

Abstract
Carboxylation of vitamin K-dependent (VKD) proteins is required for their activity and depends upon reduced vitamin K generated by vitamin K oxidoreductase (VKOR) and a redox protein that regenerates VKOR activity. VKD protein carboxylation is inefficient in mammalian cells, and to understand why carboxylation becomes saturated we developed an approach that directly measures intracellular VKD protein carboxylation. Analysis of factor IX (fIX)-expressing BHK cells indicated that slow fIX egress from the endoplasmic reticulum and preferential secretion of the carboxylated form contribute to secreted fIX being more fully-carboxylated. The analysis also revealed the first reported in vivo VKD protein turnover, which was 14-fold faster than occurs in vitro, suggesting facilitation of this process in vivo. r-VKORC1 expression increased the rate of fIX carboxylation and extent of carboxylated fIX ~2-fold, which shows that carboxylation is the rate-limiting step in fIX turnover and which was surprising because turnover in vitro is limited by release of carboxylated fIX. Interestingly, the increases were significantly less than the amount of VKOR overexpression (15-fold). However, when cell extracts were tested in single turnover experiments in vitro, where redox protein is functionally substituted by dithiothreitol, VKOR overexpression increased the fIX carboxylation rate 14-fold, showing r-VKORC1 is functional for supporting fIX carboxylation. These data indicate that the effect of VKOR overexpression is limited in vivo, possibly because a carboxylation component like the redox protein becomes saturated or because another step is now rate-limiting. The studies illustrate the complexity of carboxylation and potential importance of component stoichiometry to overall efficiency.
INTRODUCTION
Vitamin K-dependent (VKD) proteins require carboxylation of Glus to carboxylated Glus (Glas) to become active in different physiologies that include hemostasis, apoptosis, calcium homeostasis, bone development, signal transduction and growth control (1). Multiple Glus are modified, and full carboxylation results in calcium coordination and consequent interaction of the VKD proteins with either anionic phospholipids exposed on cell surfaces or with hydroxyapatite in the extracellular matrix. Efficient carboxylation of the multiple Glus is due in part to the fact that the VKD carboxylase that modifies these proteins is processive: VKD proteins contain a high-affinity domain, usually a propeptide, that tethers them to the carboxylase so that the multiple modifications occur as a consequence of a single binding event (2, 3).
The vitamin K cofactor form used by the carboxylase to drive Glu to Gla conversion is reduced vitamin K (i.e. vitamin K hydroquinone or KH2), and the generation of this form involves reductive reactions in tissue. Thus, vitamin K in the diet exists in the quinone form and therefore requires reduction to KH2. Most KH2, however, comes from the recycling of the vitamin K product of carboxylation, i.e. vitamin K epoxide (KO) (Fig. 1). Recycling is accomplished by vitamin K oxidoreductase (VKOR) which, like the carboxylase, is located in the endoplasmic reticulum (ER) where VKD proteins are modified during their secretion (4). The gene for VKORC1 was recently identified (5, 6), and currently it is unknown whether VKORC1 is the only functional isoform. The carboxylation of a single VKD protein requires multiple KO reductions by VKOR (e.g. 12 for factor IX (fIX)), and so the efficiency of VKOR reduction in supplying KH2 to the carboxylase has the potential to regulate carboxylation. VKOR itself must also be continually reduced to be active because each cycle of KO reduction results in the oxidation of active site thiols and consequent inactivation (7)(Fig. 1). Active VKOR is regenerated by a redox protein whose identity is currently unknown.
Figure 1. Vitamin K reduction is required for VKD protein carboxylation.
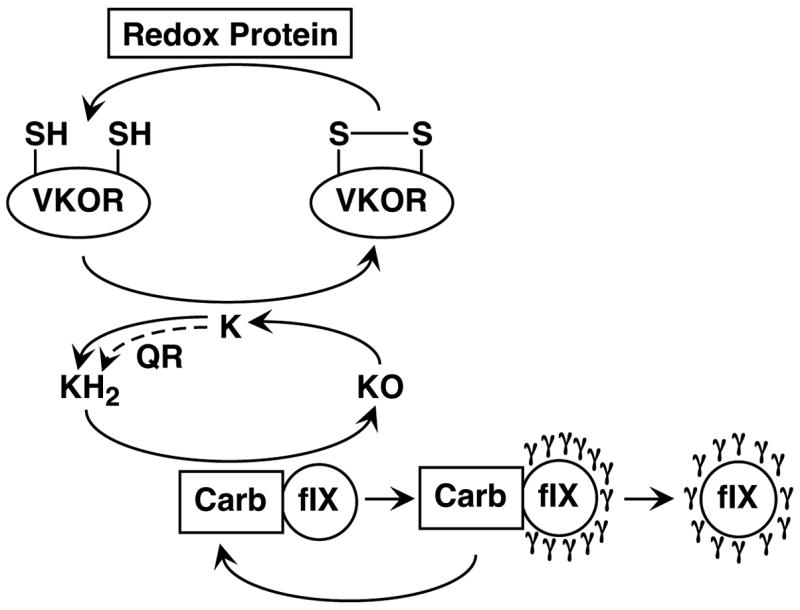
The carboxylase (Carb) converts 12 Glus to Glas (γ) and 12 vitamin K hydroquinones (KH2) to vitamin K epoxide (KO) in the carboxylation of a single molecule of factor IX (fIX). The vitamin K oxidoreductase (VKOR) recycles KO to KH2, which results in oxidation of active site thiols and inactivation of this enzyme. Reduction by an unknown redox protein regenerates VKOR activity. The dashed arrow shows a second route for KH2 generation, via one or more quinone reductase(s) (QR) that convert vitamin K quinone (K) to KH2.
VKD protein carboxylation is disrupted during therapy with coumarins like availability lowers the rate warfarin that inhibit VKOR. The consequent decrease in KH2 of catalysis of VKD protein-carboxylase complexes undergoing carboxylation. This slower rate increases the probability of dissociation of incompletely carboxylated VKD proteins from the carboxylase, and warfarin leads to the secretion of inactive un- and undercarboxylated VKD proteins, as shown for prothrombin (8, 9). Warfarin inhibition also results in the buildup of a precursor pool of VKD proteins because the proteins are still synthesized but not turned over as rapidly by the carboxylase. Vitamin K coadministration during warfarin therapy counteracts the effect of warfarin due to a second pathway for generating KH2: liver contains a high Km quinone reductase that converts the quinone (but not epoxide) form of vitamin K to KH2 and that therefore can support VKD protein carboxylation when vitamin K concentrations are high (Fig. 1) (7).
A limitation in the supply of KH2 may explain why mammalian cells are not efficient for the carboxylation of VKD proteins. Thus, the VKD proteins are fully carboxylated when their expression levels are low but carboxylation becomes saturated with higher levels of expression (10–12). This response presents a major impediment for producing recombinant VKD proteins, several of which have therapeutic potential, for example in hemophilia and sepsis (13, 14). Coexpression of r-carboxylase with VKD proteins does not improve carboxylation even though overexpression of carboxylase protein results in a proportional increase in activity (15, 16), and the change in VKD protein/carboxylase stoichiometry due to carboxylase overexpression actually results in decreased secretion of the VKD protein (i.e. fIX)(16). Intracellular analysis of fIX BHK and fIX 293 cells that contain endogenous carboxylase or that also express r-carboxylase suggested that the availability of KH2 cofactor may limit carboxylation (16). We therefore initiated studies to directly analyze the intracellular events in fIX carboxylation in mammalian cells. Our results indicate that KH2 availability regulates the rate of carboxylation and that r-VKORC1 expression increases this rate but that the effect of r-VKORC1 is limited, possibly due to saturation of at least one additional factor that is also required for VKD protein carboxylation.
EXPERIMENTAL PROCEDURES
Construction of the r-VKORC1 expressing plasmid
The human VKORC1 ORF was amplified from the EST clone BC000828 (Open Biosystems) using the primers VKRC1S (AGATCTACCATGGGCAGCACCTGGGGG) and VKRC1AS1 (TCTAGATCTTCAGTGCCTCTTAGCCTTG) with Hot Master Taq DNA polymerase (Fisher). The resulting 510 bp PCR product was cloned into the vector pCR2.1-Topo (Invitrogen) and the entire VKORC1 ORF was sequenced on both strands, which showed that it was identical to BC000828. The Bgl II fragment containing the VKORC1 ORF was then isolated and ligated to Bam HI-digested ZEM229, which is a vector containing a cassette for dihydrofolate reductase expression (17), to generate r-VKORC1/ZEM229.
Generation of BHK cell lines stably-expressing fIX or fIX and VKORC1
BHK cell lines that secrete low (i.e. 10 pmol/d/106 cells) or high (200 pmol/d/106 cells) levels of fIX were used in these studies. The low-producing fIX BHK cells were generated by transfecting BHK cells with a human fIX cDNA (18) in the expression vector ZEM228, which contains a cassette conferring resistance to G418 (19), as previously described (20). The high-producing fIX BHK cells were generated by transfecting BHK cells with the fIX cDNA in the expression vector ZEM229. Transfected cells were selected in methotrexate (1 μM) and clonal isolates were screened for fIX by a western (ECL, Amersham) using polyclonal anti-fIX antibody. VKOR·fIX cells were generated by transfecting the low-producing fIX BHK cells with VKORC1/ZEM229, followed by selection in methotrexate (1 μM), or by cotransfecting the high-producing fIX BHK cells with VKORC1/ZEM229 and pPUR (Becton Dickinson), followed by selection in puromycin (5 μg/ml). Clonal isolates (two dozen from each transfection) were screened for VKORC1 expression in a western using antibody generated against a peptide from the C-terminus of VKORC1 (FRKVQEPQGKAKRH). The antibody was affinity-purified on a column containing the peptide covalently-coupled to Sepharose. Those isolates showing the highest levels of expression by western analysis were then tested for VKOR activity by measuring KO reduction (as described in the following section). Two independent isolates from each r-VKORC1 transfection were used in subsequent analysis.
Analysis of VKOR, carboxylase and fIX in fIX BHK and VKOR·fIX BHK cell lines
VKOR activity was determined by measuring KO reduction. KO was prepared by oxidation of phylloquinone (Sigma), as described (21), and then purified by HPLC using a preparative C18 column (Vydac) and isocratic elution with methanol. Lysates (400 μl, ~1–2 mg) prepared as before (22) were incubated with this KO preparation (giving a final concentration of 15 μM KO and 1% methanol) for 1 min, followed by the addition of DTT (to 5 mM) and incubation for an additional 20 min, all at 20°C. The reaction was quenched by the addition of 800 μl of isopropanol and hexane (3:2 vol:vol), followed by vortexing and centrifugation for 5 min at 3000 rpm. Most (75%) of the organic phase was transferred to a new tube and the sample was taken to dryness under nitrogen. Samples were resuspended in ethanol and vitamin K was analyzed by HPLC as before (23), along with vitamin K standards (250 ng), to determine the per cent conversion of vitamin K epoxide to quinone. A control reaction with heat-treated extracts showed that KO reduction was not simply due to DTT. The specific activity of VKOR in the lysates was determined after quantitation of protein concentrations by BCA (Pierce). For those experiments that compared the specific activity in cells containing endogenous- or recombinant VKOR, all relevant lysates were analyzed in a single experiment.
Carboxylase levels were quantitated by an assay that measures [14C]-CO2 incorporation into a peptide substrate (Boc-Glu-Glu-Leu-OMe, Bachem), as previously described (22). A similar measurement performed on pure carboxylase quantitated by amino acid analysis (23) allowed this assay to be used for determining the concentration of carboxylase.
Secreted and intracellular fIX levels were measured using a quantitative western, which gave values similar to our previously-established ELISA (16). Samples were gel-electrophoresed along with a standard curve of pure plasma fIX (5–50 ng, Enzyme Research Laboratories). Protein was transferred to nitrocellulose and the membranes were probed with affinity-purified polyclonal anti-fIX antibody (0.4 μg/ml), which we previously showed recognizes both carboxylated and uncarboxylated fIX (16), and goat anti-rabbit antibody conjugated to alkaline phosphatase (Biorad). Substrate development was with Attophos (Promega), used according to the manufacturer’s instructions. In those experiments comparing fIX and carboxylase levels in cell lines expressing endogenous- versus recombinant VKOR, all relevant lysates were analyzed in a single experiment.
Secreted fIX was also analyzed for Gla content. FIX was isolated from fIX BHK and VKOR·fIX BHK cells as before (22) but with an additional step that was introduced because of our observation that fIX secretion is slow (described in the Results). Cells, which were cultured in the absence of vitamin K, were fluid-changed into vitamin K (2 μM)-containing serum free media and then fluid-changed again into the same media 8 hr later to remove uncarboxylated fIX. The cells were then maintained in this media for 3 days, and media was collected and fIX affinity-purified using a monoclonal antibody (ESN1, American Diagnostica) that we previously found is capable of recognizing both carboxylated and uncarboxylated fIX (16). The samples were first analyzed by SDS-PAGE and silver-staining to verify purity, and then quantitated for Gla content by base hydrolysis followed by resolution of the amino acids by HPLC on a C18 column and fluorimetric detection, as previously described (22).
In vitro carboxylation of fIX
Microsomes were prepared from fIX BHK or VKOR·fIX BHK cells cultured in the absence of vitamin K as previously described (24) except that after the 100,000xg centrifugation step, the pellets were resuspended in 25 mM Tris-HCl pH 7.4, 250 mM sucrose and 2 mM phenylmethylsulfonyl fluoride (PMSF) before storage at −80°C. The preparations were assayed for carboxylase activity and for the amount of fIX-carboxylase complex prior to being tested for fIX carboxylation. A microsomal aliquot (500 μl) was adjusted to 0.5 M NaCl and then solubilized with CHAPS (0.5% final concentration), followed by centrifugation at 100,000xg for 1 hr. All steps were at 4°C. The supernatant was incubated with anti-fIX Sepharose (100 μl, 2 mg/ml affinity-purified polyclonal anti-fIX coupled to CNBr-activated Sepharose) by overnight nutation. Aliquots (2×10 μl) of this starting slurry were reserved and the remaining sample was centrifuged (1000xg, 1 min) and aliquots (2×10 μl) of the unbound material were saved. The resin was then washed by four successive rounds of centrifugation (1000xg, 1 min) and incubation for 1 min in 1 ml of 50 mM Tris-HCl pH 7.4, 500 mM NaCl, 0.25% CHAPS, 0.25% phosphatidyl choline and 5 mM dithiothreitol. Aliquots (2×10 μl) of the 1.1 ml slurry were removed on the final wash. Starting slurry, unbound material and washed resin were then assayed for carboxylase activity, which showed that 88–90% of the carboxylase was present as a fIX-carboxylase complex and which is similar to values previously obtained for cell lines (25). This characterization allowed a determination of the amount of [14C]-CO2 incorporation into fIX (using a specific activity of 50 cpm/pmol for 14CO2).
In vitro carboxylation of fIX was then performed by incubating the microsomes (11 mg) in a reaction mixture (2.9 ml) containing 0.5 M ammonium sulfate, 0.06% phosphatidyl choline, 0.06% sodium cholate, 0.14% CHAPS, 2 mM [14C]-CO2, 12 mM DTT and 100 mM Tris-HCl pH 7.4. These optimal reaction conditions for VKOR-supported carboxylation were established by permuting the pH (over the range 6.6–7.4), type of salt (KCl, NaCl and ammonium sulfate) and salt concentration (0.1–1 M), type of detergent (Triton X, CHAPS and sodium cholate) and ratio of phospholipid to detergent (from 1:2 to 2:1). The samples were incubated at 20°C for 5 min, an aliquot (450 μl) was removed (the zero time point) and the reaction was then initiated by the addition of KO or KH2 (both at a final concentration of 16 μM; the KH2 was generated as described (26) and dissolved in the same solvent (methanol) as KO). Aliquots (450 μl) were removed at varying times, as indicated in the Results, and were quenched by the addition of 50 μl of 10% Triton X and stored on ice. The fIX in the samples was then immunoprecipitated prior to gel electrophoresis. The immunoprecipitation method was validated in pilot experiments: microsomes from fIX BHK and r-carboxylase·fIX cells ((24); used because they gave a much higher signal) were in vitro carboxylated using KH2 and duplicate aliquots were then either gassed with CO2 (to remove [14C]-CO2) and directly analyzed on SDS-PAGE followed by PhosphorImager quantitation or were first immunoprecipitated, using a range (5–20 μg) of affinity-purified polyclonal anti-fIX antibody, before gel electrophoresis. This test showed that fIX was quantitatively recovered in the immunoprecipitation, even at the lowest antibody concentration used, and that both methods gave the same results. Immunoprecipitation was therefore used in subsequent experiments because more sample could be analyzed, affording a substantial increase in signal.
For the immunoprecipitation of the in vitro carboxylation time course, the samples were centrifuged (15,000xg, 15 min, 4°C) and 450 μl of the supernatant was diluted with 450 μl of 1% Triton X, followed by the addition of anti-fIX antibody (20 μg) and protein A agarose (50 μl, Sigma). After overnight incubation, the resins were washed by four successive rounds of centrifugation (1000xg, 1 min) and resuspension in 1 ml of buffer A (300 mM NaCl, 0.05% Tween 20, 0.1% SDS, and 20 mM Tris-HCl pH 7.4). All of the immunoprecipitation steps were performed at 4°C. After the final centrifugation, SDS-PAGE loading dye (100 μl) was added to the resins, followed by boiling and centrifugation for 5 min at 5000xg. Aliquots (75 μl) were then gel-electrophoresed along with 14C-BSA standards used for quantitating the amount of [14C]-CO2 incorporation into fIX. Protein was transferred to nitrocellulose (to increase the sensitivity of detection) and the membranes were then exposed to PhosphorImager screens for approximately one month, followed by quantitation using ImageQuant software (Amersham).
Pulse-chase analyses of fIX BHK and VKOR·fIX BHK cell lines
Cell lines were analyzed in pulse-chase studies to follow the fate of newly-synthesized fIX. Cells cultured without vitamin K (at 50% confluence) were fluid changed 24 hr prior to the pulse into culturing media that either lacked or contained vitamin K (2 μM). Both the quinone form (i.e. phylloquinone, Sigma) and epoxide form (prepared as described above) were tested, as indicated in the Results, and both forms were dissolved in the same solvent (methanol). Control BHK cells were processed in parallel. One hr before the pulse the cells were rinsed with prewarmed PBS and then starved in media that lacked Met and Cys (GibcoBRL) and contained 1 mM pyr, 2 mM Gln and 0 or 2 μM vitamin K. Cells were then fluid changed with prewarmed PBS and pulsed for 10 min in the same media except for the addition of fetal calf serum (charcoal-treated (Atlanta Biologicals) and present at a final concentration of 1%) and 35S-Cys/Met (20 μCi/ml, ICN). Cells were then fluid changed and incubated in nonradiolabeled chase media (DMEM/F12 (Sigma), 1% fetal calf serum, 1 mM pyr, 2 mM Gln and 0 or 2 μM vitamin K) for varying lengths of time, as indicated in the Results. Media was harvested and adjusted to 1 mM PMSF. Cell lysates were prepared by rinsing the cells with cold PBS, followed by the addition of lysis buffer (1% Triton X100, 0.15 M NaCl, 10 μg/ml aprotinin, 40 μg/ml bestatin, 10 μg/ml leupeptin, 0.8 μg/ml pepstatin, 2 mM PMSF, 10 mM iodoacetamide, and 50 mM Tris-HCl pH 7.4; all inhibitors were from Roche) and incubation on ice for 20 min. The lysates were then centrifuged at 14,000xg, 10 min, 4°C and the pellet was discarded. FIX was immunoprecipitated from cell lysates and media (using 20–50% of the sample) with affinity-purified polyclonal anti-fIX antibody. Control experiments were first performed with increasing concentrations of anti-fIX antibody (2–20 μg), which showed that the amount (i.e. 10 μg) used in subsequent analyses resulted in quantitative recovery of fIX. Samples were incubated with anti-fIX antibody and protein A agarose (50 μl, Sigma) overnight at 4°C and then washed by four successive rounds of centrifugation (1 min, 1000xg) and incubation for 1 min in 1 ml buffer A, all at 4°C. After the last centrifugation, 100 μl of SDS-PAGE loading buffer was added and the samples were boiled and then recentrifuged for 5 min at 5000xg. Aliquots (75 μl) were then gel-electrophoresed and the gel was dried and processed by PhosphorImager (Amersham).
To compare the effect of different vitamin K forms on fIX carboxylation, cells were analyzed as above except that vitamin K was not added to the cells until the time of the chase. During the chase, cells were incubated in either the quinone or epoxide form of vitamin K, both at final concentrations of 2 μM. Pulse-chase analysis was also performed in the presence or absence of warfarin. Cells were pulsed and chased as above except that all of the media (i.e. for starving, pulsing and chasing the cells) contained varying concentrations of warfarin (0–2 μM).
RESULTS
Analysis of fIX carboxylation and secretion in fIX BHK cells
We previously found that pulse-chase analysis could detect fIX carboxylation in vivo because the conversion of Glus to Glas caused a shift in fIX migration on SDS-PAGE (16). In that study, the cells were pulsed in the absence of vitamin K and then chased in the presence of vitamin K. The lack of vitamin K had the potential to cause precursor pool buildup, as observed during warfarin treatment (27), and we therefore adapted the pulse-chase approach in the present studies for analysis under steady state conditions of vitamin K. A BHK cell line secreting relatively low levels of fIX (10 pmol/d/106 cells) was incubated for 24 hr in vitamin K and then radiolabeled for a time sufficiently short (10 min) so that most of the fIX was uncarboxylated by the end of the pulse. The cells were then chased in nonradiolabeled media for varying times to follow the fate of the newly-synthesized fIX (Fig. 2). A parallel pulse-chase was performed in the absence of vitamin K. FIX carboxylation was observed only in cells containing vitamin K, as evidenced by the characteristic shift in intracellular fIX migration on SDS-PAGE that occurs upon carboxylation (Fig. 2B). Subsequent secretion of fIX was observed both in cells that contained or lacked vitamin K, showing that carboxylation was not obligatory for secretion (Fig. 2A). Quantitation showed that by 5 hr, 70% of the newly-synthesized fIX in the vitamin K-containing BHK cells was secreted (Fig. 3A), similar to results previously obtained with this cell line (16). That pulse-chase study also included a 22 hr time point, which showed that 100% of the fIX is ultimately secreted into media (16).
Figure 2. Analysis of fIX carboxylation and secretion in a low-producing fIX BHK cell line.
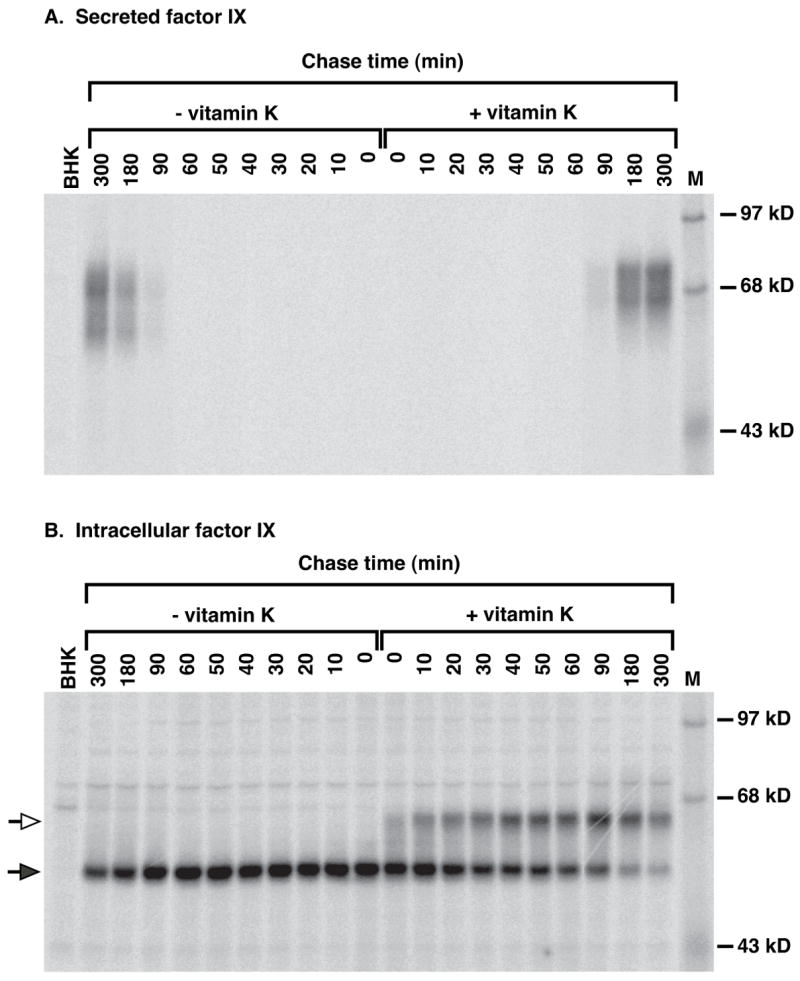
FIX BHK cells were radiolabeled (10 min) in media containing 35S-Met/Cys and then chased for the indicated times in unlabeled media, all in the absence or presence of vitamin K (2 μM). FIX in media (A) or cell lysates (B) was then immunoprecipitated with saturating amounts of anti-fIX antibody, followed by SDS-PAGE and PhosphorImager analysis. The filled and hollow arrows indicate uncarboxylated or carboxylated fIX, respectively, as previously shown by Gla quantitation of the two forms (16). Undercarboxylated (i.e. 1–11 mol Gla/mol fIX) fIX migrates as a smear of forms. M indicates molecular weight markers and BHK the untransfected progenitor cell line, which was radiolabeled for 10 min and then immediately harvested. The intracellular fIX is located in the ER whereas the secreted fIX has undergone modifications in the Golgi, which accounts for the difference in molecular weights between the intracellular and secreted fIX.
Figure 3. Quantitation of fIX carboxylation and secretion in a low-producing fIX BHK cell line.
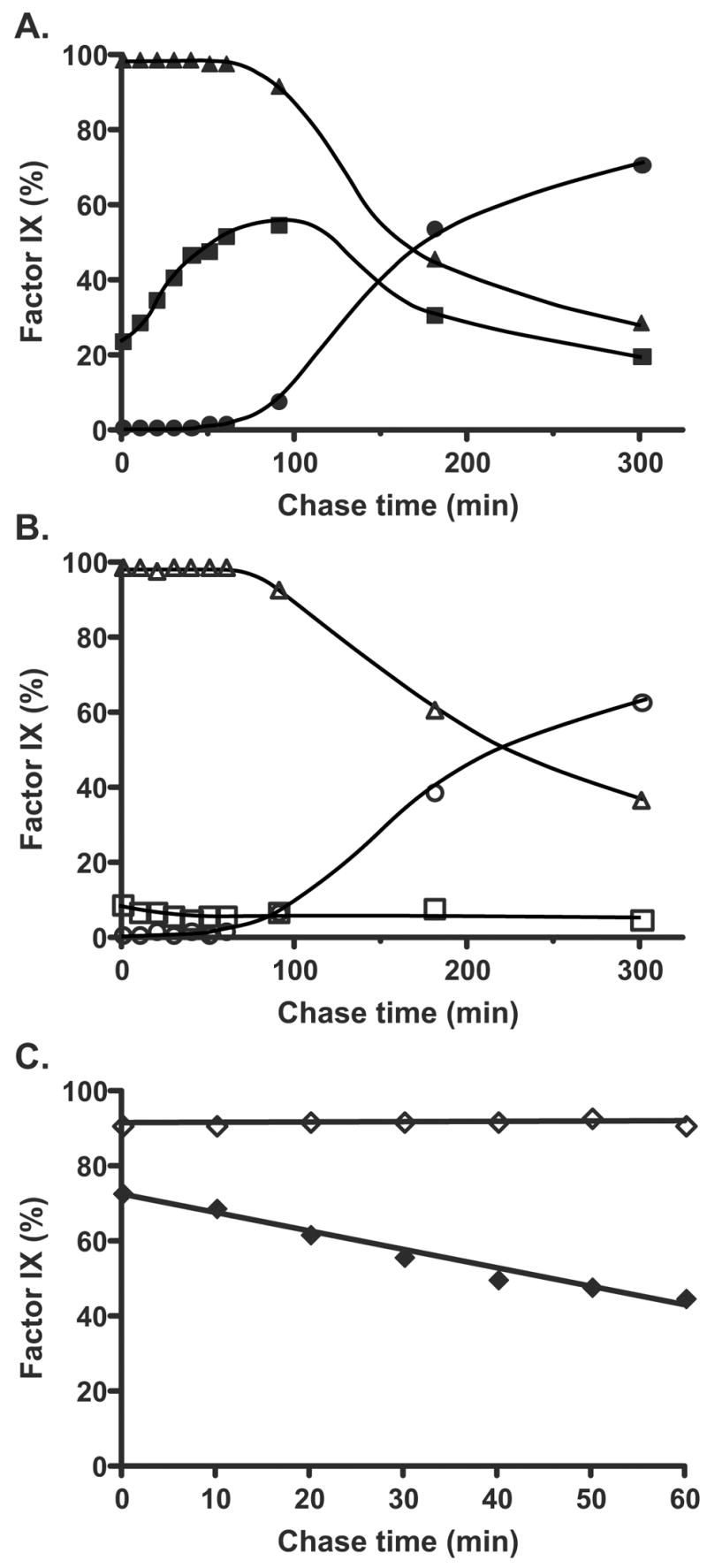
The gels shown in Fig. 2 were quantitated by PhosphorImager to determine the amount of intracellular (▲, △) or secreted (●, ○) fIX present in cells containing (A) or lacking (B) vitamin K. The amount of intracellular carboxylated fIX (i.e. both partially and fully carboxylated fIX, ■, □) was also quantitated. Part C shows the rate of disappearance of uncarboxylated fIX over the first hr, when all of the fIX was still intracellular, in cells containing (◆) or lacking (◇) vitamin K. The entire experiment was performed twice and gave nearly identical results.
The rate of fIX secretion was significantly slower than the rate of carboxylation over the course of the experiment. By the 90 min chase time over half (56%) of the fIX in vitamin K containing cells was carboxylated but only a small fraction (7%) was secreted from the cell (Fig. 3A). By the time fIX secretion was substantial (180 min), 80% of the uncarboxylated fIX had disappeared (Fig. 2B). The intracellular fIX was endoglycosidase H sensitive (data not shown) as previously observed (16)), indicating its localization to either the ER or cis Golgi. Transit to the medial and trans Golgi results in subsequent processing of the N-glycosyl groups as well as O-glycosylation of fIX, which together generate a secreted fIX with a molecular weight that is larger than that of the intracellular form (Fig. 2A versus B). The medial/trans Golgi forms are present in trace amounts that can only be detected with longer exposures (data not shown). Thus, both carboxylated and uncarboxylated fIX reside for an unusually long time in the ER/cis Golgi, followed by rapid translocation through the Golgi and exit from the cell.
Interestingly, the fIX BHK cells contained a precursor pool of fIX even under steady state conditions of vitamin K. Thus, western analysis revealed uncarboxylated fIX (Fig. 4), and a quantitative western showed that the amount of uncarboxylated fIX represented 65% of the intracellular population and corresponded to 7.04 pmol/mg lysate. The intracellular carboxylase concentration could also be quantitated because the specific activity of the carboxylase is known (determined by comparing carboxylase activity of pure enzyme to the amount of protein quantitated by amino acid analysis, (23)). An activity assay on the lysates indicated a value of 0.16 pmol/mg, showing that the intracellular stoichiometry of the carboxylase to uncarboxylated fIX was 1:44.
Figure 4. Low-producing fIX BHK cells contain an intracellular precursor pool of uncarboxylated fIX.
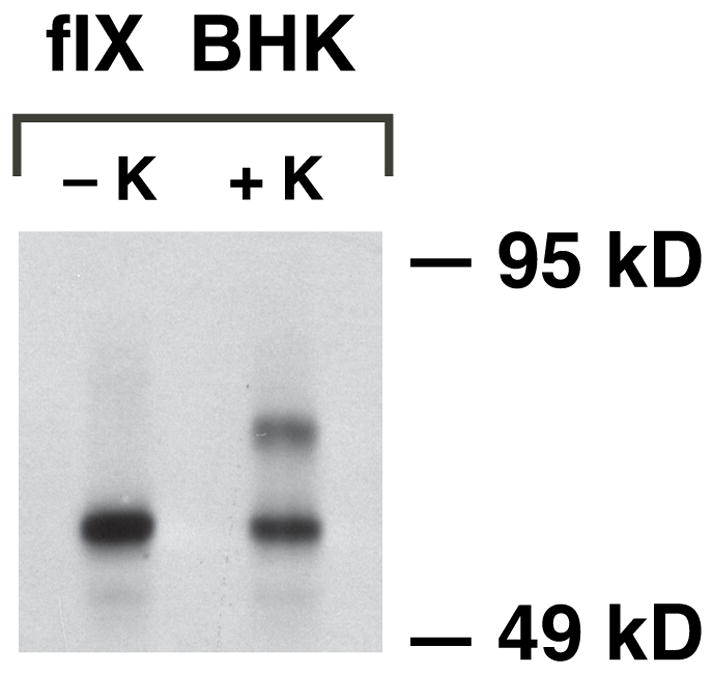
Lysates were prepared from fIX BHK cells cultured in the absence (−K) or presence (+K) of vitamin K and equivalent amounts of protein (50 μg) were analyzed in a western using polyclonal anti-fIX antibody that detects both carboxylated and uncarboxylated fIX. Quantitative western analysis (not shown) confirmed the larger amount of uncarboxylated fIX (lower band) in cells cultured without vitamin K, which presumably is due to precursor pool buildup (as occurs with warfarin treatment).
Gla quantitation showed that fIX secreted from the fIX BHK cells was extensively carboxylated. Thus, when quantitation was performed on fIX purified from the media of cells cultured in vitamin K, the secreted fIX was found to have a Gla content of 11.2 +/− mol Gla per mol fIX (versus <0.2 mol Gla per mol fIX for fIX secreted from cells cultured in the absence of vitamin K). The value for the carboxylated fIX is slightly higher than previously reported (11) due to the use of steady-state vitamin K conditions in the present study (see Experimental Procedures). The observations that most (93%) of the fIX secreted from the cells is carboxylated even though uncarboxylated fIX can be secreted (Fig. 2A) indicates that the events that occur during fIX secretion impact the extent of carboxylation of secreted VKD protein. As presented in more depth in the Discussion, this impact is likely due to the slower rate of secretion compared to carboxylation (described above) as well as preferential secretion of carboxylated fIX. Thus, in the presence of vitamin K almost all of the newly-synthesized fIX in this low-producing fIX BHK cell line was ultimately carboxylated and secreted. These data indicate that carboxylation and the processes preceding and following carboxylation are not saturated in these cells.
The intracellular analysis allows a determination of the rate of in vivo fIX turnover
The combined pulse-chase analysis and quantitation of intracellular fIX and carboxylase levels allowed a determination of the rate of in vivo fIX turnover. The turnover was measured using early chase times (0–60 min), when the rate was linear. Turnover was determined by using either the rate of disappearance of uncarboxylated fIX (Fig. 3C) or the rate of appearance of carboxylated fIX (not shown), which was possible because all of the fIX was intracellular over this time frame. Both measurements gave equivalent values and indicated that 32% of the fIX was turned over in the first 60 min. This per cent corresponded to 14 fIX molecules/hr/carboxylase based on the 44:1 stoichiometry of carboxylase and fIX (previous section).
Two other factors that might affect the turnover determination were considered. The possibility that synthesis of nonradiolabeled fIX that occurs during the chase might significantly change the pool size was tested by performing pulse-chase analyses with cells chased in the presence or absence of cycloheximide. We found that the rate of radiolabeled fIX turnover was the same under both conditions (data not shown). Presumably, the pool size is much larger than the amount of fIX synthesized during the chase, which indicates that fIX synthesized during the chase did not affect the rate determination. A second consideration was the amount of vitamin K used in the pulse-chase analyses. The fIX BHK cells analyzed in Fig. 2 were maintained in 2 μM vitamin K; however, analysis performed with vitamin K concentrations as high as 50 μM gave similar amounts of fIX carboxylation (Supplemental Figure 1).
VKOR supports most if not all fIX carboxylation in BHK cells
The vitamin K used in cell culture media is the quinone rather than reduced (i.e. KH2) form because KH2 is rapidly oxidized in media, and intracellular reduction is therefore required. As discussed in the Introduction (Fig. 1), in liver the quinone form of vitamin K can be reduced either by VKOR or by a warfarin-resistant high Km quinone reductase that can support VKD protein carboxylation when vitamin K concentrations are high (28). The identity of the warfarin-resistant quinone reductase is not known, nor is it known whether it is present in other tissues. Kidney, from which BHK cells are derived, contains at least one known warfarin-resistant quinone reductase, i.e. the flavoprotein QR2 (29), which has been implicated in metabolism of the vitamin K isoform menadione (30). Kidney also contains the warfarin-sensitive QR1 (or DT-diaphorase) (31), which was originally proposed to be important to carboxylation but more recently is thought to have a role in quinone detoxification (32). A high Km quinone reductase could be functionally significant in cultured cells because of the high concentrations of vitamin K that are typically used; however, whether quinone reductases other than VKOR contribute to VKD protein carboxylation in mammalian cells has not been investigated.
To determine whether the BHK cells contain a warfarin-resistant quinone reductase that can support carboxylation, pulse-chase analysis was performed on fIX BHK cells treated with increasing amounts of warfarin. As shown in Fig. 5, warfarin totally inhibited fIX carboxylation at concentrations as low as 0.1 μM, indicating that in BHK cells a warfarin-resistant quinone reductase does not contribute significantly to generation of KH2. This interpretation was consistent with results obtained from a second experiment, in which the quinone and epoxide forms of vitamin K were compared for their ability to support fIX carboxylation. FIX BHK cells were pulsed in the absence of vitamin K and then chased in either KO, which can be reduced only by VKOR, or K, which can be reduced by both VKOR and quinone reductase(s) (that are either warfarin sensitive or resistant). This analysis showed that the rate of fIX carboxylation was the same for both vitamin K forms (Fig. 6). As presented below, the availability of KH2 limits carboxylation, and so the observation that the rate was not any faster in the K-containing cells indicates that quinone reductase(s) did not significantly contribute to carboxylation and supports the conclusion that VKOR is responsible for generating most or all of the KH2 that drives VKD protein carboxylation in the fIX BHK cells.
Figure 5. FIX carboxylation in fIX BHK cells is abolished by warfarin.
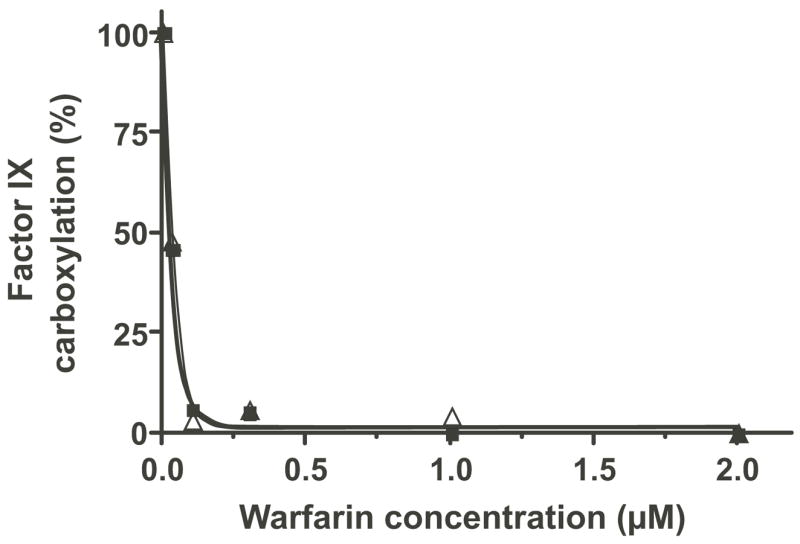
FIX BHK cells were pulsed (1 hr) in media lacking vitamin K and then chased (3 hr) in media containing the quinone (△) form of vitamin K (2 μM), all in the presence of increasing concentrations of warfarin. A control pulse-chase with KO (2 μM, ■), which can only be reduced by VKOR, was processed in parallel. FIX immunoprecipitated from cell lysates and subjected to SDS-PAGE was quantitated for the amount of carboxylated fIX by PhosphorImager analysis. The amount of carboxylation in the absence of warfarin is defined as 100%.
Figure 6. Vitamin K quinone and epoxide forms effect similar rates of fIX carboxylation in fIX BHK cells.
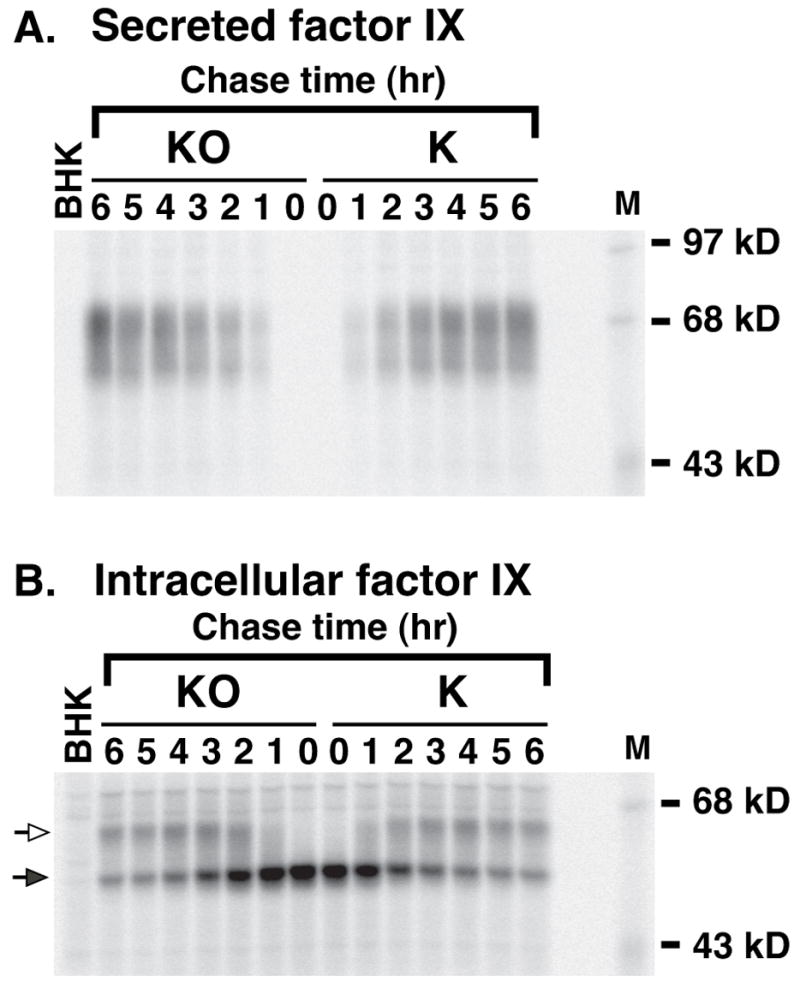
FIX BHK cells were radiolabeled (1 hr) in media lacking vitamin K and then chased for the indicated times in media containing either the epoxide (KO) or quinone (K) form of vitamin K (2 μM). FIX immunoprecipitated from media or cell lysates was then subjected to SDS-PAGE and PhosphorImager analysis. The hollow and filled arrows indicate, respectively, carboxylated and uncarboxylated fIX. Cells lacking vitamin K contain a larger fIX precursor pool (Fig. 4), which accounts for the different 35S-fIX turnover in this experiment than that shown in Fig. 2. M indicates molecular weight markers and BHK is the untransfected cell line, which was pulsed for 1 hr and then immediately harvested.
VKOR limits the rate of in vitro fIX carboxylation
The ability of VKOR to support VKD protein carboxylation in vitro was tested by comparing the dependence of fIX carboxylation on KH2, the cofactor form used directly by the carboxylase, to that of KO which requires VKOR reduction. Microsomes were prepared from fIX BHK cells (i.e. those analyzed in Figs. 2–6), which were cultured in the absence of vitamin K so that all of the fIX was uncarboxylated. Carboxylation was initiated by the addition of vitamin K (KH2 or KO) and [14C]-CO2 incorporation into fIX was monitored by SDS-PAGE and PhosphorImager, which showed that both vitamin K forms effected multiple Glu to Gla conversions in fIX as evidenced by the retarded migration of fIX (Fig. 7). KO-, but not KH2-, supported fIX carboxylation was inhibited by warfarin, as would be expected for a carboxylation reaction mediated by VKOR. The reaction mixtures with KO also contained DTT, which functionally substitutes for the reductant product of the redox protein that regenerates VKOR activity (33), and in the absence of DTT fIX carboxylation was substantially decreased (Fig. 7, lanes 5 vs. 8). The extent of fIX carboxylation was significantly higher with KH2 than KO, suggesting large rate differences between the two cofactor forms, and so the rate of fIX carboxylation was determined using shorter incubation times.
Figure 7. KO versus KH2-dependent in vitro fIX carboxylation.
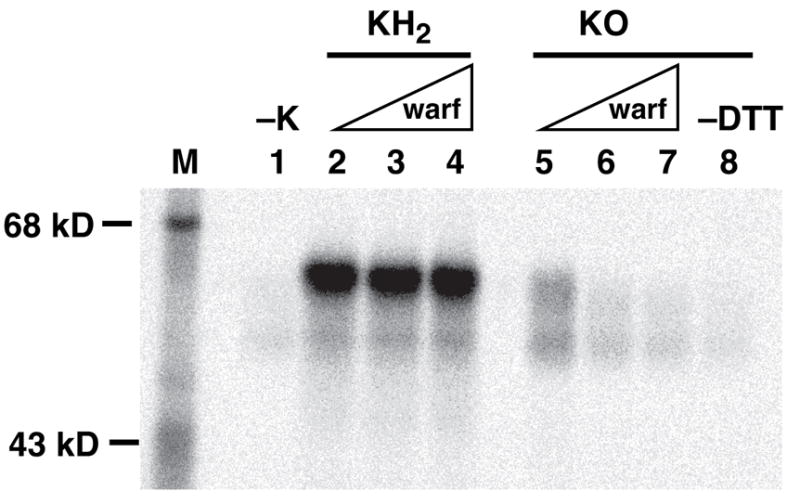
Microsomes prepared from fIX BHK cells cultured in the absence of vitamin K were incubated for 30 min in the absence (−K) or presence of the hydroquinone (KH2) or epoxide (KO) form of vitamin K, followed by SDS-PAGE and PhosphorImager analysis to measure 14CO2 incorporation into fIX. The reaction mixtures either lacked (lanes 2, 5, 8) or contained warfarin (warf) at final concentrations of 10 μM (lanes 3, 6) or 50 μM (lanes 4, 7). The reactions with KO also contained DTT, with the exception of the sample shown in lane 8. The residual activity in the −K sample is due to trace amounts of vitamin K present in the fIX BHK cells.
KH2-dependent fIX carboxylation was nonlinear (Fig. 8A), which is likely due to rapid carboxylation and slow VKD product release. Thus, previous analyses showed that most of the carboxylase in the fIX BHK cells exists in a complex with fIX (2, 16, 25). In the experiment shown in Fig. 8, a single turnover of fIX-carboxylase complex would be predicted to yield 3.1×107 counts of [14C]-CO2 incorporated into fIX based on the amount of fIX-carboxylase complex used in the assay (1.2 pmol per time point), which is similar to the value that was observed. Quantitation of the amounts of fIX and carboxylase in microsomes showed that the stoichiometry of fIX to carboxylase was 50:1, indicating that free fIX was available for multiple turnovers. The combined data, then, indicate that the kinetics of carboxylation of the preexisting fIX-carboxylase complex in microsomes is similar to that of purified fIX-carboxylase complex, which undergoes rapid fIX carboxylation (i.e. min) but slow release (i.e. hrs) of the carboxylated fIX product (2, 16).
Figure 8. VKOR activity limits in vitro fIX carboxylation.

Microsomes prepared from fIX BHK cells cultured in the absence of vitamin K, so that fIX was uncarboxylated, were incubated in reaction mixtures containing [14C]-CO2 and either KH2 (▲) (panel A) or KO (■) and DTT (panel B), and aliquots withdrawn at the indicated times were subjected to fIX immunoprecipitation, followed by SDS-PAGE and PhosphorImager quantitation (after a 30 day exposure) to analyze [14C]-CO2 incorporation into fIX. The reactions were performed in duplicate and the values varied < 10%.
In contrast to KH2-dependent carboxylation, KO-dependent fIX carboxylation was linear and substantially slower (Fig. 8B). The amount of [14C]-CO2 incorporation at the end of the reaction (4×106 counts) was less than would be predicted for a single fIX turnover, suggesting that the fIX-carboxylase complexes did not result in full carboxylation. The most likely explanation for the linearity is therefore that the observed rate of carboxylation measures only a single turnover, i.e. of the preexisting fIX-carboxylase complex. The early phase of KH2-dependent carboxylation was ~400-fold faster than the rate of KO-dependent carboxylation (Fig. 8), and may be even greater because the KH2-mediated reaction was rapid and consequently nonlinear with the time points measured. One possible explanation for the large difference in KO versus KH2-dependent rates of carboxylation is that the in vitro conditions were more optimal for carboxylase than VKOR, and we therefore repeated these experiments under varying conditions of pH (6.6–7.4) and ionic strength (0.1–1M KCl, NaCl or ammonium sulfate). These analyses all showed much lower KO than KH2-dependent fIX carboxylation, suggesting that VKOR reduction of KO to KH2 limits the rate of fIX carboxylation. We therefore tested whether VKOR might also be limiting in vivo.
VKOR overexpression causes a small increase in the rate of in vivo fIX carboxylation
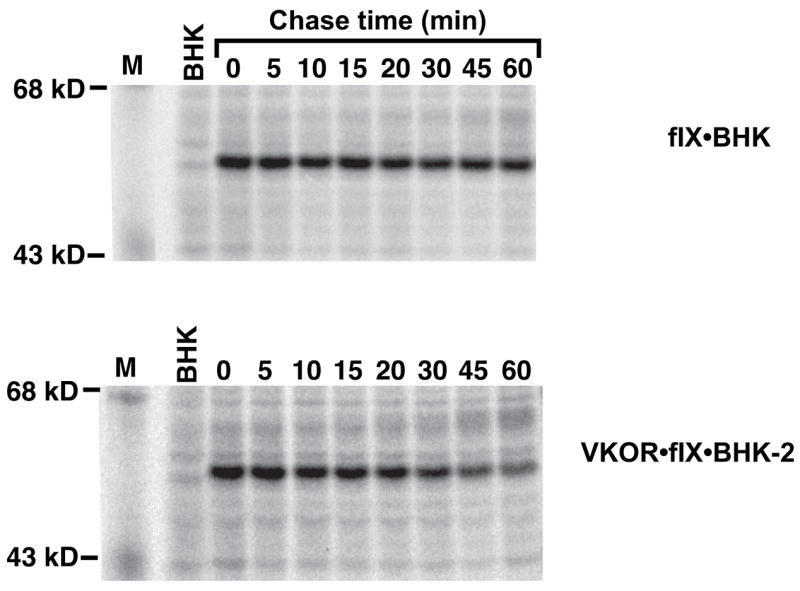
The characteristics of the fIX BHK cell line analyzed in Figs. 2–6 made it an excellent candidate for testing whether VKOR overexpression improves carboxylation in vivo. First, the fact that these cells contain a fIX precursor pool reveals that fIX turnover by the carboxylase is rate-limiting rather than protein synthesis and other processes preceding carboxylation, and offers the opportunity to test whether VKOR overexpression increases the ability of the carboxylase to turnover this fIX precursor pool more rapidly. Second, an important feature is that almost all of the newly-synthesized fIX is ultimately carboxylated and secreted, which means that the cellular components required for carboxylation and the secretory processes following carboxylation are not saturated and that the only variable introduced into the overall process is VKOR overexpression. We therefore established fIX BHK cells stably-transfected with the cDNA encoding a catalytic subunit of VKOR, r-VKORC1. Western analysis using an antibody against the C terminus of VKORC1 identified a protein (Supplemental Figure 2A) with a mass consistent with the 17 kD size predicted by the VKORC1 sequence (5, 6). The r-VKORC1 was functional, as the VKOR·fIX BHK cells showed 14–15-fold increases over the progenitor fIX BHK cells in ability to reduce KO (Table 1). Intracellular fIX and carboxylase levels were also measured in these cell lines, which showed that the concentrations were the same for fIX BHK and VKOR·fIX BHK cells. Thus, the stoichiometry of fIX and carboxylase, which had the potential to affect the carboxylation process (16), was unchanged by VKOR overexpression.
Table 1. Levels of fIX, carboxylase and VKOR in fIX BHK and VKOR·fIX BHK cells.
Lysates were prepared from BHK cells, cultured in the absence of vitamin K, that express low levels of fIX (fIX BHK) or that also express r-VKORC1 (VKOR·fIX BHK). FIX levels were measured in a quantitative western and carboxylase levels were determined by an activity assay and reference to the known specific activity (23). VKOR levels were quantitated by an activity assay that measures KO reduction. The values shown are for 1 mg of lysate.
| Cell line | Factor IX (pmol) | Carboxylase (pmol) | VKOR activity (pmol KO reduced/hr) | VKOR Activity Fold Increase |
|---|---|---|---|---|
| fIX BHK | 19.2 | 0.16 | 0.08 | 1 |
| VKOR·fIX BHK-1 | 17.8 | 0.20 | 1.12 | 14 |
| VKOR·fIX BHK-2 | 20.1 | 0.14 | 1.20 | 15 |
FIX turnover in BHK and VKOR·fIX BHK cells was analyzed by pulsing the cells for a short time (10 min) and then chasing for up to 30 min, all in the presence of vitamin K. This analysis showed that fIX turnover was faster in cells overexpressing VKOR (Fig. 9). FIX turnover could be quantitated by measuring either the rate of disappearance of uncarboxylated fIX or the rate of appearance of carboxylated fIX, which was possible because all of the fIX was intracellular during the short chase time used. Both determinations gave a rate that was 2.2-fold faster in cells overexpressing VKOR (shown for the rate of disappearance of uncarboxylated fIX in Fig. 10). The difference in rates was small but reproducible, as it was observed in four separate experiments like that shown in Fig. 9. This pulse-chase analysis was performed with the quinone form of vitamin K, which was used because it is the normal dietary and therefore physiological form. The rate of fIX carboxylation in fIX BHK and VKOR·fIX BHK cells was also tested using the epoxide form of vitamin K, which gave similar results to that of the quinone form (Fig. 11). These analyses indicate, then, that carboxylation is rate limiting in the fIX BHK cells and that VKOR activity regulates fIX turnover.
Figure 9. VKOR overexpression causes a 2.2-fold increase in the rate of in vivo fIX carboxylation.
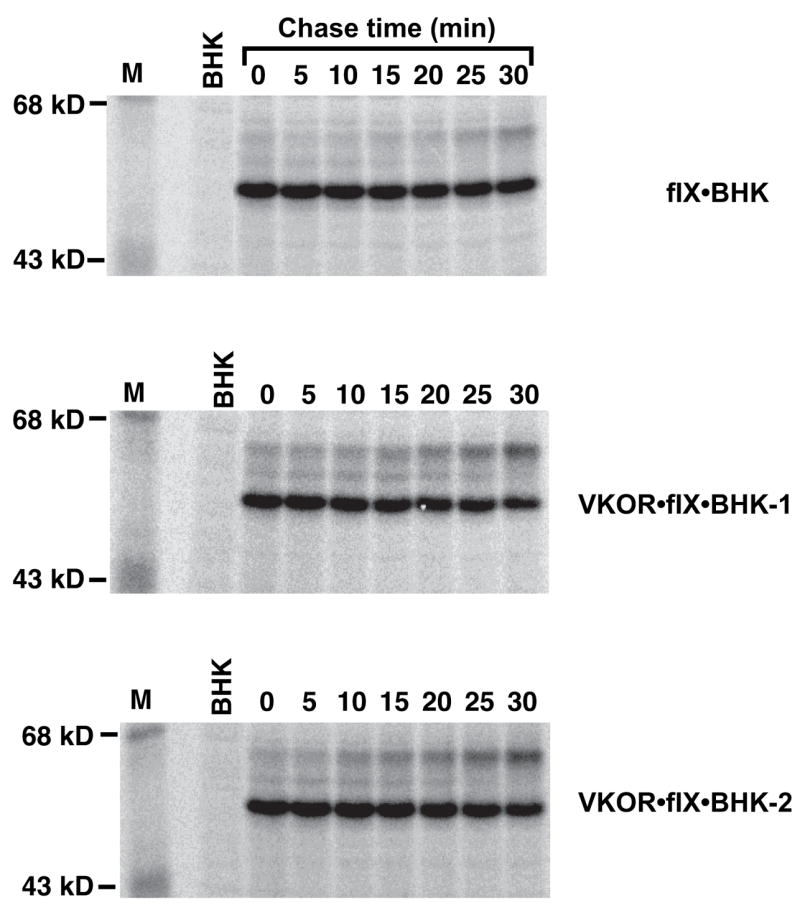
BHK cells expressing r-fIX (fIX BHK) or also expressing r-VKORC1 (VKOR·fIX BHK; two clonal isolates were analyzed) were tested for fIX turnover in pulse-chase studies. The pulse-chase was as described in the legend to Fig. 2 except that it was performed only in the presence of vitamin K quinone. All three cell lines were analyzed in a single experiment, using an equivalent number of cells for each cell line. FIX was not detected in the media (data not shown), indicating that all of the fIX was intracellular throughout the chase time used. BHK is a control untransfected cell line.
Figure 10. Quantitation of fIX turnover in fIX BHK and VKOR·fIX BHK cell lines.
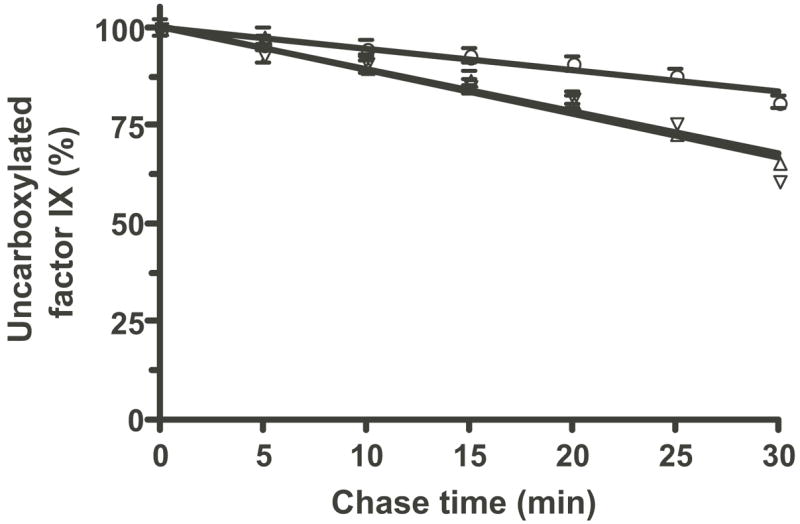
Duplicate gels that included the ones shown in Fig. 9 were quantitated by PhosphorImager and averaged (giving a 2% standard deviation as indicated by the error bars) to determine the rate of disappearance of uncarboxylated fIX in fIX BHK cells (○) or in two different VKOR·fIX BHK cell lines (△, ▽).
Figure 11. The epoxide and quinone forms of vitamin K both give the same effect of VKOR overexpression on the rate of fIX carboxylation.
Pulse-chase analysis on fIX BHK cells or VKOR·fIX BHK cells was performed as described in the legend to Figure 9 except for the use of the epoxide rather than quinone form of vitamin K.
VKOR overexpression causes a large increase in the rate of in vitro fIX carboxylation
In vitro carboxylation was performed because it allowed a test of the effect of VKOR overexpression on fIX carboxylation that is independent of the redox protein required for VKOR activity. Microsomes prepared from fIX BHK and VKOR·fIX BHK-2 cells were incubated in a reaction mixture containing [14C]-CO2 and either KH2 or KO and DTT, which functionally substitutes for the redox protein. KH2-dependent in vitro carboxylation of the VKOR·fIX BHK-2 microsomes gave similar kinetics to that observed with fIX BHK microsomes (i.e. as in Fig. 8). Both reactions started to plateau at a value (3.3×107 counts) that indicated only a single turnover of fIX (data not shown), consistent with the fact that the carboxylase levels (and therefore the amount of fIX-carboxylase complex) were the same in both of the cell lines (Table 1). In contrast, KO-dependent in vitro fIX carboxylation did not plateau (Fig. 12), consistent with the amount of carboxylation observed by the final time point (i.e. 2.5×107 counts for the VKOR·fIX BHK-2 microsomes) being less than that predicted for a single turnover. The rate of fIX carboxylation was 14-fold higher in the microsomes from BHK cells overexpressing VKOR. This increase was similar to the increase in KO reduction caused by VKOR overexpression (Table 1) and was significantly higher than the fold-increase in rate of fIX carboxylation observed in the pulse-chase analysis (Fig. 10).
Figure 12. VKOR overexpression in fIX BHK cells causes a 14-fold increase in the rate of KO-dependent in vitro fIX carboxylation.
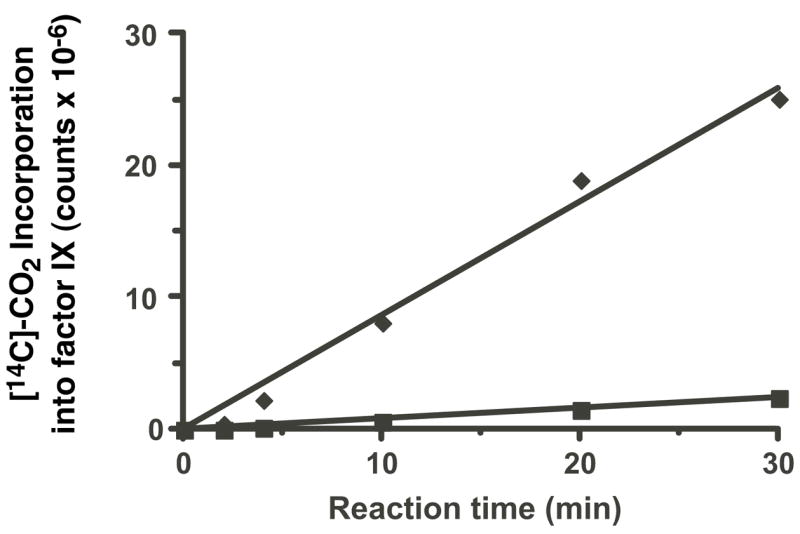
Microsomes were prepared from fIX BHK (■) and VKOR·fIX-2 BHK (◆) cells (i.e. those analyzed in Figs. 9–11) cultured in the absence of vitamin K, so that fIX was uncarboxylated. As described in the Experimental Procedures, an aliquot of the microsomes was quantitated to determine the amount of fIX-carboxylase complex, which showed equivalent amounts in the two preparations (i.e. 1.2 pmol per time point). The microsomes were incubated in a reaction mixture containing KO, DTT and [14C]-CO2 and aliquots were withdrawn at the indicated times, followed by fIX immunoprecipitation and SDS-PAGE and PhosphorImager quantitation (after a 32 day exposure) to measure [14C]-CO2 incorporation into fIX.
VKOR overexpression increases the extent of carboxylation of fIX secreted from high-producing fIX BHK cells
The acceleration in rate of fIX carboxylation caused by r-VKORC1 expression (Fig. 10) raised the possibility that similar overexpression in cells that are saturated for carboxylation might increase the extent of carboxylation of the secreted fIX. To test this possibility, r-VKORC1 was stably-transfected into a high-producing fIX BHK cell line that secretes fIX with a Gla content of 2.4 mol Gla per mol fIX (Table 2). This value was determined by purifying the secreted fIX using a MAb that recognizes both carboxylated and uncarboxylated forms (16), and performing Gla quantitation on the purified protein. r-VKORC1 expression in the high-producing fIX BHK cells (Supplemental Figure 2B) resulted in a 15-fold increase in VKOR activity but no change in the levels of carboxylase or intracellular fIX (data not shown). The amount of secreted fIX in the fIX BHK and VKOR·fIX BHK cell lines was also quantitatively similar but showed qualitative differences, with r-VKORC1 expression resulting in a 2.6-fold increase in Gla content, from 2.4 to 6–6.2 (Table 2). Thus, VKOR overexpression partially corrected the saturation in fIX carboxylation in the high-producing fIX BHK cells.
Table 2. VKOR overexpression increases the extent of carboxylation in fIX secreted from high-producing fIX BHK cells.
A fIX BHK cell line secreting fIX at 200 pmol/d/106 was stably-transfected with r-VKORC1 and clones expressing the highest VKOR levels were chosen for further analysis. FIX secreted from cells cultured in vitamin K was purified and Gla quantitation was performed by base hydrolysis and amino acid analysis, as before (22).
| Protein source | Gla content (mol Gla/mol factor IX) |
|---|---|
| fIX BHK | 2.4+/−0.2 |
| VKOR·fIX BHK-A | 6.0+/−0.3 |
| VKOR·fIX BHK-B | 6.2+/−0.3 |
| Plasma factor IX | 11.8+/−0.3 |
DISCUSSION
Our previous studies (16) suggested that the availability of reduced vitamin K is limiting and is the reason why carboxylation of VKD proteins becomes saturated when their expression levels are high, and we therefore analyzed the effect of VKOR overexpression in fIX BHK cells. Pulse-chase analyses in the presence or absence of warfarin or using different forms of vitamin K indicated that VKOR accomplishes most, if not all, vitamin K reduction that supports fIX carboxylation in BHK cells. This cell line contained endogenous VKOR activity (Table 1) that can account for KO-dependent in vivo fIX carboxylation (Figs. 6 and 11). The rate of carboxylation was not any faster when the quinone form of vitamin K was used (Fig. 6), indicating that quinone reductases other than VKOR do not augment carboxylation. VKOR is inhibited by warfarin, which totally abolished fIX carboxylation (Fig. 5) and which demonstrates that the alternative pathway present in liver which uses a warfarin-resistant quinone reductase to generate reduced vitamin K is not operational in the kidney-derived cells. This observation suggests that during warfarin therapy, kidney may respond similarly to that of bone, which does not show restored carboxylation of the VKD protein osteocalcin when vitamin K is coadministered with warfarin (34). This difference in response between liver and bone (and possible kidney) could be due to either the lack of the quinone reductase or to the accumulation of sufficiently high amounts of intracellular vitamin K for activity.
To analyze fIX carboxylation we used both low- and high-producing fIX BHK cell lines. We found that the low-producing fIX BHK cells contained a fIX precursor pool (Fig. 4) that was ultimately almost all carboxylated (93%) and secreted (Fig. 3 and (16)). This cell line was therefore used to test the effect of VKOR overexpression on intracellular fIX turnover because it provided the advantage that the components that facilitate carboxylation and secretion are not saturated and so the only variable introduced into the system is VKOR overexpression. The effect of r-VKORC1 on a high-producing fIX BHK cell line that inefficiently carboxylates fIX was also tested to determine whether VKOR overexpression improved the extent of carboxylation of secreted fIX. Combined in vitro and in vivo analyses showed that vitamin K reduction regulates the rate of fIX carboxylation. Thus, the rate of in vitro fIX carboxylation was at least 400-fold faster when driven by chemically reduced KH2 than by KO that requires VKOR reduction (Fig. 8) and was substantially increased by VKOR overexpression (Fig. 12). r-VKORC1 expression resulted in a 2.2-fold increase in rate of fIX carboxylation in low-producing fIX BHK cells (Figs. 9–11) and a similar increase in extent of carboxylation of secreted fIX in high-producing fIX BHK cells (Table 2). Notably, these increases were small compared to the amount of VKOR activity overexpression (15-fold, Table 1). When microsomes from the fIX BHK and VKOR·fIX BHK cells were tested for fIX carboxylation in vitro, r-VKORC1 caused a much larger increase (14-fold, Fig. 12) in the rate of fIX carboxylation that was nearly equivalent to the amount of VKOR activity overexpression (Table 1). Thus, r-VKORC1 is fully functional for supporting VKD protein carboxylation but its effect is limited in vivo.
One explanation for the limited effect of r-VKOR is that VKOR overexpression saturates another component of the carboxylation system, and a likely saturable component is the redox protein required for VKOR activity. VKOR is oxidized and inactivated during each conversion of KO to KH2 and active enzyme is regenerated by a redox protein (Fig. 1). DTT that functionally substitutes for the reductant generated by the redox protein (33) was used in the in vitro fIX carboxylation assay, which may explain why the increase in rate of fIX carboxylation in vitro was proportional to the amount of VKOR overexpression. In cultured cells, then, VKOR overexpression and consequent change in stoichiometry of VKOR to redox protein may saturate VKOR activity regeneration. The identity of the redox protein is currently unknown. Also unknown is whether other cell lines contain similar amounts of this protein, which could impact the ability of r-VKORC1 to increase carboxylation in these lines. Two recent papers (35, 36), which examined the effect of r-VKORC1 on VKD proteins secreted from BHK or 293 cells, also reported only 2–3-fold increases in the extent of carboxylated, secreted protein. Our determinations of the rates of in vitro and in vivo carboxylation suggest the possibility that the redox protein is critical to efficiency of carboxylation and that larger increases in carboxylation may not be possible until it is identified and coexpressed with VKORC1.
The effect of r-VKORC1 could also be limiting, however, if the increase in rate of carboxylation results in another step becoming rate limiting. As discussed in the Results, the in vitro carboxylation assays (Figs. 8, 12) likely measure the rate of fIX carboxylation in only a single turnover of fIX. In contrast, the in vivo rate determinations (Figs. 3, 10) measure multiple fIX turnovers and therefore the rate is the sum of fIX binding, multiple Glu carboxylations, and product release. If the rate of Glu carboxylation is only marginally slower than that of another step, then VKORC1 expression may cause a large increase in carboxylation but the contribution of this other step could result in there being only a small increase in overall turnover.
The observation that carboxylation was the rate limiting step in fIX turnover was surprising because previous in vitro studies indicated that release was rate limiting. Thus, in vitro carboxylation of a preformed fIX-carboxylase complex results in full carboxylation of the fIX within ~2 min but release of this fIX product takes hrs (2, 16). Likewise, the off-rate of a fully-carboxylated propeptide-containing fIX peptide is 50-fold slower than the rate of Glu carboxylation (37). The combined results indicate, then, that there is something different about release in vivo. This difference may account for the contrast in rates of fIX turnover observed in vivo and in vitro. Thus, our determination of the in vivo rate of fIX turnover (14/hr/carboxylase), which is the first such reported value, showed that the turnover is 14–28-fold faster than that observed in vitro with propeptide-containing fIX and purified carboxylase (2, 3). The fold difference may be even greater because the in vitro assays are performed using saturating amounts of KH2 while in the fIX BHK cells the generation of KH2 is rate limiting. The more rapid turnover of fIX in vivo suggests that fIX release from the carboxylase is more efficient in the fIX BHK cells, possibly because the interaction of VKD proteins and carboxylase is facilitated as observed with other multiprotein complexes assembled in the ER (38).
The intracellular analyses were revealing with regard to how the extent of carboxylation of secreted VKD proteins can be impacted by the events that occur during VKD protein secretion. The studies showed that low-producing fIX BHK cells contain a precursor pool of uncarboxylated fIX (Fig. 4) present in 44-fold excess over the carboxylase, which pulse-chase analysis showed is ultimately all secreted (Fig. 3 and (16)). Gla quantitation showed that almost all (93%) of the fIX secreted from the cells is carboxylated, which is of interest because pulse-chase analysis in the absence of vitamin K showed that carboxylation is not obligatory for secretion (Fig. 2A). One mechanism that can reconcile these observations was revealed by pulse-chase analysis which showed that ER egress of fIX was slow: 93% of the fIX was still present in the ER after 90 min (Figs. 2, 3), and this residence time is much longer than typically observed with other secreted proteins (39). During this time, 56% of the fIX was carboxylated (Fig. 3). Thus, the slow exit of fIX from the ER allowed more time for multiple fIX turnovers. A second mechanism that can help explain the extent of carboxylation of the secreted fIX is preferential secretion of carboxylated fIX. Thus, the population of uncarboxylated to carboxylated fIX was ~2:1 (Fig. 4), which would predict a value of 4 mol Gla per mol of secreted fIX rather than the observed value (11.2) in the absence of any selection. One possible step where preferential secretion of carboxylated fIX may occur is during exit from the ER: egress is mediated by cargo receptors (not yet identified for fIX) that may show specificity for carboxylated over uncarboxylated protein. An interesting question, then, is how/whether these mechanisms are affected by high production of VKD proteins, where the extent of carboxylation of secreted protein is decreased (e.g. Table 2). Presumably, the balance between secretion and carboxylation is disrupted, resulting in a secreted VKD protein population that comprises a mixture of fully carboxylated and uncarboxylated (and possibly undercarboxylated) protein.
The observation of a VKD protein precursor pool in a cell line raises the question of whether such pools normally exist in tissue. Precursor pool buildup is known to occur when vitamin K is limiting, e.g. during warfarin treatment, however whether such pools exist when vitamin K is plentiful is unknown. This issue is important because tissues express multiple VKD proteins (e.g. at least seven in liver) and the VKD proteins exhibit wide differences in propeptide affinity (40). Consequently, the presence of precursor pools could potentially result in competition between different VKD proteins for the carboxylase.
Secretory events can also impact the carboxylation status of secreted VKD proteins by a filtering mechanism which our results indicate is not the same for all VKD proteins. Thus, substantially less protein C and protein Z are secreted in the absence than presence of vitamin K (41, 42). For protein C, this difference has been shown to be due to preferential degradation of the poorly carboxylated protein via the proteasomal pathway (42). FIX did not show such a response: pulse-chase analysis showed that the newly-synthesized fIX was ultimately all secreted even in the absence of vitamin K (Fig. 2 and (16)). This difference was not cell line specific because all three of these VKD proteins were analyzed in BHK cells. These observations indicate, then, that secretory processing is not necessarily the same for all VKD proteins.
Supplementary Material
Supplemental Figures 1 and 2 are included as supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Rebecca Shtofman for her help in generating r-VKORC1 and Dr. Mark Rishavy for helpful comments during the preparation of the manuscript.
Abbreviations
- VKD
vitamin K-dependent
- VKOR
vitamin K oxidoreductase
- VKORC1
vitamin K oxidoreductase component 1
- fIX
factor IX
- KH2
vitamin K hydroquinone
- KO
vitamin K epoxide
- ER
endoplasmic reticulum
- K
vitamin K quinone
Footnotes
Funded by NIH HL55666
References
- 1.Berkner KL, Runge KW. The physiology of vitamin K nutriture and vitamin K-dependent protein function in atherosclerosis. J Thromb Haemost. 2004;2:2118–2132. doi: 10.1111/j.1538-7836.2004.00968.x. [DOI] [PubMed] [Google Scholar]
- 2.Stenina O, Pudota BN, McNally BA, Hommema EL, Berkner KL. Tethered processivity of the vitamin K-dependent carboxylase: factor IX is efficiently modified in a mechanism which distinguishes Gla’s from Glu’s and which accounts for comprehensive carboxylation in vivo. Biochemistry. 2001;40:10301–10309. doi: 10.1021/bi0107039. [DOI] [PubMed] [Google Scholar]
- 3.Morris DP, Stevens RD, Wright DJ, Stafford DW. Processive post-translational modification. Vitamin K-dependent carboxylation of a peptide substrate. J Biol Chem. 1995;270:30491–30498. doi: 10.1074/jbc.270.51.30491. [DOI] [PubMed] [Google Scholar]
- 4.Berkner KL. The vitamin K-dependent carboxylase. Annu Rev Nutr. 2005;25:127–149. doi: 10.1146/annurev.nutr.25.050304.092713. [DOI] [PubMed] [Google Scholar]
- 5.Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hortnagel K, Pelz HJ, Lappegard K, Seifried E, Scharrer I, Tuddenham EG, Muller CR, Strom TM, Oldenburg J. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427:537–541. doi: 10.1038/nature02214. [DOI] [PubMed] [Google Scholar]
- 6.Li T, Chang CY, Jin DY, Lin PJ, Khvorova A, Stafford DW. Identification of the gene for vitamin K epoxide reductase. Nature. 2004;427:541–544. doi: 10.1038/nature02254. [DOI] [PubMed] [Google Scholar]
- 7.Suttie JW. Vitamin K-dependent carboxylase. Annu Rev Biochem. 1985;54:459–477. doi: 10.1146/annurev.bi.54.070185.002331. [DOI] [PubMed] [Google Scholar]
- 8.Malhotra OP. Purification and characterization of dicoumarol-induced prothrombins. II. Barium oxalate atypical (5-Gla) variant. Thromb Res. 1979;15:439–448. doi: 10.1016/0049-3848(79)90150-6. [DOI] [PubMed] [Google Scholar]
- 9.Malhotra OP. Purification and characterization of dicoumarol-induced prothrombins. III Alumina pH 4.6 atypical (2-Gla) variant. Thromb Res. 1979;15:449–463. doi: 10.1016/0049-3848(79)90151-8. [DOI] [PubMed] [Google Scholar]
- 10.Suttie JW. Report of Workshop on expression of vitamin K-dependent proteins in bacterial and mammalian cells, Madison, Wisconsin, USA, April 1986. Thromb Res. 1986;44:129–134. doi: 10.1016/0049-3848(86)90189-1. [DOI] [PubMed] [Google Scholar]
- 11.Berkner KL. Expression of recombinant vitamin K-dependent proteins in mammalian cells: Factors IX and VII. Methods Enzymol. 1993;222:450–477. doi: 10.1016/0076-6879(93)22029-f. [DOI] [PubMed] [Google Scholar]
- 12.Kaufman RJ, Wasley LC, Furie BC, Furie B, Shoemaker CB. Expression, purification, and characterization of recombinant gamma-carboxylated factor IX synthesized in Chinese hamster ovary cells. J Biol Chem. 1986;261:9622–9628. [PubMed] [Google Scholar]
- 13.Fourrier F. Recombinant human activated protein C in the treatment of severe sepsis: an evidence-based review. Crit Care Med. 2004;32:S534–541. doi: 10.1097/01.ccm.0000145944.64532.53. [DOI] [PubMed] [Google Scholar]
- 14.Hedner U. Recombinant factor VIIa (Novoseven) as a hemostatic agent. Semin Hematol. 2001;38:43–47. doi: 10.1016/s0037-1963(01)90147-7. [DOI] [PubMed] [Google Scholar]
- 15.Rehemtulla A, Roth DA, Wasley LC, Kuliopulos A, Walsh CT, Furie B, Furie BC, Kaufman RJ. In vitro and in vivo functional characterization of bovine vitamin K-dependent gamma-carboxylase expressed in Chinese hamster ovary cells. Proc Natl Acad Sci USA. 1993;90:4611–4615. doi: 10.1073/pnas.90.10.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hallgren KW, Hommema EL, McNally BA, Berkner KL. Carboxylase overexpression impairs factor IX secretion: implications for the release of vitamin K-dependent proteins. Biochemistry. 2002;41:15045–15055. doi: 10.1021/bi026016e. [DOI] [PubMed] [Google Scholar]
- 17.Foster DC, Holly RD, Sprecher CA, Walker KM, Kumar AA. Endoproteolytic processing of the human protein C precursor by the yeast Kex2 endopeptidase coexpressed in mammalian cells. Biochemistry. 1991;30:367–372. doi: 10.1021/bi00216a009. [DOI] [PubMed] [Google Scholar]
- 18.Kurachi K, Davie EW. Isolation and characterization of a cDNA coding for human factor IX. Proc Natl Acad Sci USA. 1982;79:6461–6464. doi: 10.1073/pnas.79.21.6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Busby SJ, Mulvihill E, Rao D, Kumar AA, Lioubin P, Heipel M, Sprecher C, Halfpap L, Prunkard D, Gambee J, Foster DC. Expression of recombinant human plasminogen in mammalian cells is augmented by suppression of plasmin activity. J Biol Chem. 1991;266:15286–15292. [PubMed] [Google Scholar]
- 20.Busby S, Kumar A, Joseph M, Halfpap L, Insley M, Berkner K, Kurachi K, Woodbury R. Expression of active human factor IX in transfected cells. Nature. 1985;316:271–273. doi: 10.1038/316271a0. [DOI] [PubMed] [Google Scholar]
- 21.Fieser LF, Tishler M, Sampson WL. VITAMIN K ACTIVITY AND STRUCTURE. J Biol Chem. 1941;137:659–692. [Google Scholar]
- 22.Berkner KL, McNally BA. Purification of vitamin K-dependent carboxylase from cultured cells. Methods Enzymol. 1997;282:313–333. doi: 10.1016/s0076-6879(97)82117-9. [DOI] [PubMed] [Google Scholar]
- 23.Pudota BN, Miyagi M, Hallgren KW, West KA, Crabb JW, Misono KS, Berkner KL. Identification of the vitamin K-dependent carboxylase active site: Cys- 99 and Cys-450 are required for both epoxidation and carboxylation. Proc Natl Acad Sci USA. 2000;97:13033–13038. doi: 10.1073/pnas.97.24.13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berkner KL, Pudota BN. Vitamin K-dependent carboxylation of the carboxylase. Proc Natl Acad Sci USA. 1998;95:466–471. doi: 10.1073/pnas.95.2.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lingenfelter SE, Berkner KL. Isolation of the human gamma-carboxylase and a gamma-carboxylase- associated protein from factor IX-expressing mammalian cells. Biochemistry. 1996;35:8234–8243. doi: 10.1021/bi9523318. [DOI] [PubMed] [Google Scholar]
- 26.Sadowski JA, Esmon CT, Suttie JW. Vitamin K-dependent carboxylase. Requirements of the rat liver microsomal enzyme system. J Biol Chem. 1976;251:2770–2775. [PubMed] [Google Scholar]
- 27.Roncaglioni MC, Soute BA, de BvdBM, Vermeer C. Warfarin-induced accumulation of vitamin K-dependent proteins. Comparison between hepatic and non-hepatic tissues. Biochem Biophys Res Commun. 1983;114:991–997. doi: 10.1016/0006-291x(83)90658-7. [DOI] [PubMed] [Google Scholar]
- 28.Hirsh J, Dalen J, Anderson DR, Poller L, Bussey H, Ansell J, Deykin D. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest. 2001;119:8S–21S. doi: 10.1378/chest.119.1_suppl.8s. [DOI] [PubMed] [Google Scholar]
- 29.Long DJ, 2nd, Jaiswal AK. NRH:quinone oxidoreductase2 (NQO2) Chem Biol Interact. 2000;129:99–112. doi: 10.1016/s0009-2797(00)00200-3. [DOI] [PubMed] [Google Scholar]
- 30.Long DJ, 2nd, Iskander K, Gaikwad A, Arin M, Roop DR, Knox R, Barrios R, Jaiswal AK. Disruption of dihydronicotinamide riboside:quinone oxidoreductase 2 (NQO2) leads to myeloid hyperplasia of bone marrow and decreased sensitivity to menadione toxicity. J Biol Chem. 2002;277:46131–46139. doi: 10.1074/jbc.M208675200. [DOI] [PubMed] [Google Scholar]
- 31.Eickelmann P, Ebert T, Warskulat U, Schulz WA, Sies H. Expression of NAD(P)H:quinone oxidoreductase and glutathione S-transferases alpha and pi in human renal cell carcinoma and in kidney cancer-derived cell lines. Carcinogenesis. 1994;15:219–225. doi: 10.1093/carcin/15.2.219. [DOI] [PubMed] [Google Scholar]
- 32.Ross D, Siegel D. NAD(P)H:quinone oxidoreductase 1 (NQO1, DT-diaphorase), functions and pharmacogenetics. Methods Enzymol. 2004;382:115–144. doi: 10.1016/S0076-6879(04)82008-1. [DOI] [PubMed] [Google Scholar]
- 33.Whitlon DS, Sadowski JA, Suttie JW. Mechanism of coumarin action: significance of vitamin K epoxide reductase inhibition. Biochemistry. 1978;17:1371–1377. doi: 10.1021/bi00601a003. [DOI] [PubMed] [Google Scholar]
- 34.Price PA, Kaneda Y. Vitamin K counteracts the effect of warfarin in liver but not in bone. Thromb Res. 1987;46:121–131. doi: 10.1016/0049-3848(87)90212-x. [DOI] [PubMed] [Google Scholar]
- 35.Sun YM, Jin DY, Camire RM, Stafford DW. Vitamin K epoxide reductase significantly improves carboxylation in a cell line over-expressing factor X. Blood. 2005;106 doi: 10.1182/blood-2005-06-2495. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 36.Wajih N, Hutson SM, Owen J, Wallin R. Increased production of functional recombinant human clotting factor IX by baby hamster kidney cells engineered to overexpress VKORC1, the vitamin K 2,3-epoxide-reducing enzyme of the vitamin K cycle. J Biol Chem. 2005;280:31603–31607. doi: 10.1074/jbc.M505373200. [DOI] [PubMed] [Google Scholar]
- 37.Lin PJ, Straight DL, Stafford DW. Binding of the factor IX gamma-carboxyglutamic acid domain to the vitamin K-dependent gamma-glutamyl carboxylase active site induces an allosteric effect that may ensure processive carboxylation and regulate the release of carboxylated product. J Biol Chem. 2004;279:6560–6566. doi: 10.1074/jbc.M312239200. [DOI] [PubMed] [Google Scholar]
- 38.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 39.Lodish HF, Kong N, Snider M, Strous GJ. Hepatoma secretory proteins migrate from rough endoplasmic reticulum to Golgi at characteristic rates. Nature. 1983;304:80–83. doi: 10.1038/304080a0. [DOI] [PubMed] [Google Scholar]
- 40.Stanley TB, Jin DY, Lin PJ, Stafford DW. The propeptides of the vitamin K-dependent proteins possess different affinities for the vitamin K-dependent carboxylase. J Biol Chem. 1999;274:16940–16944. doi: 10.1074/jbc.274.24.16940. [DOI] [PubMed] [Google Scholar]
- 41.Souri M, Koseki-Kuno S, Iwata H, Kemkes-Matthes B, Ichinose A. A naturally occurring E30Q mutation in the Gla domain of protein Z causes its impaired secretion and subsequent deficiency. Blood. 2005;105:3149–3154. doi: 10.1182/blood-2004-06-2250. [DOI] [PubMed] [Google Scholar]
- 42.Tokunaga F, Wakabayashi S, Koide T. Warfarin causes the degradation of protein C precursor in the endoplasmic reticulum. Biochemistry. 1995;34:1163–1170. doi: 10.1021/bi00004a009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figures 1 and 2 are included as supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.