

Abstract
A recombinantly produced murine leptin analog (MLA) antagonizes leptin-induced signaling in cell lines that express the long form of the leptin receptor. However, the effects of MLA on the activity of leptin-sensitive neurons and on central neural controls of food intake have not been reported. Here we report effects of MLA on food intake and body weight in adult rats and on the activity of cultured rat vagal afferent neurons. Daily intracerebroventricular coinjection of MLA with exogenous leptin significantly attenuated leptin-induced reduction of 48-h food intake and body weight. Coinjection of MLA with leptin also reduced leptin-induced phosphorylation of signal transducer and activator of transcription 3 (STAT3) in the hypothalamus. In addition, chronic intracerebroventricular MLA infusion over 14 d via osmotic minipumps significantly increased daily food intake, rate of body weight gain, fat-pad mass, and circulating plasma leptin concentrations. Surprisingly, however, MLA did not antagonize leptin-evoked increases in cytosolic calcium concentrations in vagal afferent neurons in primary culture. Rather, MLA itself produced acute activation selectively in leptin-responsive vagal afferent neurons. These data suggest that MLA is an antagonist for the central effects of leptin on food intake and body weight but an agonist at sites where leptin induces acute neuronal activation. This mixed antagonist/agonist action suggests either 1) that the coupling of a single leptin receptor (ObRb) to acute activation of neurons occurs by a signaling mechanism different from those that mediate centrally evoked reductions in food intake and body weight or 2) that acute neuronal activation and centrally induced reductions of food intake and body weight are mediated by different leptin receptor subtypes.
The adipokine leptin is produced by white adipose tissue (WAT) (1) and released into the systemic circulation where the resulting plasma concentration of leptin is positively correlated with total WAT mass (2–4). Administration of exogenous leptin, either systemically or directly into the brain, produces loss of body fat and reduction of food intake (1, 5), whereas mutations that result in the absence of leptin secretion, or defective leptin receptor signaling, result in obesity in rodents (6) and humans (7). These observations suggest that leptin serves as a negative feedback signal controlling food intake and energy expenditure in relation to energy stores in the form of WAT mass.
Many of leptin’s effects on food intake and body fat storage have been attributed to its actions on hypothalamic neurons that coordinate behavioral and metabolic controls of energy balance (8). Leptin’s influence on these neurons is mediated, at least in part, by coupling of the leptin receptor (ObR) with Janus kinase 2 (JAK2) leading the phosphorylation and activation of the transcription factor signal transducer and activator of transcription 3 (STAT3) (9). Additional signaling cascades have been implicated in leptin-induced changes in feeding and body weight control as well as other leptin actions. Specifically, the phosphoinositide 3-kinase/phosphodiesterase 3B/cAMP pathway has been implicated in leptin-induced changes in food intake (10, 11); whereas the MAPK cascade may participate in leptin effects on cellular proliferation (12). However, some leptin-evoked responses occur too rapidly to be accounted for by changes in gene transcription. Such responses, detected in vitro, include leptin-induced changes in membrane voltage or in intracellular calcium concentrations in hypothalamic (13, 14), hindbrain (15), and vagal afferent neurons (16, 17). In vivo, very rapid leptin-evoked responses include increased discharge of vagal afferent fibers (18, 19), a vagally mediated enhancement of meal termination (satiation) when leptin is infused via the celiac artery (18), and reduced transport of glucose across the small intestinal epithelium within minutes of leptin application (20). These acute actions of leptin are likely mediated by nontranscriptional effects on existing membrane proteins that result in changed membrane conductances (17). However, the mechanisms by which they are coupled to the leptin receptor, or even whether they are all mediated by the same leptin receptor splice variant, are unclear.
Although substantial progress has been made in our understanding of the mechanisms of leptin’s actions, progress has occurred in the absence of effective leptin receptor antagonists. Recently, Salomon et al. (21) reported the development of a series of recombinant leptin analogs in which alanine was substituted for amino acids expressed in positions 39, 40, and 41 of wild-type leptin. These substitutions resulted in compounds with antagonist effects on leptin-induced transcriptional changes observed in a heterologous cell system expressing the long form of the leptin receptor (ObRb). Promising as these initial findings appear, there are no reports on the actions of this putative leptin antagonist in vivo or on primary cultures of leptin-sensitive neurons.
Here we present the results from a series of experiments with the murine form of a leptin analog developed by Salomon et al. (21) that possesses putative antagonistic properties against leptin signaling. We refer to this compound as MLA, for murine leptin analog. When we acutely coadministered MLA together with exogenous leptin via intracerebroventricular (icv) cannula, it attenuated the magnitude of leptin-induced weight loss and suppression of food intake as well as leptin-induced phosphorylation of STAT3. We also found that chronic central administration of MLA into rats over a 14-d period via osmotic minipump resulted in increased food intake, greater weight gain, increased WAT mass, and increased circulating leptin concentrations. These findings are consistent with pharmacological antagonism of exogenous, as well as endogenous, leptin effects. However, when we compared the effects of MLA with those of leptin on vagal afferent neurons in culture, we found that MLA triggered rapid increases in cytoplasmic calcium concentration selectively in those neurons that also responded to leptin. These results suggest that MLA has both leptin antagonistic and agonistic actions depending on the specific system examined and may serve as a pharmacological tool to delineate mechanisms responsible for mediation of chronic and acute actions of leptin.
Materials and Methods
Animals
Adult male Sprague Dawley rats (320–360 g), purchased from Simonsen Laboratories (Gilroy, CA), were subjects in all experiments. The animals were housed under a 12-h light, 12-h dark cycle and provided with ad libitum pelleted chow and tap water. Animals were housed in quarters accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. All procedures performed were approved by the Washington State University Institutional Animal Care and Use Committee. For all surgical procedures, the rats were deeply anesthetized (ketamine 25 mg/100 g, xylazine 2.5 mg/100 g), and surgeries were performed under aseptic conditions.
Placement of icv cannula
In both the chronic minipump infusion and icv injection experiments, drug was delivered into the lateral ventricle. For both experiments, cannulas were implanted with stereotaxic methods. Briefly, after exposing the skull, a 23-gauge guide cannula was placed into the lateral ventricle via a trefine hole placed at 1 mm caudal to Bregma and 1.5 mm lateral to the midline. The guide cannula was then lowered to a depth of 4 mm beneath the dural surface. The cannula was secured to the skull surface using stainless steel screws and methacrylic cement (Patterson Dental Supply, Spokane, WA). The guide cannulae were sized to accept insertion of a 30-gauge injection cannula, the tip of which was sharpened and extended 0.5 mm beyond the end of the guide cannula. Cannula patency and placement in the ventricles in the animals used for the acute infusion experiment was tested 5–7 d after surgery and was considered acceptable if the rats drank more than 5 ml water within 15 min after an infusion of angiotensin II (1 μg/3 μl).
Osmotic minipumps
To test whether MLA could antagonize the effects of endogenous leptin on body weight maintenance and food intake, we infused MLA (1 μg/μl at 0.5 μl/h, or 12 μg/d) into the lateral ventricle via an osmotic minipump. Weight-matched control animals also were infused with either saline vehicle (0.9% at 0.5 μl/h) or leptin (0.25 μg/ml at 0.5 μl/h, or 3 μg/d). After recovery from cannula implantation, animals were equipped with osmotic minipumps (model 2002, 200-μl reservoir; Alzet, Cupertino, CA). Pumps were filled with saline, leptin, or MLA solutions, and pumping was verified before implantation. Animals were lightly anesthetized, and pumps were implanted sc between the scapulas and connected to the icv cannula. The minipumps pumped at a rate of 0.5 μl/h (12 μl/d) with drug concentrations adjusted such that animals receiving leptin were exposed to 3 μg/d, whereas animals receiving MLA were exposed to 12 μg/d. Leptin and MLA have the same molecular weight, so MLA was administered at four times the molar dose of exogenous leptin.
Acute icv injections
In this preparation, animals received injections of saline, leptin, MLA, or both leptin and MLA in combination using a 10-μl Hamilton syringe (Hamilton, Reno, NV) connected to the 30-gauge injection cannula via PE10 polyethylene tubing. The infusion volume for all groups was 3 μl and was administered 3 h after lights on.
Body weight and food intake
In both the icv injection and minipump icv infusion studies, body weight and food intake were measured daily 3 h after lights on. Animals were fed a preweighed amount of food on the cage floor, with the remainder and spillage collected and measured to determine the amount consumed.
Isolation of fat pads
At the completion of the chronic infusion study, animals were killed and both the epididymal and retroperitoneal fat pads were collected and weighed. All dissections were performed by a single experimenter blind to the treatment received. Leptin-treated animals had no discernable fat pads.
Plasma leptin measurements
Plasma leptin concentrations were measured in systemic blood plasma collected at the time of killing, 14 d after minipump placement. Plasma for these measurements was prepared from trunk blood collected after decapitation. The blood samples were mixed with 20 μl aprotinin to inhibit protease activity and 1.5 mg EDTA to prevent coagulation. After collection, the samples were centrifuged (6000 × g) for 10 min at 4 C. The plasma was then removed and stored at −80 C until leptin was assayed using a rat leptin RIA (LINCO Research, Inc., St. Charles, MO).
Measurements of STAT3 and phosphorylated STAT3 (pSTAT3)
Rats were killed by decapitation, brains were rapidly removed, and the hypothalami were dissected on an ice-chilled plate. The dissected tissue was frozen in liquid nitrogen and stored at −80 C until the Western blotting. Hypothalami were homogenized on ice in a cell lysis buffer containing protease and phosphatase inhibitors. The homogenates were then centrifuged, and protein concentrations were measured spectrophotometrically. Samples containing 13.5 μg protein were loaded onto polyacrylamide gels for electrophoresis, after which protein was transferred onto nitrocellulose membranes. Membranes were blocked in 5% nonfat milk and incubated with a mouse anti-pSTAT3 antibody specific to phosphorylation of tyrosine 705 (1:2000; Cell Signaling, Boston, MA; lot 6) overnight at 4 C. Membranes were then washed and incubated with a goat antimouse Ig/horseradish peroxidase secondary antibody (1:4000; DakoCytomation, Glostrup, Denmark; lot 00003918) at room temperature for 1 h. After a final wash, blots were developed and bands were imaged using chemiluminescence detection (Kodak M35 X-OMAT Processor). For determination of total STAT3, the membranes were stripped with stripping buffer (62.5 mM Tris-HCl, 0.2% SDS, 0.78% 2-mercaptoethanol) and reprobed for total STAT3 protein with a mouse anti-STAT3 antibody (1:200; Santa Cruz Biotechnology, Santa Cruz, CA; lot J0306). Incubation with secondary antibody and detection were as described above for pSTAT3.
Collection of vagal afferent neurons
Dissociated nodose neurons were obtained as previously described (22). Nodose ganglia were isolated and digested for 90 min at 37 C in Ca2+- and Mg2+-free Hanks’ balanced salt solution containing 1 mg/ml dispase II (Roche Molecular Biochemicals, Indianapolis, IN) and 1 mg/ml collagenase type Ia (Sigma Chemical Co., St. Louis, MO). Cells were dispersed by gentle trituration through silanized Pasteur pipettes, washed two times with HEPES-buffered DMEM (Life Technologies, Grand Island, NY) containing 10% fetal calf serum (Life Technologies) supplemented with antibiotic (100 U/ml penicillin/100 μg/ml streptomycin) and then plated onto poly-L-lysine-coated coverslips and maintained in HEPES-buffered DMEM with 10% fetal calf serum at 37 C in a 5% CO2 atmosphere. All experiments were performed within 48 h of collecting the nodose ganglia.
Calcium measurements
Ratiometric measurements were made using the fluorescent calcium indicator fura-2-AM (Molecular Probes, Eugene, OR) at room temperature (22 C) in a physiological saline (in mM: 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 6 glucose, and 10 HEPES, pH adjusted to 7.4 with NaOH). Image pairs (340- and 380-nm excitation, 510 emissions) were collected every 6 sec, and ratios of fluorescence intensity were converted to calcium concentrations using a standard curve. Data collection and manipulations were performed with MetaFluor Software (Universal Imaging Inc., West Chester, PA).
Statistical analyses
The chronic and acute infusion experiments were a between-subjects design with separate groups of animals for each treatment. The measurements of body weight and food intake were analyzed with a two-way ANOVA (day vs. treatment) followed with comparisons with controls with the Holm-Sidak method (SigmaStat 3.5; Systat Software Inc., Point Richmond, CA). Differences in fat pad mass and circulating leptin concentrations were tested using unpaired t tests against control. The overlap in distribution of MLA- and leptin-sensitive cultured vagal afferent neurons was determined using a χ2 analysis. In all tests, P < 0.05 was considered significant.
Materials
MLA was purchased from Protein Laboratories Rehovot (Rehovot, Israel), and leptin was purchased from PeproTech (Rocky Hills, NJ). All other agents not specified above were purchased from Sigma.
Results
Infusion of leptin and MLA via icv cannula
We found that acute icv administration of MLA alone at 24 μg in 3 μl on two consecutive days had no significant effect on 24-h food intake and a small but significant effect on body weight gain (Fig. 1). In contrast to MLA, acute icv microinjection of 3 μg leptin on two consecutive days produced large and robust reductions of both 24-h food intake and body weight (Fig. 1). The addition of 24 μg MLA to the 3-μg leptin microinjection significantly attenuated leptin-induced reduction of food intake and loss of body weight in response to exogenous icv leptin (Fig. 1). These results are consistent with an antagonist effect of MLA on the actions of leptin in the brain.
Fig. 1.

The effects of leptin (○, 3 μg/d, n = 5), MLA (□, 24 μg/d, n = 7), leptin plus MLA (▲, 3 μg/d leptin, 24 μg/d MLA, n = 11), and saline controls (●, 3 μl, n = 5) on body weight and 24-h food intake when administered into the lateral ventricle. In A and C, both body weight and food intake have been normalized and expressed as the percent change relative to d 0. The arrows indicate the 2 d of infusions. Summary graphs (B and D) represent the average daily percent change during both days of infusions. Leptin infused alone into the lateral ventricle produced significant decreases in both body weight (B) (P = 0.0006) and food intake (D) (P = 0.0007) relative to saline controls (*). The presence of MLA in combination with leptin attenuated the leptin-induced decreases in body weight (B) (P = 0.010 compared with saline; P = 0.0047 compared with leptin) and food intake (D) (P = 0.037 compared with saline; P = 0.021 compared with leptin) (**).
In a separate group of rats, we also examined the effects of MLA on leptin-induced phosphorylation of STAT3 in the hypothalamus. Leptin (3 μg) induced a robust increase in pSTAT3 30 min post injection (Fig. 2). However, neither MLA alone (24 μg) nor MLA (24 μg) coinjected with leptin (3 μg) induced a significant increase in STAT3 phosphorylation compared with controls 30 min post injection (Fig. 2), suggesting that MLA antagonizes JAK2 activation in response to leptin binding.
Fig. 2.
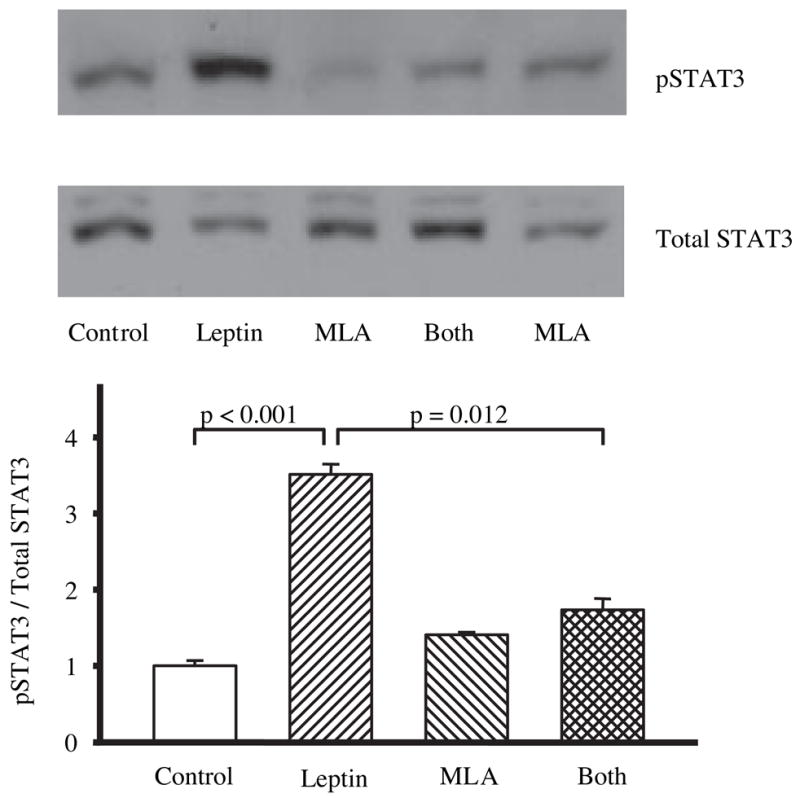
The effects of leptin (3 μg, n = 5), MLA (24 μg, n = 7), leptin plus MLA (3 μg leptin, 24 μg MLA, n = 11), and saline controls (3 μl, n = 5) on pSTAT3/STAT3 expression in the hypothalamus 30 min after lateral ventricle infusions. The top panel shows a representative Western blot gel. Western blots were analyzed, and the data have been expressed in arbitrary units as the ratio of pSTAT3 to STAT3 and then normalized to saline controls (lower panel). Leptin, but not MLA, infused alone into the lateral ventricle produced a significant increase in hypothalamic pSTAT3 relative to saline controls. The presence of MLA in combination with leptin attenuated the leptin-induced increase in pSTAT3.
Chronic infusion of MLA via minipumps increases body weight
Animals were continuously infused with either leptin, MLA, or a saline vehicle into the lateral ventricle for 14 d using osmotic minipumps. Compared with vehicle-infused rats, the leptin-treated animals significantly reduced their food intake within 48 h of the start of infusion (Fig. 3C) and weighed significantly less than vehicle controls beginning 48 h after the start of infusion (Fig. 3B). Neither food intake nor body weight of MLA-treated rats differed significantly from that of vehicle-infused control rats for the first 5 d of infusion. However, beginning on the sixth day of infusion, the MLA-infused animals exhibited increased food intake relative to control infused rats (Fig. 3C). Nine days after the start of infusion, increased body weight gain also was apparent in MLA-infused rats (Fig. 3B). Although the average absolute increase in body weight was relatively small (~11 g; Fig. 3A), when expressed as a percentage of the starting weight, the MLA-treated animals gained weight at almost twice the rate as saline controls (Fig. 3B). As expected, at the end of the 14-d treatment period, the leptin-treated rats weighed significantly less than controls or MLA-infused rats (Fig. 3, A and B).
Fig. 3.
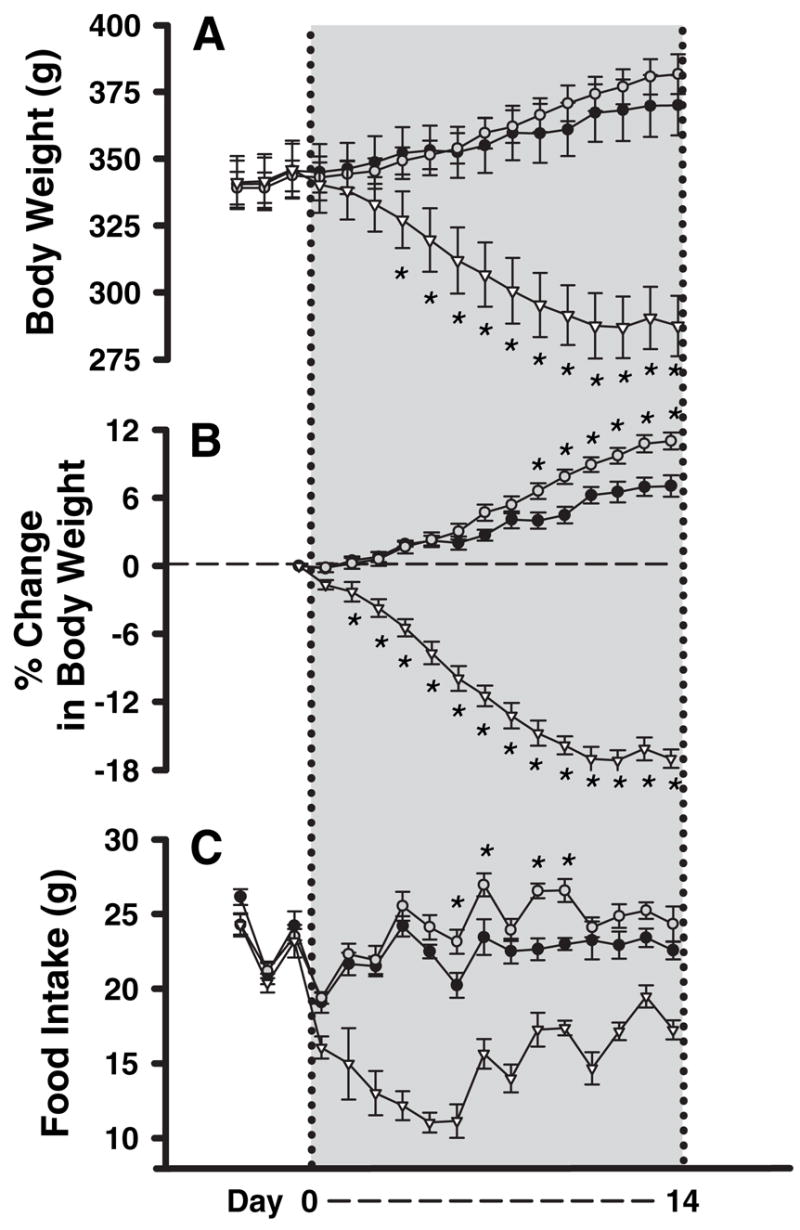
Effects of chronic infusion of saline, leptin (3 μg/d), or MLA (12 μg/d) via osmotic minipump into the lateral ventricle on body weight and food intake: ●, saline; ▽, leptin; ○, MLA. The gray shading delineates the duration of infusions. Measurements were made daily starting 2 d before the start of the infusion. A, Absolute body weight; B, body weight gain for each rat has been normalized to the preinfusion body for each rat. MLA significantly increased (P = 0.006), whereas leptin significantly decreased (n = 6, P < 0.001) body weight when compared with saline-treated controls (n = 6). C, Daily food intake (pelleted chow) is shown. Average daily food intake was significantly higher in MLA-infused animals (P = 0.009) and significantly lower in leptin-infused animals (P < 0.001) compared with saline-treated controls. *, days in which there was a significant difference from saline controls.
Although daily food intake of control and MLA-treated rats (Fig. 3C) did not differ significantly over the first few days of the treatment, the 14-d cumulative food intake by the MLA-infused rats was significantly greater than that of control infused rats. As expected, the cumulative food intake of the leptin-treated rats was much lower than that of either control infused or MLA-infused rats (values in g ± SEM: saline 313 ± 6, MLA 339 ± 7, leptin 211 ± 6; P = 0.02 for saline vs. MLA; P < 0.001 for saline vs. leptin). Average daily intake of MLA-treated rats was not significantly greater than control until infusion d 7. During this period of time, the MLA-infused rats consumed about 10% more food per day than control rats, whereas leptin-treated rats consumed about one third less than the control rats (values in g ± SE: saline 22.6 ± 0.4, MLA 24.2 ± 0.6, leptin 15.1 ± 0.7; P < 0.009 for saline vs. MLA; P < 0.001 for saline vs. leptin). Thus, MLA increased both body weight and food intake over the 14-d experimental period.
At the end of the 14-d infusion, rats were killed, blood samples were collected for leptin RIA, and epididymal and retroperitoneal fat pads were dissected and weighed. We found that average weights of both the retroperitoneal and epididymal fat pads were significantly increased in MLA-infused rats compared with those of saline-infused rats (P = 0.03 and P = 0.005, respectively) (Fig. 4A). Together, the increased weight of these two fat pads accounted for about 2.5 g of the approximately 11-g increase in body weight of the MLA-infused rats (Fig. 4A). In fact, the retroperitoneal fat pads in the MLA-treated animals were more than twice as heavy as those of the saline control animals (Fig. 4A). Neither the retroperitoneal nor the epididymal fat pads were detectable after 14 d of leptin infusion. Thus, the addition of WAT made a significant contribution to the increase in body weight produced by the MLA treatment.
Fig. 4.
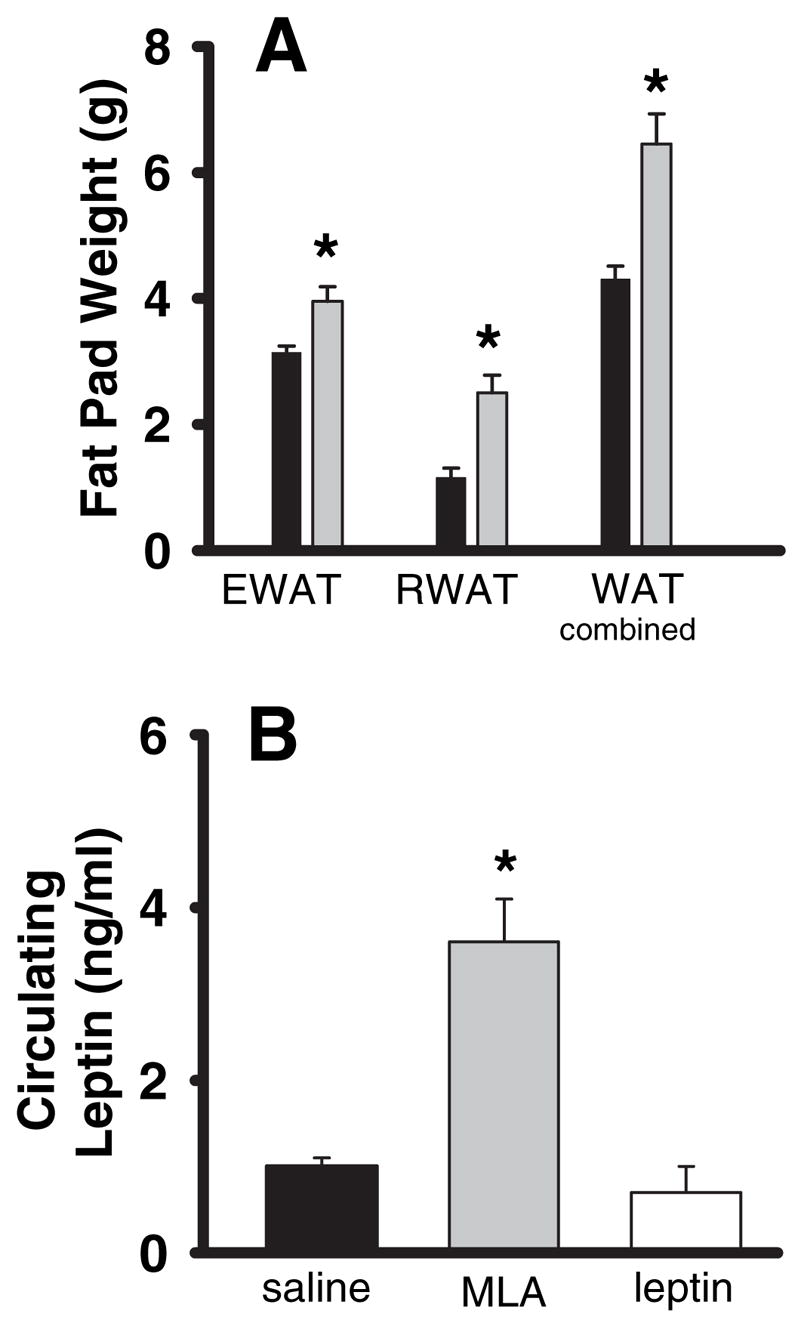
Measurement of fat pad masses and circulating leptin concentrations after chronic icv infusion of saline, leptin, or MLA (same animals as those shown in Fig. 3). A, Absolute weight of fad pads are shown (black bars, saline; gray bars, MLA; leptin-treated animals had no discernable fat pads). Both epididymal (EWAT) and retroperitoneal (RWAT) fat pad masses were significantly larger in the MLA-treated animals (P = 0.03 and P = 0.005, respectively) compared with saline-treated controls. The bars labeled WAT combined are the sums of the EWAT and RWAT (P < 0.0076 compared with controls). B, Plasma leptin levels were significantly elevated in MLA-infused animals (P = 0.002) but did not differ in leptin-treated animals (P = 0.40) compared with saline-treated controls. *, significant difference from saline controls.
A final parameter we hypothesized that would change as a result of the chronic MLA treatment was circulating plasma leptin concentrations. MLA infusion resulted in greater than a 2-fold increase in circulating leptin concentrations compared with controls (Fig. 4B). Although there was not a significant decrease in the average circulating leptin concentration in the leptin-treated rats (Fig. 4B), it is worth noting that three of the six leptin-treated rats had undetectable levels of leptin, whereas in all other groups, circulating leptin was within the detectable limits of the assay (data not shown).
MLA mimics leptin actions on cultured vagal afferent neurons
To explore putative antagonist actions of MLA on acute activation of leptin-sensitive neurons, we examined the effects of MLA on adult rat vagal afferent neurons in primary culture. We previously demonstrated that leptin at physiological concentrations (<40 ng/ml, or 2.5 nM) depolarizes a subpopulation of vagal afferent neurons in primary culture (16). We also found that leptin evokes an increase in cytosolic calcium concentrations in these leptin-sensitive vagal afferent neurons (16). Surprisingly, in the current study, we found that MLA did not antagonize the leptin-evoked increases in cytosolic calcium in cultured nodose ganglion neurons. In fact, when leptin-sensitive vagal afferents were exposed to MLA, they exhibited a dose-related increase in cytoplasmic calcium (n = 6–10 cells per concentration) (Fig. 5, A and B). Furthermore, MLA appeared to be equipotent with leptin in this regard. Similar to leptin, the calcium response to repeated challenges with MLA sometimes exhibited desensitization (Fig. 5A; see challenge to 6.0 nM MLA after 0.60 nM). However, despite the potential for desensitization, when cells were repeatedly challenged with leptin and MLA (order of application alternated), we observed that only cells responsive to leptin responded to MLA and that no cells that responded to MLA failed to respond to leptin (Fig. 5, C and D).
Fig. 5.
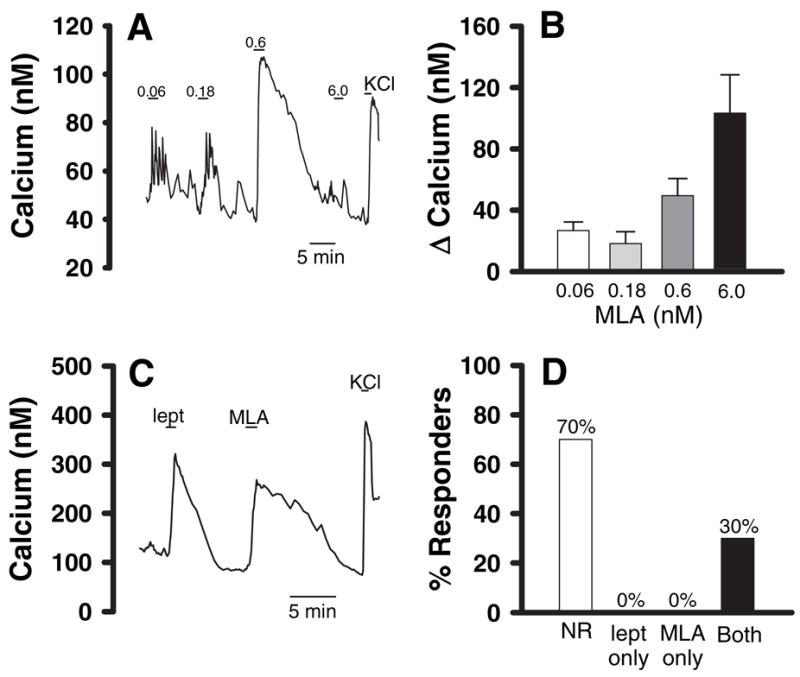
MLA and leptin both increase cytosolic calcium levels in cultured vagal afferent neurons. A and C, Changes in calcium concentration in two representative cultured vagal afferent neurons. A, Increased calcium concentration in response to three concentrations of MLA. The numbers over the bars in A indicate the MLA concentrations in nanomolar. Note that desensitization occurred after the 0.60 nM concentration but that the cell was still alive, as indicated by the response to KCl-induced depolarization. In C, the bars above the trace indicate when the neuron was challenged with 6.00 nM leptin (lept) or 6.00 nM MLA. The bar labeled KCl indicates when the neuron was challenged with a bath containing 55 mM KCl as a positive control for cell viability. B, A summary of the dose-response characteristics of MLA-induced calcium responses. Only MLA-responsive cells are included (n = 6–10 cells at each concentration). D, Coincidence between MLA responses and leptin responses (total cells tested = 53). Note that 70% of nodose neurons have no response to leptin (NR). Also, note that we found no neurons that responded to MLA that did not respond to leptin, and vice versa. However, we found that 30% of the neurons tested responded to both leptin and MLA, which is in agreement with our prior reports that approximately 30% of vagal afferents are leptin sensitive. The coincidence of leptin and MLA responses in the same neurons was highly significant by a ξ2 analysis (P < 0.001).
Discussion
The central findings of this study are that icv MLA acts as an antagonist to endogenous and exogenous leptin but mimics the action of leptin in activating vagal afferent neurons in culture. Our observations indicating that intracerebral MLA antagonizes leptin-induced reduction of food intake and body weight is consistent with the in vitro results of Salomon et al. (21) in which they demonstrated that MLA and other mutated leptins, with alanine substitutions at amino acid positions 39, 40, and 41, are antagonists of ObRb signaling in transfected cells. The unexpected finding that MLA mimicked leptin in evoking acute responses from cultured vagal afferent neurons indicates that MLA has mixed antagonist/agonist properties and suggests that leptin receptor signaling may be even more complex than currently considered.
MLA antagonism of exogenous leptin effects
The icv injection of 3 μg recombinant murine leptin, once a day for 2 d, evoked reductions of food intake and body weight, both of which were significant within 24 h after the first leptin administration. Although cumulative changes in food intake over the 48-h period did not differ between the MLA and saline control group, MLA did produce a small but significant lack of body weight gain relative to saline controls. Although this effect may simply reflect variability between groups, it also is possible that this short-term effect of MLA might indicate that it had some brief leptin-agonist effect on central neurons, similar to what we demonstrated for cultured nodose neurons (see below). A determination of whether MLA shares leptin’s ability to excite populations of brain neurons awaits future investigation. When we coadministered MLA (24 μg) with daily leptin (3 μg), leptin-induced reductions of 48-h food intake and body weight were attenuated although not completely abolished. These results suggest that MLA acts as an antagonist to the effects of exogenous leptin in the brain. Our finding that MLA attenuated leptin-induced increase in phosphorylation of STAT3 in the hypothalamus supports this interpretation and is consistent with results of experiments with transfected cells (21) and leptin-responsive tumor cell lines (23), indicating that Site III leptin mutants, such as MLA, antagonize phosphorylation of STAT3 (23) by exogenous leptin.
Despite the 8-fold ratio of MLA to leptin, MLA’s reversal of exogenous leptin actions was incomplete. The relatively low efficacy of MLA is consistent with previous reports suggesting 100- to 1000-fold excesses of MLA are required to block leptin-induced effects in transfected cell systems (21, 23). Nevertheless, there are several possible explanations for the relatively low competitive efficacy of MLA vs. leptin in our experimental setting. First, it is possible that the response to leptin involves a spare-receptor mechanism, wherein very high concentrations of ligand are needed to saturate all of the leptin binding sites, but sufficient intracellular signaling may be mediated by agonist occupancy of relatively few sites. Thus, very high ratios of antagonist to agonist are required for complete antagonism of receptor-mediated effects. An alternative explanation, which is supported by our observations on the effect of MLA on cultured vagal afferent neurons, is that MLA may antagonize some of leptin’s STAT3-mediated transcriptional actions but be an ineffective antagonist or even an agonist for other leptin-evoked responses. We found that MLA does not antagonize leptin-evoked activation of cultured vagal afferent neurons and in fact acts as a leptin agonist on these neurons (see below for additional discussion). Others have reported that leptin evokes depolarization as well as STAT3 phosphorylation in hypothalamic proopiomelanocortin neurons (9, 13). If the mechanisms of leptin-evoked neuronal excitation in the hypothalamus are similar to those for vagal afferents, then perhaps the delayed attenuation of leptin-induced reduction of food intake in our experiment was due to a combination of MLA acting as a leptin agonist for acute neuronal membrane depolarization while acting as an antagonist for leptin-induced STAT3 phosphorylation. Additional studies will be needed to resolve these questions.
Chronic MLA and leptin infusions
We found that icv infusion of MLA over a 14-d period, using osmotic minipumps, resulted in a significant increase in body weight gain and daily food intake. At the end of the 14-d infusion period, a dramatic and significant increase in fat pad weight and an elevation of circulating leptin were also observed. These data suggest that chronic infusion of MLA antagonizes the central effects of endogenous leptin, leading to increased food intake and increased body fat deposition. Although a significant reduction in 24-h food intake and body weight was evident after the first 48 h in rats infused with wild-type murine leptin, significant increases in food intake and body weight were not detected in the MLA-infused group until the sixth to ninth days of infusion. It is not entirely clear why the kinetics of MLA-induced increases in food intake and body weight differed from those of leptin-induced reductions in these parameters. Some possibilities for the delayed MLA response are that several days may be required to build adequate MLA levels to block endogenous leptin and/or because many of the actions of leptin are transcriptionally mediated, it requires several days of antagonist treatment before expression of critical protein components change to a degree required to translate into a behavioral or metabolic response. This would be in contrast to the relatively rapid increase in protein levels (<24 h) that could occur after agonist treatment. However, it also is conceivable that the relatively late onset of MLA-associated increases in food intake and body weight may be additional support for the hypothesis that MLA has mixed antagonist/agonist properties and that additional time was required for the net antagonist effects of MLA to emerge.
The significantly increased epididymal and retroperitoneal fat pad weights of MLA-infused rats at the end of the 14-d infusion period are consistent with other reports in which defective central leptin signaling results in increased fat deposition (24). Increased fat mass likely accounts for the increased plasma leptin immunoreactivity that we detected in MLA-infused rats. However, because MLA is not distinguishable from rat leptin using the rat leptin RIA, it also is possible that some of the increased plasma leptin immunoreactivity represented detection of MLA leaking to the systemic circulation from the brain. Al-Barazanji et al. (25) have reported substantial leakage of murine leptin to the systemic circulation of rats during icv infusion over a 7-d period. On the other hand, if this were occurring in our system, we would also have expected to see elevated circulating leptin in the leptin-treated animals, but we did not. Therefore, it seems most plausible that increased immunoreactive leptin in our samples from MLA-infused rats arose from increased endogenous leptin secretion as a result of their increased WAT mass.
Agonistic effects of MLA on cultured vagal afferents
We previously reported that approximately 30–40% of adult rat vagal afferents in culture exhibit prompt increases of cytosolic calcium accompanied by membrane depolarization when they are exposed to leptin (16, 17). These responses to leptin occur within seconds of leptin application, so are too rapid to be attributed to STAT3-mediated by transcriptional changes. In this regard, it is very interesting that we found MLA to be equipotent with wild-type murine leptin in evoking increases in cytosolic calcium in cultured vagal afferent neurons. Furthermore, MLA evoked calcium responses only in neurons that responded to leptin. Such a high coincidence of leptin- and MLA-triggered responses suggests that MLA activates vagal afferent neurons by acting at the same receptor as leptin. We obtained similar results with an analog of human leptin substituted with alanines at the same three sites (Leu 39, Asp 40, and Phe 41; data not shown). These results suggest that the amino acid substitutions that enable MLA to antagonize chronic leptin signaling in the brain do not alter its ability to trigger rapid signaling events when it binds to leptin receptors on vagal afferents. Other investigators have reported that leptin evokes rapid-onset increases in the activity of some hypothalamic neurons (13, 14). Therefore, it is conceivable that MLA, and similar leptin mutants, might antagonize the action of leptin on some neurons but act as leptin agonists on other neurons. The fact that MLA behaves as a leptin antagonist for some leptin-mediated responses and an agonist for others also raises the possibility that leptin signaling may be mediated by more than one splice variant of the receptor.
Conclusions
Our results indicate that chronic icv infusion of MLA increases food intake and fat deposition. The icv MLA also attenuates hypothalamic STAT3 phosphorylation and weight loss induced by injection of exogenous leptin. These actions are consistent with a leptin receptor antagonist action of MLA. Our results are consistent with those of Zhang et al. (26), which were reported while our paper was under review. They reported that MLA antagonizes leptin-induced reduction of food intake and body weight in F344 × Brown Norway rats. In addition to our confirmation of the findings of Zhang et al., our results also indicate that MLA does not antagonize all leptin-evoked responses. Specifically, we found that MLA did not antagonize, and in fact mimicked, leptin’s ability to evoke rapid increases in intracellular calcium in cultured vagal afferent neurons. The basis for mixed antagonist/agonist effects of MLA is unknown at present. However, binding of leptin to ObRb triggers intracellular signaling via several pathways distinct from the JAK/STAT mechanism (for review, see Fruhbeck, Ref. 27). Conceivably, MLA might mimic leptin by activating some intracellular signaling pathways while antagonizing leptin-induced activation of the JAK2/STAT3 pathway. Alternatively, multiple splice variants of ObR exist (24), and it is possible that the mutated leptin analogs interact differently with different receptor variants. We do not know whether leptin-evoked increases in intracellular calcium require leptin binding to the long variant of the leptin receptor (ObRb). Conceivably, this response to leptin might be mediated by a shorter splice variant (ObRa). It is known that both the ObRb and ObRa are expressed in neurons of the nodose ganglia (28, 29). Although MLA may antagonize leptin-induced activation of the JAK2/STAT3 pathway via ObRb, it might mimic leptin-evoked increases in intracellular calcium and membrane depolarization by acting as an agonist at ObRa. Which of these explanations is correct will require additional experimentation, perhaps in systems in which ObRa is expressed in the absence of ObRb expression. Nevertheless, our current results suggest that agents can be developed that selectively activate specific aspects of ObR signaling and that the portion of the leptin molecule responsible for evoking acute increases in intracellular calcium in neurons may be distinct from the site that triggers JAK2/STAT3 signaling.
Acknowledgments
We acknowledge Dallas Kinch for help with the care and maintenance of the rats and assistance with the calcium measurements, Tricia Duffy for assistance with the leptin RIA, and Michael Wiater for isolation of the fat pads.
This work was funded by National Institutes of Health Grants NS-20561 to R.C.R. and DK-67146 to S.M.S.
Abbreviations
- icv
Intracerebroventricular
- JAK2
Janus kinase 2
- MLA
murine leptin analog
- ObR
leptin receptor
- pSTAT3
phosphorylated STAT3
- STAT3
signal transducer and activator of transcription 3
- WAT
white adipose tissue
Footnotes
Disclosure: J.H.P., S.M.S., and R.C.R. have nothing to declare.
References
- 1.Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 2.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D., Jr Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med. 1996;2:589–593. doi: 10.1038/nm0596-589. [DOI] [PubMed] [Google Scholar]
- 4.Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 5.Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci USA. 1997;94:8878–8883. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 7.Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, Cheetham CH, Earley AR, Barnett AH, Prins JB, O’Rahilly S. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest. 1996;98:1101–1106. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaisse C, Halaas JL, Horvath CM, Darnell JE, Jr, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet. 1996;14:95–97. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 10.Zhao AZ, Huan JN, Gupta S, Pal R, Sahu A. A phosphatidylinositol 3-kinase phosphodiesterase 3B-cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat Neurosci. 2002;5:727–728. doi: 10.1038/nn885. [DOI] [PubMed] [Google Scholar]
- 11.Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr, Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–795. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- 12.Schneider R, Bornstein SR, Chrousos GP, Boxberger S, Ehninger G, Breidert M. Leptin mediates a proliferative response in human gastric mucosa cells with functional receptor. Horm Metab Res. 2001;33:1–6. doi: 10.1055/s-2001-12617. [DOI] [PubMed] [Google Scholar]
- 13.Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 14.Glaum SR, Hara M, Bindokas VP, Lee CC, Polonsky KS, Bell GI, Miller RJ. Leptin, the obese gene product, rapidly modulates synaptic transmission in the hypothalamus. Mol Pharmacol. 1996;50:230–235. [PubMed] [Google Scholar]
- 15.Williams KW, Smith BN. Rapid inhibition of neural excitability in the nucleus tractus solitarii by leptin: implications for ingestive behaviour. J Physiol. 2006;573:395–412. doi: 10.1113/jphysiol.2006.106336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peters JH, Karpiel AB, Ritter RC, Simasko SM. Cooperative activation of cultured vagal afferent neurons by leptin and cholecystokinin. Endocrinology. 2004;145:3652–3657. doi: 10.1210/en.2004-0221. [DOI] [PubMed] [Google Scholar]
- 17.Peters JH, Ritter RC, Simasko SM. Leptin and CCK modulate complementary background conductances to depolarize cultured nodose neurons. Am J Physiol Cell Physiol. 2006;290:C427–C432. doi: 10.1152/ajpcell.00439.2005. [DOI] [PubMed] [Google Scholar]
- 18.Peters JH, McKay BM, Simasko SM, Ritter RC. Leptin-induced satiation mediated by abdominal vagal afferents. Am J Physiol Regul Integr Comp Physiol. 2005;288:R879–R884. doi: 10.1152/ajpregu.00716.2004. [DOI] [PubMed] [Google Scholar]
- 19.Wang YH, Tache Y, Sheibel AB, Go VL, Wei JY. Two types of leptin-responsive gastric vagal afferent terminals: an in vitro single-unit study in rats. Am J Physiol. 1997;273:R833–R837. doi: 10.1152/ajpregu.1997.273.2.R833. [DOI] [PubMed] [Google Scholar]
- 20.Ducroc R, Guilmeau S, Akasbi K, Devaud H, Buyse M, Bado A. Luminal leptin induces rapid inhibition of active intestinal absorption of glucose mediated by sodium-glucose cotransporter 1. Diabetes. 2005;54:348–354. doi: 10.2337/diabetes.54.2.348. [DOI] [PubMed] [Google Scholar]
- 21.Salomon G, Niv-Spector L, Gussakovsky EE, Gertler A. Large-scale preparation of biologically active mouse and rat leptins and their L39A/D40A/F41A muteins which act as potent antagonists. Protein Exp Purif. 2006;47:128–136. doi: 10.1016/j.pep.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 22.Lancaster E, Oh EJ, Weinreich D. Vagotomy decreases excitability in primary vagal afferent somata. J Neurophysiol. 2001;85:247–253. doi: 10.1152/jn.2001.85.1.247. [DOI] [PubMed] [Google Scholar]
- 23.Niv-Spector L, Gonen-Berger D, Gourdou I, Biener E, Gussakovsky EE, Benomar Y, Ramanujan KV, Taouis M, Herman B, Callebaut I, Djiane J, Gertler A. Identification of the hydrophobic strand in the A-B loop of leptin as major binding site. III. Implications for large-scale preparation of potent recombinant human and ovine leptin antagonists. Biochem J. 2005;391:221–230. doi: 10.1042/BJ20050457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 25.Al-Barazanji KA, Buckingham RE, Arch JR, Briscoe C, Jenkins O, Tadayyon M. Effects of chronic murine and human leptin infusion on plasma leptin and corticosterone levels and energy balance in lean Zucker rats. Diabetes Obes Metab. 2001;3:435–442. doi: 10.1046/j.1463-1326.2001.00162.x. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Matheny MK, Tumer N, Mitchell MK, Scarpace PJ. Leptin antagonist reveals that the normalization of caloric intake and the thermic effect of food following high-fat feeding are leptin dependent. Am J Physiol Regul Integr Comp Physiol. 2007;292:R868–R874. doi: 10.1152/ajpregu.00213.2006. [DOI] [PubMed] [Google Scholar]
- 27.Fruhbeck G. Intracellular signalling pathways activated by leptin. Biochem J. 2006;393:7–20. doi: 10.1042/BJ20051578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buyse M, Ovesjo ML, Goiot H, Guilmeau S, Peranzi G, Moizo L, Walker F, Lewin MJ, Meister B, Bado A. Expression and regulation of leptin receptor proteins in afferent and efferent neurons of the vagus nerve. Eur J Neurosci. 2001;14:64–72. doi: 10.1046/j.0953-816x.2001.01628.x. [DOI] [PubMed] [Google Scholar]
- 29.Peiser C, Springer J, Groneberg DA, McGregor GP, Fischer A, Lang RE. Leptin receptor expression in nodose ganglion cells projecting to the rat gastric fundus. Neurosci Lett. 2002;320:41–44. doi: 10.1016/s0304-3940(02)00023-x. [DOI] [PubMed] [Google Scholar]