

Abstract
We previously showed that angiocidin, a tumor and vascular associated protein, is a potent inhibitor of angiogenesis and tumor growth [J Cellular Biochem 2004;92(1):125-46]. Angiocidin is a multidomain protein that exerts its anti-angiogenic activity through multiple mechanisms, including effects on cell matrix interaction [Exp Cell Research 2006; 312(13):2443-53]. Here we describe another activity of angiocidin that may contribute to its anti-tumor activity. We show that angiocidin activates monocytes to secrete a mixture of pro-inflammatory cytokines and induces them to differentiate into a macrophage-like cell. Using the monocytic cell line THP-1, we demonstrate that angiocidin induces the cells to become adherent and phagocytic, express macrophage markers, and secret matrix matelloproteinase-9 (MMP-9). Microarray analysis of control and angiocidin-treated THP-1 cells revealed that angiocidin up-regulated p105/p50, p100/p52, and rel B, components of the NFκB pathway. We confirmed the microarray data and showed that angiocidin induced phosphorylation of Iκβ, p50, and p65 and translocation of p50 and p65 to the nucleus. We also showed that angiocidin activated up-stream mediators of NFκB, such as the mitogen activated protein kinase pathway (MAPK) and phosphoinositide-3 kinase (PI3-kinase). Blockage of NFκB and MAPK activation with small molecule inhibitors completely prevented angiocidin-mediated secretion of cytokines from THP-1 cells, but did not inhibit their adhesive phenotype. Blocking PI3-kinase inhibited both secretion of cytokines as well as the adhesive phenotype. These data suggest that angiocidin activates monocytes to secrete cytokines and differentiates them to a macrophage-like phenotype through at least two pathways mediated by MAPK and NFκB as well as PI3-kinase.
Keywords: angiocidin, monocytes, macrophages, differentiation, angiogenesis, tumor progression, immunotherapy, cytokines
Introduction
Solid human tumors are often infiltrated by host immune cells, which can have a diverse effect on tumor progression. Among other cells types, macrophages are known to be a major component of the leukocyte infiltrate of tumors. These tumor-associated macrophages, or TAMs, have a complex dual function in their interactions with neoplastic cells (1). Cytotoxic T-cells, when presented the appropriate tumor antigen by macrophages, can kill tumor cells. In contrast, TAMs can also promote angiogenesis and metastasis. The contradictory reports that have surfaced can best be explained in terms of the “macrophage balance hypothesis” which asserts that the outcome of the interaction between macrophages and neoplastic cells depends on the number of macrophages recruited to the tumor microenvironment, and their state of activation (2). The number and distribution of TAMs is linked to prognosis in different types of human malignancies (3,4). Furthermore, ex vivo-grown tumor cytotoxic macrophages are effective against murine models of metastasizing tumors (5). Immunomodulation, therefore, is a therapeutic strategy worthy of continued investigation.
Our laboratory has been studying the potential anti-tumor activity of a protein originally isolated from lung carcinoma (6). We cloned the protein and identified matrix protein binding domains within the molecule that were essential for its anti-tumor activity (7). The cloned recombinant protein, termed angiocidin, or it's matrix binding domain peptide, when injected systemically into tumor bearing mice, significantly inhibited tumor growth and angiogenesis (7-9). Since the matrix is important in binding cytokines that enable leukocytes to home to the tumor, and angiocidin binds important tumor matrix proteins such as thrombospondin-1, we postulated that angiocidin might function to activate leukocytes.
In this report, we show that angiocidin potently stimulates monocytes and the monocytic cell line THP-1 to secrete a mixture of inflammatory cytokines. Angiocidin-treated THP-1 cells become adherent and differentiate into cells that morphologically resemble macrophages. We further show that this pro-inflammatory activity of angiocidin is mediated through a pathway involving the activation of nuclear factor kappa B (NFκB), MAPK, and PI3-kinase. These macrophage-like cells are capable of phagocytosis and antigen presentation. These newly discovered activities of angiocidin likely contribute to its anti-tumor activity.
Materials and Methods
Chemicals and Antibodies
Roswell Park Memorial Institute (RPMI)-1640 media was purchased from MediaTech (Herndon, VA). Trizol Reagent was purchased from Invitrogen (Carlsbad, CA). Ficoll Paque was purchased from Amersham Biosciences (Piscataway, NJ). Brefeldin A solution and the MCP-1 ELISA kit were purchased from eBioscience (San Diego, CA). The Cytokine Antibody Array was obtained from RayBiotech, Inc. (Norcross, GA). Fluorescent microparticles were purchased from Polysciences, Inc. (Warrington, PA). The Nuclear Extraction Kit was purchased from Panomics (Fremont, CA). The NF-κB p65 inhibitor 6-Amino-4-(4-phenoxyphenylethylamino)quinazoline was purchased from Biomol International (Plymouth Meeting, PA). The MEK 1/2 inhibitor U0126 and the PI3-K inhibitor LY294002 were purchased from Cell Signaling (Danvers, MA). The anti-MMP-9 antibody was purchased from R&D Systems (Minneapolis, MN). The TNF-α antibody was obtained from BD Biosciences (San Jose, CA). The anti-phospho-FAK, anti-phospho-paxillin, anti-phospho-p44/42 and anti-p44/42 antibodies, as well as the NF-κB Pathway Sampler Kit, were purchased from Cell Signaling (Danvers, MA). Anti-phospho-p50 and anti-p50 antibodies were obtained from Santa Cruz (Santa Cruz, CA).
Expression, and Purification of His-Tagged Recombinant Angiocidin
Expression and purification of his-tagged angiocidin was performed as previously described (7). Endotoxin was removed from the preparation by Triton X-114 phase separation as previously described (10). Purified protein contained less than 3 international units of endotoxin per mg of protein.
Cell Culture of THP-1 Cells
Human pro-monocytic THP-1 cells were obtained from American Type Culture Collection and cultured in RPMI-1640 media supplemented with 10% heat-inactivated fetal bovine serum, 2% glutamate, 0.5% penicillin/streptamycin, and 0.1% gentamycin. Cells were maintained at 37° C in a humidified growth chamber supplemented with 5% CO2.
Angiocidin Treatment Conditions
Cells were treated for 24 hours with or without 10 μg/ml angiocidin at a density of 2 X 106 cells/ml in serum free RPMI media supplemented with 0.1% BSA for 24 hours.
Immunoblotting
25 μg of lysate was run on a 10% SDS polyacrylamide gel under reducing conditions and transferred to PVDF membrane. Membranes were blocked with 5% milk/TBST, then probed with the appropriate antibody at a 1:1000 dilution. The following day, after washing, membranes were incubated with a 1:10:000 dilution of goat anti-rabbit IgG/HRP and developed chemiluminescently with ECL Plus. To assess cytoplasmic to nuclear shuttling of the various NF-κB proteins, cytoplasmic and nuclear fractions were isolated using a Nuclear Extraction Kit according to the manufacturer's protocol.
Microarray Analysis
Total RNA was collected from cells using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Microarray analysis was then performed by Fox Chase Cancer Center using an Agilent gene chip array.
Flow Cytometry
Peripheral blood mononuclear cells were harvested from whole blood of a healthy human donor and isolated by density gradient centrifugation using Ficoll Paque according to the manufacturer's protocol. Peripheral blood monocytes were isolated from lymphocytes with anti-CD14-conjugated magnetic beads using a MidiMACS separator according to the manufacturer's instructions. After treatment with various concentrations of angiocidin for 6 hours, purified monocytes were stained with a PE-conjugated anti-TNF-α antibody for 30 minutes in the dark prior to fixation with 4% paraformaldehyde. Cells were treated with brefeldin A solution in order to inhibit intracellular transport of the accumulated TNF-α protein according to the manufacturer's suggestions.
Cytokine Antibody Array
A cytokine antibody array was used according to the manufacturer's instructions.
MCP-1 ELISA
A capture ELISA was used according to the manufacturer's protocol.
Phagocytosis Assay
Fluorescent green 0.5 μm microparticles diluted to a density of 5 X 1011 particles/ml sterile PBS were added to cells at a concentration of 25 μl microparticles/ml media for 2 hours at 37° C. Phagocytosis was stopped with the addition of 1 ml ice cold sterile PBS. Cells were washed in ice cold PBS five times to remove microparticles that remained in suspension, or were loosely adherent to the tissue culture plastic or the surface of the cells. Cells were covered with a glass coverslip and observed and photographed using fluorescence microscopy.
Gelatin Zymography
Gelatin zymography was performed as previously described (11). Immunoblot confirmation of zymogram results was performed as described above using a 1 μg/ml solution of mouse monoclonal anti-MMP-9 antibody.
Inhibitor Studies
THP-1 cells were pre-treated with 1 μg/ml 6-Amino-4-(4-phenoxyphenylethylamino)quinazoline, 10 μM U0126, or 50 μM LY294002 at 37° C for 60 minutes. Angiocidin was then added to the appropriate wells. The volume of DMSO (vehicle control) added to the cells was equivalent to the volume of inhibitor used.
Results
Angiocidin Induces THP-1 Adhesion/Differentiation
Angiocidin-treated THP-1 cells, a suspension cell line, became more adherent to the plastic substratum in a dose-dependent manner as shown in Figure 1A. While untreated cells remained spherical and completely in suspension, angiocidin-treated cells became flattened and elongated, extending numerous “processes.” The dose required to obtain optimum adhesion/differentiation was 10 μg/ml.
Figure 1. Angiocidin Induces Focal Adhesion-Dependent Changes in THP-1 Cell Adhesion and Morphology.
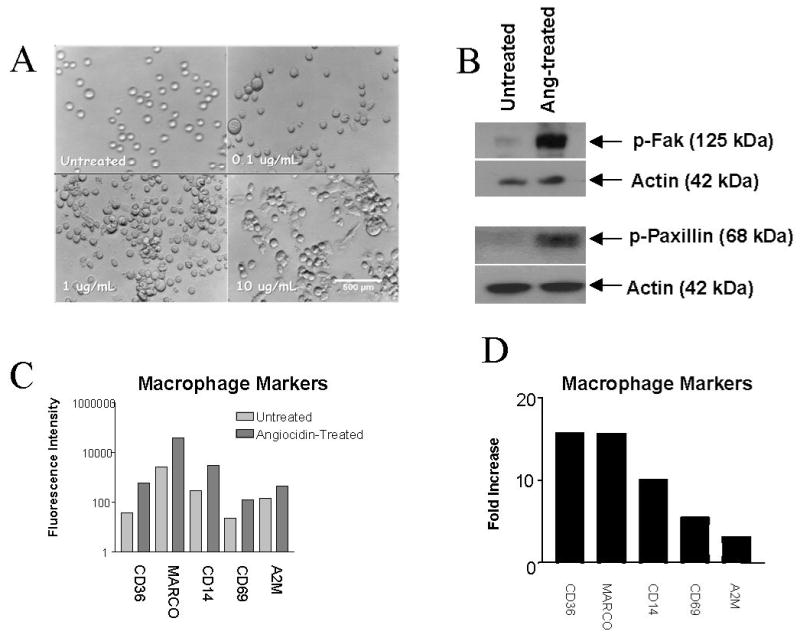
THP-1 cells were treated in a dose-dependent manner with concentrations of angiocidin ranging from 0 – 10 μg/ml. After 24 hours, wells were examined using Hoffman optics and photographed. For Immunoblot analysis, THP-1 cells were treated with or without 10 μg/ml angiocidin and left to activate for 24 hours. Blots were probed with an anti-phospho-FAK antibody or an anti-phospho-paxillin antibody at a 1:1000 dilution in 5% BSA/PBS and developed as described in the Materials and Methods section. Molecular weights in the blots were estimated from molecular weight markers run simultaneously on the same gel. For microarray analysis, cells were treated with or without 10 μg/ml angiocidin for 24 hours. The following day, total RNA was collected from cells using Trizol Reagent according to the manufacturer's protocol. (A) Photomicrographs of THP-1 cells treated with different concentrations of angiocidin, 40X magnification. The experiment was repeated numerous times, and the data presented represents a typical experiment. (B) Angiocidin-induced phosphorylation of focal adhesion kinase (FAK) and paxillin. The experiment was repeated three times, and the data presented represents a typical experiment. (C) Total fluorescence intensity values for macrophage markers as assessed by microarray analysis. The data represent an average of at least two determinations obtained from the Agilent Chip. (D) Fold increase in fluorescence intensity for each macrophage marker as assessed by microarray analysis
Angiocidin Induces Focal Adhesion Kinase and Paxillin Phosphorylation
Untreated cells showed barely detectable levels of phosphorylated FAK protein, consistent with their non-adherent morphology, while treatment of THP-1 cells with angiocidin resulted in a dramatic increase in levels of phosphorylated FAK protein, in accordance with these cells' newly-acquired adhesive phenotype (Figure 1B). Angiocidin treatment also resulted in significant phosphorylation of paxillin protein, while untreated cells again contained barely detectable levels of the phosphorylated form of this molecule (Figure 1B).
Angiocidin-treated THP-1 Cells Express Macrophage Markers
We next sought to analyze angiocidin-induced changes in gene expression in THP-1 cells. We hypothesized that various markers of monocyte activation and differentiation would become upregulated. We found several important genes upregulated at the mRNA level in angiocidin-treated cells, namely CD36, macrophage receptor with collagenous structure (MARCO), CD14, CD69, and alpha 2 macroglobulin (A2M) (Figure 1C and 1D). There is evidence in the literature that upregulation of each of these genes, discussed below, occurs upon differentiation of monocytes into macrophage-like cells.
Angiocidin Activates Peripheral Blood Mononuclear Cells
To ascertain whether angiocidin is able to induce activation of freshly isolated peripheral blood monocytes, we used flow cytometric analysis to assay intracellular accumulation of TNF-α. Angiocidin activated mononuclear cells in a dose-dependent manner at angiocidin levels as low as 1.0 μg/ml after only 6 hours of treatment (Figure 2A). This figure also demonstrates a population of cells expressing low levels of CD14, which are not activated by angiocidin to produce TNF-α. This result is likely due to the presence of low levels of granulocytes present in our monocyte preparation, since granulocytes are known to express low levels of this cell surface marker.
Figure 2. Angiocidin Induces Activation of Human Peripheral Blood Mononuclear and THP-1 Cells.

Peripheral blood mononuclear cells (PBMCs) and monocytes were harvested as described in the Materials and Methods section. For MCP-1 quantitation, a capture ELISA was utilized according to the manufacturer's instructions. Culture supernatants for the Cytokine Antibody Array and the MCP-1 ELISA were obtained as described in the Materials and Methods section. (A) Flow cytometric analysis of cellular activation induced by angiocidin in human peripheral blood monocytes. Percent activation is shown in each plot. The experiment was repeated three times and the data presented represents a typical experiment. (B) Integrated Density Values (IDVs) for each inflammatory mediator secreted by angiocidin-treated or untreated THP-1 cells as assessed by the Cytokine Antibody Array. Each bar represents an average of two replicates. Error bars represent the standard error of the mean. (C) The percentage by which each cytokine increased in concentration in culture supernatants upon treatment with angiocidin (D) Concentration of MCP-1 in the culture supernatants of angiocidin-treated and untreated THP-1 cells. Each bar represents an average of three replicates. The experiment was repeated three times, and the data presented represents a typical experiment.
Angiocidin Induces Secretion of a Specific Mixture of Inflammatory Mediators
As monocyte activation is associated with the secretion of pro-inflammatory cytokines/chemokines, we sought to determine whether angiocidin-induced monocyte differentiation was concurrent with cytokine/chemokine secretion (12, 13). We used densitometry scanning of cytokine array membranes to evaluate changes in relative cytokine/chemokine secretion levels between angiocidin-treated and untreated cell supernatants. We found that 18 of the 42 cytokines/chemokines assayed by this array membrane were secreted at increased levels by angiocidin-treated THP-1 cells (Figures 2B/2C). The largest increase in secretion between angiocidin-treated and untreated cells occurred with MCP-1. We confirmed these results with our microarray analysis, which also showed that various inflammatory cytokines/chemokines, as well as their cognate receptors, became upregulated at the mRNA level upon treatment with angiocidin (Supplemental Data). For example, angiocidin treatment results in a nearly 800-fold increase in mRNA expression levels of MCP-1 (Supplemental Data). We sought to confirm these results and quantitate the amount of MCP-1 secreted into culture supernatants upon treatment of THP-1 cells with angiocidin. Untreated cells secreted barely detectable levels of MCP-1, while treatment with angiocidin resulted in secretion of approximately 1.3 ng/ml MCP-1(Figure 2D). These results confirm the data obtained with our cytokine antibody array and microarray, and demonstrate that angiocidin is indeed able to induce the secretion of important pro-inflammatory mediators.
Angiocidin Increases the Phagocytic Ability of THP-1 Cells
Monocyte to macrophage differentiation has been demonstrated to be accompanied by an increase in the phagocytic activities of these cells (14). We therefore analyzed whether angiocidin-treated THP-1 cells acquired increased phagocytic activity toward fluorescent microparticles designed specifically for this purpose. Angiocidin-treated cells displayed an increased phagocytic activity, with greater than 90% of the cells internalizing these fluorescent microparticles, while THP-1 cells not treated with angiocidin did not phagocytose microparticles as efficiently (Figure 3A). These results indicate that, upon treatment with angiocidin, THP-1 cells acquire macrophage-like functions.
Figure 3. Angiocidin Induces THP-1 Cells to Become Phagocytic and Secrete MMP-9.

Angiocidin-treated THP-1 cells were assayed for phagoctic activity and MMP-9 secretion as described in the Materials and Methods section. (A-TOP) Microparticle phagocytosis by angiocidin-treated THP-1 cells (A-BOTTOM) Microparticle phagocytosis by untreated THP-1 cells. The experiment was repeated three times, and the data presented represents a typical experiment. (B-TOP) Zymogram of culture supernatants from angiocidin-treated or untreated THP-1 cells and PBMCs (B–BOTTOM) Immunoblot of the same culture supernatants. The experiment was repeated three times, and the data presented represents a typical experiment.
Angiocidin Induces THP-1 Cells to Secrete MMP-9
Activated macrophages secrete several enzymes that cause changes in the molecular and mechanical structure of the extracellular matrix. They are a rich source of metalloproteases (MMPs) (15). MMP-9 expression typically falls into one of two distinct patterns. Some cell types, such as macrophages, only produce MMP-9 in response to prolonged exposure to an inflammatory stimulus. Other cell types constitutively produce MMP-9 and can rapidly release it from intracellular stores (16). Given the activating effect of angiocidin on both THP-1 and human peripheral blood monocytes, we next wanted to characterize the effect angiocidin treatment would have on the secretion of MMPs. Angiocidin stimulated THP-1 cells and PBMCs to secrete enzymatically active MMP-9 (Figure 3B).
Angiocidin Induces Activation of MAPK and NF-κB Pathways
We performed a series of immunoblots investigating key signaling molecules known to become activated by cytokine treatment (17). Angiocidin treatment resulted in activation of MAPK p44/42, as well as the upstream kinase c-Raf, as assessed by the phosphorylation state of these molecules (Figure 4A).
Figure 4. Angiocidin Induces Activation of MAPK and NF-κB Signaling Molecules.
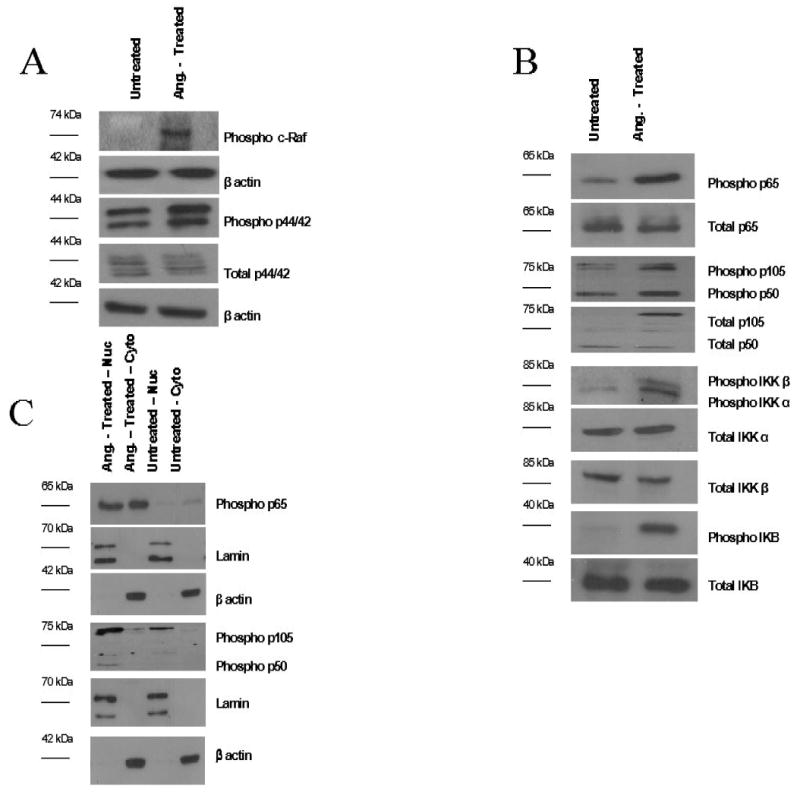
Cell lysates and cytoplasmic/nuclear extracts from angiocidin-treated and untreated THP-1 cells were prepared and analyzed as described in the Materials and Methods section. (A) Angiocidin-induced phosphorylation of various players in the MAPK p44/42 pathway (B) Angiocidin-induced phosphorylation of various players in the NF-κB pathway (C) Angiocidin-induced cytoplasmic to nuclear shuttling of NF-κB p65 and p50. Each line indicates the molecular weight of the immunoblotted factor. Each experiment was repeated three times, and the data presented represents a typical experiment.
Our microarray experiment demonstrated that various members of the NF-κB signaling pathway became upregulated at the mRNA level upon treatment with angiocidin (Supplemental Data). We therefore decided to analyze the involvement of the NF-κB pathway in angiocidin-induced signaling. Both NF-κB p65 and p50 showed increased phosphorylation upon treatment with angiocidin, while total protein levels remained the same. Angiocidin-treated THP-1 cells also expressed much higher levels of phosphorylated IKB-α and IKK proteins (Figure 4B). The IKK complex is responsible for phosphorylation of the inhibitory IKB-α protein, which results in polyubiquitination and degradation of this inhibitor, subsequently releasing the p65/p50 heterodimer to translocate into the nucleus and initiate DNA transcription.
We also performed immunoblots on isolated nuclear and cytoplasmic lysates to determine whether p65 and p50 showed increased nuclear localization in angiocidin-treated THP-1 cells. Levels of phosphorylated p65 are significantly increased in the nuclear fractions of angiocidin-treated THP-1 cells, while untreated nuclear lysates contain barely detectable levels of these proteins (Figure 4C). Figure 4C also shows a similar result was achieved when analyzing cytoplasmic and nuclear lysates for phosphorylated p50. These data indicate that, once the inhibitory IKB-α molecule is phosphorylated and degraded, the p50/p65 heterodimer is released and able to translocate into the nucleus and initiate transcription of its target genes.
MAPK, NF-κB, and PI3-K Pathways are Involved in Angiocidin-Induced Cytokine and MMP-9 Secretion
It has been demonstrated in the literature that various MAPK pathways are involved in the induction of cytokine secretion (18-20). Likewise, activation of the NF-κB pathway has been shown to initiate the secretion of a wide variety of cytokines and immunomodulatory molecules. Therefore, we wanted to determine whether inhibition of the MAPK and NF-κB pathways would minimize angiocidin-induced cytokine secretion. We chose to assay for secreted levels of MCP-1, due to the high abundance of this chemotactic cytokine in the conditioned media of angiocidin-treated THP-1 cells (Figure 2D).
THP-1 cells treated with angiocidin alone secreted approximately 1.4 ng/ml MCP-1, while untreated cells secreted undetectable levels of this chemokine. Cells treated with both angiocidin and the U0126 MAPK MEK 1/2 inhibitor secreted only 77 pg/ml MCP-1, demonstrating that this MAPK pathway is involved in angiocidin-induced cytokine secretion (Figure 5A). Although cells treated with both U0126 and angiocidin secreted extremely low levels of MCP-1, the cells were observed to undergo dramatic morphological changes. These results suggest that the adhesive phenotype induced by angiocidin is separate from angiocidin-induced cytokine/chemokine secreting activity.
Figure 5. Inhibition of MAPK p44/42, NF-κB p65, and PI3-K pathways Blocks Angiocidin-Induced MCP-1 and MMP-9 Secretion.
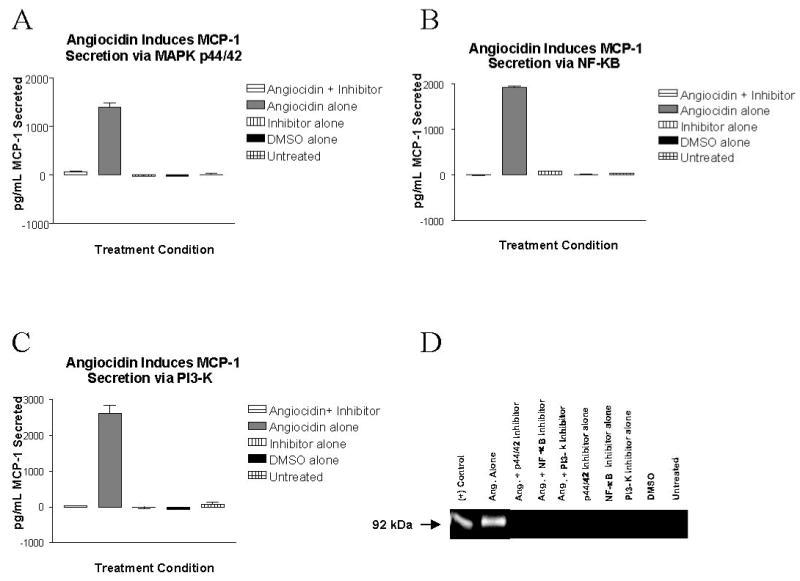
THP-1 cells were pre-treated with 1 μg/ml 6-Amino-4-(4-phenoxyphenylethylamino)quinazoline, 10 μM U0126, or 50 μM LY294002 at 37° C for 60 minutes. After this time, 10 μg/ml angiocidin was added to the appropriate wells. After 24 hours, culture supernatants were collected, centrifuged to remove any cellular contamination, and analyzed for the presence of secreted MCP-1 using a capture ELISA, or the matrix metalloproteinase MMP-9 using gelatin zymography, as described in the Materials and Methods section. (A) Inhibition of MAPK MEK 1/2 abrogates angiocidin-induced MCP-1 secretion (B) Inhibition of NF-κB p65 abrogates angiocidin-induced MCP-1 secretion (C) Inhibition of PI3-kinase abrogates angiocidin-induced MCP-1 secretion. In experiments A-C, each bar represents an average of three replicates. Error bars indicate the standard error of the mean. The experiment was repeated three times, and the data presented represents a typical experiment. (D) Inhibition of MAPK MEK 1/2, NF-κB p65, and PI3-kinase abrogates angiocidin-induced MMP-9 secretion. The experiment was repeated three times, and the data presented represents a typical experiment.
Similarly, angiocidin-treated THP-1 cells pre-treated with an NF-κB inhibitor molecule known to abrogate NF-κB p65 transcriptional activity did not secrete any detectable levels of MCP-1 suggesting that the NF-κB pathway is also involved in angiocidin-induced cytokine/chemokine secretion (Figure 5B). As with U0126, cells treated with the NF-κB inhibitor were also observed to adhere/differentiate to the same extent as cells treated with angiocidin alone. These results once again suggest that the processes of adhesion/differentiation and cytokine/chemokine secretion are initiated by separate pathways in our system.
Use of both the MAPK MEK 1/2 and NF-κB inhibitors indicated that an additional signaling pathway was responsible for angiocidin-induced cellular adhesion/differentiation. We therefore sought to determine whether PI3-kinase was responsible for the observed adhesive effect. PI3-kinase has been implicated in the control of cytokine release from a variety of cell types (21-24). In addition, various studies have demonstrated crosstalk between PI3-kinase and the MAPK and NF-κB pathways, and that PI3-kinase signals upstream of both these pathways in various cell types (25-27). Treatment of THP-1 cells with angiocidin and the PI3-kinase inhibitor LY294002 reduced the levels of MCP-1 in the conditioned media to the level present in the conditioned media of unreated cells (Figure 5C). These data demonstrate that, in addition to the MAPK and NF-κB pathways, PI3-kinase also plays a role in angiocidin-induced cytokine/chemokine secretion. Of note, cells treated with both angiocidin and the PI3-kinase inhibitor did not become adherent to the tissue culture plastic. Therefore, PI3-kinase may be playing a dual role in our system, controlling cytokine/chemokine release as well as adhesion/differentiation of the THP-1 cells.
We also sought to clarify whether inhibition of MAPK MEK 1/2, NF-κB, and PI3-kinase would inhibit angiocidin-induced MMP-9 secretion. Treatment with angiocidin elicited secretion of this matrix metalloproteinase. However, pre-treatment of cells with the MEK 1/2 inhibitor, the NF-κB inhibitor, or the PI3-kinase inhibitor, prior to treatment with angiocidin, completely abrogated angiocidin-induced MMP-9 secretion (Figure 5D). As expected, cells left untreated, or those treated with the inhibitor alone or the DMSO vehicle control, also did not secrete any detectable levels of MMP-9.
MAPK and PI3-K Function Upstream of NF-κB
We have shown that inhibition of MAPK MEK 1/2 or NF-κB p65 elicit similar effects in our system. These results indicated the possibility that MAPK p44/42 (whose phosphorylation is prevented by the MEK 1/2 inhibitor) is acting upstream of NF-κB p65. There is extensive literature citing a relationship between the MAPK and NF-κB pathways (28-30). We therefore hypothesized that inhibition of MEK 1/2, and therefore p44/42, would result in inhibition of the NF-κB pathway. THP-1 cells treated with angiocidin showed increased levels of phospho-p65 compared to untreated cells, as previously demonstrated, while pre-treatment of cells with the U1026 inhibitor reduced angiocidin-induced NF-κB p65 phosphorylation to control levels (Figure 6A). These results indicate that MAPK p44/42 is indeed acting upstream of the NF-κB pathway.
Figure 6. In Angiocidin-Treated THP-1 Cells, Both MAPK p44/42 and PI3-kinase Function Upstream of NF-κB p65.

THP-1 cells were treated with the appropriate inhibitors, and cell extracts analyzed for the activation of signaling factors as described in the Materials and Methods section. (A) Inhibition of MAPK MEK 1/2 abrogates angiocidin-induced phosphorylation of NF-κB p65 (B) Inhibition of PI3-kinase abrogates angiocidin-induced phosphorylation of NF-κB p65. Each experiment was repeated three times, and the data represents a typical experiment. (C) Proposed signal transduction mechanisms induced by angiocidin in THP-1 cells.
As inhibition of PI3-K elicited similar results in our system as inhibition of MAPK MEK 1/2 and NF-κB, we next hypothesized that perhaps PI3-kinase was functioning upstream of the MAPK - NF-κB pathway. We again used the LY294002 PI3-kinase inhibitor to reduce PI3-kinase activity prior to angiocidin treatment. We then probed these cell lysates for phosphorylated p65 protein. In agreement with our hypothesis, treatment with angiocidin alone once again resulted in phosphorylation of the NF-κB molecule, while treatment of the cells with LY294002 prior to angiocidin treatment reduced the levels of phosphorylated p65 protein (Figure 6B). These results therefore indicate that PI3-kinase is indeed functioning upstream of our MAPK - NF-κB pathway.
As a final step to confirm this hypothesis, we then probed our LY294002-pre-treated cell lysates for the phosphorylated form of the MAPK p44/42 protein. We hypothesized that levels of phosphorylated MAPK p44/42 would be significantly reduced when the PI3-K pathway was inhibited. However, we obtained unexpected results. There was virtually no difference in phospho-p44/42 levels, whether or not the cells were pre-treated with LY294002 (data not shown). These results therefore indicate that angiocidin is inducing the activation of at least two separate pathways in THP-1 cells. In the first pathway, MAPK p44/42 is functioning upstream of the NF-κB pathway. In the second scenario, NF-κB is also activated, however, it is functioning downstream of PI3-K, without utilizing MAPK p44/42 as an intermediary signaling molecule. A diagram describing these proposed angiocidin-induced signal transduction pathways is depicted in Figure 6C.
Discussion
Angiocidin was first isolated from lung carcinoma by thrombospondin-1 peptide affinity chromatography (6). Angiocidin was demonstrated to be over-expressed by invasive cancer cells and capillary endothelial cells (31-33). This protein was also found in the blood of patients with advanced melanoma, as well as cancers of the colon, prostate and breast. Patients with more advanced cancers present with higher levels of angiocidin in their sera than healthy individuals, or patients with less advanced disease (34). These results indicated that angiocidin may regulate tumor progression.
Consistent with its regulatory role in tumorigenesis, we found that angiocidin inhibited critical steps of tumor progression in vitro, including angiogenesis and invasion. Systemically injected angiocidin also exhibited potent anti-tumor activity in vivo (7).
A counterintuitive observation previously set forth is that removal of a primary tumor stimulates growth of secondary tumors, suggesting that the primary tumor produces factors that inhibit the development of these secondary metastases. Folkman and colleagues (3) have identified a number of these factors, including endostatin and angiostatin. Our data suggest that angiocidin belongs to this family of molecules that suppress tumor growth. By regulating the levels of secreted, anti-tumorigenic molecules such as angiocidin, we can take advantage of the therapeutic potential of these molecules in the treatment of cancer.
Angiocidin binds the matrix proteins TSP-1 and collagen, as well as the collagen receptor α2β1 (7, 8). This integrin may be a potential cell surface receptor for angiocidin in THP-1 cells as well. A 20 amino acid domain in the N-terminal domain of angiocidin mediates its matrix and integrin-binding activities (7, 8). This matrix-and integrin binding domain of angiocidin is required for its anti-tumor activity. A mutant angiocidin, missing the high-affinity matrix-binding site, lacked inhibitory activity in tumor cell invasion of collagen and angiogenesis. Furthermore, a 25 amino acid peptide containing the matrix integrin binding domain of angiocidin inhibited progression of Lewis Lung and human colon carcinoma in vivo (8, 9). These data support the hypothesis that the anti-tumor activity of angiocidin depends strongly on its capacity to interact with matrix and integrin receptors.
Here we demonstrate that angiocidin has the capacity to potently induce the monocytic cell line THP-1 to differentiate into adherent cells resembling tissue macrophages (Figure 1A). We have demonstrated this adherence phenotypically, as well as by probing angiocidin-treated and untreated lysates for the phosphorylated form of the focal adhesion proteins focal adhesion kinase (FAK) and paxillin (Figure 1B).
We have also demonstrated genotypically that THP-1 cells become differentiated upon treatment with angiocidin. Our microarray data shows that angiocidin induces mRNA upregulation of various molecules known to be “differentiation state dependent” (Figure 1C/1D and Supplemental Data). As demonstrated in these figures, CD36 expression increased nearly 16-fold. CD36 is an adhesive receptor for TSP-1 and collagen, as well as a scavenger receptor expressed during the late stages of monocyte differentiation. The steady-state mRNA levels of CD36 increase during monocyte-to-macrophage differentiation (35, 36). We also observed a nearly 16-fold increase in mRNA expression of MARCO (macrophage receptor with collagenous structure). MARCO is a novel macrophage-specific receptor structurally related to scavenger receptors. It is believed that expression of MARCO has a direct effect in generating the phenotype of activated macrophages necessary for the trapping and removal of pathogens and other foreign substances (37). It is possible that the phagocytic activities of angiocidin-treated THP-1 cells may be attributable to increased expression of scavenger receptors such as CD36 or MARCO. Another molecule whose mRNA expression increased significantly upon treatment with angiocidin was the cell surface molecule CD14. This pattern recognition receptor has been used as a macrophage-specific differentiation antigen whose expression increases in response to differentiation inducers such as PMA and Vitamin D3 (38-42). CD69, which is also rapidly induced after leukocyte activation and is not present on the surface of resting cells, also increased nearly 6-fold in angiocidin-treated THP-1s (43). We also observed an increase in the mRNA expression levels of alpha 2 macroglobulin (A2M), the synthesis of which has been demonstrated to be differentiation state-dependent in THP-1 cells (44). These results help support the hypothesis that angiocidin is activating THP-1 cells to become macrophage-like.
Angiocidin treatment also induces cellular activation. Flow cytometric analysis revealed a nearly 40% activation of monocytes with a dose of 1 μg/ml angiocidin (Figure 2A). Additionally, our cytokine antibody array data demonstrate that, of the 42 inflammatory mediators assayed, 18 become increasingly secreted upon angiocidin treatment (Figures 2B and 2C). We demonstrate that angiocidin treatment results in secretion of ng/ml quantities of MCP-1 into culture supernatants (Figure 2D).
Angiocidin treatment also results in secretion of MMP-9 (Figure 3B). This protease may aid tumorigenic leukocytes in their extravasation from the blood compartment into the tumor microenvironment. Angiocidin-treated THP-1 cells also presented with phagocytic activity (Figure 3A). Phagocytosis by macrophages aids these cells in ingesting debris shed by tumor cells, which is then presented on the macrophage cell surface to immune effector cells, particularly tumoricidal CD8+ T cells. Angiocidin-activated peripheral blood mononuclear cells are able to effectively present antigen to cytotoxic T lymphocytes, and angiocidin-treated THP-1 cells upregulate proteins important to the process of antigen presentation, such as various MHC class I and class II molecules (Kremlev et al. in press).
Angiocidin treatment results in an increase in the phosphorylation of MAPK p44/42, as well as its upstream kinase c-Raf (Figure 4A). In addition, various players in the NF-κB pathway become phosphorylated and translocate into the nucleus (Figures 4B and 4C). As shown in Figure 5 A-D, these signaling molecules are responsible for angiocidin-induced cytokine secretion and MMP-9 secretion. We also demonstrate the involvement of PI3-K in angiocidin-induced MCP-1 and MMP-9 secretion and cell adhesion.
MAPK p44/42 is likely functioning upstream of NF-κB in our system, as is PI3-K (Figures 6A and 6B). Interestingly, inhibition of PI3-K had no effect on angiocidin-induced phosphorylation of MAPK p44/42 (Data Not Shown), indicating that two separate pathways may be at work in our system. The first pathway places MAPK p44/42 upstream of NF-κB. The second places PI3-K also upstream of NF-κB, however, MAPK p44/42 is bypassed. A diagram describing these proposed angiocidin-induced signal transduction pathways is depicted in Figure 6C.
Given its matrix-binding ability, angiocidin becomes localized to the matrix of the tumor microenvironment, where it is easily accessible and able to exert its pro-inflammatory effects on the immune system. Matrix-bound angiocidin, behaving in a “cytokine-like manner,” would be readily available to induce differentiation and activation of monocytes into phagocytic, tumor antigen-presenting macrophages. The specific “mixture” of inflammatory cytokines and chemokines inducibly released from these activated macrophages would further this immune response and allow this matrix-bound angiocidin to serve as the “soil” to which other activated, potentially tumoricidal leukocytes would be recruited.
Activated macrophages can be tumoricidal. Macrophages can present tumor antigens to T cells, or directly kill tumor cells following activation with cytokines such as IL-2, INF-y and IL-12. The so-called Macrophage-Mediated Tumor Cytotoxicity (or MTC) involves the secretion of lytic factors into neoplastic cells, resulting in tumor cell lysis. Thus, monocyte-derived macrophages have powerful constitutive anti-neoplastic properties even in the absence of specific immunity. Tumor formation has been demonstrated to depend on the level of MCP-1 secretion and macrophage infiltration. Cells producing high levels of MCP-1 invoked a massive TAM infiltrate and subsequent tumor destruction within a few days after injection into mice (4). As angiocidin has been demonstrated to produce MCP-1 upregulation at both the mRNA and protein levels of expression, it is conceivable that systemically injected angiocidin could inhibit tumor growth through the upregulation of this important chemotactic cytokine in the tumor microenvironment.
We believe that angiocidin, and its 25 amino acid matrix-binding peptide, present a therapeutic advantage in immunomodulatory cancer therapy. Not only does angiocidin exert potent anti-tumor effects on a variety of cell types, but it does so without any cytotoxicity. Tumor-bearing mice injected systemically with angiocidin present with no evidence of necrosis in any of the organs examined, including liver, kidney, intestine, lung, and brain (7). Angiocidin's matrix-binding activity suggests an additional therapeutic advantage, that of being localized to the matrix surrounding the tumor where it is readily available and accessible to exert a direct anti-tumorigenic effect, or additionally through elicitation of an immune response.
An additional significance of our results lies in the fact that the primary cell type we are using in our investigations is the monocytic leukemia cell line THP-1. Our results therefore suggest that angiocidin may have therapeutic potential in the treatment of this hematological cancer. In recent years, the question has arisen as to whether the malignant phenotype of leukemia can be reverted to a non-malignant phenotype without correcting any genetic abnormalities. This reversion can be achieved by reprogramming tumor cells by epigenetic changes which induce differentiation. If leukemic tumor cells can be forced to differentiate and to cease proliferation, then their malignant potential will be controlled. Thus the epigenetic suppression of malignancy by the induction of differentiation bypasses the genetic abnormalities present in tumor cells (45-48). Differentiation therapy has aroused great interest because its mechanism is different from the chemotherapy currently in use, which is based on the theory of “total cell kill,” and because it is expected to be less toxic than such cytotoxic chemotherapies (49, 50). We believe that our angiocidin protein may have therapeutic potential in differentiation therapy.
Supplementary Material
Acknowledgments
The work was supported in part by NIH Grant RO1 CA 88931 (GPT) and the Temple University Bridge Funding Program.
References
- 1.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 2.Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The origin and function of tumor-associated macrophages. Immunol Today. 1992;13:265–70. doi: 10.1016/0167-5699(92)90008-U. [DOI] [PubMed] [Google Scholar]
- 3.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–65. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 4.Nesbit M, Schaider H, Miller TH, Herlyn M. Low-level monocyte chemoattractant protein-1 stimulation of monocytes leads to tumor formation in nontumorigenic melanoma cells. J Immunol. 2001;166:6483–90. doi: 10.4049/jimmunol.166.11.6483. [DOI] [PubMed] [Google Scholar]
- 5.Andreesen R, Hennemann B, Krause SW. Adoptive immunotherapy of cancer using monocyte-derived macrophages: rationale, current status, and perspectives. J Leukocyte Biol. 1998;64:419–26. doi: 10.1002/jlb.64.4.419. [DOI] [PubMed] [Google Scholar]
- 6.Tuszynski GP, Rothman VL, Papale M, Hamilton BK, Eyal J. Identification and characterization of a tumor cell receptor for CSVTCG, a thrombospondin adhesive domain. J Cell Biol. 1993;120:513–21. doi: 10.1083/jcb.120.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou J, Rothman VL, Sargiannidou I, et al. Cloning and characterization of angiocidin, a tumor cell binding protein for thrombospondin-1. J Cell Biochem. 2004;92:125–46. doi: 10.1002/jcb.20076. [DOI] [PubMed] [Google Scholar]
- 8.Sabherwal Y, Rothman VL, Dimitrov S, et al. Integrin alpha2beta1 mediates the anti-angiogenic and anti-tumor activities of angiocidin, a novel tumor-associated protein. Exp Cell Res. 2006;312:2443–53. doi: 10.1016/j.yexcr.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Liebig C, Agarwal N, Ayala GE, Verstovsek G, Tuszynski GP, Albo D. Angiocidin inhibitory peptides decrease tumor burden in a murine colon cancer model. J Surg Res. 2007;142:320–6. doi: 10.1016/j.jss.2007.02.036. [DOI] [PubMed] [Google Scholar]
- 10.Aida Y, Pabst MJ. Removal of endotoxin from protein solutions by phase separation using Triton X-114. J Immunol Meth. 1990;132:191–5. doi: 10.1016/0022-1759(90)90029-u. [DOI] [PubMed] [Google Scholar]
- 11.Qian X, Wang TN, Rothman VL, Nicosia RF, Tuszynski GP. Thrombospondin-1 modulates angiogenesis in vitro by up-regulation of matrix metalloproteinase-9 in endothelial cells. Exp Cell Res. 1997;235:403–12. doi: 10.1006/excr.1997.3681. [DOI] [PubMed] [Google Scholar]
- 12.Araujo F, Slifer T, Li S, Kuver A, Fong L, Remington J. Gemifloxacin inhibits cytokine secretion by lipopolysaccharide stimulated human monocytes at the post-transcriptional level. Clin Microbiol Infect. 2004;10:213–9. doi: 10.1111/j.1198-743x.2004.00824.x. [DOI] [PubMed] [Google Scholar]
- 13.Micouin A, Rouillard D, Bauvois B. Induction of macrophagic differentiation and cytokine secretion by IgG1 molecules in human normal monocytes and myelogenous leukemia cells. Leukemia. 1997;11:552–60. doi: 10.1038/sj.leu.2400602. [DOI] [PubMed] [Google Scholar]
- 14.Reyes L, Davidson MK, Thomas LC, Davis JK. Effects of Mycoplasma fermentans incognitus on differentiation of THP-1 cells. Infect Immun. 1999;67:3188–92. doi: 10.1128/iai.67.7.3188-3192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Worley JR, Baugh MD, Hughes DA, et al. Metalloproteinase expression in PMA-stimulated THP-1 cells. Effects of peroxisome proliferator-activated receptor-gamma (PPAR gamma) agonists and 9-cis-retinoic acid. J Biol Chem. 2003;278:51340–6. doi: 10.1074/jbc.M310865200. [DOI] [PubMed] [Google Scholar]
- 16.Wiehler S, Cuvelier SL, Chakrabarti S, Patel KD. p38 MAP kinase regulates rapid matrix metalloproteinase-9 release from eosinophils. Biochem Biophys Res Comm. 2004;315:463–70. doi: 10.1016/j.bbrc.2004.01.078. [DOI] [PubMed] [Google Scholar]
- 17.Rao KM. MAP kinase activation in macrophages. J Leukocyte Biol. 2001;69:3–10. [PubMed] [Google Scholar]
- 18.Zhou HR, Islam Z, Pestka JJ. Rapid, sequential activation of mitogen-activated protein kinases and transcription factors precedes proinflammatory cytokine mRNA expression in spleens of mice exposed to the trichothecene vomitoxin. Toxicol Sci. 2003;72:130–42. doi: 10.1093/toxsci/kfg006. [DOI] [PubMed] [Google Scholar]
- 19.Hedges JC, Singer CA, Gerthoffer WT. Mitogen-activated protein kinases regulate cytokine gene expression in human airway myocytes. Am J Resp Cell Mol Biol. 2000;23:86–94. doi: 10.1165/ajrcmb.23.1.4014. [DOI] [PubMed] [Google Scholar]
- 20.Means TK, Pavlovich RP, Roca D, Vermeulen MW, Fenton MJ. Activation of TNF-alpha transcription utilizes distinct MAP kinase pathways in different macrophage populations. J Leukocyte Biol. 2000;67:885–93. doi: 10.1002/jlb.67.6.885. [DOI] [PubMed] [Google Scholar]
- 21.Gibbs BF, Grabbe J. Inhibitors of PI 3-kinase and MEK kinase differentially affect mediator secretion from immunologically activated human basophils. J Leukocyte Biol. 1999;65:883–90. doi: 10.1002/jlb.65.6.883. [DOI] [PubMed] [Google Scholar]
- 22.Kalinina N, Agrotis A, Antropova Y, et al. Increased expression of the DNA-binding cytokine HMGB1 in human atherosclerotic lesions: role of activated macrophages and cytokines. Arterio Thromb Vascular Biol. 2004;24:2320–5. doi: 10.1161/01.ATV.0000145573.36113.8a. [DOI] [PubMed] [Google Scholar]
- 23.Ishizuka T, Chayama K, Takeda K, et al. Mitogen-activated protein kinase activation through Fc epsilon receptor I and stem cell factor receptor is differentially regulated by phosphatidylinositol 3-kinase and calcineurin in mouse bone marrow-derived mast cells. J Immunol. 1999;162:2087–94. [PubMed] [Google Scholar]
- 24.Chandrasekar B, Mummidi S, Perla RP, et al. Fractalkine (CX3CL1) stimulated by nuclear factor kappaB (NF-kappaB)-dependent inflammatory signals induces aortic smooth muscle cell proliferation through an autocrine pathway. Biochem J. 2003;373:547–58. doi: 10.1042/BJ20030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gingery A, Bradley E, Shaw A, Oursler MJ. Phosphatidylinositol 3-kinase coordinately activates the MEK/ERK and AKT/NFkappaB pathways to maintain osteoclast survival. J Cell Biochem. 2003;89:165–79. doi: 10.1002/jcb.10503. [DOI] [PubMed] [Google Scholar]
- 26.Gustin JA, Ozes ON, Akca H, et al. Cell type-specific expression of the IkappaB kinases determines the significance of phosphatidylinositol 3-kinase/Akt signaling to NF-kappa B activation. J Biol Chem. 2004;279:1615–20. doi: 10.1074/jbc.M306976200. [DOI] [PubMed] [Google Scholar]
- 27.Ouyang W, Li J, Ma Q, Huang C. Essential roles of PI-3K/Akt/IKKbeta/NFkappaB pathway in cyclin D1 induction by arsenite in JB6 Cl41 cells. Carcinogenesis. 2006;27:864–73. doi: 10.1093/carcin/bgi321. [DOI] [PubMed] [Google Scholar]
- 28.Lee FS, Hagler J, Chen ZJ, Maniatis T. Activation of the IkappaB alpha kinase complex by MEKK1, a kinase of the JNK pathway. Cell. 1997;88:213–22. doi: 10.1016/s0092-8674(00)81842-5. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Q, Lee FS. Mitogen-activated protein kinase/ERK kinase kinases 2 and 3 activate nuclear factor-kappaB through IkappaB kinase-alpha and IkappaB kinase-beta. J Biol Chem. 1999;274:8355–8. doi: 10.1074/jbc.274.13.8355. [DOI] [PubMed] [Google Scholar]
- 30.Nakano H, Shindo M, Sakon S, et al. Differential regulation of IkappaB kinase alpha and beta by two upstream kinases, NF-kappaB-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc Natl Acad Sci U S A. 1998;95:3537–42. doi: 10.1073/pnas.95.7.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tuszynski GP, Nicosia RF. Localization of thrombospondin and its cysteine-serine-valine-threonine-cysteine-glycine-specific receptor in human breast carcinoma. Lab Invest. 1994;70:228–33. [PubMed] [Google Scholar]
- 32.Arnoletti JP, Albo D, Jhala N, et al. Computer-assisted image analysis of tumor sections for a new thrombospondin receptor. Am J Surg. 1994;168:433–6. doi: 10.1016/s0002-9610(05)80093-5. [DOI] [PubMed] [Google Scholar]
- 33.Wakiyama T, Shinohara T, Shirakusa T, John AS, Tuszynski GP. The localization of thrombospondin-1 (TSP-1), cysteine-serine-valine-threonine-cysteine-glycine (CSVTCG) TSP receptor, and matrix metalloproteinase-9 (MMP-9) in colorectal cancer. Histol Histopathol. 2001;16:345–51. doi: 10.14670/HH-16.345. [DOI] [PubMed] [Google Scholar]
- 34.Sabherwal Y, Rothman VL, Poon RT, Tuszynski GP. Clinical significance of serum angiocidin levels in hepatocellular carcinoma. Cancer Lett. 2007;251:28–35. doi: 10.1016/j.canlet.2006.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huh HY, Pearce SF, Yesner LM, Schindler JL, Silverstein RL. Regulated expression of CD36 during monocyte-to-macrophage differentiation: potential role of CD36 in foam cell formation. Blood. 1996;87:2020–8. [PubMed] [Google Scholar]
- 36.Alessio M, De Monte L, Scirea A, Gruarin P, Tandon NN, Sitia R. Synthesis, processing, and intracellular transport of CD36 during monocytic differentiation. J Biol Chem. 1996;271:1770–5. doi: 10.1074/jbc.271.3.1770. [DOI] [PubMed] [Google Scholar]
- 37.Pikkarainen T, Brannstrom A, Tryggvason K. Expression of macrophage MARCO receptor induces formation of dendritic plasma membrane processes. J Biol Chem. 1999;274:10975–82. doi: 10.1074/jbc.274.16.10975. [DOI] [PubMed] [Google Scholar]
- 38.Meier CA, Chicheportiche R, Juge-Aubry CE, Dreyer MG, Dayer JM. Regulation of the interleukin-1 receptor antagonist in THP-1 cells by ligands of the peroxisome proliferator-activated receptor gamma. Cytokine. 2002;18:320–8. doi: 10.1006/cyto.2002.1945. [DOI] [PubMed] [Google Scholar]
- 39.Vey E, Zhang JH, Dayer JM. IFN-gamma and 1,25(OH)2D3 induce on THP-1 cells distinct patterns of cell surface antigen expression, cytokine production, and responsiveness to contact with activated T cells. J Immunol. 1992;149:2040–6. [PubMed] [Google Scholar]
- 40.Takashiba S, Van Dyke TE, Amar S, Murayama Y, Soskolne AW, Shapira L. Differentiation of monocytes to macrophages primes cells for lipopolysaccharide stimulation via accumulation of cytoplasmic nuclear factor kappaB. Infect Immun. 1999;67:5573–8. doi: 10.1128/iai.67.11.5573-5578.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwende H, Fitzke E, Ambs P, Dieter P. Differences in the state of differentiation of THP-1 cells induced by phorbol ester and 1,25-dihydroxyvitamin D3. J Leukocyte Biol. 1996;59:555–61. [PubMed] [Google Scholar]
- 42.Park EK, Jung HS, Yang HI, Yoo MC, Kim C, Kim KS. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm Res. 2007;56:45–50. doi: 10.1007/s00011-007-6115-5. [DOI] [PubMed] [Google Scholar]
- 43.Sancho D, Gomez M, Sanchez-Madrid F. CD69 is an immunoregulatory molecule induced following activation. Trends Immunol. 2005;26:136–40. doi: 10.1016/j.it.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 44.Lysiak JJ, Hussaini IM, Gonias SL. alpha 2-Macroglobulin synthesis by the human monocytic cell line THP-1 is differentiation state-dependent. J Cell Biochem. 1997;67:492–7. [PubMed] [Google Scholar]
- 45.Lotem J, Sachs L. Cytokine control of developmental programs in normal hematopoiesis and leukemia. Oncogene. 2002;21:3284–94. doi: 10.1038/sj.onc.1205319. [DOI] [PubMed] [Google Scholar]
- 46.Lotem J, Sachs L. Epigenetics wins over genetics: induction of differentiation in tumor cells. Seminars Cancer Biol. 2002;12:339–46. doi: 10.1016/s1044-579x(02)00054-8. [DOI] [PubMed] [Google Scholar]
- 47.Sachs L. The control of hematopoiesis and leukemia: from basic biology to the clinic. Proc Natl Acad Sci U S A. 1996;93:4742–9. doi: 10.1073/pnas.93.10.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sell S. Stem cell origin of cancer and differentiation therapy. Critical Reviews Oncology/hematology. 2004;51:1–28. doi: 10.1016/j.critrevonc.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 49.Fujii Y, Yuki N, Takeichi N, Kobayashi H, Miyazaki T. Differentiation therapy of a myelomonocytic leukemia (c-WRT-7) in rats by injection of lipopolysaccharide and daunomycin. Cancer Res. 1987;47:1668–73. [PubMed] [Google Scholar]
- 50.Spira AI, Carducci MA. Differentiation therapy. Current Opinion Pharm. 2003;3:338–43. doi: 10.1016/s1471-4892(03)00081-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.