

Abstract
Heme oxygenase regiospecifically oxidizes heme at the α-meso position to give biliverdin IXα, CO, and iron. The heme orientation within the active site, which is thought to determine the oxidation regiospecificity, is shown here for the human enzyme (hHO1) to be largely determined by interactions between the heme carboxylic acid groups and residues Arg183 and Lys18 but not Tyr134. Mutation of either Arg183 or Lys18 individually does not significantly alter the NADPH-cytochrome P450 reductase-dependent reaction regiochemistry, but partially shifts the oxidation to the β/δ-meso positions in the reaction supported by ascorbic acid. Mutation of Glu29 to a lysine, which places a positive charge where it can interact with a heme carboxyl if the heme rotates by ~90°, causes a slight loss of regiospecificity, but combined with the R183E and K18E mutations results primarily in β/δ-meso oxidation of the heme under all conditions. NMR analysis of heme binding to the triple K18E/E29K/R183E mutant confirms rotation of the heme in the active site. Kinetic studies demonstrate that mutations of Arg183 greatly impair the rate of the P450 reductase-dependent reaction, in accord with the earlier finding that Arg183 is involved in binding of the reductase to hHO1, but have little effect on the ascorbate reaction. Mutations of Asp140 and Tyr58 that disrupt the active site hydrogen bonding network, impair catalytic rates but do not influence the oxidation regiochemistry. The results indicate both that the oxidation regiochemistry is largely controlled by ionic interactions of the heme propionic acid groups with the protein and that shifts in regiospecificity involve rotation of the heme about an axis perpendicular to the heme plane.
Heme oxygenase is a microsomal enzyme that catalyzes the oxidation of heme1 to biliverdin IXα, carbon monoxide and free iron. Biliverdin is subsequently reduced to bilirubin IXα, glucuronidated, and excreted in the bile (1, 2). Heme oxygenases modulate diverse biological functions through the action of one or more of their heme degradation products, including angiogenesis (3, 4), anti-inflammation (5, 6) and anti-apoptosis (7–9). Both biliverdin and bilirubin are potential antioxidants (10), while carbon monoxide is a gaseous signaling molecule akin to nitric oxide (11–13). The free iron is reutilized and is the major source of this metal in the body (14), so that heme oxygenase is important for maintaining iron homeostasis.
Three isoforms of heme oxygenase, HO1, HO2, and HO3, have been reported in humans, although the catalytic activity of HO3 is very low and its physiological role, if any, remains unclear (15, 16). The two major isoforms differ not only in primary sequence, but also in organ distribution. Thus, HO1 is primarily localized within the liver and spleen, and can be induced by a wide variety of stimuli, including UV, heavy metals, and hyperoxia (17, 18). One common characteristic of most of these stimuli is their ability to cause oxidative stress. Characterization of human HO1 has been greatly facilitated by expression in Escherichia coli of a water soluble but fully active truncated form (amino acids 1–265, denoted as hHO1) that lacks the 23 C-terminal amino acids (19). HO2 exists predominantly in the brain and testis and may be involved in CO signaling pathways (11, 20). It has also recently been reported to function as an oxygen sensor for a calcium-sensitive potassium channel in carotid cells (21).
The oxidation of heme by heme oxygenases is a multi-step reaction (Fig. 1). First, heme is hydroxylated to α-meso hydroxyheme employing O2 and electrons provided by NADPH-cytochrome P450 reductase. In the second step, hydroxyheme is oxidized to verdoheme in a reaction that only requires O2 and that results in release of the α-meso carbon as a molecule of CO. Finally, verdoheme is converted to ferric biliverdin in a reaction that requires NADPH-cytochrome P450 reductase and O2. The iron is finally reduced to the ferrous state and dissociates from the enzyme, followed by release of biliverdin itself (22).
FIGURE 1.

Oxidation of heme by heme oxygenases. The α, β, γ, and δ-meso positions are labeled.
The analogous oxidation of heme can be observed in model systems in which heme is incubated under aerobic conditions with ascorbic acid, a process that is termed coupled oxidation (23). Under these conditions, the heme is oxidized at approximately the same rate at all of its four meso-positions, resulting in the formation of an approximately equimolar mixture of the four biliverdin isomers (IXα, IXβ, IXγ and IXδ). The oxidation of heme by heme oxygenase can also be supported by ascorbic acid in the absence of cytochrome P450 reductase, but in this system the oxidation only occurs at the α-meso carbon, just as it does in the normal reaction supported by cytochrome P450 reductase (Fig. 1). The product in this instance is exclusively biliverdin IXα.
In the crystal structure of the complex of hHO1 with heme (24, 25), the heme is sandwiched between two helices, one of which is termed the “proximal” helix and the other the “distal” helix. The proximal helix contains the axial iron ligand, His25, whereas the distal ligand is a water molecule. The binding site for O2 as well as the site of catalysis is the pocket above the heme defined by the distal helix. Analysis of the currently available structures of complexes of various ligands with heme-hHO1 suggest that steric interactions between the distal helix and heme-bound dioxygen promote oxidation at the α-meso carbon. In addition, the structure of a dioxygen-bound heme oxygenase from Corynebacterium diphtheriae demonstrates that steric constraints direct the dioxygen ligand towards the α-meso carbon (26). Nevertheless, it is remarkable that a substituent on the α-meso carbon can alter the reaction regiospecificity (27, 28). Although α-meso-methylheme is still exclusively cleaved at the α-meso carbon despite the presence of the substituent on the same carbon, α-meso formylheme is oxidized only at the non-substituted meso-positions. The fact that methyl and formyl groups differentially direct the reaction regiospecificity suggests that electronic factors may contribute to control of the regiospecificity.
Direct interactions between the protein and the active oxidizing species, Fe(III)·OOH, may also play a role in controlling the regiospecificity. Asp140 on the distal helix of human HO1 forms a hydrogen bond through a water molecule with the distal oxygen of the Fe(III)·OOH. The crystal structure of the Fe(III)-NO hHO1 complex, in which NO mimics oxygen binding, clearly shows that the distal hydrogen-bonding network includes Asp140, Tyr58, Thr135 and Arg136 (29, 30) (Fig. 2B). This network has been proposed to stabilize the Fe(III)·OOH reactive species (29–32).
FIGURE 2.
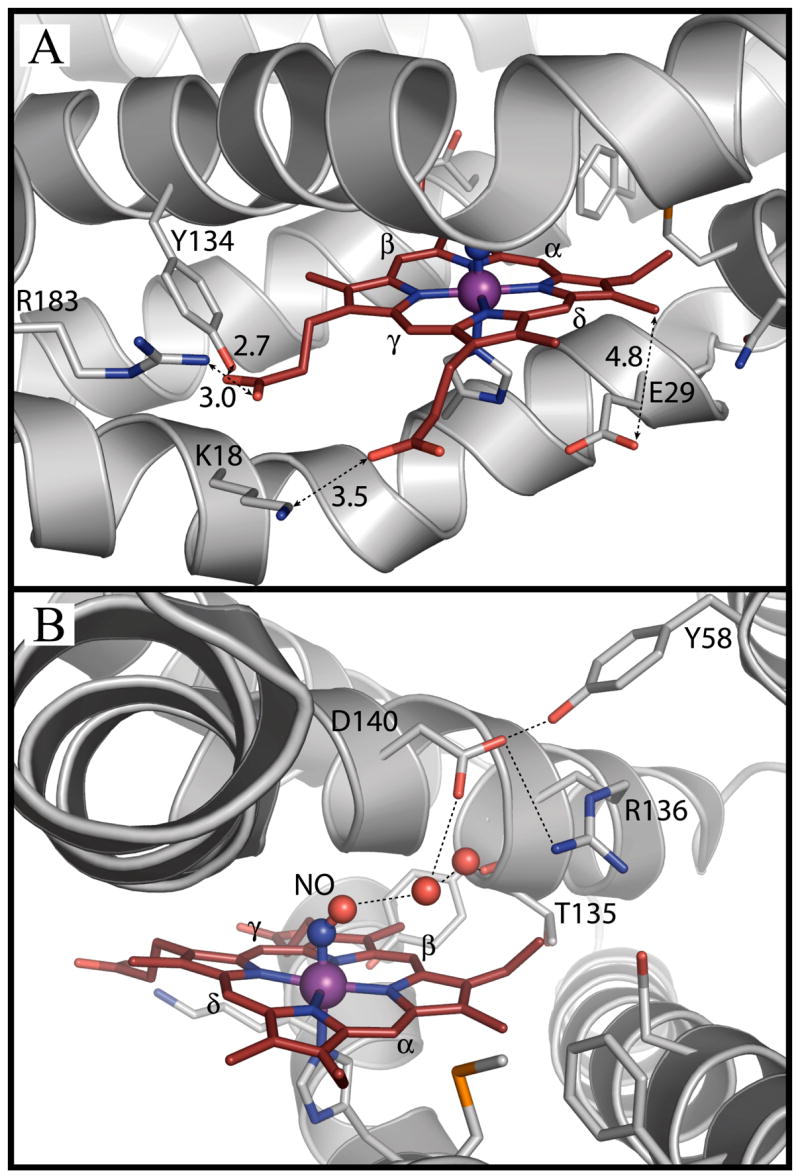
Active site structure of the human heme-hHO-1 complex with bound NO (PDB accession code 1OZW, ref 28), showing; A) Interaction of hHO1 with the heme propionate groups; B) Distal hydrogen bonding network.
A number of bacterial heme oxygenases have been characterized, including HmuO from Corynebacterium diphtheriae (33), HemO from Neisseria meningitides (34), PigA and BphO from Pseudomonas aeruginosa (35, 36), and HemS from Yersinia enterocolitica (37). Like the mammalian heme oxygenases, all of these enzymes, except for PigA, oxidize heme exclusively at the α-meso position. However, PigA, which has 37% amino acid sequence identity with HemO, oxidizes heme to biliverdin IXδ (70%) and IXβ (30%) (35, 38). This novel δ/β-regiospecificity of PigA is due to an unusual seating of the heme in which it is rotated by ~100° counter-clockwise relative to its position in other heme oxygenases. This rotation places the δ-meso carbon in the position normally occupied by the α-meso carbon (39). Furthermore, a 180° flip of the heme about the α,γ-meso axis places the β-meso carbon at the same position. NMR data clearly demonstrate that this 180° flip of the heme can occur even in mammalian HO, but it has no consequences on the regiospecificity of the reaction so long as it occurs at the α-meso carbon (40). Disruption of the interactions between the heme propionate groups and heme oxygenase by mutagenesis can alter the regiospecificity of the PigA reaction to favor the formation of α-biliverdin (41). The interactions of the heme propionate groups with the protein thus appear to play an important role in correct alignment of the heme.
In human HO1, three amino acids (Arg183, Lys18, and Tyr134) interact via hydrogen bonds with the heme propionate groups (Fig. 2A). In previous work, we have demonstrated that mutation of Arg183 to a glutamic acid (42) does not alter the α-regiospecificity of the normal reaction supported by cytochrome P450 reductase. However, in the coupled oxidation reaction supported by ascorbate, up to 20% biliverdin IXδ and a trace of IXβ were produced (42). Here we have mutated Arg183, Lys18 and Tyr134 in efforts to completely remove the interactions between the protein and the propionate groups that anchor the heme. Furthermore, if the heme were to rotate to an orientation that would place the δ- or β-meso carbon at the site originally occupied by the α-meso carbon, one of its propionate groups would be located close to Glu29. We have therefore also mutated Glu29 to a Lys in order to introduce a stabilizing rather than repulsive interaction and thus to favor rotation of the heme (Fig. 2). Finally, to evaluate the contribution of the distal hydrogen bonding network to control of the regiospecificity, we have mutated Asp140 and Tyr58 to disrupt this network. Analysis of the oxidation regiochemistry and catalytic rates of these mutants in the reactions supported by both cytochrome P450 reductase and ascorbate, in conjunction with NMR analysis of the heme seating in a triple mutant with almost completely altered regiochemistry, clarify the determinants of the regiospecificity of human heme oxygenase.
EXPERIMENTAL PROCEDURES
Materials
Ampicillin, sodium ascorbate, isopropyl-β-D-thiogalactopyranoside, adenosine, riboflavin, hemin, 2,5-AMP, deferoxamine mesylate, and NADPH were purchased from Sigma or Aldrich (St. Louis, MO). Sodium dithionite was from Mallinckrodt Baker, Inc (Phillipsburg, NJ) and methanol (HPLC grade) from Fisher Scientific. Quaternary ammonium cellulose (QA52) was purchased from Whatman International Ltd. (England). Hydroxyapatite Bio-Gel® HTP Gel was obtained from Bio-Rad Laboratories (Hercules, CA). The QIAprep® Spin Miniprep Kit was from QIAGEN Sciences (Valencia, CA). The Quikchange® site-directed mutagenesis kit and DH5α competent cells were purchased from Stratagene (La Jolla, CA). The YMC ODS-AQ™ column (S-5, 120 Å, 4.6 ×250 mm) was from Waters Corporation (Milford, MA).
Enzymes
Truncated human heme oxygenase-1 (hHO1) lacking the 23 C-terminal residues was used in these studies. Mutations in this protein were generated with the Quikchange® site-directed mutagenesis kit. The required oligonucleotides were synthesized by Life Technologies, Inc., through the Cell Culture Facility of the University of California, San Francisco. Bacterial transformation was carried out by a standard procedure (43). Transformants were initially screened by ampicillin resistance and the mutations were then confirmed by sequencing. Plasmids were extracted using a QIAprep® spin miniprep kit. Purification of the enzyme and reconstitution with heme were performed by published procedures (44). The heme oxygenase concentration was determined from the extinction coefficient (140 mM−1.cm−1) at the Soret absorption maximum. Human NADPH-cytochrome P450 reductase (45) and rat biliverdin reductase (46) were purified as reported. SOD and catalase were from Sigma-Aldrich (St. Louis, MO).
UV-Visible Spectra of heme-hHO1 in the Ferric, Ferrous and Ferrous-CO States
Sodium dithionite powder was added to a heme-hHO1 solution after purging with argon to generate ferrous hHO1. The CO-ferrous hHO1 complex was generated by passing CO gas through the ferrous hHO1 solution. All spectra were performed on a Hewlett Packard 8452A Diode Array Spectrophotometer in 0.1 M potassium phosphate buffer (pH = 7.4) at room temperature (~25°C). Since all the reactions in this study were carried out in 0.1 M potassium phosphate buffer (pH = 7.4), this buffer is referred to as the standard buffer.
HPLC Analysis of Biliverdin Isomers
A solution of heme-hHO1 (10 nmol), cytochrome P450 reductase (1 nmol), and NADPH (0.8 μmol) in standard buffer was incubated for 3 h at room temperature. The reaction volume was 500 μL. To extract the biliverdin products, one drop of hydrochloric acid (37%) plus a few drops of acetic acid were added before extracting the reaction mixture with 500 μL of CH2Cl2. The organic phase was washed with 300 μL of water and was then evaporated under a stream of air. The resulting residue was dissolved in 500 μL of methanol (5% H2SO4 v/v) and was maintained at 4 °C for at least 16 h in the dark. The dimethyl esterified biliverdins were extracted with 200 μL of CHCl3. The organic phase was washed with water and was then evaporated to dryness under a stream of air. The residue was dissolved in 70% methanol for further analysis.
Coupled oxidations were carried out by incubating heme-hHO1 (10 nmol) with sodium ascorbate (4.5 mg) in standard buffer for 3 h at room temperature. Some reactions were performed in the presence of 150 units of catalase and SOD. The reaction volume was 500 μL. The biliverdin products were extracted as described above.
The biliverdin dimethyl esters were loaded onto a YMC ODS-AQ™ column (S-5, 120 Å, 4.6 × 250 mm). Solvents A and B were water and methanol, respectively. The HPLC running conditions were as follows: flow rate, 1.0 mL/min; 30% B for 5 min, 30–70% B in 0.1 min, 70–95% B in 25 min, 95% B for 7 min, 9530% B in 1 min, and finally 30% B for 20 min. The eluent was monitored at 375 nm and was referenced against the absorption at 598 nm. The standards of the biliverdin dimethyl esters were prepared as published elsewhere (42). The ratio of biliverdin isomers produced was calculated from the areas of the corresponding peaks in the HPLC traces.
Bilirubin Formation in the Presence of Biliverdin Reductase
The activity assays were performed in triplicate at room temperature in standard buffer. Each assay (100 μL) contained 5 μM hHO1, 30 μM heme, 4 μM biliverdin reductase, 2 mM deferoxamine, 500 μM NADPH, and either 1 μM NADPH-cytochrome P450 reductase or 10 mM sodium ascorbate. The initial rate of bilirubin formation was monitored at 468 nm and calculated using an extinction coefficient of 43.5 mM−1 cm−1.
NMR Analysis
The protoheme (PH) complex of hHO1 K18E/E29K/R183E in 1H2O, was exchanged to 2H2O, 57 mM KPi pD=7.23, 13 mM KCN using a Sephadex G-25 column. The resulting solution was concentrated to 270 μM using an Amicon ultrafiltration cell, and placed in a Shigemi tube before the NMR measurements. The final protein concentration was 1 mM. 1H NMR data were collected on a Bruker AVANCE 600 spectrometer operating at 600.16 MHz. Reference spectra were collected in 2H2O over the temperature range of 12–30 °C at a repetition rate of 1 s−1 using a standard one-pulse sequence with saturation of the water solvent signal. Chemical shifts are referenced to 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) through the water resonance calibrated at each temperature. Non-selective spin-lattice relaxation rates were estimated in 2H2O at 12–30 °C from the null point of a hard-pulse inversion-recovery pulse sequence. NOESY spectra (47) (mixing time 40 ms; 13–25 °C) and Clean-TOCSY spectrum (48) (spin-lock duration 25 ms; 25 °C) using MLEV-17 (49) were recorded over the a bandwidth of 22–65 kHz (NOESY) and 12.5 kHz (TOCSY) with a recycle rate of 1–2 s−1, using 512 t1 blocks of 224–456 scans, each consisting of 2048 or 4096 t2 points. Two-dimensional data sets were processed using Bruker XWIN software on a PC running the Hummingbird Exceed X Window emulator. The processing consisted of 30 or 45°-sine-squared-bell apodization in both dimensions, and zero-fill to 2048 × 2048 or 4096 × 2048 data points, prior to Fourier transformation.
RESULTS
UV-Visible Spectra of Wild-type and Mutant Heme-hHO1 Complexes
Wild-type heme-hHO1 complex in the ferric state has a Soret absorption at 404 nm. The absorption red-shifts to 430 nm in the ferrous state and to 418 nm in the ferrous-CO complex. Most of the mutants have similar Soret absorptions with shifts of less than 4 nm, suggesting that the mutations have an insignificant effect on the heme environment (Table 1). The biggest change is observed for the D140H mutant in the ferric state, which has a Soret absorption at 412 nm. Resonance Raman spectroscopy studies of the D140H mutant have shown that His140 replaces the distal water ligand and becomes the sixth ligand to the heme iron atom (46).
Table 1.
Soret Absorption of Wild-type and Mutant hHO1 in the Ferric, Ferrous, and Ferrous-CO States.
| enzyme | Fe(III) | Fe(II) | Fe(II)-CO |
|---|---|---|---|
| wild-type | 404 | 430 | 418 |
| Y58A | 404 | 430 | 420 |
| Y58F | 404 | 428 | 420 |
| D140A | 404 | 430 | 420 |
| D140H | 412 | 426 | 420 |
| D140K | 408 | 426 | 420 |
| D140AY58A | 406 | 430 | 420 |
| K18A | 406 | 430 | 420 |
| K18E | 404 | 428 | 418 |
| E29K | 404 | 424 | 418 |
| R183A | 404 | 430 | 418 |
| R183E* | 404 | 430 | 418 |
| Y134F | 406 | 430 | 418 |
| K18A/R183E | 404 | 430 | 418 |
| Y134F/R183E | 404 | 428 | 418 |
| K18A/Y134F/R183E | 402 | 428 | 418 |
| K18E/R183E | 404 | 430 | 418 |
| K18E/E29K/R183E | 404 | 430 | 418 |
Reference (42).
Regiospecificity of hHO1 with Modified Heme Seating Interactions
To facilitate isolation and identification of the biliverdin isomers, they were first dimethyl esterified. The HPLC conditions employed in this study readily separate the four biliverdin dimethyl esters. In the reaction supported by cytochrome P450 reductase (termed the normal reaction), wild-type hHO1 exclusively produces biliverdin IXα (Fig. 3, Table 2). The single mutations K18A, K18E, R183A, and Y134F have no discernible effect on this α-regiospecificity. Combining two of the mutations in the K18A/R183E double mutant results in production of 84.3 ± 3% of biliverdin IXα, 14.0 ± 2.6% of biliverdin IXδ, and 1.7 ± 1.5% of biliverdin IXβ. Simultaneous mutation of the positively charged Lys18 and Arg183 to negatively charged Glu residues results in equal production of biliverdin IXα and IXδ, with no IXβ isomer being detected. However, in another double mutant, Y134F/R183E, the regiospecificity is only slightly altered, yielding 96.0 ± 0.1% of biliverdin IXα and 4.0 ± 0.6% of biliverdin IXδ. This clearly suggests that Lys18 and Arg183 are the more important residues for proper seating of the heme. Tyr134 plays a minor role even though it forms a hydrogen bond with one of the heme propionate groups. As might be expected from these results, the triple mutant K18A/Y134F/R183E gives results very similar to those of the double K18A/R183E mutant. Interestingly, the E29K mutant can oxidize heme at not only the α but also the δ and γ positions, even though only traces of biliverdin IXγ and IXδ were observed (~0.5%). In comparison with the K18E/R183E mutant, the triple K18E/E29K/R183E mutant significantly increases the production of biliverdin IXδ and IXβ to 76.4 ± 0.9% and 7.3 ± 0.3%, respectively, and decreases biliverdin IXα formation to 16.3 ± 1.2%. No IXγ isomer was observed.
FIGURE 3.

Regiospecificity of mutant hHO1 involved in unseating heme. HPLC analysis of biliverdin dimethyl esters isolated from the reactions supported by P450 reductase. The retention times are biliverdin IXα (26.8 min), IXβ (28.2 min), IXδ (28.7 min) and IXγ (30.6 min).
Table 2.
Regiospecificity of Wild-type and Mutant hHO1 in the Reaction Supported by Cytochrome P450 Reductase.a
| enzyme | biliverdin isomers (%) | ||
|---|---|---|---|
| hHO1 + P450 reductase+ NADPH | |||
| α | β | δ | |
| wild-type | 100 | – | – |
| D140A | 100 | – | – |
| D140H | 81.8 ± 6.7 | 9.3 ± 1.0 | 8.9 ± 6.6 |
| D140K | 96.5 ± 0.4 | 1.2 ± 0.1 | 2.3 ± 0.4 |
| Y58F | 100 | – | – |
| Y58A | 100 | – | – |
| Y58A/D140A | 100 | – | – |
| K18A | 100 | – | – |
| K18E | 100 | – | – |
| E29Kb | 98.9 ± 0.5 | – | 0.6 ± 0.3 |
| R183A | 100 | – | – |
| R183Ec | 100 | – | – |
| Y134F | 100 | – | – |
| Y134F/R183E | 96.0 ± 0.1 | – | 4.0 ± 0.6 |
| K18A/R183E | 84.3 ± 3 | 1.7 ± 1.5 | 14.0 ± 2.6 |
| K18E/R183E | 48.5 ± 2.6 | – | 51.5 ± 2.6 |
| K18A/Y134F/R183E | 85.3 ± 6.4 | 0.7 ± 1.2 | 13.7 ± 4.7 |
| K18E/E29K/R183E | 16.3 ± 1.2 | 7.3 ± 0.3 | 76.4 ± 0.9 |
All experiments were repeated at least three times.
E29K mutant also generated 0.5 ± 0.3% of biliverdin IXγ, which was not observed for the other mutants.
Reference (42).
Wild-type hHO1 cleaves heme only at the α-meso carbon in the coupled oxidation process. In coupled oxidation, all the mutations had similar effects on the regiospecificity of the reaction as was observed in the normal reaction, except to a greater degree (Fig. 4, Table 3). The single mutants, K18A, Y134F and R183A only had small effects on the regiospecificity, with biliverdin IXα representing >95% of the products in all cases. Mutation of the positively charged Lys18 to a negatively charged Glu produced 91.6 ± 4.7% of biliverdin IXα, 1.2 ± 0.2% of IXβ, and 7.2 ± 4.7% of IXδ. Coupled oxidation of heme by the single E29K mutant occurs at all the four meso carbons, yielding 83.8 ± 0.8% biliverdin IXα, 6.7 ± 0.4% IXβ, 8.4 ± 0.3% IXδ and 1 ± 0.1% IXγ. A greater shift in regiospecificity is observed with the Y134F/R183E double mutant, which produces 73.3 ± 6.4% of biliverdin IXα and 25.0 ± 5.3% of biliverdin IXδ, and for the K18A/R183E double mutant and the K18A/Y134F/R183E triple mutant, which generates approximately equal amounts of biliverdin IXα and biliverdin IXδ. Even greater change is seen with the K18E/R183E double mutant, which increases biliverdin IXδ to 76.9 ± 3.6% and decreases biliverdin IXα to 20.5 ± 3.7%. Combination of this latter double mutation with an E29K mutation significantly increases biliverdin IXβ to 12.9 ± 3.9% while decreasing biliverdin IXα to 8.0 ± 2.7%. Thus, in coupled oxidation of the K18E/E29K/R183E triple mutant, cleavage at the α-meso carbon becomes a very minor process relative to cleavage at the δ- and β-positions. As already noted, the overall results are consistent with those obtained for the normal reaction except that the effects of the mutations on regiospecificity are amplified in the reactions under coupled oxidation conditions.
FIGURE 4.

Regiospecificity of mutant hHO1 involved in unseating heme. HPLC analysis of biliverdin dimethyl esters isolated from the reactions supported by sodium ascorbate in the asbsence of superoxide dismutase and catalase.
Table 3.
Regiospecificity of Wild-type and Mutant hHO1 in the Reaction Supported by Sodium Ascorbate in the Absence or Presence of SOD and Catalase.a
| enzyme | biliverdin isomers (%) | biliverdin isomers (%) | ||||
|---|---|---|---|---|---|---|
| hHO1 + sodium ascorbate | hHO1 + sodium ascorbate + SOD + catalase | |||||
| α | β | δ | α | β | δ | |
| wild-type | 100 | – | – | 100 | – | – |
| D140A | 94.0 ± 1.5 | 2.7 ± 0.6 | 3.3 ± 1.5 | 94.7 ± 2.3 | 2.0 ± 0.1 | 3.3 ± 1.7 |
| D140H | 83.0 ± 10.5 | 7.0 ± 4.2 | 10.0 ± 6.0 | 90.4 ± 4.4 | 4.1 ± 1.5 | 5.5 ± 4.2 |
| D140K | 91.7 ± 1.5 | 3.3 ± 2.1 | 5.0 ± 2.6 | 96.5 ± 0.6 | 1.3 ± 0.6 | 2.2 ± 1.0 |
| Y58F | 99.3 ± 0.6 | 0.3 ± 0.5 | 0.4 ± 0.3 | N.D.d | N.D. | N.D. |
| Y58A | 100 | – | – | N.D. | N.D. | N.D. |
| Y58A/D140A | 96.7 ± 1.5 | 1.3 ± 0.6 | 2.0 ± 0.6 | N.D. | N.D. | N.D. |
| K18A | 99.5 ± 0.6 | – | 0.5 ± 0.5 | N.D. | N.D. | N.D. |
| K18E | 91.6 ± 4.7 | 1.2 ± 0.2 | 7.2 ± 4.7 | 97.5 ± 0.2 | 1.5 ± 0.2 | 1.5 ± 0.1 |
| E29Kb | 83.8 ± 0.8 | 6.7 ± 0.4 | 8.4 ± 0.3 | 95.6 ± 0.4 | 2.3 ± 0.3 | 2.2 ± 0.2 |
| R183A | 96.7 ± 2.5 | – | 3.3 ± 2.5 | N.D. | N.D. | N.D. |
| R183Ec | 80.0 ± 3.6 | 0.7 ± 0.5 | 19.3 ± 3.1 | 91.2 ± 1.0 | 0.8 ± 1.0 | 8.0 ± 0.8 |
| Y134F | 99.7 ± 0.6 | – | 0.4 ± 0.5 | N.D. | N.D. | N.D. |
| Y134F/R183E | 73.3 ± 6.4 | 1.7 ± 1.2 | 25.0 ± 5.3 | 76.7 ± 3.8 | 4.0 ± 2.0 | 19.3 ± 2.9 |
| K18A/R183E | 51.0 ± 1.7 | 2.3 ± 0.6 | 46.7 ± 1.5 | 55.7 ± 0.6 | 2.0 ± 0.1 | 42.3 ± 0.6 |
| K18E/R183E | 20.5 ± 3.7 | 2.6 ± 0.2 | 76.9 ± 3.6 | 21.1 ± 1.8 | 2.5 ± 0.0 | 76.4 ± 1.8 |
| K18A/Y134F/R183E | 49.0 ± 4.5 | 3.0 ± 1.0 | 48.0 ± 5.1 | 52.7 ± 1.5 | 3.0 ± 0.1 | 44.3 ± 1.5 |
| K18E/E29K/R183E | 8.0 ± 2.7 | 12.9 ± 3.9 | 79.1 ± 5.7 | 10.2 ± 0.5 | 3.4 ± 0.3 | 86.4 ± 0.5 |
All experiments were repeated at least three times.
E29K mutant also generated 1.1 ± 0.1% of biliverdin IXγ, which was not observed for the other mutants.
Reference (42).
N.D. = not determined.
Our previous study of the R183E mutant suggested that hydrogen peroxide contributes to the generation of biliverdin IXβ and IXδ isomers in the coupled oxidation reaction (42). Therefore, for the mutants which produce significant amounts of biliverdin IXβ and IXδ, the reactions were also carried out in the presence of catalase and SOD (Table 3). SOD and catalase were only found to modestly alter the regiospecificity of two single mutants, E29K and K18E. In the case of E29K, the formation of both biliverdin IXβ and IXδ was reduced three-fold and no biliverdin IXγ was observed. For the K18E mutant, biliverdin IXδ formation was decreased at least two-fold but no significant effects were observed on the formation of biliverdin IXα and IXβ.
Regiospecificity of hHO1 with Mutants that Disrupt the Distal Hydrogen Bonding Network
Asp140, through a water molecule, can form hydrogen bonds with the terminal oxygen atom in the Fe(III)·OOH active species. It also forms hydrogen bonds with Tyr58. To disrupt the resulting hydrogen bonding network, we mutated Asp140 to His, Lys and Ala. The crystal structure of D140A shows that the solvent structure in the distal pocket is altered although Tyr58 can still interact through a water molecule with the oxygen atom of the Fe(III)-OOH. To eliminate this interaction, we mutated Tyr58 to an Ala or Phe, and simultaneously mutated both Asp140 and Tyr58 to an Ala. Among these single and double mutants, the regiospecificity of the reaction supported by NADPH-cytochrome P450 reductase was only slightly altered by the D140H and D140K mutations. The D140H mutant produced 81.8 ± 6.7% of biliverdin IXα, 9.3 ± 1.0% of IXβ and 8.9 ± 6.6% of IXδ, and the D140K mutant generated 96.5 ± 0.4% of biliverdin IXα, 1.2 ± 0.1% of IXβ and 2.3 ± 0.4% of IXδ (Fig. 5, Table 2). Under coupled oxidation conditions, the D140H and D140K mutants generated roughly the same ratio of biliverdin IXα, βand δ isomers as in the normal reaction (Table 3). For the other mutations, only small amounts of biliverdin IXβ and δ were observed (Fig. 5, Table 3). To investigate the involvement of hydrogen peroxide and superoxide, coupled oxidations of D140H, D140K and D140A were also performed in the presence of SOD and catalase. These two antioxidant enzymes had no significant effect on the ratios of the biliverdin isomers, suggesting that hydrogen peroxide is not involved in the alteration of regiospecificity (Table 3).
FIGURE 5.

Regiospecificty of mutant hHO1 involved in disrupting the distal hydrogen-bonding network. HPLC analysis of biliverdin dimethyl esters isolated from the reactions supported by P450 reductase (left) and sodium ascorbate (right)
NMR Studies of Heme Seating in K18E/E29K/R183E hHO1
We have used 1H NMR to directly determine the orientation of the heme in the active site of the K18E/E29K/R183E triple mutant of hHO1, the protein that shows the greatest shift in regiospecificity of oxidation from the α- to the β/δ-meso-positions. The position of the heme within the protein matrix can be established in solution by NMR of diamagnetic or low-spin ferric paramagnetic derivatives (50). The potential presence of four isomers for the complex of interest requires the ferric cyanide complex because of the superior resolution due to hyperfine shifts. The resolved portions of the 600 MHz 1H NMR spectrum of K18E/E29K/R183E-hHO1-PH-CN in 2H2O are presented in Fig. 6A–6C. Approximately three times as many resonances are resolved to low-field as for the wild-type complex (41, 51), with most exhibiting temperature-dependent NOESY cross peak intensities due to chemical exchange (52) as shown in Figs. 6G and 6K. Four sets of resonances, corresponding to four isomers, exist that exclusively show pairwise chemical exchange. The obvious methyls for the four complexes are labeled Mi, mi, m* and m+ (i represents the position on heme, if assigned) and represent relative equilibrium populations of ~50:05:25:20 for M:m:m*:m+. The exchange cross peaks for the pair Mi↔mi are labeled by #, while those between m* ↔ m+ are labeled by §. For the pair Mi, mi, it is possible to locate all four methyls in both isomers. For the pair m*, m+, only a single cross peak is observed between resolved resonances (i.e., m#, m+ in Figs. 6A, 6K). Since the populations for mi+ and mi# are quite similar, it is not even possible to unequivocally differentiate m* from m+ peaks in all cases. However, exchange is so strong between the m* and m+ peaks that it precludes detection of sufficient NOESY cross peaks for assignment of resonances. The exchange cross peaks are considerably weaker for the Mi↔mi than mi*↔mi+ pairs. For the Mi-mi pairs, moreover, NOESY cross peaks are quite strong and sufficient to allow unambiguous determination of the seating of the heme within the protein matrix. Heme structural characterization is restricted to the major isomers with methyl peaks Mi in Fig. 6. The chemical shifts for the heme methyl for isomers Mi and mi are compared to those for wild-type hHO1-PH-CN in Table 4.
FIGURE 6.

Resolved portion (A, B, C) of the 600 MHz 1H NMR spectra of K18E/E29K/R183E-hHO1-PH-CN in 2H2O, 57 mM in phosphate, pH 7.23 at 25°C. The resonances m1, H4α, M5, M3 are shown at 18 °C as inset A′ that allows estimation of the population of the isomer with peaks mi. NOESY data were collected over a 46 ppm window for all panels except H, for which a 26 ppm window was used. The chemical exchange peaks between the two methyl resonance sets Mi↔mi, are labeled # in (K), while those between the two methyl sets mi*↔mi+, are labeled by § (Fig. 2G, 2K). Peaks are labeled Mi, (Hi) mi where i represents the pyrrole methyl (single proton) position in the Fisher notation. Intraheme contacts for the major isomer (Mi, Hi) are illustrated in panels (K, N), intra-residue cross peaks are shown in panel (F, H, K (H25β-H25β′)), inter-residue contacts are shown in panels (E, G, I), and heme-residue contacts are labeled in panels (D, G, J, L and M). Dotted circles indicate cross peaks observed either at lower contour or at other temperatures.
Table 4.
Heme Methyl Chemical Shifts in K18E/E29K/R183E-hHO1-PH-CN and Wild-type hHO1-PH-CN.a
| isomer Bb (Mi) | isomer Ab (mi) | wild-typec (Isomer C)b | isomers C/Db,d (m*,m+) |
|---|---|---|---|
| 12.69 (M1) | 17.00 (m1) | 4.43 (M1) | 8.94/26.23 |
| 15.95 (M3) | 10.89 (m3) | 19.63 (M3) | 5.22/25.03 |
| 16.37 (M5) | 18.33 (m5) | 9.04 (M5) | 7.08/4.19 |
| 25.21 (M8) | 12.69 (m8) | 10.48 (M8) | 22.11/13. 33 (mi*, mi+) |
Chemical shifts, in ppm, referenced to DSS in 2H2O solution ~60 mM in phosphate, pH 7.2 at 25°C.
A, B, C, D represent the protohemin seatings described in Figures 7A–7D.
Due to spectral overlap that prevents accurate intensity determination, the four methyl peaks for the individual isomer could not be uniquely determined.
All four heme methyls, Mi (i = 1, 3, 5, 8) are resolved for the major isomer, as are the single low-field (H4α) and two upfield (H4βs) single protons of a vinyl. A weak NOESY cross peak between the methyls at 25.2 and 12.3 ppm (Fig. 6K) identifies them uniquely as the 1-CH3/8-CH3 pair, but does not distinguish between them. The resolved vinyl exhibits moderate intensity Hβ NOESY cross peaks to one methyl at 16.0 ppm and a weak Hα cross peak to the methyls at 16.8 ppm, dictating that the two methyls and one vinyl must originate from either the set 3-CH3/4-vinyl/5-CH3 or 1-CH3/2-vinyl/3-CH3. Since 1-CH3 is identified as one of the other pair at 12.6 or 25.2 ppm, we uniquely identify 3-CH3 and 5-CH3 at 16.0 and 16.3 ppm, with 4Hα at 16.8 ppm. The failure to assign either the expected low-field shifted 2-vinyl or propionate CαH peaks precludes a direct differentiation of 1-CH3 and 8-CH3. However, contacts between the heme methyls/vinyls and the protein matrix lead to the distinction between 1-CH3 and 8-CH3 and uniquely seat the heme in the protein matrix.
To characterize the isomers with peaks Mi, we take advantage of a highly conserved HO fold with electronic/magnetic properties that invariably result in characteristic hyperfine shifts for residues in direct contact with substrate in not only hHO1 (41, 53), but also the analogous bacterial HO complexes (54, 55). The position of residues relative to PH in the wild-type crystal structure are depicted in Fig. 7A. Characteristic upfield shifted and resolved resonances are expected (53–55) for the proximal helix Ala28 CβH3, the distal helix Thr135 CγH3 and Ser142 CβHs, in contact with pyrrole A, the pyrrole A/D junction and the pyrrole C/D junctions, respectively, the latter with strong relaxation (T1, ~30–80 ms). The only characteristic downfield shifted non-labile resonances arise from the axial His CβHs (53–55). Three additional key residues are a moderately upfield shifted Phe207 ring in contact with the axial His25, Ala28 and Thr135, and the pyrrole A/D junction, a weakly shifted Phe214 in contact with Ala28 and pyrrole A, and a weakly shifted Tyr134 ring in contact with both Thr135 and pyrrole D (53). These expected contacts in the wild-type complex are depicted in Fig. 7A. Each of these residues is readily identified for the mutant by the TOCSY connectivity (not shown; see Supporting Information), characteristic hyperfine shift (Table S1 in Supporting Information), paramagnetic relaxation (not shown), and inter-residue contacts (2D data in panels D-N of Fig. 6, and schematic summary in Fig 7A and 7B). Also assigned by TOCSY and NOESY are the ring protons of a prominent aromatic cluster in the distal pocket (53, 56). The hyperfine shifts for these key residues, which are sensitive indicators of both molecular and electronic structures, are very similar in the mutant (Table S1) and wild-type complexes.
FIGURE 7.
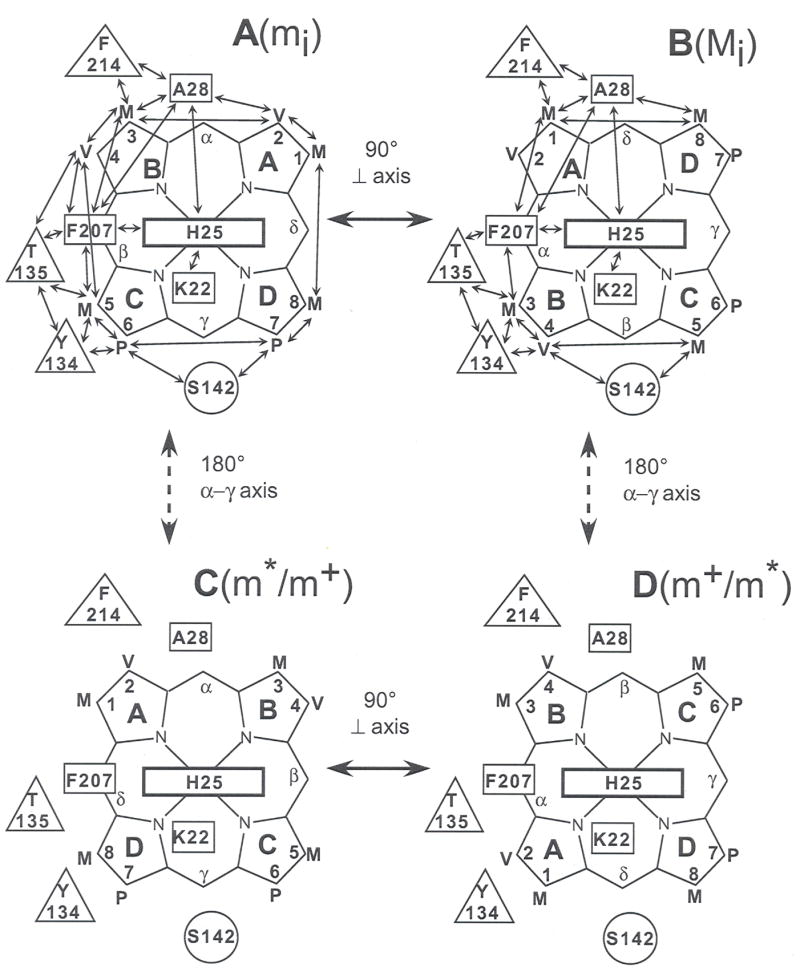
Schematic depiction of the potential seating of protoheme, PH, in the active site of hHO1, where proximal and distal residues in contact with PH or axial His are shown as rectangles and circles, respectively. The double-sided arrows indicate expected, and observed, dipolar contacts. (A) PH seated as found in the wild-type crystal structure (24), the orientation giving rise to methyl peaks m; (B) PH in the crystal is rotated counter-clockwise about the PH normal by approximately 90°, as found in the major isomer of the triple mutant complex, the species gives rise to methyl peak M; (C) the alternate seating of PH in the wild-type which is rotated 180° about the α-γ-meso axis; this is the major isomer in solution (40, 51) and gives rise to methyl peaks m+ (or m*); (D) The PH orientation rotated counter-clockwise by ~90° about the PH normal relative to that in (C); the species gives rise to methyl resonances m* (or m+).
The assigned 3-CH3 at 16.0 ppm in the mutant exhibits NOESY cross peaks to the rings of Phe207 and Tyr134 (Fig. 6L) as well as the CγH3 of Thr135 (Fig. 6M), dictating that it occupies the position held by 5-CH3 in the wild-type. Similarly, the 5-CH3 (Fig. 6M) and 4Hα (not shown; observed at lower contour) exhibits NOESY cross peaks to Ser142 CβHs, the position occupied by the propionate CαHs in the wild-type. Hence pyrroles B and C (and hence pyrroles A and B) of the major solution isomer are necessarily oriented in the HO matrix as shown in Fig. 7B. The 1-CH3/8-CH3 peaks are differentiated in that only 1-CH3 can be close to both the Phe214 ring (Fig. 6L) and Ala28 CβH3 (Fig. 6M). The NOESY cross peak pattern to Phe214/Ala28 for the 1-CH3 and 8-CH3 peaks in the mutant is essentially the same as reported for the 2-vinyl and 3-CH3, respectively, of the wild-type complex (41, 53). Thus, the NOESY contacts between the heme and conserved active site residues establish that the heme seating in the major isomer form (with methyl Mi in Fig. 6A) of the mutant is ~90° in-plane rotated counter-clockwise (Fig. 7B) relative to that in the wild-type crystal (24) (Fig. 7A). It is known that a 90° in-plane rotation of the heme in low spin hemin results in interchange between methyls with large and small hyperfine shifts (38, 41, 57, 58). Hence a strongly off-diagonal exchange cross peak to the minor isomer, mi, argues for a ~90° difference in the heme in-plane with orientation as in wild-type in Fig. 7A. The strong exchange contribution between methyls in isomers m*, m+ preclude determination of the seating. However, the strongly off-diagonal shift differences are consistent in originating from two in-plane ~90° interconverting heme seatings with the heme rotated ~180° about the α-β-meso axis relative to that in the crystal, as depicted in Fig. 7C and 7D.
Relative Catalytic Activities of the Isoforms
The coupled assay with biliverdin reductase was used to compare relative rates of biliverdin formation among the mutants. A previous study showed that rat liver biliverdin reductase has a much higher affinity for biliverdin IXα than for the other isomers, resulting in an apparent rate of biliverdin IXδ reduction that is only 7% that of biliverdin IXα (59). In our assays we used a significant excess of biliverdin reductase to ensure complete conversion of the biliverdin isomers into their corresponding bilirubin isomers. Controls were performed under the conditions described with sodium ascorbate in which biliverdin reductase was added to the assays after significant biliverdin had been produced (10 minutes). The maximal apparent rate of bilirubin formation from reduction of the biliverdin isomers produced by the K18E/E29K/R183E triple mutant, in the approximate ratio of 8:13:79 biliverdin IXα:IXβ:IXδ, was 20% that of wild-type, but was still five-fold greater than the maximal apparent rate observed in assays containing biliverdin reductase. As shown in Table 5, the mutants catalyze the formation of biliverdin with varying degrees of efficiency. A constant concentration of the ferric iron chelator deferroxamine (desferrioxamine) was added to the assays to compare relative rates of biliverdin formation in the normal cytochrome P450 reductase-dependent reaction and the ascorbate catalyzed coupled oxidation reaction. This was necessary to liberate the biliverdin that accumulates in the Fe(III)-biliverdin intermediate (60), not only in the ascorbate catalyzed reactions but also in the P450 reductase catalyzed reaction with a number of mutants, namely E29K, and all of the R183E containing mutants. Wild-type hHO1 activity in the P450 reductase catalyzed reaction was 30% higher in the presence of deferroxamine than in its absence.
Table 5.
Relative Bilirubin Formation Rates for Wild-type and Mutant hHO1.
| NADPH/P450 reductase | sodium ascorbate | |||
|---|---|---|---|---|
| Enzyme | rate(nmol bilirubin min−1mg−1) | relative rate % | rate(nmol bilirubin min−1mg−1) | relative rate % |
| wild-type | 60.2 ± 1.5 | 100 ± 4 | 7.4 ± 0.7 | 100 ± 13 |
| Y58A | 27.5 ± 0.6 | 46 ± 2 | 6.9 ± 0.1 | 94 ± 9 |
| Y58F | 22.6 ± 1.6 | 38 ± 3 | 4.9 ± 0.3 | 66 ± 7 |
| D140A | 12.9 ± 1.5 | 21 ± 3 | 1.5 ± 0.3 | 20 ± 4 |
| D140H | 0.30 ± 0.06 | 0.5 ± 0.1 | 0.12 ± 0.10 | 2 ± 1 |
| D140K | 4.3 ± 0.5 | 7.1 ± 0.8 | 0.5 ± 0.2 | 7 ± 2 |
| Y58A/D140A | 8.0 ± 1.2 | 13 ± 2 | 2.0 ± 0.2 | 27 ± 4 |
| K18A | 68.9 ± 0.1 | 114 ± 3 | 8.5 ± 0.1 | 115 ± 10 |
| K18E | 55.2 ± 2.9 | 92 ± 5 | 8.2 ± 1.5 | 111 ± 22 |
| E29K | 15.6 ± 0.6 | 26 ± 1 | 3.0 ± 0.2 | 41 ± 4 |
| R183A | 14.2 ± 0.9 | 24 ± 2 | 7.1 ± 0.9 | 96 ± 15 |
| R183E | 1.7 ± 0.2 | 2.8 ± 0.4 | 9.2 ± 0.4 | 125 ± 13 |
| Y134F | 46.0 ± 9.7 | 76 ± 16 | 10.8 ± 1.1 | 146 ± 20 |
| K18A/R183E | 1.8 ± 0.3 | 2.0 ± 0.1 | 6.9 ± 1.2 | 93 ± 18 |
| Y134F/R183E | 1.7 ± 0.1 | 2.8 ± 0.2 | 8.6 ± 1.0 | 116 ± 17 |
| K18A/Y134F/R183E | 1.2 ± 0.4 | 2.0 ± 0.7 | 6.4 ± 0.7 | 87 ± 12 |
| K18E/R183E | 1.8 ± 0.3 | 3.0 ± 0.6 | 6.0 ± 0.4 | 81 ± 9 |
| K18E/E29K/R183E | 0.7 ± 0.1 | 1.2 ± 0.2 | 5.8 ± 1.1 | 78 ± 16 |
The mutants involved in the distal hydrogen bonding network that behave like the wild-type are Y58A and Y58F. The Asp140 mutants disrupt the distal hydrogen bonding network and heme oxygenase activity and promote peroxidase activity (46). In our assays, their heme oxygenase activities with either reductant were equally disrupted; D140A, shows 20% of wild-type activity, D140K, 7%, and D140H was the least active, with as little as 0.5%.
Of the mutations to residues involved in ionic interactions with the heme, the single mutants, K18A, K18E, and Y134F, showed activity similar to wild-type. The mutants containing R183E had an activity of ≤3% of wild-type activity in the P450 reductase catalyzed reaction, but retained wild-type levels of activity in the coupled oxidation reaction. The triple K18E/E29K/R183E mutant is the least active with 1.2 ± 0.2% wild-type activity. In the crystal structure, Glu29 is in close proximity, and is hydrogen bonded, to the proximal histidine ligand to the heme iron. Thus mutation of this residue to Lys shows a significant loss in activity of 26 ± 1% and 41 ± 4% for the P450 reductase and ascorbate catalyzed reactions, respectively.
DISCUSSION
The heme in heme oxygenases is sandwiched between two helices. In human hHO1, three positively charged residues, Lys18, Tyr134 and Arg183, form hydrogen bonds with the propionate groups of the heme. These ionic and hydrogen-bonding interactions help to hold the heme in the correct position and orientation. We have recently reported that mutation of Arg183 to Glu183 does not alter the regiospecificity of heme oxidation in the cytochrome P450 reductase supported reaction but, as reported earlier by others (61), some loss of specificity is observed in the coupled oxidation reaction (42). The crystal structure of the R183E mutant shows that Lys18 and Tyr134 still form hydrogen bonds with the propionates and sufficiently anchor the heme to preserve the normal regiochemistry of the reaction (42). In the present study we therefore mutated these two residues to alanine/glutamic acid and alanine/phenylalanine, respectively, to investigate their role in controlling the reaction regiochemistry. In the cytochrome P450 reductase-dependent reaction, none of the single mutations significantly altered the regiospecificity (Table 2). However, mutation of all three residues simultaneously (K18A/Y134F/R183E), which completely suppresses the normal interactions between the protein and the heme propionate groups, results in the formation of 13.7% biliverdin IXδ and a trace of IXβ in addition to the usual IXα isomer. The K18A/R183E double mutant has the same effects as the above triple mutant (Table 2), indicating that Lys18 and Arg183 are important for maintaining the orientation of the heme whereas the contribution of Tyr134 is relatively minor. Mutation of both Lys18 and Arg183 to glutamic acids, which introduces negative charges in the region adjacent to the heme propionic acid groups, further increases the formation of biliverdin IXδ to 51.5%. It appears that the repulsive force between the negatively charged Glu and the propionate carboxylate groups promotes rotation of the heme about an axis through the iron and perpendicular to the heme plane.
In order to complement the repulsive interactions introduced by the K18E and R183E mutations, we have mutated Glu29 to a lysine, thus introducing a positive charge into the region occupied by a heme carboxylate group if the heme rotates by approximately 90° (Fig. 2). The E29K mutation by itself only resulted in the formation of a trace of biliverdins IXδ and IXγ, but in combination with the K18E/R183E mutations, it increased the formation of biliverdin IXδ to 76.4% and IXβ to 7.3% in the P450 reductase dependent reaction (Table 2). These results suggest that electrostatic interactions, both positive and negative, contribute critically to orientation of the heme in the hHO1 active site.
Our previous study of the R183E mutant demonstrated that the normal heme oxygenase reaction supported by NADPH-cytochrome P450 reductase has a more stringent regiospecificity than the reaction that occurs under coupled oxidation conditions (42). This difference is not detected with the wild-type enzyme, but becomes apparent when the R183E mutation is introduced, as this mutation results in a small loss of regiochemical specificity in the coupled oxidation but not in the normal reaction. The same trend is observed in these more extensive studies. In the coupled oxidation process, the influence of the mutations on the reaction regiospecificity is similar to that observed in the normal reaction, but the magnitude of the changes is greater (compare Tables 2 and 3). In a few instances, this increased loss of specificity is caused by hydrogen peroxide generated in the ascorbate-supported reaction. Thus, for the single K18E and R183E mutants, SOD and catalase reduce the amount of biliverdin IXδ that is formed by 2–4 fold. However, for the multiple mutants including mutations of either Lys18 or Arg183, SOD and catalase have little effect on the biliverdin isomer ratio. The crystal structure of R183E shows that the predominant orientation of the heme in this mutant is the same as that in wild-type (42). These results suggest that the ascorbate-dependent transfer of electrons to the reaction center of hHO1 is more permissive and may occur with protein conformations/heme orientations that do not allow electron transfer from cytochrome P450 reductase.
Analysis by NMR of the orientation of the heme in the K18E/E29K/R183E hHO1 active site indicates the existence in solution of a 50:05:25:20 mixture of heme orientations in the protein. The conserved hyperfine shifts and inter-residue contacts observed for the active site confirm that the active site of the triple mutant is largely conserved and closely resembles that of the wild-type enzyme. This is supported by the crystal structure of the R183E mutant, which also shows little difference from that of the wild-type enzyme (42). Detailed examination of the NMR data establishes that in the predominant isomer the heme has rotated from its position in the wild-type crystal structure by approximately 90° about an axis through the iron and perpendicular to the heme.
The orientation of a heme relative to the axial His can also be estimated from the heme methyl hyperfine shift pattern itself (38, 57). The data for the major isomer Mi in Table 4 suggest a protoheme rotation of 60±15°, relative to wild-type, a value that is only qualitatively consistent with the ~90° based on protohemin to protein matrix. The exchange is too rapid for the minor isomers with methyl probes m* and m+ to allow determination of the heme orientation in those isomers. However, on the basis of strongly off-diagonal exchange cross peaks reflecting ~90° in-plane rotation relative to the axial His, the two isomers most likely originate from the two heme seatings illustrated in Fig. 7C and 7D. In these orientations, the heme is flipped by 180° relative to its orientation in the wild-type and 90°-rotated orientations (Fig. 7A and B).
If the distribution of heme orientations observed by NMR was to directly determine the distribution of biliverdin isomers formed, one would expect a selectivity of 50%δ, 25–30%α, and 25–20%β, in contrast to the 76:16:07, respectively, actually observed with P450 reductase (Table 2) and 86:10:03 observed with ascorbate in the presence of SOD and catalase (Table 3). These product regioisomer differences are not surprising for three reasons. First, it is known that the equilibrium protoheme seating preference in the wild-type about the α-γ-meso axis differs significantly in the cyanide relative to the aquo complexes (62), such that the equilibrium isomer distribution for the cyanide complex will not necessarily correspond to that for the resting state aquo complex, nor to that for the activated hydroperoxy species. Second, the “rotated” position of the protohemin in the different isomers of the mutant may not correspond precisely to a 90° difference, so that not all heme seatings may place a meso-position in the “most reactive” position relative to the blockage of three meso-Hs and the steric directing of the Fe-OOH toward the fourth meso position. Finally, as illustrated by comparison of the data for the P450 reductase-and ascorbate-dependent reactions, the small differences in the active site associated with electron delivery by these two different agents are sufficient in some instances to significantly alter the regiochemistry of the oxidation. Thus, the NMR results clearly demonstrate that the principal orientation of the heme involves an in-plane rotation, even if they cannot precisely predict the ratio of heme oxidation at the four meso-positions.
Arg183 and a number of other positively charged residues lining the edge of the heme binding crevice play a role in the binding of P450 reductase to hHO1 (63, 64). Replacement of Arg183 by a glutamic acid or even alanine is shown here to have a marked effect on the P450 reductase-dependent but not ascorbate-dependent catalytic activity (Table 5). Therefore, Arg183 not only plays a role in properly orienting the heme, but also facilitates electron transfer between cytochrome P450 reductase and the heme iron atom. In contrast to the wild-type reaction, the K18E/R183E double and K18E/E29K/R183E triple mutants produced a greater amount of biliverdin in the coupled oxidation process than in the normal reaction (Table 5). Our previous studies have established that the binding of hHO1 to cytochrome P450 reductase is determined, at least in part, by electrostatic charge interactions (63). The binding site identified on heme oxygenase for this interaction includes Arg183 and Lys18 (63). Mutation of each of these two positively charged residues to negatively charged glutamic acids introduces a potentially repulsive interaction in binding of the reductase. The decreased yield in the reductase-dependent reaction for the R183E-containing mutants may reflect this lowered binding affinity of hHO1 for cytochrome P450 reductase. Furthermore, from the crystal structure, Glu29 is hydrogen bonded to the proximal histidine (His25) through its side chain, and Gln38 through its backbone (Fig. 2). These interactions may contribute to formation of the heme pocket that keeps the heme in a tightly bound position. Loss of Glu29 may prevent formation of this tightly bound state and facilitate unwanted side reactions such as peroxidase activity, accounting for the somewhat lower activity of the E29K mutant under both normal and coupled oxidation conditions.
Additional basic residues within the vicinity of the heme propionates include Lys22 and Lys179. Their role in binding cytochrome P450 reductase is evident from the elevation in their Kd values observed when these residues are mutated to alanines. However, their specific role in binding the propionates has not been investigated. In the structure of the heme-hHO-1 depicted in Figure 2, K22 and K179 are 10.6 and 6.3 Å, respectively, from the heme 7-propionate group. In fact, the crystal structure of R183E hHO-1 shows Lys179 occupying a position within 4 Å of the propionates. The remaining alpha specificity of the triple mutant may originate from this favorable electrostatic interaction. However, additional structures also show Lys179 facing away and at greater distance from the propionates. It remains to be seen what whether this residue contributes to the maintenance of alpha regiospecificity.
In the distal heme oxygenase site, an extensive hydrogen bonding network stabilizes the Fe(III)·OOH species that actually hydroxylates the heme. The residues involved are Asp140, Tyr58, Thr135 and Arg136 (Fig. 2). Asp140 is known to be essential for activation of the iron bound dioxygen and hydroperoxide, and its mutation to other residues largely converts heme oxygenase into a peroxidase (31,46). In the D140A crystal structure, a water molecule takes the place of the missing Asp140, so that a distal hydrogen-bonding network is preserved even though it differs from that in wild-type enzyme (29). However, simultaneous mutation of both Asp140 and Tyr58 should greatly disrupt the network, resulting, as observed (Table 5), in a much lower heme oxygenase activity of the Y58A/D140A double mutant with both cytochrome P450 reductase and ascorbate. Nevertheless, the double Y58A/D140A mutation, like the single D140A/K/H or Y58A mutations, has no significant effects on the regiospecificity of heme oxygenase (Tables 2 and 3). The distal hydrogen bonding network thus appears to play a key role in the magnitude and nature of the catalytic activity, but not in controlling the regiospecificity of heme cleavage.
CONCLUSIONS
The regiochemistry of heme oxidation by mammalian heme oxygenases, as demonstrated here with hHO1, is strongly controlled by the orientation of the heme in the active site cavity, and this orientation is largely determined by ionic interactions of the heme propionic acid substituents with ionic residues lining the active site cavity. Particularly important in this context are interactions with Lys18 and Arg183, as mutations of these two cationic residues to anionic ones results in a major shift of the oxidation to the β- and δ-meso positions. The shift in sterochemistry reflects a ~90° rotation of the heme about the iron-ligand axis, which places the β-(or by a 180° flip, the δ-) meso-carbon at the position normally occupied by the α-meso-carbon. The shift in the regiochemistry of oxidation can be enhanced by introducing a cationic residue at a position that can interact with the heme carboxyls if the heme undergoes the ~90° rotation. NMR studies of the triple K18E/E29K/R183E mutant, in which the β/δ-oxidation regiochemistry is optimized, directly confirm the proposed rotation of the heme. In view of the fact that ionic interactions of the propionate carboxyl groups with protein residues determine the heme oxidation regiochemistry not only by human hHO1, as demonstrated here, but also by the analogous enzymes from Pseudomonas aeruginosa (38) and Corynebacterium diphtheriae (26), indicates that this is a general property of this class of enzymes presumably associated with the basic protein fold.
As we found in our earlier study (42), stereospecificity is more readily lost in the coupled oxidation supported by ascorbate than in the normal P450 reductase-dependent reaction. In the case of Arg183 mutants, the rate of the ascorbate reaction is little affected, but that of the P450 reductase dependent reaction is severely diminished, a finding consistent with our earlier demonstration (63) that Arg183 is involved in docking of P450 reductase to the hHO1 protein surface. The finding of heme rotation as a mechanism for shifting the oxidation regiospecificity is consistent with studies of the modified regiospecificity of the heme oxygenase from Pseudomonas aeruginosa (38), and Corynebacterium diphtheriae (65), and a chimeric heme oxygenase from Neisseria meningitides (66).
Supplementary Material
One Table (chemical shifts of assigned residues) and one figure (TOCSY spectra). This information is available free of charge via the internet at http://pubs.acs.org
Footnotes
Abbreviations: heme, iron protoporphyrin IX regardless of oxidation and ligation state; PH, iron protoporphyrin IX; hHO1, truncated wild-type human heme oxygenase-1; heme-hHO1, complex of iron protoporphyrin IX with hHO1; NADPH-P450 reductase, NADPH-cytochrome P450 reductase; biliverdin reductase, biliverdin IXα reductase; SOD, superoxide dismutase. NOESY, two-dimensional nuclear Overhauser spectroscopy; TOCSY, two-dimensional total correlation spectroscopy; DSS, 2,2-dimethyl-2-silapentane-5-sulfonate.
This work was supported by National Institutes of Health Grants DK30297 (PROM) and GM62830 (GLM).
References
- 1.Tehunen R, Marver HS, Schmid R. Microsomal heme oxygenase. Characterization of the enzyme. J Biol Chem. 1969;244:6388–6394. [PubMed] [Google Scholar]
- 2.Auclair K, Ortiz de Montellano PR. Heme oxygenase structure and mechanism. In: Kadish K, Smith K, Guilard R, editors. Porphyrin Handbook. Vol. 12. Academic Press; New York: 2002. pp. 183–210. [Google Scholar]
- 3.Bussolati B, Ahmed A, Pemberton H, Landis RC, Carlo FD, Haskard DO, Mason JC. Bifunctional role for VEGF-induced heme oxygenase-1 in vivo: induction of angiogenesis and inhibition of leukocytic infiltration. Blood. 2004;103:761–766. doi: 10.1182/blood-2003-06-1974. [DOI] [PubMed] [Google Scholar]
- 4.Józkowicz A, Huk I, Nigisch A, Weigel G, Dietrich W, Motterlini R, Dulak J. Heme oxygenase and angiogenic activity of endothelial cells: stimulation by carbon monoxide and inhibition by tin protoporphyrin-IX. Antioxid Redox Signal. 2003;5:155–162. doi: 10.1089/152308603764816514. [DOI] [PubMed] [Google Scholar]
- 5.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nature Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 6.Otterbein L, Bach FH, Alam J, Soares MP, Lu HT, Wysk M, Davis RJ, Flavell R, Choi AM. Carbon monoxide mediates anti-inflammatory effects via the mitogen activated protein kinase pathway. Nature Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 7.Tobiasch E, Gunther L, Bach FH. Heme oxygenase-1 protects pancreatic beta cells from apoptosis caused by various stimuli. J Investig Med. 2001;49:566–571. doi: 10.2310/6650.2001.33721. [DOI] [PubMed] [Google Scholar]
- 8.Lang D, Reuter S, Buzescu T, August C, Heidenreich S. Heme-induced heme oxygenase-1 (HO-1) in human monocytes inhibits apoptosis despite caspase-3 up-regulation. Int Immunol. 2005;17:155–165. doi: 10.1093/intimm/dxh196. [DOI] [PubMed] [Google Scholar]
- 9.Liu XM, Chapman GB, Wang H, Durante W. Adenovirus-mediated heme oxygenase-1 gene expression stimulates apoptosis in vascular smooth muscle cells. Circulation. 2002;105:79–84. doi: 10.1161/hc0102.101369. [DOI] [PubMed] [Google Scholar]
- 10.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 11.Zakhary R, Poss KD, Jaffrey SR, Ferris CD, Tonegawa S, Snyder SH. Targeted gene deletion of heme oxygenase 2 reveals neural role for carbon monoxide. Proc Natl Acad Sci USA. 1997;94:14848–14853. doi: 10.1073/pnas.94.26.14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duckers HJ, Boehm M, True AL, Yet SF, San H, Park JL, Webb RC, Lee ME, Nabel GJ, Nabel EG. Heme oxygenase-1 protects against vascular constriction and proliferation. Nature Med. 2001;7:693–698. doi: 10.1038/89068. [DOI] [PubMed] [Google Scholar]
- 13.Barañano DE, Ferris CD, Snyder SH. Atypical neural messengers. Trends Neurosci. 2001;24:99–106. doi: 10.1016/s0166-2236(00)01716-1. [DOI] [PubMed] [Google Scholar]
- 14.Uzel C, Conrad ME. Absorption of heme iron. Semin Hematol. 1998;35:27–34. [PubMed] [Google Scholar]
- 15.McCoubrey WK, Jr, Huang TJ, Maines MD. Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur J Biochem. 1997;247:725–732. doi: 10.1111/j.1432-1033.1997.00725.x. [DOI] [PubMed] [Google Scholar]
- 16.Hayashi S, Omata Y, Sakamoto H, Higashimoto Y, Hara T, Sagara Y, Noguchi M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene. 2004;336:241–250. doi: 10.1016/j.gene.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 17.Maines MD. Heme Oxygenase: Clinical Applications and Functions. CRC Press; Boca Raton, FL: 1992. p. 145. [Google Scholar]
- 18.Choi AM, Alam J. Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am J Respir Cell Mol Biol. 1996;15:9–19. doi: 10.1165/ajrcmb.15.1.8679227. [DOI] [PubMed] [Google Scholar]
- 19.Wilks A, Black SM, Miller WL, Ortiz de Montellano PR. Expression and characterization of truncated human heme oxygenase (hHO-1) and a fusion protein of hHO-1 with human cytochrome P450 reductase. Biochemistry. 1995;34:4421–4427. doi: 10.1021/bi00013a034. [DOI] [PubMed] [Google Scholar]
- 20.McCoubrey WK, Jr, Ewing JF, Maines MD. Human heme oxygenase-2: characterization and expression of a full-length cDNA and evidence suggesting that the two HO-2 transcripts may differ by choice of polyadenylation signal. Arch Biochem Biophys. 1992;295:13–20. doi: 10.1016/0003-9861(92)90481-b. [DOI] [PubMed] [Google Scholar]
- 21.Williams SEJ, Wooton P, Mason HS, Bould J, Elles D, Riccardi D, Peers C, Kemp PJ. Heme oxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004;306:2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Ortiz de Montellano PR. Reaction intermediates and single turnover rate constants for the oxidation of heme by human heme oxygenase-1. J Biol Chem. 2000;275:5297–5307. doi: 10.1074/jbc.275.8.5297. [DOI] [PubMed] [Google Scholar]
- 23.Lemberg R. The chemical mechanism of bile pigment formation. Rev Pure Appl Chem. 1956;6:1–23. [Google Scholar]
- 24.Schuller DJ, Wilks A, Ortiz de Montellano PR, Poulos TL. Crystal structure of human heme oxygenase-1. Nature Struct Biol. 1999;6:860–867. doi: 10.1038/12319. [DOI] [PubMed] [Google Scholar]
- 25.Sugishima M, Omata Y, Kakuta Y, Sakamoto H, Noguchi M, Fukuyama K. Crystal structure of rat heme oxygenase-1 in complex with heme. FEBS Lett. 2000;471:61–66. doi: 10.1016/s0014-5793(00)01353-3. [DOI] [PubMed] [Google Scholar]
- 26.Unno M, Matsui T, Chu GC, Couture M, Yoshida T, Rousseau DL, Olson JS, Ikeda-Saito M. Crystal structure of the dioxygen-bound heme oxygenase from Corynebacterium diphtheriae: implications for heme oxygenase function. J Biol Chem. 2004;279:21055–21061. doi: 10.1074/jbc.M400491200. [DOI] [PubMed] [Google Scholar]
- 27.Torpey J, Ortiz de Montellano PR. Oxidation of the meso-methylmesoheme regioisomers by heme oxygenase. Electronic control of the reaction regiospecificity. J Biol Chem. 1996;271:26067–26073. doi: 10.1074/jbc.271.42.26067. [DOI] [PubMed] [Google Scholar]
- 28.Torpey J, Ortiz de Montellano PR. Oxidation of alpha-meso-formylmesoheme by heme oxygenase. Electronic control of the reaction regiospecificity. J Biol Chem. 1997;272:22008–22014. doi: 10.1074/jbc.272.35.22008. [DOI] [PubMed] [Google Scholar]
- 29.Lad L, Wang J, Li H, Friedman J, Bhaskar B, Ortiz de Montellano PR, Poulos TL. Crystal structures of the ferric, ferrous, and ferrous-NO forms of the Asp140Ala mutant of human heme oxygenase-1: catalytic implications. J Mol Biol. 2003;330:527–538. doi: 10.1016/s0022-2836(03)00578-3. [DOI] [PubMed] [Google Scholar]
- 30.Sugishima M, Sakamoto H, Noguchi M, Fukuyama K. Crystal structures of ferrous and CO-, CN(-)-, and NO-bound forms of rat heme oxygenase-1 (HO-1) in complex with heme: structural implications for discrimination between CO and O2 in HO-1. Biochemistry. 2003;42:9898–9905. doi: 10.1021/bi027268i. [DOI] [PubMed] [Google Scholar]
- 31.Fujii H, Zhang X, Tomita T, Ikeda-Saito M, Yoshida TA. Role for highly conserved carboxylate, aspartate-140, in oxygen activation and heme degradation by heme oxygenase-1. J Am Chem Soc. 2001;123:6475–6484. doi: 10.1021/ja010490a. [DOI] [PubMed] [Google Scholar]
- 32.Davydov R, Kofman V, Fujii H, Yoshida T, Ikeda-Saito M, Hoffman BM. Catalytic Mechanism of heme oxygenase through EPR and ENDOR of cryoreduced oxy-heme oxygenase and its Asp 140 mutants. J Am Chem Soc. 2002;124:1798–1808. doi: 10.1021/ja0122391. [DOI] [PubMed] [Google Scholar]
- 33.Wilks A, Schmitt MP. Expression and characterization of a heme oxygenase (Hmu O) from Corynebacterium diphtheriae. Iron acquisition requires oxidative cleavage of the heme macrocycle. J Biol Chem. 1998;273:837–841. doi: 10.1074/jbc.273.2.837. [DOI] [PubMed] [Google Scholar]
- 34.Wenming Zhu, Angela Wilks, Igor Stojiljkovic. Degradation of heme in gram-negative bacteria: the product of the hemO gene of Neisseriae is a heme oxygenase. J Bacteriol. 2000;182:6783–6790. doi: 10.1128/jb.182.23.6783-6790.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ratliff M, Zhu W, Deshmukh R, Wilks A, Stojiljkovic I. Homologues of Neisserial heme oxygenase in gram-negative bacteria: degradation of heme by the product of the pigA gene of Pseudomonas aeruginosa. J Bacteriol. 2001;183:6394–6403. doi: 10.1128/JB.183.21.6394-6403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wegele R, Tasler R, Zeng Y, Rivera M, Frankenberg-Dinkel N. The heme oxygenase(s)-phytochrome system of Pseudomonas aeruginosa. J Biol Chem. 2004;279:45791–45802. doi: 10.1074/jbc.M408303200. [DOI] [PubMed] [Google Scholar]
- 37.Stojiljkovic I, Hantke K. Transport of haemin across the cytoplasmic membrane through a haemin-specific periplasmic binding-protein-dependent transport system in Yersinia enterocolitica. Mol Microbiol. 1994;13:719–732. doi: 10.1111/j.1365-2958.1994.tb00465.x. [DOI] [PubMed] [Google Scholar]
- 38.Caignan GA, Deshmukh R, Wilks A, Zeng Y, Huang HW, Moenne-Loccoz P, Bunce RA, Eastman MA, Rivera M. Oxidation of heme to beta- and delta-biliverdin by Pseudomonas aeruginosa heme oxygenase as a consequence of an unusual seating of the heme. J Am Chem Soc. 2002;124:14879–14892. doi: 10.1021/ja0274960. [DOI] [PubMed] [Google Scholar]
- 39.Friedman J, Lad L, Li H, Wilks A, Poulos TL. Structural basis for novel delta-regioselective heme oxygenation in the opportunistic pathogen Pseudomonas aeruginosa. Biochemistry. 2004;43:5239–5245. doi: 10.1021/bi049687g. [DOI] [PubMed] [Google Scholar]
- 40.La Mar GN, Asokan A, Espiritu B, Yeh dC, Auclair K, Ortiz de Montellano PR. Solution 1H NMR of the active site of substrate-bound, cyanide-inhibited human heme oxygenase. Comparison to the crystal structure of the water-ligated form. J Biol Chem. 2001;276:15676–15687. doi: 10.1074/jbc.M009974200. [DOI] [PubMed] [Google Scholar]
- 41.Fujii H, Zhang X, Yoshida T. Essential amino acid residues controlling the unique regioselectivity of heme oxygenase in Pseudomonas aeruginosa. J Am Chem Soc. 2004;126:4466–4467. doi: 10.1021/ja031791i. [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Lad L, Poulos TL, Ortiz de Montellano PR. Regiospecificity determinants of human heme oxygenase: differential NADPH- and ascorbate-dependent heme cleavage by the R183E mutant. J Biol Chem. 2005;280:2797–2806. doi: 10.1074/jbc.M411229200. [DOI] [PubMed] [Google Scholar]
- 43.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 44.Wilks A, Medzihradszky KF, Ortiz de Montellano PR. Heme oxygenase active-site residues identified by heme-protein cross-linking during reduction of CBrCl3. Biochemistry. 1998;37:2889–2896. doi: 10.1021/bi972720x. [DOI] [PubMed] [Google Scholar]
- 45.Shen AL, Porter TD, Wilson TE, Kasper CB. Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site–dicrected mutagenesis. J Biol Chem. 1989;264:7584–7589. [PubMed] [Google Scholar]
- 46.Lightning LK, Huang H, Moenne-Loccoz P, Loehr TM, Schuller DJ, Poulos TL, Ortiz de Montellano PR. Disruption of an active site hydrogen bond converts human heme oxygenase-1 into a peroxidase. J Biol Chem. 2001;276:10612–10619. doi: 10.1074/jbc.M010349200. [DOI] [PubMed] [Google Scholar]
- 47.Jeener J, Meier BH, Bachmann P, Ernst RR. Investigation of exchange processes by two dimensional NMR spectroscopy. J Chem Phys. 1979;71:4546–4553. [Google Scholar]
- 48.Griesinger C, Otting G, Wüthrich K, Ernst RR. Clean TOCSY for 1H spin system identification in macromolecules. J Am Chem Soc. 1988;110:7870–7872. [Google Scholar]
- 49.Bax A, Davis DG. Mlev-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J Magn Reson. 1985;65:355–360. [Google Scholar]
- 50.La Mar GN, Satterlee JD, de Ropp JS. In: The Porphyrins Handbook. Kadish KM, Smith KM, Guilard R, editors. Academic Press; San Diego: 2000. pp. 185–298. [Google Scholar]
- 51.Gorst CM, Wilks A, Yeh DC, Ortiz de Montellano PR, La Mar GN. Solution 1H NMR investigation of the molecular and electronic structure of the active site of substrate-bound human heme oxygenase: the nature of the distal hydrogen bond donor to bound ligands. J Am Chem Soc. 1998;120:8875–8884. [Google Scholar]
- 52.Sandström J. Dynamic NMR Spectroscopy. Academic Press; New York: 1982. [Google Scholar]
- 53.Li Y, Syvitski RT, Auclair K, Wilks A, Ortiz de Montellano PR, La Mar GN. Solution NMR characterization of an unusual distal H-bond network in the active site of the cyanide-inhibited, human heme oxygenase complex of the symmetric substrate, 2,4-dimethyldeuterohemin. J Biol Chem. 2002;277:33018–33031. doi: 10.1074/jbc.M204216200. [DOI] [PubMed] [Google Scholar]
- 54.Li Y, Syvitski RT, Chu GC, Ikeda-Saito M, La Mar GN. Solution 1H NMR investigation of the active site molecular and electronic structures of the substrate-bound, cyanide-inhibited bacterial heme oxygenase from C. diphtheriae. J Biol Chem. 2003;279:6651–6663. doi: 10.1074/jbc.M211249200. [DOI] [PubMed] [Google Scholar]
- 55.Liu Y, Zhang X, Yoshida T, La Mar GN. 1H NMR characterization of the solution active site structure of substrate-bound, cyanide-inhibited heme oxygenase from Neisseria meningitidis; comparison to crystal structures. Biochemistry. 2004;43:10112–10126. doi: 10.1021/bi049438s. [DOI] [PubMed] [Google Scholar]
- 56.Li Y, Syvitski RT, Auclair K, Ortiz de Montellano PR, La Mar GN. Solution 1H, 15N NMR spectroscopic characterization of substrate-bound cyanide-inhibited, human heme oxygenase: water occupation of the distal cavity. J Am Chem Soc. 2003;125:13392–13403. doi: 10.1021/ja036176t. [DOI] [PubMed] [Google Scholar]
- 57.Shokhirev NV, Walker FA. The effect of axial ligand plane orientation on the contact and pseudocontact shifts of low-spin ferriheme proteins. J Biol Inorg Chem. 1998;3:581–594. [Google Scholar]
- 58.Zhang J, Osborne JP, Gennis RB, Wang X. Proton NMR study of the heme environment in bacterial quinol oxidases. Arch Biochem Biophys. 2004;421:186–191. doi: 10.1016/j.abb.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 59.Noguchi M, Yoshida T, Kikuchi G. Purification and properties of biliverdin reductases from pig spleen and rat liver. J Biochem. 1979;86:833–848. doi: 10.1093/oxfordjournals.jbchem.a132615. [DOI] [PubMed] [Google Scholar]
- 60.Yoshida T, Kikuchi G. Features of the reaction of heme degradation catalyzed by the reconstituted microsomal heme oxygenase system. J Biol Chem. 1978;253:4230–4236. [PubMed] [Google Scholar]
- 61.Zhou H, Migita CT, Sato M, Sun DY, Zhang XH, Ikeda-Saito M, Fujii H, Yoshida T. Participation of carboxylate amino acid side chain in regiospecific oxidation of heme by heme oxygenase. J Am Chem Soc. 2000;122:8311–8312. [Google Scholar]
- 62.Zhu W, Li Y, Wang J, Ortiz de Montellano PR, La Mar GN. Solution NMR study of environmental effects on substrate seating in human heme oxygenase; Influence of polypeptide truncation, substrate modification and axial ligand. J Inorg Biochem. 2005 doi: 10.1016/j.jinorgbio.2005.08.010. in press. [DOI] [PubMed] [Google Scholar]
- 63.Wang J, Ortiz de Montellano PR. The binding sites on human heme oxygenase-1 for cytochrome P450 reductase and biliverdin reductase. J Biol Chem. 2003;278:20069–20076. doi: 10.1074/jbc.M300989200. [DOI] [PubMed] [Google Scholar]
- 64.Higashimoto Y, Sakamoto H, Hayashi S, Sugishima M, Fukuyama K, Palmer G, Noguchi M. Involvement of NADP(H) in the interaction between heme oxygenase-1 and cytochrome P450 reductase. J Biol Chem. 2005;280:729–737. doi: 10.1074/jbc.M406203200. [DOI] [PubMed] [Google Scholar]
- 65.Zeng Y, Deshmukh R, Caigan GA, Bunce RA, Rivera M, Wilks A. Mixed regioselectivity in the Arg-177 mutants of Corynebacterium diphtheriae heme oxygenase as a consequence of in-plane heme disorder. Biochemistry. 2004;43:5222–5238. doi: 10.1021/bi035970o. [DOI] [PubMed] [Google Scholar]
- 66.Deshmukh R, Zeng Y, Furci LM, Huang HW, Morgan BN, Sander S, Alontaga AY, Bunce RA, Moenne-Loccoz P, Rivera M, Wilks A. Heme oxidation in a chimeric protein of the α-selective Neisseriae meningitides heme oxygenase with the distal helix of the δ-selective Pseudomonas aeruginosa. Biochemistry. 2005;44:13713–13723. doi: 10.1021/bi050810t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
One Table (chemical shifts of assigned residues) and one figure (TOCSY spectra). This information is available free of charge via the internet at http://pubs.acs.org