinant expression of NAT2 alleles/haplotypes and individual SNPs using both mammalian and bacterial cell systems. These studies confirm that different combinations of these SNPs in the NAT2 coding region result in proteins with altered stability, degradation, and/or kinetic characteristics. These effects of SNPs on NAT2 proteins are the basis for slow, intermediate, and rapid acetylator phenotypes. With these associations between NAT2 genotype and acetylator phenotype identified, studies have documented the role that human NAT2 acetylation polymorphisms play in modifying individual cancer risk and drug efficacy and/or toxicity.
In addition, molecular homology modeling techniques have enhanced understanding of NAT2 protein structure-function relationships. The recent release of a human NAT2 crystal structure has made it possible to evaluate NAT2 structure-function relationships without the limitations inherent in molecular homology modeling. We presented a structural evaluation of the residues affected by individual human NAT2 SNPs, and in each case attempted to correlate NAT2 structural changes induced by NAT2 SNPs with their functional effects. The structural features of amino acid substitutions R64Q, R64W, I114T, D122N, L137F, Q145P, E167K, R197Q, K268R, K282T, and G286E are summarized in Table 2 providing insight into how slight changes in the NAT2 protein sequence can have substantial effects on NAT2 structure and function.
Table 2.
Summary of Non-Synonymous SNP-Induced Changes in NAT2 Function & Structure
| SNP/Amino Acid Change (rs identifier) | Domain | Secondary Structure | Side-Chain Location | Type of Change(s) | Functional Effect(s) | Structural Effect(s) |
|---|---|---|---|---|---|---|
| G191A
R64Q (rs1801279) |
I | Coil | Core | ↑Hydrophobicity
↓H-bond Capacity (+) Charge Lost ↓Size |
↓Stability | Loss of Electrostatic Interactions |
| C190T
R64W (rs1805158) |
I | Coil | Core | ↓Flexibility
↓H-bond Capacity ↑Hydrophobicity ↑Aromaticity |
↓Stability | Loss of Electrostatic Interactions |
| T341C
I114T (rs1801280) |
II | β-Barrel | Hydrophobic Core | ↑H-bond Capacity
↓Hydrophobicity ↑Polarity ↓Size |
↑Protein Degradation | Conformational Change |
| G364A
D122N (rs4986996) |
II | Active Site Loop | Core | Conservative | ↓Protein | Catalytic Triad Disruption |
| A411T
L137F (rs4986997) |
II | β-Barrel | 6.1% Surface Exposed | ↑Aromaticity
↑Size |
↓Protein | Conformational Change |
| A434C
Q145P |
II | β-Barrel | 11.1% Surface Exposed | ↓Flexibility
H-bond Capacity Lost Polarity Lost |
↓Protein | Conformational Change |
| G499A
E167K |
II | Coil | Domain II Loop | ↓H-bond Capacity
↓Hydrophobicity Different Charge ↑Size |
↓Protein | Conformational Change |
| G590A
R197Q (rs1799930) |
II | Coil | 16.6% Surface Exposed | ↑Hydrophobicity
↓H-bond Capacity (+) Charge Lost ↓Size |
↓Stability | Loss of Electrostatic Interactions |
| A803G
K268R (rs1208) |
III | α-Helix | Surface | Conservative | None Detected | None Expected |
| A845C
K282T |
III | Coil | C-Terminal Tail | ↓Flexibility
↑ Hydrophobicity (+) Charge Lost ↓Size |
Slight ↓Stability | Loss of Electrostatic Interactions |
| G857A
G286E (rs1799931) |
III | Coil | C-Terminal Tail | ↑H-bond Capacity
(−) Charge ↓Hydrophobicity ↑Size |
↓Protein | Altered Active Site Size/Shape |
Acknowledgments
This work represented partial fulfillment for the Ph.D. degrees in Pharmacology and Toxicology to Yu Zang and Jason Walraven at the University of Louisville School of Medicine. The studies were partially supported by National Institutes of Health grants R01-CA034627 (DWH), T32-ES011564 (DWH to support JMW), and P30-ES014443, and a dissertation research award (DISS0403147) from the Susan G. Komen Breast Cancer Foundation (DWH to support YZ). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Weber WW, Hein DW. Clin Pharmacokinet. 1979;4(6):401–422. doi: 10.2165/00003088-197904060-00001. [DOI] [PubMed] [Google Scholar]
- 2.Weber WW, Hein DW. Pharmacol Rev. 1985;37(1):25–79. [PubMed] [Google Scholar]
- 3.Hein DW, Doll MA, Fretland AJ, Leff MA, Webb SJ, Xiao GH, Devanaboyina US, Nangju NA, Feng Y. Cancer Epidemiol Biomarkers Prev. 2000;9(1):29–42. [PubMed] [Google Scholar]
- 4.Hein DW. Biochim Biophys Acta. 1988;948(1):37–66. doi: 10.1016/0304-419x(88)90004-2. [DOI] [PubMed] [Google Scholar]
- 5.Hein DW, Rustan TD, Doll MA, Bucher KD, Ferguson RJ, Feng Y, Furman EJ, Gray K. Toxicol Lett. 1992;64-65:123–130. doi: 10.1016/0378-4274(92)90181-i. [DOI] [PubMed] [Google Scholar]
- 6.Hanna PE. Adv Pharmacol. 1994;27:401–430. doi: 10.1016/s1054-3589(08)61041-8. [DOI] [PubMed] [Google Scholar]
- 7.Sinclair JC, Sandy J, Delgoda R, Sim E, Noble ME. Nat Struct Biol. 2000;7(7):560–564. doi: 10.1038/76783. [DOI] [PubMed] [Google Scholar]
- 8.Wang HQ, Vath GM, Gleason KJ, Hanna PE, Wagner CR. Biochemistry. 2004;43(25):8234–8246. doi: 10.1021/bi0497244. [DOI] [PubMed] [Google Scholar]
- 9.Hein DW, Doll MA, Rustan TD, Gray K, Feng Y, Ferguson RJ, Grant DM. Carcinogenesis. 1993;14(8):1633–1638. doi: 10.1093/carcin/14.8.1633. [DOI] [PubMed] [Google Scholar]
- 10.Kawamura A, Graham J, Mushtaq A, Tsiftsoglou SA, Vath GM, Hanna PE, Wagner CR, Sim E. Biochem Pharmacol. 2005;69(2):347–359. doi: 10.1016/j.bcp.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 11.Goodfellow GH, Dupret JM, Grant DM. Biochem J. 2000;348(Pt 1):159–166. [PMC free article] [PubMed] [Google Scholar]
- 12.Deguchi T. J Biol Chem. 1992;267(25):18140–18147. [PubMed] [Google Scholar]
- 13.Blum M, Demierre A, Grant DM, Heim M, Meyer UA. Proc Natl Acad Sci USA. 1991;88(12):5237–5241. doi: 10.1073/pnas.88.12.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vatsis KP, Martell KJ, Weber WW. Proc Natl Acad Sci USA. 1991;88(14):6333–6337. doi: 10.1073/pnas.88.14.6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blum M, Grant DM, McBride W, Heim M, Meyer UA. DNA Cell Biol. 1990;9:193–203. doi: 10.1089/dna.1990.9.193. [DOI] [PubMed] [Google Scholar]
- 16.Ebisawa T, Deguchi T. Biochem Biophys Res Commun. 1991;177(3):1252–1257. doi: 10.1016/0006-291x(91)90676-x. [DOI] [PubMed] [Google Scholar]
- 17.Ohsako S, Deguchi T. J Biol Chem. 1990;265(8):4630–4634. [PubMed] [Google Scholar]
- 18.Grant DM, Blum M, Demierre A, Meyer UA. Nucleic Acids Res. 1989;17(10):3978. doi: 10.1093/nar/17.10.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hickman D, Sim E. Biochem Pharmacol. 1991;42(5):1007–1014. doi: 10.1016/0006-2952(91)90282-a. [DOI] [PubMed] [Google Scholar]
- 20.Hein DW, Ferguson RJ, Doll MA, Rustan TD, Gray K. Hum Mol Genet. 1994;3(5):729–734. doi: 10.1093/hmg/3.5.729. [DOI] [PubMed] [Google Scholar]
- 21.Hein DW, Doll MA, Rustan TD, Ferguson RJ. Cancer Res. 1995;55(16):3531–3536. [PubMed] [Google Scholar]
- 22.Hickman D, Palamanda JR, Unadkat JD, Sim E. Biochem Pharmacol. 1995;50(5):697–703. doi: 10.1016/0006-2952(95)00182-y. [DOI] [PubMed] [Google Scholar]
- 23.Fretland AJ, Leff MA, Doll MA, Hein DW. Pharmacogenetics. 2001;11(3):207–215. doi: 10.1097/00008571-200104000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Svensson CK, Hein DW. In: Drug Metabolism and Transport: Molecular Methods and Mechanisms. Lash LH, editor. Humana Press; Totowa, NJ: 2005. pp. 173–195. [Google Scholar]
- 25.Hein DW. Oncogene. 2006;25(11):1649–1658. doi: 10.1038/sj.onc.1209374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hein DW, Fretland AJ, Doll MA. Int J Cancer. 2006;119(5):1208–1211. doi: 10.1002/ijc.21957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zang Y, Zhao S, Doll MA, States JC, Hein DW. Functional characterization of the A411T (L137F) and G364A (D122N) genetic polymorphisms in human N-acetyltransferase 2. Pharmacogenet Genomics. 2007;17(1):37–45. doi: 10.1097/01.fpc.0000236325.73186.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zang Y, Doll MA, Zhao S, States JC, Hein DW. Carcinogenesis. 2007;28(8):1665–1671. doi: 10.1093/carcin/bgm085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hickman D, Risch A, Camilleri JP, Sim E. Pharmacogenetics. 1992;2(5):217–226. doi: 10.1097/00008571-199210000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Lin HJ, Han CY, Lin BK, Hardy S. Am J Hum Genet. 1993;52(4):827–834. [PMC free article] [PubMed] [Google Scholar]
- 31.Ferguson RJ, Doll MA, Rustan TD, Gray K, Hein DW. Drug Metab Dispos. 1994;22(3):371–376. [PubMed] [Google Scholar]
- 32.Abe M, Deguchi T, Suzuki T. Biochem Biophys Res Commun. 1993;191(3):811–816. doi: 10.1006/bbrc.1993.1289. [DOI] [PubMed] [Google Scholar]
- 33.Cascorbi I, Drakoulis N, Brockmoller J, Maurer A, Sperling K, Roots I. Am J Hum Genet. 1995;57(3):581–592. [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez C, Agundez JA, Olivera M, Martin R, Ladero JM, Benitez J. Pharmacogenetics. 1995;5(4):207–214. [PubMed] [Google Scholar]
- 35.Agundez JA, Olivera M, Ladero JM, Rodriguez-Lescure A, Ledesma MC, Diaz-Rubio M, Meyer UA, Benitez J. Pharmacogenetics. 1996;6(6):501–512. doi: 10.1097/00008571-199612000-00003. [DOI] [PubMed] [Google Scholar]
- 36.Agundez JA, Olivera M, Martinez C, Ladero JM, Benitez J. Pharmacogenetics. 1996;6(5):423–428. [PubMed] [Google Scholar]
- 37.Leff MA, Fretland AJ, Doll MA, Hein DW. ) J Biol Chem. 1999;274(49):34519–34522. doi: 10.1074/jbc.274.49.34519. [DOI] [PubMed] [Google Scholar]
- 38.Woolhouse NM, Qureshi MM, Bastaki SM, Patel M, Abdulrazzaq Y, Bayoumi RA. Pharmacogenetics. 1997;7(1):73–82. doi: 10.1097/00008571-199702000-00010. [DOI] [PubMed] [Google Scholar]
- 39.Anitha A, Banerjee M. Int J Mol Med. 2003;11(1):125–131. [PubMed] [Google Scholar]
- 40.Tanira MOM, Simsek M, Al Balushi K, Al Lawatia K, Al Barawani H, Bayoumi RA. SQU J Sci Res Med Sci. 2003;5:125–131. [PMC free article] [PubMed] [Google Scholar]
- 41.Patin E, Barreiro LB, Sabeti PC, Austerlitz F, Luca F, Sajantila A, Behar DM, Semino O, Sakuntabhai A, Guiso N, Gicquel B, McElreavey K, Harding RM, Heyer E, Quintana-Murci L. Am J Hum Genet. 2006;78(3):423–436. doi: 10.1086/500614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patin E, Harmant C, Kidd KK, Kidd J, Froment A, Mehdi SQ, Sica L, Heyer E, Quintana-Murci L. Hum Mutat. 2006;27(7):720. doi: 10.1002/humu.9438. [DOI] [PubMed] [Google Scholar]
- 43.Sabbagh A, Langaney A, Darlu P, Gerard N, Krishnamoorthy R, Poloni ES. BMC Genet. 2008;9(1):21. doi: 10.1186/1471-2156-9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Agundez J, Golka K, Martinez C, Selinski S, Blaszkewicz M, Garcia-Martin E. Clin Chem. 2008 doi: 10.1373/clinchem.2008.105569. in press. [DOI] [PubMed] [Google Scholar]
- 45.Dandara C, Masimirembwa CM, Magimba A, Kaaya S, Sayi J, Sommers de K, Snyman JR, Hasler JA. Pharmacogenetics. 2003;13(1):55–58. doi: 10.1097/00008571-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 46.Sekine A, Saito S, Iida A, Mitsunobu Y, Higuchi S, Harigae S, Nakamura Y. J Hum Genet. 2001;46(6):314–319. doi: 10.1007/s100380170065. [DOI] [PubMed] [Google Scholar]
- 47.Cascorbi I, Brockmoller J, Bauer S, Reum T, Roots I. Pharmacogenetics. 1996;6(3):257–259. doi: 10.1097/00008571-199606000-00009. [DOI] [PubMed] [Google Scholar]
- 48.Bolt HM, Selinski S, Dannappel D, Blaszkewicz M, Golka K. Arch Toxicol. 2005;79(4):196–200. doi: 10.1007/s00204-004-0622-8. [DOI] [PubMed] [Google Scholar]
- 49.Agundez JA, Martinez C, Olivera M, Ledesma MC, Ladero JM, Benitez J. Clin Pharmacol Ther. 1994;56(2):202–209. doi: 10.1038/clpt.1994.124. [DOI] [PubMed] [Google Scholar]
- 50.Bell DA, Taylor JA, Butler MA, Stephens EA, Wiest J, Brubaker LH, Kadlubar FF, Lucier GW. Carcinogenesis. 1993;14(8):1689–1692. doi: 10.1093/carcin/14.8.1689. [DOI] [PubMed] [Google Scholar]
- 51.Delomenie C, Sica L, Grant DM, Krishnamoorthy R, Dupret JM. Pharmacogenetics. 1996;6(2):177–185. doi: 10.1097/00008571-199604000-00004. [DOI] [PubMed] [Google Scholar]
- 52.Lin HJ, Han CY, Lin BK, Hardy S. Pharmacogenetics. 1994;4(3):125–134. doi: 10.1097/00008571-199406000-00003. [DOI] [PubMed] [Google Scholar]
- 53.Shishikura K, Hohjoh H, Tokunaga K. Hum Mutat. 2000;15(6):581. doi: 10.1002/1098-1004(200006)15:6<581::AID-HUMU17>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 54.Lee SY, Lee KA, Ki CS, Kwon OJ, Kim HJ, Chung MP, Suh GY, Kim JW. Clin Chem. 2002;48(5):775–777. [PubMed] [Google Scholar]
- 55.Zhu Y, Doll MA, Hein DW. Biol Chem. 2002;383(6):983–987. doi: 10.1515/BC.2002.105. [DOI] [PubMed] [Google Scholar]
- 56.Hein DW, Boukouvala S, Grant DM, Minchin RF, Sim E. Pharmacogenet Genomics. 2008;18(4):367–368. doi: 10.1097/FPC.0b013e3282f60db0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yuliwulandari R, Sachrowardi Q, Nishida N, Takasu M, Batubara L, Susmiarsih TP, Rochani JT, Wikaningrum R, Miyashita R, Miyagawa T, Sofro AS, Tokunaga KJ. J Hum Genet. 2008;53(3):201–209. doi: 10.1007/s10038-007-0237-z. [DOI] [PubMed] [Google Scholar]
- 58.Grant DM, Hughes NC, Janezic SA, Goodfellow GH, Chen HJ, Gaedigk A, Yu VL, Grewal R. Mutat Res. 1997;376(1–2):61–70. doi: 10.1016/s0027-5107(97)00026-2. [DOI] [PubMed] [Google Scholar]
- 59.Butcher NJ, Boukouvala S, Sim E, Minchin RF. Pharmacogenomics J. 2002;2(1):30–42. doi: 10.1038/sj.tpj.6500053. [DOI] [PubMed] [Google Scholar]
- 60.Hein DW. Mutat Res. 2002;506-507:65–77. doi: 10.1016/s0027-5107(02)00153-7. [DOI] [PubMed] [Google Scholar]
- 61.Boukouvala S, Fakis G. Drug Metab Rev. 2005;37(3):511–564. doi: 10.1080/03602530500251204. [DOI] [PubMed] [Google Scholar]
- 62.Sim E, Westwood I, Fullam E. Expert Opin Drug Metab Toxicol. 2007;3(2):169–184. doi: 10.1517/17425255.3.2.169. [DOI] [PubMed] [Google Scholar]
- 63.Garcia-Closas M, Malats N, Silverman D, Dosemeci M, Kogevinas M, Hein DW, Tardon A, Serra C, Carrato A, Garcia-Closas R, Lloreta J, Castano-Vinyals G, Yeager M, Welch R, Chanock S, Chatterjee N, Wacholder S, Samanic C, Tora M, Fernandez F, Real FX, Rothman N. Lancet. 2005;366(9486):649–659. doi: 10.1016/S0140-6736(05)67137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deitz AC, Rothman N, Rebbeck TR, Hayes RB, Chow WH, Zheng W, Hein DW, Garcia-Closas M. Cancer Epidemiol Biomarkers Prev. 2004;13(9):1543–1546. [PubMed] [Google Scholar]
- 65.Vatsis KP, Weber WW, Bell DA, Dupret JM, Evans DA, Grant DM, Hein DW, Lin HJ, Meyer UA, Relling MV, Sim E, Suzuki T, Yamazoe Y. Nomenclature for N-acetyltransferases. Pharmacogenetics. 1995;5(1):1–17. doi: 10.1097/00008571-199502000-00001. [DOI] [PubMed] [Google Scholar]
- 66.Grant DM, Morike K, Eichelbaum M, Meyer UA. J Clin Invest. 1990;85(3):968–972. doi: 10.1172/JCI114527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zang Y, Zhao S, Doll MA, States JC, Hein DW. Pharmacogenetics. 2004;14(11):717–723. doi: 10.1097/00008571-200411000-00002. [DOI] [PubMed] [Google Scholar]
- 68.Gross M, Kruisselbrink T, Anderson K, Lang N, McGovern P, Delongchamp R, Kadlubar F. Cancer Epidemiol Biomarkers Prev. 1999;8(8):683–692. [PubMed] [Google Scholar]
- 69.Cascorbi I, Roots I. Pharmacogenetics. 1999;9(1):123–127. doi: 10.1097/00008571-199902000-00016. [DOI] [PubMed] [Google Scholar]
- 70.Parkin DP, Vandenplas S, Botha FJ, Vandenplas ML, Seifart HI, van Helden PD, van der Walt BJ, Donald PR, van Jaarsveld PP. Am J Respir Crit Care Med. 1997;155(5):1717–1722. doi: 10.1164/ajrccm.155.5.9154882. [DOI] [PubMed] [Google Scholar]
- 71.Cribb AE, Isbrucker R, Levatte T, Tsui B, Gillespie CT, Renton KW. Pharmacogenetics. 1994;4(3):166–170. [PubMed] [Google Scholar]
- 72.Lorenzo B, Reidenberg MM. Br J Clin Pharmacol. 1989;28(2):207–208. doi: 10.1111/j.1365-2125.1989.tb05420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Neil WM, Drobitch RK, MacArthur RD, Farrough MJ, Doll MA, Fretland AJ, Hein DW, Crane LR, Svensson CK. Pharmacogenetics. 2000;10(2):171–182. doi: 10.1097/00008571-200003000-00009. [DOI] [PubMed] [Google Scholar]
- 74.Rodrigues-Lima F, Delomenie C, Goodfellow GH, Grant DM, Dupret JM. Biochem J. 2001;356(Pt 2):327–334. doi: 10.1042/0264-6021:3560327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rodrigues-Lima F, Dupret JM. Biochem Biophys Res Commun. 2002;291(1):116–123. doi: 10.1006/bbrc.2002.6414. [DOI] [PubMed] [Google Scholar]
- 76.Lau EY, Felton JS, Lightstone FC. Chem Res Toxicol. 2006;19(9):1182–1190. doi: 10.1021/tx0600999. [DOI] [PubMed] [Google Scholar]
- 77.Walraven JM, Trent JO, Hein DW. Drug Metab Dispos. 2007;35(6):1001–1007. doi: 10.1124/dmd.107.015040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sandy J, Mushtaq A, Kawamura A, Sinclair J, Sim E, Noble M. J Mol Biol. 2002;318(4):1071–1083. doi: 10.1016/S0022-2836(02)00141-9. [DOI] [PubMed] [Google Scholar]
- 79.Holton SJ, Dairou J, Sandy J, Rodrigues-Lima F, Dupret JM, Noble ME, Sim E. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61(Pt 1):14–16. doi: 10.1107/S1744309104030659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Westwood IM, Holton SJ, Rodrigues-Lima F, Dupret JM, Bhakta S, Noble ME, Sim E. Biochem J. 2005;385(Pt 2):605–612. doi: 10.1042/BJ20041330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Westwood IM, Kawamura A, Fullam E, Russell AJ, Davies SG, Sim E. Curr Top Med Chem. 2006;6(15):1641–1654. doi: 10.2174/156802606778108979. [DOI] [PubMed] [Google Scholar]
- 82.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 83.Payton M, Mushtaq A, Yu TW, Wu LJ, Sinclair J, Sim E. Microbiology. 2001;147(Pt 5):1137–1147. doi: 10.1099/00221287-147-5-1137. [DOI] [PubMed] [Google Scholar]
- 84.Hubbard SJ, Thornton JM. ‘NACCESS’, Computer Program, Department of Biochemistry and Molecular Biology. University College London; 1993. [Google Scholar]
- 85.McDonald IK, Thornton JM. J Mol Biol. 1994;238(5):777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 86.Wu H, Dombrovsky L, Tempel W, Martin F, Loppnau P, Goodfellow GH, Grant DM, Plotnikov AN. J Biol Chem. 2007;282(41):30189–30197. doi: 10.1074/jbc.M704138200. [DOI] [PubMed] [Google Scholar]
- 87.Sandy J, Mushtaq A, Holton SJ, Schartau P, Noble ME, Sim E. Biochem J. 2005;390(Pt 1):115–123. doi: 10.1042/BJ20050277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Butcher NJ, Arulpragasam A, Minchin RF. J Biol Chem. 2004;279(21):22131–22137. doi: 10.1074/jbc.M312858200. [DOI] [PubMed] [Google Scholar]
- 89.Li SC, Goto NK, Williams KA, Deber CM. Proc Natl Acad Sci USA. 1996;93(13):6676–6681. doi: 10.1073/pnas.93.13.6676. [DOI] [PMC free article] [PubMed] [Google Scholar]