

Abstract
Shiga toxins (Stx), released into the intestinal lumen by enterohemorrhagic E. coli (EHEC), are major virulence factors responsible for gastrointestinal and systemic illnesses. These pathologies are believed to be due to the action of the toxins on endothelial cells, which express the Stx receptor, the glycosphingolipid Gb3. To reach the endothelial cells, Stx must translocate across the intestinal epithelial monolayer. This process is poorly understood. We investigated Stx1 movement across the intestinal epithelial T84 cell model and the role of actin turnover in this transcytosis. We showed that changes in the actin cytoskeleton due to latrunculin B, but not cytochalasin D or jasplakinolide, significantly facilitate toxin transcytosis across T84 monolayers. This trafficking is transcellular and completely inhibited by tannic acid, a cell impermeable plasma membrane fixative. This indicates that actin turnover could play an important role in Stx1 transcellular transcytosis across intestinal epithelium in vitro. Since EHEC attachment to epithelial cells causes an actin rearrangement, this finding may be highly relevant to Stx-induced disease.
Keywords: Shiga toxin, intestinal epithelial cells, transcytosis, actin turnover, latrunculin B
1. Introduction
Shiga toxins 1 and 2 (Stx1, Stx2), are major virulence factors in foodborne illnesses caused by enterohemorrhagic E. coli (EHEC). Stx1 and 2 belong to a class of so-called AB5 bacterial toxins [1], which consist of a single catalytic A-subunit and a pentamer of identical B-subunits, the latter responsible for receptor binding and intracellular trafficking. The A-subunit of Stx1 possesses N-glycosidase activity and inhibits protein synthesis through specific removal of the adenine base at position 4324 of the 28S ribosomal RNA of the 60S subunit of the eukaryotic ribosome.
Individuals who become infected with EHEC often develop watery diarrhea, which then becomes bloody, followed by recovery. However, up to 10% of infected individuals, mostly children, develop life-threatening systemic complications including hemolytic uremic syndrome (HUS) [1,2]. Antibiotic use against the EHEC infections has proven to be harmful, increasing the risks of HUS [3,4].
The principal gastrointestinal and systemic manifestations of Stx-related diseases are believed to be due to the action of the toxins on vascular endothelial cells, which express the glycosphingolipid Gb3, the only well characterized Stx receptor [5,6]. EHEC strains are not generally invasive like the prototypic Stx-producing Shigella dysenteriae type 1. The question of how Stx gains access from the gut lumen to the underlying endothelial cells thus is crucial to understand the systemic complications of EHEC.
Stx1 might traverse intestinal epithelial layer by transcellular or paracellular pathways. It has been shown that Gb3 is absent from human intestinal epithelial cells [7–11]. This excludes a Gb3-dependent route of toxin transcytosis and adds more complexity to the mechanism of interaction between the toxin and intestinal epithelial cells.
Several studies have been performed to elucidate the pathways of Stx transcytosis across intestinal epithelial cells. Translocation of Stx1 was examined [12,13] in highly polarized intestinal epithelial CaCo-2A cells (Gb3 positive) and T84 cells (Gb3 negative). Biologically active Stx1 was translocated from the apical to the basolateral surfaces of both cell lines, which indicates that toxin transcytosis does not require Gb3 receptor binding. Stx1 transcytosis did not affect either transepithelial electrical resistance (TER) or movement of [3H]inulin, suggesting that apical-to-basolateral toxin movement was through the cells rather than the paracellular pathway [12, 13]. The T84 cells have been shown to be resistant to Stx1-induced cytotoxicity over 24h of exposure [9,12,13]. However, the efficiency of Stx1 transcytosis was relatively low and only ~ 2% of the apical amount of Stx1 was transferred across the cell monolayer in 24h.
We speculated that the process of transcellular toxin transcytosis might be facilitated in EHEC infection. During the course of infection, EHEC subvert the host cell signaling machinery to rearrange the actin cytoskeleton and produce characteristic attaching and effacing lesions on the surface of intestinal epithelial cells [14,15]. This actin rearrangement might facilitate the toxin movement across the monolayer. Therefore, the goal of these studies was to determine the effect of actin polymerization-depolymerization on toxin transcytosis across the intestinal epithelial barrier.
Here using an in vitro intestinal epithelial T84 cell model, we demonstrated that latrunculin B (LatB), significantly increases the transcytosis of the Stx1 B-subunit (Stx1B). Complete inhibition of LatB-induced Stx1B transcytosis by basolateral application of tannic acid demonstrates that this toxin movement occurs through the cells, rather than through tight junctions (TJ). The effect of LatB on Stx1 transcytosis is specific, and neither cytochalasin D (CytD) nor jasplakinolide (Jasp), which both affect filamentous actin turnover and similarly to LatB decrease TER of intestinal epithelial monolayers, did not influence Stx1B transport across the T84 cells. This LatB-facilitated Stx1B transcellular transcytosis is associated with changes in amount of at least one actin-binding protein, such as phospho-cofilin-1. We suggest that changes in actin cytoskeleton similar to these caused by LatB might occur during EHEC interaction with the intestinal epithelium.
2. Materials and Methods
2.1. Materials
Purified Stx1 and recombinant Stx1B were prepared by Dr. A. Kane (GRASP Center, Tufts New England Medical Center, Boston, MA) as previously described [16,17]. The quality of Stx1 and Stx1B has been checked on 15% SDS-PAGE followed by GelCode Blue staining (Pierce, Rockford, IL) and two bands (~ 32kDa for A-subunit and) or one band (< 10kDa for B-subunit) respectively, were detected. B-subunit of Cholera toxin (CTB) was from List Biological Laboratories, Inc. LatB was from Axxora (San Diego, CA). The following antibodies were rabbit polyclonals: anti-cofilin, a generous gift from Dr. J.R.Bamburg (Colorado State University, CO), anti-phospho-cofilin and anti-profilin from Cell Signaling Technology (Danvers, MA). Rabbit monoclonal anti-GAPDH was from US Biological (Swampscott, MA), mouse monoclonal anti-cathepsin D was a gift from Dr. A. Hubbard (Johns Hopkins University, Baltimore, MD). All fluorescent secondary antibodies, phalloidin, and 40kDa dextran conjugated to Alexa 488 fluorescent dye were from Molecular Probes-Invitrogen (Carslbad, CA). Stx1, CTB, and recombinant Stx1B were conjugated with Alexa fluor 680, Alexa fluor 568, or Alexa fluor 488 reactive dyes according to the manufacturer’s protocol (Molecular Probes–Invitrogen). All other chemical reagents were purchased from Sigma (St. Louis, MO).
2.2. Cell culture
Human colonic epithelial T84 cells (A.T.C.C., Manassas, VA) were grown and maintained in culture in DMEM (Dulbecco's modified Eagle's medium)/Ham's F-12 medium (1:1) supplemented with 10% fetal bovine serum, 100 units/ml penicillin and 100 µg/ml streptomycin. All media ingredients were obtained from Invitrogen (Carlsbad, CA). Monolayers (passage 27–45) were grown on polycarbonate inserts with 0.4-µm pore size (Costar, Cambridge, MA) for 7–12 days. The experiments were performed on confluent monolayers with transepithelial electrical resistance (TER) ~ 1,500 Ω·cm2.
2.3. Detection of Stx1, Stx1B, CTB and dextran transcytosis
Cells grown on polycarbonate inserts were incubated with indicated concentrations of Stx1, Stx1B, CTB or dextran in the presence or absence of LatB over the indicated times. At the end of incubation, the inserts were removed and the relative fluorescence intensity in the lower chamber was measured by Odyssey Fluorescence Imaging System (Li-Cor Biosciences, Lincoln, NE).
2.4. Detection of Intracellular Stx1B
Confluent T84 cells were incubated over indicated time with indicated concentrations of Stx1B conjugated to Alexa 680 fluorescent dye (Stx1B-680). Cells then were washed three times with cold PBS to remove extracellular toxin and lysed in RIPA buffer (1% Triton X100, 0.5% deoxycholic acid, 0.1% SDS, 50mM TRIS HCL, pH 7.4, 150mM NaCl), containing 0.5mM Na3VO4 and Protease inhibitor cocktail 1:1000 (Sigma P8340) and sonicated. Lysates were centrifuged at 20,000g at 4°C for 15min. Equal amounts of collected proteins were loaded into the 96-well plates and the fluorescence intensity of Stx1B-680, which corresponds to the amount of intracellular Stx1B, was measured and normalized by the autofluorescence of an equal amount of protein from cells not exposed to Stx1B.
For experiments with Brefeldin A (BFA), T84 cells were pretreated with 0.1 µM BFA for 1h and incubated with Stx1B in the presence of BFA for additional 1, 4 or 24h. The amount of Stx1B inside the BFA-treated cells and in lower chambers relative to these in controls was measured by Odyssey.
2.5. Immunoblotting
Equal amounts of collected proteins from total cell lysates were separated by SDS-PAGE, transblotted onto nitrocellulose membranes, and analyzed by Western blot. Proteins of interest were detected by either ECL Western Blotting Detection reagents (Amersham, Princeton, NJ) or by Odyssey Fluorescence Imaging System.
2.6. Immunofluorescence
For F-actin detection, the confluent T84 monolayers were incubated over indicated times with 0.5 µg/ml Stx1B conjugated to Alexa 568 fluorescent dye (Stx1B-568), fixed with 3% formaldehyde in PBS for 10 min., washed extensively in PBS, non-permeabilized or permeabilized with 0.1% saponin, blocked with 2% BSA and 15% FBS for 30 min. and incubated 1h with phalloidin and Hoechst for nuclear staining. For Stx1B and CTB co-localization experiments, cells were incubated over the indicated time with 0.5 µg/ml Stx1B-568 and 0.5 µg/ml CTB conjugated to Alexa fluor 488 (CTB-488) and fixed as above. For Stx1B and lysosomal co-localization, cells were incubated over the indicated time with 0.5 µg/ml Stx1B-568, fixed, permeabilized and blocked as above, incubated with anti-cathepsin D antibody at room temperature for 1 h, washed 3 times in PBS and incubated with secondary fluorescent antibodies for an additional 1h. All cell preparations were mounted on glass slides for confocal microscopy examination. Fluorescent image acquisition of tissue was performed using a Zeiss 410 LSM confocal imaging system. Collected 8-bit fluorescence images of confocal optical sectioning with 0.5 µm steps were stored on disc and fluorescence intensity was quantified by MetaMorph software (Molecular Devices Corporation, Downingtown, PA), as we previously described [18].
2.7. Quantification of G-actin and F-actin
Fractionation of G-actin and F-actin was performed by Triton X-100 extraction of intracellular actin as described [19]. Briefly, T84 cells were washed with HBSS and G-actin was extracted by gentle shaking for 5 min at room temperature in HBSS containing 1% Triton X-100, proteinase inhibitor cocktail and 1 µg/ml phalloidin to prevent filament disassembly. The Triton X-100–soluble G-actin fraction was mixed with an equal volume of 2x SDS sample buffer and boiled. Cells were then briefly washed with HBSS and the Triton X-100–insoluble F-actin fraction was collected by scraping cells in two volumes of SDS sample buffer, and boiled. The amount of actin in each fraction was determined by gel electrophoresis, Western blotting, and Odyssey Fluorescence Imaging System as described above.
2.8. Statistical analysis
Values are presented as mean ± SEM. Normal distribution for each dataset was tested using statistical package available at http://home.ubalt.edu/ntsbarsh/. Statistical significance was determined using Student’s unpaired t- test and p-value <0.05 was considered significant.
3. Results
3.1. Stx1 and Stx1B are both transcytosed across T84 cells
It has been previously shown that ~ 2% of apically added active Stx1 holotoxin translocates across the T84 monolayer in 24h [12]. We repeated these experiments using both fluorescently labeled holotoxin (Stx1-680) and B-subunit of Stx1 (Stx1B-680). Confluent T84 cells were exposed apically either to Stx1B-680 (0.1 µg), or to increasing amount of Stx1B-680 (0.06 µg, 0.125 µg, 0.25 µg and 0.5µg) in 0.5 ml of apical media. The relative amount of transcytosed Stx1-680 or Stx1B-680 was measured in the lower chamber media 24 h later by quantifying the toxin 680 nm relative fluorescence intensity. The absolute amount of transcytosed Stx1-680 or Stx1B-680 was calculated from fluorescence intensity by comparison to the Stx1-680 or Stx1B-680 fluorescence intensity calibration curve generated from three different concentrations of Stx1-680 or seven different concentrations of Stx1B in a range between 1µg/ml and 5ng/ml. Similarly to the non- fluorescent holotoxin [12], Stx1-680 and Stx1B-680 transcytosis across confluent T84 cells is a slow process. Only ~ 4 ng of Stx1-680, which corresponds to 3.9 ±0.6 % of total apical Stx1-680 (n= 6 filters) was transcytosed across the monolayer in 24h. In case of Stx1B, only ~ 4 ng, ~ 8ng, ~ 13ng and ~ 28ng of toxin respectively was recovered from lower chamber in 24h, which corresponds to 6.0 % ± 0.4 of total apical amount of toxin (n= 6 filters per each conditions).
We also measured the TER of T84 monolayers treated for 24h with Stx1B-680 to ensure that there were no Stx1B-induced changes in the TJ. Similarly to the previously studied holotoxin [9,12,13] neither 1µg/ml of apical non-fluorescent Stx1B nor Stx1B-680 change TER over 24h of incubation. From these experiments we concluded that Stx1B is sufficient for holotoxin transcytosis across T84 monolayer and also that Stx1B-680 is a suitable tool to study the Stx1 translocation across the intestinal epithelial monolayer.
As in case of holotoxin [12], Stx1B-680 transcytosis in T84 cells was also insensitive to 0.1µM BFA, indicating a lack of involvement of the retrograde pathway in toxin transcytosis. Higher doses of BFA were toxic to the T84 cells, caused a significant drop in TER and apoptosis characterized by caspase-3 activation (data not shown).
3.2. Stx1 and Stx1B are internalized by Gb3 free intestinal epithelial T84 cells
Stx1 movement through the Gb3-negative T84 monolayers without changes in TER or paracellular permeability [9,12] suggests that in the absence of Gb3 the Stx1 is endocytosed by an as yet uncharacterized mechanism. We hypothesized, if Stx1/Stx1B uptake by Gb3-free cells is a receptor mediated process (possibly another low affinity or low expression receptor), then first order reaction kinetics should apply, which predicts a concentration and time-dependent saturated uptake due to receptor saturation [20]. To determine the toxin uptake as a function of the apical toxin concentration and as a function of time, T84 cells were incubated with Stx1-680 holotoxin or Stx1B-680 and the Stx1-680 or Stx1B-680 fluorescence intensity in total cell lysates, which corresponds to the relative amount of endocytosed toxin, was measured. Both, dose and time dependencies of toxin uptake demonstrated a linear non-saturating relationship between the amounts of endocytosed holotoxin or Stx1B and apical toxin concentration or incubation time, respectively (Fig. 1A and B). Additionally, incubation of T84 cells at 4°C with fluorescently labeled Stx1B over 3h did not result in detectable toxin binding to the apical surface (data not shown). These results demonstrate that Stx1 and Stx1B uptake by T84 cells is not a receptor mediated process.
Fig. 1.
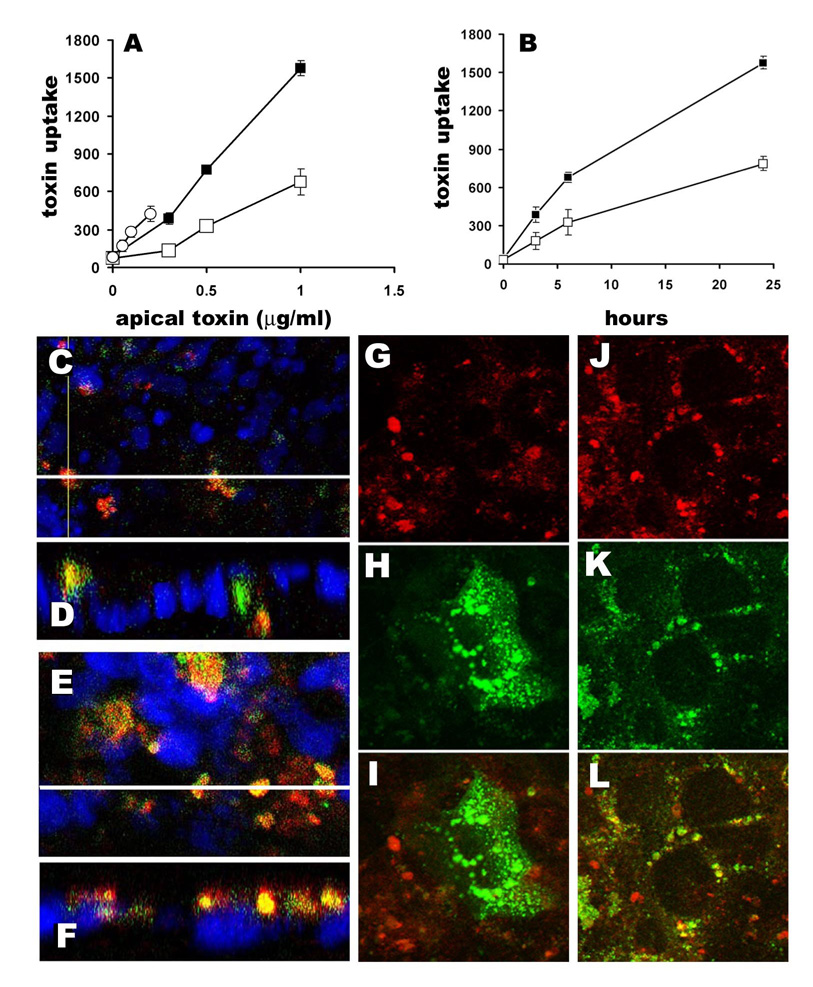
Stx1B uptake by T84 cells is not receptor-mediated, but is an actin-dependent process. A and B - concentration and time dependencies of Stx1 and Stx1B uptake by T84 cells. A – three apical concentrations of Stx1-680 (0.05, 0.1 and 0.2 µg/ml) and Stx1B (0.3, 0.5 and 1µg/ml) were used and the intracellular amount of Stx1B at 6h (open squares) and 24h (black squares) or Stx1 at 24h (open circles) were measured in total cell lysates. B - Two concentrations of Stx1B (0.5 µg/ml - open squares and 1 µg/ml - black squares) were applied to the apical surface of T84 cells. Stx1B amount in 100 µg of protein from total cell lysates was measured as fluorescence intensity of Stx1B-680 at 15 min, 3, 6 and 24, h of incubation. C–F - representative confocal images of T84 cells incubated with Stx1B-568 (red) and 40 kDa dextran-488 (green) for C – 4 h and E – 24 h. D and F are corresponding confocal microscopy XZ projections, respectively. Nuclei in all panels – blue (by Hoechst). White lines in C and E show the direction of optical z-sections. More Stx1B and dextran-positive vesicles were present inside the cells in 24 h than in 4h. The majority of Stx1B and dextran vesicles were co-localized (yellow) at 4h and 24h of incubation. G-I – Stx1B does not co-localize with B-subunit of cholera toxin. G- representative confocal image of Stx1B-568, and H- corresponding image of B-subunit of cholera toxin- 488, and I – overlay. J–L – Stx1B intacellular pattern partially overlaps with cathepsin D. J-representative confocal image of Stx1B-568, and K- corresponding image of cathepsin D, and L – overlay.
To further test the hypothesis that Stx1/Stx1B uptake into T84 cells is mainly through non-receptor mediated endocytosis, T84 cells were incubated simultaneously with fluorescently labeled Stx1B and 40 kDa dextran, the latter being a fluid phase endocytosis marker, the MW of which closely matches the MW of the Stx1B pentamer. The distribution of Stx1B relative to the dextran was compared by confocal microscopy after 4 and 24h of co-incubation (Fig. 1C–F). The co-localization function (MetaMorph), showed that ~ 78±13 % of Stx1B intracellular vesicles were co-localized with 40kDa dextran vesicles upon 24h of co-incubation. This suggests that fluid phase endocytosis may be a major pathway for Stx1 uptake in receptor-free cells. The Stx1B co-localized significantly neither with B-subunit of cholera toxin (CTB) (Fig. 1G–I), which upon uptake by GM1-receptor mediated endocytosis marks the whole retrograde pathway [21], nor with a marker for recycling endosomes, Rab11 (data not shown). However, the Stx1B intracellular pattern partially matched the pattern of the lysosomal marker cathepsin D (Fig. 1J–L).
3.3. Cortical actin is involved in Stx1/Stx1B uptake by T84 cells
Fluid phase endocytosis is an actin-driven process and in polarized cells the cargo resides inside apical actin-coated vesicles [22–24]. We found that during uptake, Stx1B resides inside spherical actin vesicles on the apical surface of T84 cells (Fig. 2A–F). Moreover, Stx1B uptake is accompanied by an increase in apical F-actin amount. However, inside the cells the Stx1B vesicles are not covered by actin (Fig. 2G), indicating that Stx1B-containing vesicles undergo some sub-apical sorting. We concluded that in receptor-free intestinal epithelial cells, actin remodeling is involved in Stx1/Stx1B uptake into some sub-apical compartment.
Fig. 2.
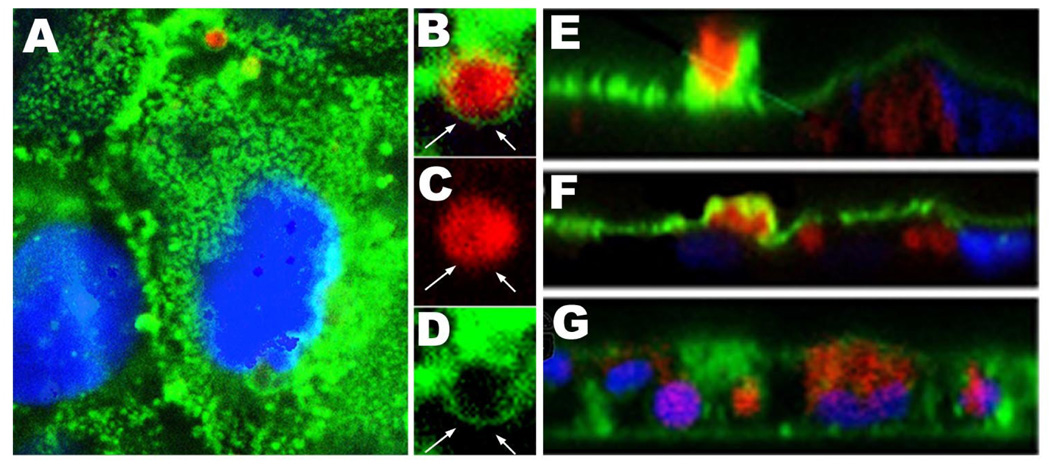
A – representative confocal image of the apical area of T84 cells which are in the process of Stx1B uptake, Stx1B-568 – red, actin (by phalloidin-Alexa 488) – green. B, C and D – zoom on Stx1B-actin vesicle (square from A). Note, that Stx1B (red) is coated by the F-actin (green) sphere (arrows). E and F - XZ projection through non-permeabilized T84 cells incubated with Stx1B for 4h. Apical actin (green) forms a sphere around Stx1B vesicle (red), which is taken up across the apical membrane. Some Stx1B vesicles are already inside the cells. G - XZ projection through permeabilized T84 cells. Stx1B vesicles (red) inside the cells are not covered by actin (green), indicating that upon internalization the Stx1B vesicles are loosing F-actin coating.
3.4. LatB significantly increased the transcytosis and uptake of Stx1B
Based on our data, we hypothesized that changes in actin cytoskeleton might facilitate toxin movement across the intestinal epithelial layer. Subsequently, the effect of actin polymerizing-depolymerizing agents, including CytD, LatB and Jasp, on Stx1/Stx1B uptake and transcytosis was studied. For these experiments, T84 cells were treated for 24h with Stx1B-680 and 1µM of Jasp or LatB were apically added for the last 4h or 1µM of CytD for the last 2 h. These treatments had been previously reported to significantly affect the F-actin pool in T84 cells [22,25].
Quantification of Stx1B in total cell lysates (Fig. 3A) showed significant increase in the amount of intracellular toxin due to LatB. CytD significantly decreased the intracellular amount of Stx1B, while Jasp had no effect on the amount of intracellular toxin. Quantification of Stx1B in the lower chamber showed that LatB significantly increased toxin transcytosis, while neither CytD nor Jasp affected transcytosis (Fig.3 B and C).
Fig. 3.
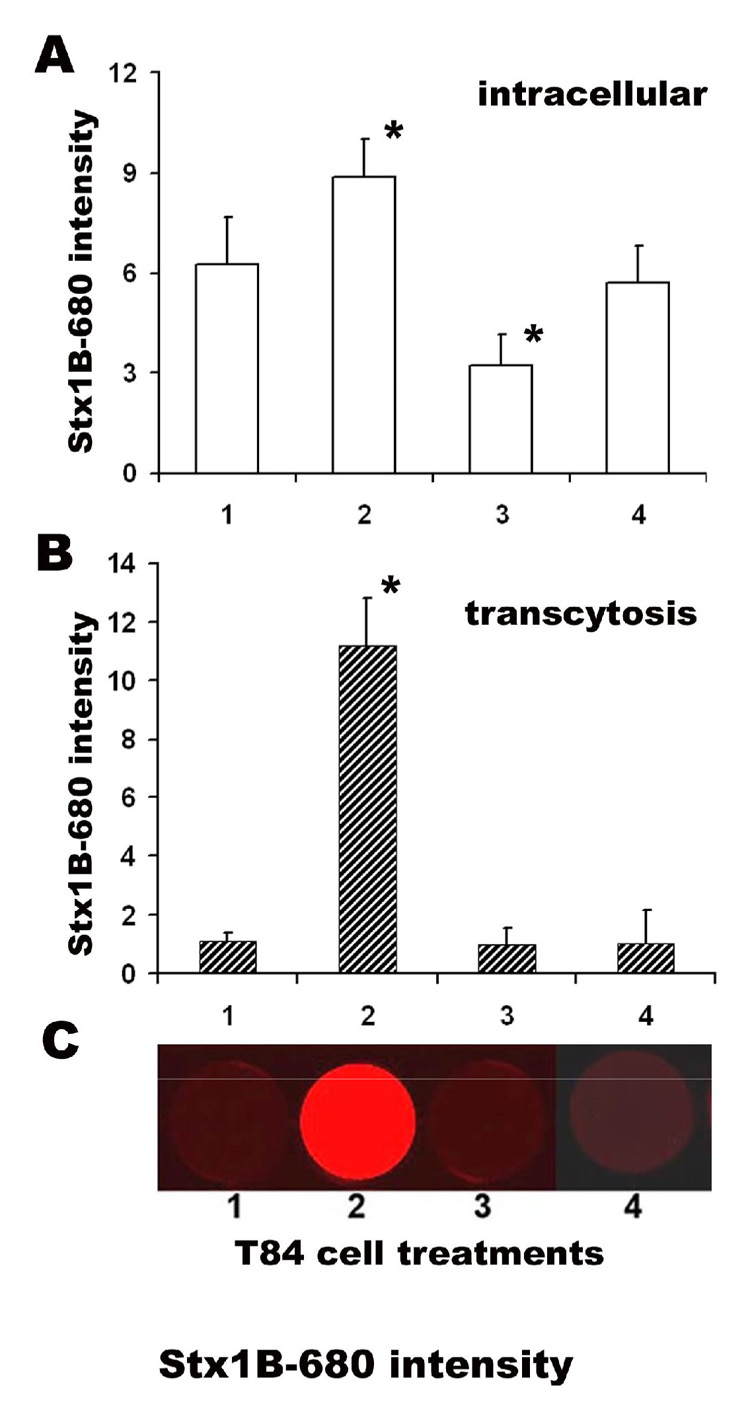
LatB significantly increase Stx1 transcellular transcytosis across T-84 monolayers. Stx1B-680 relative fluorescence intensity in A – total cell lysates from cells 1 - contols; or treated with 2 - 1µM LatB; 3 - 1µM CytD; 6 - 1µM Jasp. LatB significantly increased the intracellular Stx1B amount compared to control non-treated cells, while CytD significantly decreased it (* p<0.05 vs. control). Jasp did not affect the intracellular Stx1B content. Stx1B-680 relative fluorescence intensity in B – lower chamber (transcytosis) with C - representative images of Stx1B-680 fluorescence intensity in lower chamber. LatB significantly increased (*p<0.05 vs. control) the Stx1B transcytosis, while neither CytD nor Jasp affected the Stx1B transcytosis. The data shown are means ± SEM for six independent experiments.
These data show that LatB treatment possibly increases both the toxin uptake across the apical membrane and toxin efflux through the basolateral membrane. To test whether actin modification by LatB stimulates both processes, the effect of LatB on Stx1B uptake and transcytosis was measured over shorter periods of time than 24h. For these experiments T84 cells were exposed simultaneously to 1 µg/ml Stx1B-680 and 1µM LatB over different times between 15 min and 4h, and the amounts of toxin inside the cells and in the lower chamber were compared to control cells incubated with Stx1B only (Fig.4 A and B). Incubation with LatB over 30 min significantly increased the amount of intracellular toxin compared to control cells; 4h of LatB treatment increased the intracellular amount of Stx1B ~ 5 times. LatB also significantly increased Stx1B transcytosis, indicating that in T84 cells LatB facilitates the Stx1B transport across both apical and basolateral membranes. However, there was a time delay in LatB stimulated Stx1B transcytosis compared to its effect on Stx1B uptake, and at times shorter than 1h, LatB treatment did not affect transcytosis (Fig.4B). Treatment of T84 cells with different concentrations of LatB over 4h showed significant increase of Stx1B transcytosis starting at 0.5 µM LatB (Fig.4C).
Fig. 4.
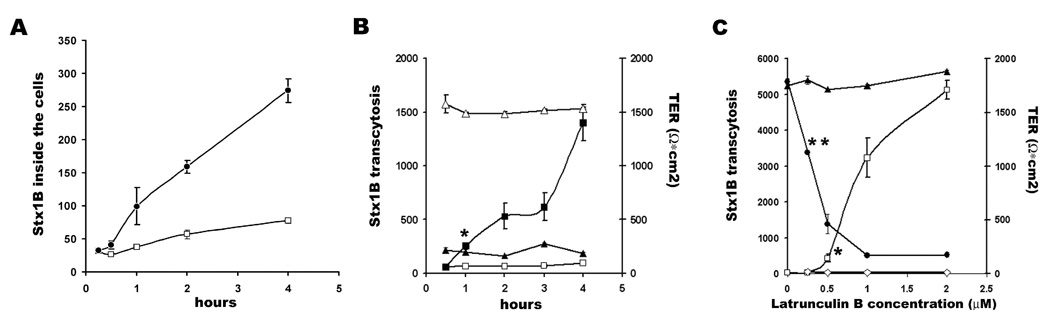
LatB time and dose effects on Stx1B uptake and transcytosis. A- Stx1B-680 relative intensity fluorescence in total cell lysates as a function of time from 1µM LatB treated (black circles) and non-treated (open squares) cells. LatB significantly increased Stx1B uptake in 30 min after its apical application (* p<0.05 vs. control). B – Relative fluorescence intensity of transcytosed Stx1B-680 (left) as a function of time from 1µM LatB treated (black squares) and non-treated (open) cells. LatB significantly increased Stx1B transcytosis 1h after application (*p<0.05 vs. control). While TER of T84 monolayers (right) dropped significantly in 30 min of LatB treatment (black triangles) compared to control monolayers (open triangles). C- Relative fluorescence intensity of transcytosed Stx1B-680 (left) as a function of LatB concentration (open squares) or LatB in combination with 0.1% tannic acid (open rhombs) measured after 4h of treatment. Starting from 0.5 µM LatB significantly increased Stx1B transcytosis (*p<0.05 vs. control). Tannic acid completely prevented Stx1B transcytosis induced by LatB. TER (right) significantly decreased from 0.25 µM LatB (**p<0.05 vs. control). The data shown are means ± SEM for four independent experiments.
It is well established that latrunculins, including LatB, significantly decrease the TER of polarized intestinal epithelial monolayers, including T84 cells [19,26,27]. To ensure that LatB induced increase in Stx1 transcytosis was not caused by changes in TER (Fig.4 B and C) and possibly changes in paracellular permeability [24,25], but is due to the actin-dependent changes in toxin uptake and transcellular trafficking, T84 cells were treated basolaterally for 4h with 0.1% tannic acid, a cell-impermeable cross-linker of cell surface carbohydrates that selectively fixes the plasma membrane but does not diffuse across the TJ [22, 28–30]. This treatment did not change the properties of the TJ in T84 monolayers [22]. Tannic acid completely inhibited the effect of LatB on Stx1B transcytosis (Fig. 4C), suggesting that LatB facilitated transcytosis of Stx1B is a transcellular process and actin polymerization-depolymerization plays an important role in its regulation. Basolateral tannic acid treatment in the presence of LatB also significantly increased the Stx1B amount in total cell lysates compared to non-treated controls. Thus, relative fluorescence intensity in control cells exposed to Stx1B-680 for 4h was 57 ± 11 units, while in cells treated with for 4h with 1µM LatB apically and tannic acid basolaterally the Stx1B-680 relative intensity was 678± 37 units (p<0.05 vs. control, 3 independent experiments). Tannic acid alone did not affect the amount of intracellular toxin compared to the control cells, and Stx1B-680 relative intensity in the presence of tannic acid was 66 ± 8 units (3 independent experiments).
The LatB-mediated changes in cytoskeleton that facilitate Stx1B uptake and transcytosis through T84 monolayers are different from these leading to decrease in TER (Fig. 3), which occur rapidly 10–15 min after treatment and at low LatB concentrations that do not affect Stx1B transcytosis [27].
3.5 LatB facilitates the transcytosis of other molecules across intestinal epithelial cells
The effect of LatB on transcellular transcytosis has not been studied in detail in polarized intestinal epithelial cells. Subsequently, we tested whether LatB stimulates the transcytosis of several molecules which use different endocytic mechanisms for uptake. For these experiments, T84 cells were incubated for 4h in the presence or the absence of 1µM LatB either with 40kDa dextran or CTB, the last binds to apical GM1 receptors. CTB can be taken up by clathrin- and caveolin-dependent and independent mechanisms and is transcytosed independently of its retrograde trafficking pathway [21,31,32]. Similarly to Stx1B, LatB increased dextran transcytosis from 19.5±4.2 relative fluorescent intensity units in control cells to 182.7±3.7 in the presence of LatB (p<0.05; n= 8 experiments). LatB also significantly increased the transcytosis of CTB from 30.2±2.8 relative fluorescent intensity units in control cells to 664.8 ±111.0 in the presence of LatB (p<0.05; n= 8 experiments).
3.6. LatB-induced change in cofilin-1 phosphorylation may be involved in Stx1B facilitated transcytosis
Additionally to the sequestration of monomeric G-actin, both Lat A and B has been shown to influence the expression or phosphorylation of several actin binding proteins [33,34]. We tested whether LatB-induced increase in G-actin is also accompanied by changes in the expression or postranslational modification of some G-actin-binding proteins in T84 cells, such as cofilin and profilin [35,36]. For these experiments, cells were treated with 1µM LatB over different time intervals between 15 min and 4h, and the expression of cofilin-1 and its phosphorylated form, and profilin, were tested in total cell lysates by immunoblotting. LatB treatment significantly increased the cofilin-1 phosphorylation, which occurred as early as 15 min and continued over 4h (Fig.5). Moreover, the increase in the amount of phospho-cofilin-1 over time of LatB treatment correlates (r= 0.93 by correlation test for independent variables from Excel) with increase in the amount of G-actin, reflected in G/F actin ratio (Fig. 5C). Neither the total amount of cofilin-1 nor profilin changed over these experimental time intervals (Fig.5). Additionally, the total amount of other actin-binding proteins such as ezrin and vinculin did not change upon LatB treatment (data not shown). These data suggest that facilitation of transcellular transcytosis by LatB may be related to LatB-induced changes in the activity of cofilin-1.
Fig. 5.
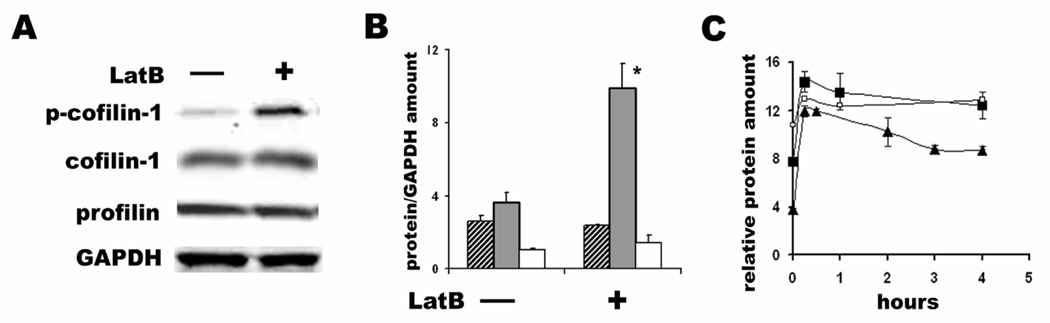
Effect of LatB on expression of actin and actin-binding proteins. A – Representative immunoblot from cells incubated with 1µM LatB for 4h and control non-treated cells. B – Quantitative analysis of cofilin-1 (striped bars), phosphocofilin-1 (black bars) and profilin (white bars) expression normalized on GAPDH in the absence and the presence of 1µM LatB for 4h. LatB significantly increase the amount of phosphocofilin-1 (*p < 0.05 vs. control). The data shown are means ± SEM for three independent experiments. C – LatB-induced changes in the relative amount of G-actin alone (open squares) or in the G/F actin ratio (black squares) are closely followed by the increase in phosphocofilin-1 (black triangles). The amount of G actin, and G/F-actin ratio and the amount of phosphocofilin-1 normalized on GAPDH are significantly higher at each timepoint of LatB treatment compared to time = 0 without LatB (p < 0.05).
4. Discussion
The Stx translocation across the intestinal epithelial layer to spread systemically is a necessary step in the development of the EHEC-induced spectrum of disease. However, the mechanisms of this translocation are unknown. The ability of fully active Stx1 and Stx2 to cross the epithelial barrier through the cells has been demonstrated [12,13, 37–39]. However, the efficiency of transcytosis was very low with only ~ 2% of apical Stx1 crossing monolayers in 24h. Although such a small amount of toxin potentially could be responsible for systemic complications, we speculated that the changes in host cell protein expression and particularly, in actin and actin-binding proteins due to EHEC attachment and effacement [14,15] might stimulate toxin uptake and transcytosis. The impact of EHEC infection on the host proteins has not been investigated in detail. However, there are some evidences that EHEC colonization changes the protein expression in intestinal epithelial cells [40]. Moreover, proteomic analysis of intestinal epithelial Caco-2 cells infected with enteropathogenic E.coli (EPEC), which form attaching/effacing lesions similar to those of EHEC, showed significant increase in the abundance of multiple actin depolymerizing proteins [41]. For instance, actin severing proteins gelsolin and cofilin were upregulated 3.57 and 2.5 times, respectively. This shows that changes in actin binding proteins occur as a part of bacterial attachment and effacement to the intestinal epithelial cells.
Here we demonstrate that actin depolymerizing drug LatB significantly increases Stx1B transcytosis in a time and a concentration-dependent manner. It is known that LatB decreases TER and increases permeability to ions and uncharged molecules with size < 10kDa [22,27]. We show that the LatB facilitated Stx1B transcytosis is not across the TJ, because Stx1B transcytosis in the presence of LatB is completely blocked by basolateral application of tannic acid. Recently the addition of tannic acid to the basolatheral cell surface has been shown to inhibit the apical to basolateral transcellular transcytosis of adeno-associated virus [39]. The selectivity of tannic acid (apical vs. basolateral) has been previously tested in T84 cells [22], and apically (but not basolaterally) applied tannic acid completely prevented cholera toxin apical uptake. In contrast, basolateral tannic acid (but not apical) effectively inhibited uptake of transferrin from the basolateral side. Taken together, our data show that LatB facilitates Stx1B transepithelial flux is transcellular and not paracellular. Additionally, both CytD and Jasp treatments have also been shown to decrease TER of intestinal epithelial monolayers [26,42,43]. However, neither drug increased Stx1B transcytosis in T84 cells, supporting our conclusion that LatB facilitates transcellular transcytosis of toxin. The increase in Stx1B amount in total cell lysates due to the tannic acid treatment in the presence of LatB additionally supports that the toxin moves across the cells.
It has been shown that CytD inhibits both receptor mediated and fluid phase endocytosis at the apical surface [44–46], while Jasp stimulates basolateral fluid phase endocytosis, but does not affect apical uptake [43]. These data are in good agreements with our findings that CytD inhibits Stx1B apical uptake, and Jasp does not change it. In general, latrunculins significantly inhibit receptor mediated endocytosis [44,47]. However, latrunculins have been shown to stimulate internalization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and the cystic fibrosis transmembrane conductance regulator (CFTR), while the mechanisms of endocytosis have not be addressed [48,49]. The increase in apical Stx1B uptake by LatB (but not by CytD or Jasp) serves as additional evidence that Gb3-free T84 cells take up Stx1B by a non-receptor mediated pathway.
These results prompted us to look for the LatB induced molecular changes which might be involved in the regulation of transcellular transcytosis. LatB affects the kinetics of actin polymerization by forming 1:1 molar complexes with G-actin [50,51]. The actin sequestration over time leads to changes in activity and expression of several actin binding proteins, including vinculin, VASP, profilin, cofilin and ADF [33,34]. The novelty in our experiments is that LatB rapidly (15 min) increased the amount of phosphorylated cofilin-1, which is inactive in terms of actin binding [52], while the total amount of this protein was unchanged. This effect was sustained over the 4h of experimental time. Cofilin-1 is a ubiquitous mediator of actin filament turnover in eukaryotes. In the phosphorylated form cofilin is unable to bind to actin, while the dephosphorylation of cofilin enables its actin severing and depolymerizing activity [33,52]. LatB-induced cofilin-1 phosphorylation should lead to F-actin destabilization and this might be important for stimulation of the transcellular pathway. A possibility that LatB influences the expression of other proteins involved in actin turnover in intestinal epithelial cells remains to be tested.
Interestingly, this LatB mediated route for transcytosis might be a common pathway for many molecules which are able to traffic across epithelial monolayers. It has been previously shown that cholera toxin (CT) and its B-subunit are transcytosed transcellularly by T84 cells, and BFA did not inhibit, but rather stimulated this process [31]. Moreover, CT transcytosis directly correlated with CT-induced Cl-secretion, indicating functional importance of this pathway. The results allow us to speculate that interaction of bacteria or bacterial products with intestinal epithelial cells might potentially recreate the LatB-induced molecular changes which significantly facilitate the transcellular transcytosis of toxins and other antigens. These data that two different AB5 toxins [53] move across the intestinal epithelial cells by a Golgi/ER independent pathway shows the importance of transcellular transcytosis in the interactions of intestinal epithelial cells with a broad variety of bacterial products, which similarly to Stx might gain access to the underlying tissues.
Acknowledgments
This work was supported by NIH grants RO1DK58928, R24DK064388 (The Hopkins Basic Research Digestive Disease Development Core Center) and P30DK 34928 (the GRASP Center). We acknowledge the editorial assistance of Ms. H. McCann.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Acheson DWK, Donohue-Rolfe A, Keusch GT. The family of Shiga and Shiga-like toxins. In: Aloufand JE, Freer JH, editors. Sourcebook of bacterial protein toxin. London, UK: Academic Press Ltd.; 1991. pp. 415–433. [Google Scholar]
- 2.Siegler R, Oakes R. Hemolytic uremic syndrome; pathogenesis, treatment, and outcome. Curr. Opin. Pediatr. 2005;17:200–204. doi: 10.1097/01.mop.0000152997.66070.e9. [DOI] [PubMed] [Google Scholar]
- 3.Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 1995;365:1073–1086. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 4.Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N. Engl. J. Med. 2000;342:1930–1936. doi: 10.1056/NEJM200006293422601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donohue-Rolfe A, Keusch GT, Edson C, Thorley-Lawson D, Jacewicz M. Pathogenesis of Shigella diarrhea. 9. Simplified high-yield purification of shigella toxin and characterization of subunit composition and function by the use of subunit-specific monoclonal and polyclonal antibodies. J Exp Med. 1984;160:1767–1781. doi: 10.1084/jem.160.6.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacewicz M, Clausen H, Nudelman E, Donohue-Rolfe A, Keusch GT. Pathogenesis of Shigella diarrhea.11. Isolation of a Shigella toxin-binding glycolipid from rabbit jejunum and Hela cells and its identification as globotriaosylceramide. J. Exp. Med. 1986;163:1391–1404. doi: 10.1084/jem.163.6.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holgersson J, Jovell PA, Breimer ME. Glycosphingolipids of the human large intestine: detailed structural characterization with special reference to blood group compounds and bacterial receptor structures. J Biochem (Tokyo) 1991;110:120–131. doi: 10.1093/oxfordjournals.jbchem.a123530. [DOI] [PubMed] [Google Scholar]
- 8.Ergonul Z, Clayton F, Fogo AB, Kohan DE. Shigatoxin-1 binding and receptor expression in human kidneys do not change with age. Pediatr. Nephrol. 2003;18:246–253. doi: 10.1007/s00467-002-1025-9. [DOI] [PubMed] [Google Scholar]
- 9.Schüller S, Frankel G, Phillips AD. Interaction of Shiga toxin from Escherichia coli with human intestinal epithelial cell lines and explants: Stx2 induces epithelial damage in organ culture. Cell. Microbiol. 2004;6:289–301. doi: 10.1046/j.1462-5822.2004.00370.x. [DOI] [PubMed] [Google Scholar]
- 10.Kovbasnjuk O, Mourtazina R, Baibakov B, Wang T, Elowsky C, Choti MA, Kane A, Donowitz M. The glycosphingolipid globotriaosylceramide in the metastatic transformation of colon cancer. Proc. Natl. Acad. Sci. U S A. 2005;102:19087–19092. doi: 10.1073/pnas.0506474102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyamoto Y, Iimura M, Kaper JB, Torres AG, Kagnoff MF. Role of Shiga toxin versus H7 flagellin in enterohaemorrhagic Escherichia coli signalling of human colon epithelium in vivo. Cell. Microbiol. 2006;8:869–879. doi: 10.1111/j.1462-5822.2005.00673.x. [DOI] [PubMed] [Google Scholar]
- 12.Acheson DW, Moore R, De Breucker S, Lincicome L, Jacewicz M, Skutelsky E, Keusch GT. Translocation of Shiga toxin across polarized intestinal cells in tissue culture. Infect. Immun. 1996;64:3294–3300. doi: 10.1128/iai.64.8.3294-3300.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sherman A, Philpott DJ, Ackerley CA, Kiliaan AJ, Karmali MA, Perdue MH, Sherman PM. Translocation of verotoxin-1 across T84 monolayers: mechanism of bacterial toxin penetration of epithelium. Am. J. Physiol. 1997;273:1349–1358. doi: 10.1152/ajpgi.1997.273.6.G1349. [DOI] [PubMed] [Google Scholar]
- 14.Goosney DL, DeVinney R, Finlay BB. Recruitment of cytoskeletal and signaling proteins to enteropathogenic and enterohemorrhagic Escherichia coli pedestals. Infect. Immun. 2001;69:3315–3322. doi: 10.1128/IAI.69.5.3315-3322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caron E, Crepin VF, Simpson N, Knutton S, Garmendia J, Frankel G. Subversion of actin dynamics by EPEC and EHEC. Curr. Opin. Microbiol. 2006;9:40–45. doi: 10.1016/j.mib.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Acheson DW, Jacewicz M, Kane AV, Donohue-Rolfe A, Keusch GT. One step high yield affinity purification of Shiga-like toxin II variants and quantitation using enzyme linked immunosorbent assays. Microbial Pathogenesis. 1993;14:57–66. doi: 10.1006/mpat.1993.1006. [DOI] [PubMed] [Google Scholar]
- 17.Acheson DW, Calderwood SB, Boyko SA, Lincicome LL, Kane AV, Donohue-Rolfe A, Keusch GT. Comparison of Shiga-like toxin I B-subunit expression and localization in Escherichia coli and Vibrio cholerae by using trc or iron-regulated promoter systems. Infect. Immun. 1993;61:1098–1104. doi: 10.1128/iai.61.3.1098-1104.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kovbasnjuk O, Edidin M, Donowitz M. Role of lipid rafts in Shiga toxin 1 interaction with the apical surface of Caco-2 cells. J.Cell Sci. 2001;114:4025–4031. doi: 10.1242/jcs.114.22.4025. [DOI] [PubMed] [Google Scholar]
- 19.Ivanov AI, Hunt D, Utech M, Nusrat A, Parkos CA. Differential roles for actin polymerization and a myosin II motor in assembly of the apithelial apical junctional complex. Mol. Biol. Cell. 2005;16:2636–2650. doi: 10.1091/mbc.E05-01-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zorko M, Langel U. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv. Drug. Deliv. Rev. 2005;57:529–545. doi: 10.1016/j.addr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 21.Chinnapen DJ, Chinnapen H, Saslowsky D, Lencer WI. Rafting with cholera toxin: endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol. Lett. 2007;266:129–137. doi: 10.1111/j.1574-6968.2006.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A. Mechanism of IFN-γ-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol. Biol. Cell. 2005;16:5040–5052. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mettlen M, Platek A, Van Der Smissen P, Carpentier S, Amyere M, Lanzetti L, de Diesbach P, Tyteca D, Courtoy PJ. Src triggers circular ruffling and macropinocytosis at the apical surface of polarized MDCK cells. Traffic. 2006;7:589–603. doi: 10.1111/j.1600-0854.2006.00412.x. [DOI] [PubMed] [Google Scholar]
- 24.Jones AT. Macropinocytosis: searching for an endocytic identity and role in the uptake of cell penetrating peptides. J. Cell. Mol. Med. 2007;11:670–684. doi: 10.1111/j.1582-4934.2007.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanov AI, McCall IC, Parkos CA, Nusrat A. Role for actin filament turnover and a myosin II motor in cytoskeleton-driven disassembly of the epithelial apical junctional complex. Mol. Biol. Cell. 2004;15:2639–2651. doi: 10.1091/mbc.E04-02-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matthews JB, Smith JA, Hrnjez BJ. Effects of F-actin stabilization or disassembly on epithelial Cl- secretion and Na-K-2Cl cotransport. Am. J. Physiol. 1997;72:254–262. doi: 10.1152/ajpcell.1997.272.1.C254. [DOI] [PubMed] [Google Scholar]
- 27.Shen L, Turner JR. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol. Biol. Cell. 2005;16:3919–3936. doi: 10.1091/mbc.E04-12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newman TM, Severs NJ. Effect of neuromimetics upon the release of atrial natriuretic peptide granules: are multiple pathways involved in secretion? J. Cell. Physiol. 1996;168:134–140. doi: 10.1002/(SICI)1097-4652(199607)168:1<134::AID-JCP16>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 29.Polishchuk R, Di Pentima A, Lippincott-Schwartz J. Delivery of raft-associated, GPI-anchored proteins to the apical surface of polarized MDCK cells by a transcytotic pathway. Nat. Cell. Biol. 2004;6:297–307. doi: 10.1038/ncb1109. [DOI] [PubMed] [Google Scholar]
- 30.Paladino S, Pocard T, Catino MA, Zurzolo C. GPI-anchored proteins are directly targeted to the apical surface in fully polarized MDCK cells. J. Cell Biol. 2006;172:1023–1034. doi: 10.1083/jcb.200507116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lencer WI, Moe S, Rufo PA, Madara JL. Transcytosis of cholera toxin subunits across model human intestinal epithelia. Proc. Natl. Acad. Sci. U S A. 1995;92:10094–10098. doi: 10.1073/pnas.92.22.10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirkham M, Fujita A, Chadda R, Nixon SJ, Kurzchalia TV, Sharma DK, Pagano RE, Hancock JF, Mayor S, Parton RG. Ultrastructural identification of uncoated caveolin-independent early endocytic vehicles. J. Cell. Biol. 2005;168:465–476. doi: 10.1083/jcb.200407078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minamide LS, Painter WB, Schevzov G, Gunning P, Bamburg JR. Differential regulation of actin depolymerizing factor and cofilin in response to alterations in the actin monomer pool. J. Biol. Chem. 1997;272:8303–8309. doi: 10.1074/jbc.272.13.8303. [DOI] [PubMed] [Google Scholar]
- 34.Quinlan MP. Vinculin, VASP, and profilin are coordinately regulated during actin remodeling in epithelial cells, which requires de novo protein synthesis and protein kinase signal transduction pathways. J. Cell. Physiol. 2004;200:277–290. doi: 10.1002/jcp.20009. [DOI] [PubMed] [Google Scholar]
- 35.Yarmola EG, Bubb MR. Profilin: emerging concepts and lingering misconceptions. Trends Biochem. Sci. 2006;31:197–205. doi: 10.1016/j.tibs.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 36.Ono S. Mechanism of depolymerization and severing of actin filaments and its significance in cytoskeletal dynamics. Int. Rev. Cytol. 2007;258:1–82. doi: 10.1016/S0074-7696(07)58001-0. [DOI] [PubMed] [Google Scholar]
- 37.Hurley BP, Jacewicz M, Thorpe CM, Lincicome LL, King AJ, Keusch GT, Acheson DW. Shiga toxins 1 and 2 translocate differently across polarized intestinal epithelial cells. Infect. Immun. 1999;67:6670–6677. doi: 10.1128/iai.67.12.6670-6677.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hurley BP, Thorpe CM, Acheson DW. Shiga toxin translocation across intestinal epithelial cells is enhanced by neutrophil transmigration. Infect. Immun. 2001;69:61, 48–55. doi: 10.1128/IAI.69.10.6148-6155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Pasquale G, Chiorini JA. AAV transcytosis through barrier epithelia and endothelium. Mol. Ther. 2006;13:506–516. doi: 10.1016/j.ymthe.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 40.Robinson CM, Sinclair JF, Smith MJ, O'Brien AD. Shiga toxin of enterohemorrhagic Escherichia coli type O157:H7 promotes intestinal colonization. Proc. Natl. Acad. Sci. USA. 2006;103:9667–9672. doi: 10.1073/pnas.0602359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hardwidge PR, Rodriguez-Escudero I, Goode D, Donohoe S, Eng J, Goodlett DR, Aebersold R, Finlay BB. Proteomic analysis of the intestinal epithelial cell response to enteropathogenic Escherichia coli. J. Biol. Chem. 2004;279:20127–20136. doi: 10.1074/jbc.M401228200. [DOI] [PubMed] [Google Scholar]
- 42.Mark MW, Walsh-Reitz MM, Chang EB. Roles of ZO-1, occludin, and actin in oxidant-induced barrier disruption. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;290:222–231. doi: 10.1152/ajpgi.00301.2005. [DOI] [PubMed] [Google Scholar]
- 43.Shurety W, Stewart NL, Stow JL. Fluid-phase markers in the basolateral endocytic pathway accumulate in response to the actin assembly-promoting drug jasplakinolide. Mol. Biol. Cell. 1998;9:957–975. doi: 10.1091/mbc.9.4.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamaze C, Fujimoto LM, Yin HL, Schmid SL. The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J. Biol. Chem. 1997;272:20332–20335. doi: 10.1074/jbc.272.33.20332. [DOI] [PubMed] [Google Scholar]
- 45.Gottlieb TA, Ivanov IE, Adesnik M, Sabatini DD. Actin microfilaments play a critical role in endocytosis at the apical but not the basolateral surface of polarized epithelial cells. J. Cell Biol. 1993;120:695–710. doi: 10.1083/jcb.120.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jackman MR, Shurety W, Ellis JA, Luzio JP. Inhibition of apical but not basolateral endocytosis of ricin and folate in Caco-2 cells by cytochalasin D. J. Cell Sci. 1994;107:2547–2556. doi: 10.1242/jcs.107.9.2547. [DOI] [PubMed] [Google Scholar]
- 47.Volovyk ZM, Wolf MJ, Prasad SV, Rockman HA. Agonist-stimulated beta-adrenergic receptor internalization requires dynamic cytoskeletal actin turnover. J. Biol. Chem. 2006;281:9773–9780. doi: 10.1074/jbc.M511435200. [DOI] [PubMed] [Google Scholar]
- 48.Ganeshan R, Nowotarski K, Di A, Nelson DJ, Kirk KL. CFTR surface expression and chloride currents are decreased by inhibitors of N-WASP and actin polymerization. Biochim. Biophys. Acta. 2007;1773:192–200. doi: 10.1016/j.bbamcr.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Q, Xiao MY, Nicoll RA. Contribution of cytoskeleton to the internalization of AMPA receptors. Proc. Natl. Acad. Sci. USA. 2001;98:1261–1266. doi: 10.1073/pnas.031573798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coue M, Brenner SL, Spector I, Korn ED. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 1987;213:316–318. doi: 10.1016/0014-5793(87)81513-2. [DOI] [PubMed] [Google Scholar]
- 51.Spector I, Shochet NR, Blasberger D, Kashman Y. Latrunculins-novel marine macrolides that disrupt microfilament organization and affect cell growth: I. Comparison with cytochalasin D. Cell. Motil. Cytoskeleton. 1989;13:127–144. doi: 10.1002/cm.970130302. [DOI] [PubMed] [Google Scholar]
- 52.Huang TY, DerMardirossian C, Bokoch GM. Cofilin phosphatases and regulation of actin dynamics. Curr. Opin. Cell. Biol. 2006;18:26–31. doi: 10.1016/j.ceb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 53.Sandvig K, van Deurs B. Transport of protein toxins into cells: pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett. 2002;529:49–53. doi: 10.1016/s0014-5793(02)03182-4. [DOI] [PubMed] [Google Scholar]