

Abstract
In a continuing structure-activity relationship study of potent anti-HIV agents, seven new triterpene derivatives were designed, synthesized, and evaluated for in vitro antiviral activity. Among them, moronic acid derivatives 19, 20 and 21 showed significant activity in HIV-1 infected H9 lymphocytes. Compounds 19 and 20 were also evaluated against HIV-1 NL4−3 and drug resistant strains in the MT-4 cell line. Compounds 19 and 20 showed better antiviral profiles than the betulinic acid analog 8 (PA-457), which has successfully completed a Phase IIa clinical trial. Compound 20 showed potent anti-HIV activity with EC50 values of 0.0085 μM against NL4−3, 0.021 μM against PI-R (a multiple protease inhibitor resistant strain), and 0.13 μM against FHR-2 (an HIV strain resistant to 8), respectively. The promising compound 20 has become a new lead for modification, and further development of 20-related compounds as clinical trial candidates is warranted.
Introduction
Acquired immunodeficiency syndrome (AIDS) remains an exceptional crisis due to both its emergent and long-term development. The epidemic remains extremely dynamic, growing and changing character as the human immunodeficiency virus (HIV) exploits new opportunities for transmission. No region of the world has been spared, and the AIDS pandemic continues to outpace the global response to HIV treatment.2 Although current anti-AIDS drugs include inhibitors of reverse transcriptase (RT), protease and fusion,3 the increasing prevalence of drug resistant HIV strains is one of the major problems for the treatment of HIV infection, driving the demand for the development of novel drugs with new mechanisms of action.
Triterpenes, including betulinic acid (1), constitute a promising class of anti-HIV agents. Two types of anti-HIV 1-derivatives have exhibited potent anti-HIV profiles. In our prior studies, we found that 3-O-(3′,3′-dimethylsuccinyl)-betulinic acid (8) inhibits HIV-1 maturation by interfering with HIV-1 P24/P25 processing, which results in a noninfectious HIV-1 particle.4-7 The second type of anti-HIV 1-analogs contain various C-28 amide modifications. IC9564 (9), a statine analog of 1, represents this compound type, and acts at an early stage of viral infection.8,9
Compound 8, designated as PA-457 by Panacos Pharmaceuticals, Inc.,10 is a maturation inhibitor directed against a novel viral target. Panacos Inc. recently concluded a successful Phase IIa clinical trial with 8.10 Because 8 has a different target than approved HIV drugs, it retains activity against virus isolates resistant to currently available treatments including RT and protease inhibitors.
In the current study, we synthesized a series of new triterpene derivatives with 3-, 28-, and both substitutions on different triterpene skeletons, including betulinic acid (BA), glycyrrhetinic acid (GLA), moronic acid (MA), oleanolic acid (OA), and ursolic acid (UA). This paper reports their design, synthesis, and SAR.
Design
From previous SAR study of 8-type analogs, the most potent derivatives contain either 3′,3′-dimethylsuccinyl or 3′,3′-dimethylglutaryl moieties at C-3.8, 9,11 Thus, these side chains were also used in the current study.
Our first aim was to investigate the SAR of the triterpene core. In order to optimize this core and gain more information about the target binding requirements, we designed and synthesized 3-substituted derivatives of triterpenes other than 1. Moreover, by replacing the betulin core with other triterpenes, we could potentially lower the cytotoxicity, improve the pharmacological profiles and obtain more SAR information. Lupeol (2) has the same skeleton as 1, but lacks the C-28 carboxylic acid. OA (3) and UA (4) have a 6-membered E ring rather than the 5-membered E ring found in 1. MA (5) and 3 differ at the double bond position. GLA (7) has a carboxylic acid at C-30 rather than C-28. Activity data with these latter compounds will provide information on the contribution and positional requirement of the carboxylic acid on the antiviral activity. Previously synthesized compounds 12 − 15 were also included in the current study.12
Among many C-28 amide BA derivatives, a relatively long C-28 amide side chain is a common structural feature that is essential for potent anti-HIV entry activity.8 These results suggest that BA derivatives may interfere at more than one stage of HIV infection, depending on the side chains on C-3 and C-28. Two compounds (10 and 11) with C-3 ester and C-28 amide side chains exhibited both anti-fusion and maturation activity.13 These observations prompted us to hypothesize that the betulin moiety may serve as a molecular scaffold or “docker”, the C-28 amide side chain is the pharmacophore for the anti-HIV entry activity, and the C-3 ester group has an important role in target interaction during HIV-1 maturation. Based on this hypothesis, a compound with both DMS on C-3 and a proper amide on C-28 will probably exhibit dual mechanisms of action, both anti-fusion and anti-maturation, to increase potency and decrease HIV-resistance profiles.
Our previous paper reported that 5, which is isolated from Brazilian propolis, an herbal medicine widely used worldwide today, exhibited significant anti-HIV activity (EC50 <0.1 μg/mL, TI >186) in H9 lymphocytes.14 In addition, the observed toxicity of MA derivatives is generally lower than that of BA derivatives. This finding implies that changing the betulin core to 5 may alter the cytotoxicity profile without impairing anti-HIV potency. In our continuing development of novel triterpene anti-HIV agents, compound 5 was dually functionalized at C-3 and C-28 based on the SAR of BA derivatives.
The β-amyrin series triterpene glycyrrhizin or glycyrrhizic acid (6) is used in Japanese clinics by intravenous administration for the treatment of chronic viral hepatitis B. In some clinical studies in Japan, the administration of 6 to AIDS patients resulted in delayed progression of HIV infection symptoms.15 Therefore, we also modified 7, the aglycone of 6, with both ester and amide side chains.
Chemistry
Compounds 2 and 7 were reacted separately with 2,2-dimethylsuccinic anhydride to yield target compounds 16 and 17, respectively. Compound 23, the acetate of 7, was then treated with oxalyl chloride, and without isolation, further reacted with leucine methyl ester hydrochloride to afford 24 in a yield of 95% over two steps. Compound 24 was hydrolyzed under basic conditions to 25 in a 95% yield. The 3β-hydroxy group of 25 was reacted with 2,2-dimethylsuccinic anhydride to yield final target product 18 in a 65% yield (Scheme 1)
Scheme 1.

Conditions: i, 2,2-dimethylsuccinyl anhydride, DMAP, pyridine, reflux 12 hr; ii, Ac2O, py, rt; iii, oxalyl chloride, CH2Cl2, rt; iv. leucine methyl ester, Et3N, CH2Cl2, rt; v. 2N KOH, THF-MeOH (1:1), rt.
The methanol extract (600g) of the propolis collected by Africanized Apis mellifera in southern Brazil, gave 5 in good yield (1.5g, 0.25%).14 Scheme 2 shows the synthetic steps to moronic acid derivatives 19 − 22. Using the same method of compound 24, compounds 26 and 27 were synthesized from 5. To reduce the C-3 keto group, sodium borohydride (NaBH4) was added to a solution of 26 or 27 in MeOH and THF to produce the corresponding reduction products 28 and 29 in yields of 33% and 85%, respectively. At the same time, 26 was also reduced to 32 in a 60% yield. Compounds 28 and 29 were hydrolyzed to 30 and 31, and yielded final target products 20 − 22 in 30 − 40% yield, respectively. Compound 32 reacted with 2,2-dimethylsuccinyl anhydride to yield crude di-succinated compound 33. Without isolation, 33 was treated with 2N KOH in MeOH and THF at 0 °C to afford target compound 19 in 70% yield (Scheme 2).
Scheme 2.

Conditions: i, oxalyl chloride, CH2Cl2, rt; ii. amino acid methyl esters, Et3N, CH2Cl2, rt; iii, NaBH4, MeOH/THF, rt; iv. 2N KOH, THF-MeOH (1:1), rt; v. 2,2-dimethylsuccinyl anhydride, DMAP, pyridine, reflux 12 hr
Results and Discussion
In our current study, newly synthesized triterpene derivatives 16 − 22 were evaluated for anti-HIV-1 replication activity against two different viral strains in two different cell lines in parallel with 8 and other previously synthesized compounds 10 − 15. Two MA derivatives 19 and 20 were also evaluated for anti-HIV-1 replication activity against three different viral strains in MT-4 cells in parallel with 8 and compound 10, which has the same C-3 and C-28 substituents as 20 but with a BA core. The results are summarized in Tables 1 and 2, respectively.
Table 1.
Anti-HIV Activities of Triterpene Derivatives Against HIV-1 Strains in H9 Lymphocytes and MT-4 Cells
| compound | HIV-1IIIB in H9 lymphocytes |
NL4−3 in MT-4 cells |
||||
|---|---|---|---|---|---|---|
| IC50 (μM) | EC50 (μM) | TI | IC50 (μM) | EC50 (μM) | TI | |
| 8 | >42.8 | 0.007 | >6,250 | >5* | 0.096 | >52 |
| 10 | NT | 12.2 | 0.010 | >1,220 | ||
| 11 | NT | 11 | 0.007 | 1,570 | ||
| 12 | >42.7 | 0.32 | >133 | 20.2 | 8.3 | 2.4 |
| 13 | NS | NS | ||||
| 14 | 28.5 | 0.079 | 360 | 21 | 8.3 | 2.5 |
| 15 | 28.5 | 0.68 | 42 | NS | ||
| 16 | >45.1 | 2.6 | >17.2 | >36.1 | 21.6 | >1.7 |
| 17 | NS | NS | ||||
| 18 | NS | NS | ||||
| 19 | >42.7 | 0.017 | 2,500 | 23.5 | 0.045 | 522 |
| 20 | >42.7 | 0.0156 | 2,735 | 22.5 | 0.0085 | 2,650 |
| 21 | 24.0 | 0.007 | 3,400 | 19.5 | 0.11 | 177.3 |
| 22 | >32.6 | 9.3 | >3.5 | NS | ||
The highest concentration tested for 8 was 5 μM; there was no observable cytotoxicity at this concentration. NT, not tested in this assay. NS, no suppression.
Table 2.
Comparison of Anti-HIV Activities of Selected Derivatives Against Several HIV-1 Strains in MT-4 Cells
| compound | EC50 (μM) in different viral strains |
IC50 (μM) | ||
|---|---|---|---|---|
| NL4−3 | PI-R | FHR-2 | ||
| 8 | 0.096 | 0.43 | NS | >5* |
| 10 | 0.010 | 0.006 | 0.05 | 12.2 |
| 19 | 0.045 | 0.088 | 2.78 | 23.5 |
| 20 | 0.0085 | 0.021 | 0.13 | 21.0 |
| AZT | 0.013 | 0.019 | 0.019 | >37.5 |
NL4−3 is a T-cell adapted HIV-1 strain, X4 wild type virus; PI-R is an HIV-1 strain, HIV-1 M46I/L63P/V82T/I84V, resistant to multiple protease inhibitors;17 and FHR-2 is an HIV-1 strain resistant to 8.7 EC50 is the concentration that inhibits HIV-1 replication by 50%; and IC50 is the concentration that decreases 50% of viable cell number.
The highest concentration tested for DSB was 5 μM; there was no observable cytotoxicity at this concentration. NS: No suppression at testing concentration, 5 μM.
First, our new results confirm prior reports that a 3′,3′-dimethylsuccinyl side chain is superior to 3′,3′-dimethylsuccinylglutaryl at the C-3 position. Accordingly, OA derivative 13 with the latter substituent was inactive, while OA analog 12with the former substituent had an EC50 value of 0.32 μM. Similarly, among comparable UA analogs, 15 was tenfold weaker than 14.*
Compounds 16 and 8 have identical structures, except at C-28, which is a methyl group in the former and a carboxylic acid in the latter compound. Compound 16 showed dramatically decreased activity in both HIV-1 infected H9 lymphocytes and MT-4 cells with EC50 values of 2.6 and 21.6 μM, respectively. OA analog 12 and UA analog 14, which also have a carboxylic acid at C-28, were 10 − 50 fold more potent than 16 in the H9 lymphocyte assay; however, they were still less active than 8. GLA analog 17, which has a C-28 methyl and a C-30 carboxylic acid, was inactive in both assays. These results indicate that the carboxylic acid at C-28 might be required for high anti-HIV activity in this compound type.
From the 3D structures of 1, 3β-hydroxymoronic acid, and 7 shown in Figure 4, the C-28 carboxylic acid is oriented in the same direction in 1 and 3β-hydroxymoronic acid, even though they have different E rings. However, the C-30 carboxylic acid of 7, is oriented in a different direction. Accordingly, a substituent on this position of 7 will create a totally different molecular shape compared with analogous BA and MA derivatives. Therefore, like 17, the disubstituted derivative 18 was inactive in both assays.
Figure 4.
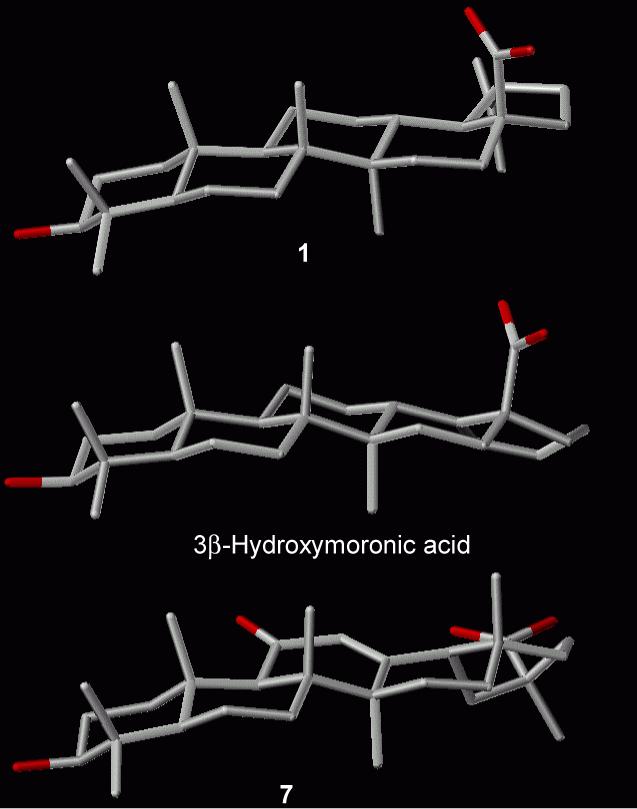
3D Structures of 1, 3β-Hydroxymoronic acid, and 7
Similarly to di-substituted BA derivatives (10 and 11), di-substituted MA derivatives (19 − 21) also exhibited high potency. Compound 21 with 3-(3′,3′-dimethylsuccinyl) and 28-undecanoic amide substituents exhibited the highest potency (EC50 = 0.007 μM; TI = 3400, in H9 lymphocytes), with similar results to those of 8 in the same assay. Compounds 19 and 20 also demonstrated high potency with EC50 values of 0.017 and 0.016 μM, respectively. Compound 22, the 3-(2′,2′-dimethylsuccinyl) isomer of 21, almost lost anti-HIV activity.
These compounds were also tested in an HIV-1 replication assay using MT-4 cells infected with the NL4−3 strain, which is a T-cell adapted X4 wild type HIV-1 virus. Most compounds exhibited weaker activity in this system. Interestingly, unlike in H9 lymphocytes, compound 20, which showed the best activity (EC50 = 0.0085 μM) in MT-4 cells, was more potent than 8 in this assay. Compound 19 also exhibited slightly better activity than 8 with an EC50 value of 0.045 μM. However, the most active compound in H9 lymphocytes, 21, showed an EC50 value of only 0.11 μM. The discrepancy might result from differences in the assay protocols, especially viral infecting dose.
Because 19 and 20 were active in both assays, these two compounds, together with 10, were also screened against other viral strains. The data are shown in Table 2. PI-R is HIV-1 M46I / L63P / V82T / I84V, an HIV-1 strain resistant to multiple protease inhibitors,17 and FHR-2 is an HIV-1 strain resistant to 8.7 Both 19 and 20 showed better activity (EC50 = 0.088 and 0.021 μM) than 8 against multi-PI resistant strain, but were less active than 10 (EC50 = 0.006 μM). As for the FHR-2 HIV-1 strain, which is resistant to 8, compound 20 retained some potency with an EC50 of 0.13 μM, but was less active than 10 (EC50 = 0.05 μM), while 19 showed only weak activity (EC50 = 2.78 μM). With a reduced carboxylic acid in the 28-side chain, 19 showed less activity against all tested viral strains than 20; thus, a carboxylic acid within the leucine side chain is more favorable for target interaction. Our results indicate that the MA derivatives 19, 20 and 21 exhibited weak anti-HIV fusion activity (Table 1). BA derivatives 10 and 11 also exhibited anti-fusion activity in previous study.13 The anti-fusion activity of these compounds are weaker than that of 9 (EC50 0.0086 μM). These data suggest that the disubstituted compounds 10, 11, 19, 20 and 21 can block HIV-1 entry.
The key structural feature of disubstituted compounds 10, 11, 19, 20 and 21 is the presence of both side chains at positions 3 and 28. This feature enables our target compounds to be active against viruses that are resistant to 8, which has a side chain only at C-3. Therefore, this modification study indicated that the BA cid scaffold can be replaced by an analogous triterpene, MA, without loss of activity. Disubstituted MA derivatives with side chains on both C-3 and C-28 are likely to have dual functions and lower cytotoxicity as well as better drug resistance profiles than their parent compound 8. However, the detailed molecular mechanisms of action of these new disubstituted MA derivatives remain to be determined. Their mechanisms of action might be different from that of 8. Additional modification and SAR studies are in progress with an aim to continually improve potency, particularly against drug-resistant viral strains. Further development of 20-related compounds as next generation clinical trial candidates is warranted.
Experimental Section
Chemistry
Melting points were measured with a Fisher-Johns melting apparatus without correction. The proton nuclear magnetic resonance (1H NMR) spectra were measured on 300MHz Varian Gemini 2000 and 500MHz Varian Inova spectrometers using TMS as internal standard. The solvent used was CDCl3 unless indicated. Mass spectra were obtained on an Agilent 1100 MSD ion trap mass spectrometer or PE-SCIEX API-3000 with turbo ion spray source. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA. All target compounds were analyzed for C, H, N and gave values within ±0.4% of the theoretical values. Thin-layer chromatography (TLC) was performed on PLC silica gel 60 F254 plates (0.5 mm, Merck). Teledyn-Isco Companion systems were used as medium pressure column chromatography. Compound 7 was provided by Tokiwa Pharmaceuticals Inc. All other chemicals were obtained from Aldrich, Inc.
3β-O(3′,3′-Dimethylsuccinyl)-lupeol (16)
Compound 2 (21.3 mg, 0.05 mmol) was heated with 2,2-dimethylsuccinic anhydride (25.6 mg, 0.2 mmol ) and 4-dimethylamino pyridine (DMAP, 12.2 mg, 0.1 mmol) in pyridine (2 mL) to reflux overnight. After addition of EtOAc (50 mL), the mixture was worked up with 2N HCl and H2O. Evaporation of EtOAc gave a mixture of starting material and succinate, which was separated by SiO2 column chromatography (CH2Cl2: MeOH 100:2). Yield 63%; white amorphous powder; mp 193 − 194 °C; MS (ESI-) m/z: 553.5 (M−-1) for C36H58O4. 1H NMR (500 MHz) δ 0.79, 0.81, 0.83, 0.84, 0.94, 1.03 (3H each, s, CH3-23, 24, 25, 26, 27 and 28), 1.29, 1.31 (3H each, s, CH3-3′×2), 1.68 (3H, s, CH3-30), 1.91 (2H, m, H-2), 2.37 (1H, m, H-19), 2.57, 2.67 (1H each, d, J = 15.5 Hz, H-2′), 4.49 (1H, dd, J = 11.0, 5.5 Hz, H-3), 5.30 (1H, s, COOH-4′), 4.68, 4.56 (1H each, d, J = 2 Hz, H-29); Anal. (C36H58O4), C 77.77, H 10.50.
3β-O-(3′,3′-Dimethylsuccinyl)-glycyrrhetinic acid (17)
Following the procedure described for 16, 17 was obtained from 7 as white amorphous powder (235 mg, 0.5 mmol) in a yield of 60%; mp >300°C; MS (ESI-) m/z: 597.4 (M−-1) for C36H54O7. 1H NMR (500 MHz, CD3OD) δ 0.83, 0.89, 0.90, 0.92, 1.14, 1.16 (3H each, s, CH3-23, 24, 25, 26, 27 and 28), 1.25, 1.26 (3H each, s, CH3-3′×2), 1.43 (3H, s, CH3-29), 2.28 (1H, m, H-18), 2.51 (2H, m, H-7), 4.48 (1H, m, H-3), 5.58 (1H, s, H-12); Anal. (C36H54NO7), C 72.30, H 8.90.
3β-O-Acetylglycyrrhetinic acid (23)
Compound 7 (935 mg, 2 mmol) was reacted > with Ac2O (0.5 mL) in pyridine (5 mL) and dichloromethane (10 mL) at rt for 1 day. The mixture was washed with 20 % HCl and H2O. Evaporation of CH2Cl2 followed by silica gel column chromatography (CH2Cl2-MeOH 100:2) gave 23. Yield 54%; white powder; mp 175 − 177 °C; MS (ESI-) m/z: 511.5 (M−-1) for C32H48O5. 1H NMR (500 MHz, DMSO-d6) δ 0.76, 0.83, 1.04, 1.07, 1.10, 1.15 (3H each, s, CH3-23, 24, 25, 26, 27 and 28), 1.37 (3H, s, CH3-29), 2.0 (3H, s, CH3CO-3), 2.07 (1H, m, H-18), 2.63 (1H, m, H-7), 4.42 (1H, dd, J = 12.0, 4.5 Hz, H-3), 5.41 (1H, s, H-12), 12.18 (1H, COOH-30).
N-(Olean-3β-O-acetyl-11-oxo-12-en-30-oyl)-L-leucine methyl ester (24)
Following the procedure described for 26, 99% yield from compound 23; white amorphous powder; mp 103 − 105 °C; MS (ESI+) m/z: 662.7 (M++Na), 640.7 (M++1) for C39H61NO6. 1H NMR (500 MHz) δ 0.81, 0.88, 1.14, 1.15, 1.17, 1.38 (3H each, s, CH3-23, 24, 25, 26, 27 and 28), 0.94, 0.95 (3H each, d, J = 6.5 Hz, -CH(CH3)2-30 side chain), 1.57 (3H, s, CH3-29), 2.05 (3H, s, CH3CO-3), 2.28 (1H, m, H-18), 2.81 (1H, m, H-7), 3.75 (3H, s, COOCH3), 4.52 (1H, dd, J = 12.0, 5.2 Hz, H-3), 4.67 (1H, m, -NHCH-), 5.77 (1H, s, H-12), 5.93 (1H, d, J = 8.5 Hz, -CONH-).
N-(Olean-3β-hydroxy-11-oxo-12-en-30-oyl)-L-leucine (25)
Following the procedure described for 30, 98% yield from 24; white amorphous powder; mp 184 − 186 °C; MS (ESI-) m/z: 582.7 (M−-1) for C36H57NO5. 1H NMR (500 MHz, CD3OD) δ 0.79, 0.80, 0.96, 1.13, 1.14, 1.25 (3H each, s, CH3-23, 24, 25, 26, 27 and 28), 0.93, 0.95 (3H each, d, J = 6.0 Hz, -CH(CH3)2-30 side chain), 1.42 (3H, s, CH3-29), 3.16 (1H, dd, J = 12.0, 5.0 Hz, H-3), 4.52 (1H, m, -NHCH-), 5.74 (1H, s, H-12).
N-[3β-O-(3′,3′-Dimethylsuccinyl)-olean-11-oxo-12-en-30-oyl]-L-leucine (18)
Yield 65% from 25; white amorphous powder; mp 145 − 147 °C; MS (ESI+) m/z: 734.5 (M++Na) for C42H65NO8. 1H NMR (300 MHz) δ 0.78, 0.84, 0.86, 1.08, 1.12, 1.14 (3H each, s, CH3-23, 24, 25, 26, 27, and 28), 0.91, 0.93 (3H each, d, J =7.2 Hz, -CH(CH3)2-30 side chain), 1.29 (6H, s, 2×CH3-3′), 1.30 (3H, s, CH3-29), 4.51 (1H, m, H-3), 4.69 (1H, m, -NHCH-), 5.68 (1H, s, H-12), 6.49 (1H, br.s, -CONH-); Anal. (C42H65NO8), C 71.14, H 9.18, N 2.22.
N-(Olean-3-oxo-18-en-28-oyl)-leucine methyl ester (26)
MA (5, 454 mg, 1 mmol) was dissolved in CH2Cl2 (5 mL) and ice-cooled. To this solution, oxalyl chloride (20 mL of 2.0M solution in CH2Cl2) was added dropwise with stirring. The mixture was stirred at rt for 2 h, then CH2Cl2 was evaporated. To the residue, CH2Cl2 (5 mL×3) was added and evaporated to give a yellow solid. This residue was dissolved in CH2Cl2 (5 mL) and a CH2Cl2 solution (10 mL) of leucine methyl ester hydrochloride (543 mg, 3 mmol) and Et3N (303 mg, 3 mmol) was added. The reaction mixture was left overnight at rt. After washing with water 3 times, CH2Cl2 was removed to give a viscous oil, which then subjected to silica gel column chromatography (CH2Cl2-MeOH 100:1) to give 26. 88% yield; mp 86 − 88 °C; MS (ESI+) m/z: 582.6 (M++1) for C37H59NO4. 1H NMR (300 MHz) δ 0.78, 0.96, 0.99, 1.01, 1.01, 1.03, 1.08 (3H each, s, CH3-23, 24, 25, 26, 27, 29 and 30), 0.93, 0.94 (3H each, d, J = 6 Hz, -CH(CH3)2-28 side chain), 3.73 (3H, s, COOCH3), 4.61 (1H, m, H-3), 5.37 (1H, s, H-19), 6.13 (1H, d, J = 7.8 Hz, -CONH-).
N-(Olean-3-oxo-18-en-28-oyl)-aminoundecanoic acid methyl ester (27)
Following the procedure described for 26, 51% yield; white amorphous powder; mp 90 − 92 °C; MS (ESI+) m/z: 652.9 (M++1) for C42H69NO4. 1H NMR (500 MHz) δ 0.77, 0.95, 0.98, 0.98, 1.01, 1.03, 1.08 (3H each, s, CH3-23, 24, 25, 26, 27, 29 and 30), 3.30 (2H, m, -NHCH2-), 3.67 (3H, s, COOCH3), 5.32 (1H, s, H-19), 5.82 (1H, t, J = 5.8 Hz, -CONH-).
N-(Olean-3β-hydroxy-18-en-28-oyl)-leucine methyl ester (28)
Compound 26 (920 mg, 1.58 mmol) was dissolved in a mixture of MeOH and THF (50 mL, 3mL) and stirred in an ice bath. To the solution, solid NaBH4 (597 mg, 15.8 mmol) was added slowly and the reaction mixture was kept overnight at rt. Water and CH2Cl2 (50 mL, 100 mL) were added to the warmed reaction mixture, and insoluble solid was filtered off. The aqueous and organic layers were separated, and evaporation of the organic solvent gave an oily residue, which was subjected to silica gel column chromatography (CH2Cl2-MeOH 200:1). Compounds 32 (300 mg, 60%) and 28 (560 mg, 33%) were isolated and crystallized from a mixture of CH2Cl2 and MeOH; mp: 145 − 146 °C; MS (ESI-) m/z: 582.7 (M−-1) for C37H61NO4; 1H NMR (300 MHz) δ 0.76, 0.76, 0.87, 0.97, 0.98, 0.99, 1.00 (3H each, s, CH3-23, 24, 25, 26, 27, 29 and 30), 0.93, 0.94 (3H each, d, J = 6.3 Hz, -CH(CH3)2-28 side chain), 3.20 (1H, dd, J = 10.6, 5.6 Hz, H-3), 3.72 (3H, s, -COOCH3-28 side chain), 4.61 (1H, td, J = 8.4, 4.5 Hz, -CH(CH3)2-28 side chain), 5.36 (1H, s, H-19), 6.12 (1H, d, J = 8.4 Hz, -CONH-).
N-(Olean-3-hydroxy-18-en-28-oyl)-aminoundecanoic acid methyl ester (29)
Following the procedure described for 28, yield 85% from 27; white amorphous powder; mp 152 − 154 °C; MS (ESI+) m/z: 676.8 (M++Na), 654.7 (M++1) for C42H71NO4. 1H NMR (300 MHz) δ 0.74, 0.75, 0.84, 0.94, 0.95, 0.96, 0.99 (3H each, s, CH3-23, 24, 25, 26, 27, 29 and 30), 3.18 (2H, m, -NHCH2-), 3.29 (1H, m, H-3), 3.65 (3H, s, COOCH3), 5.30 (1H, s, H-19), 5.80 (1H, t, J = 6.0 Hz, -CONH-).
N-(Olean-3β-hydroxy-18-en-28-oyl)-leucine (30)
Compound 28 (25 mg) was dissolved in a mixture of MeOH (2 mL) and THF (1 mL), and 2N KOH (0.5 mL) was added to this mixture at 0°C with stirring. The reaction mixture was acidified with 2N HCl and extracted with CH2Cl2 to yield compound 30 in a 96% yield after evaporation of solvent. mp 245 − 247 °C; MS (ESI-) m/z: 568.6 (M−-1) for C36H59NO4. 1H NMR (300 MHz, in CD3OD and CDCl3 mixture) δ 0.70, 0.76, 0.84, 0.90 (6H, br. s), 0.95 (br. s), 0.98 (3H each, s, CH3-23, 24, 25, 26, 27, 29 and 30), 0.90, 0.95 (3H each, br.s, -CH(CH3)2-28 side chain), 3.09 (1H, dd, J = 10.8, 5.2 Hz, H-3), 4.46 (1H, m, -CH(CH3)2-28 side chain), 5.34 (1H, s, H-19), 6.54 (1H, d, J = 8.7 Hz, -CONH-).
N-(Olean-3β-hydroxy-18-en-28-oyl)-aminoundecanoic acid (31)
Following the procedure described for 30, yield 85% from 29; white amorphous powder; mp 168 − 170 °C; MS (ESI+) m/z: 640.7 (M++1) for C H41H69NO4. 1H NMR (300 MHz) δ 0.74, 0.75, 0.85, 0.94, 0.95, 0.96, 0.97 (3H each, s, CH3-23, 24, 25, 26, 27, 29 and 30), 3.17 (2H, m, -NHCH2-), 3.30 (1H, m, H-3), 5.30 (1H, s, H-19), 5.83 (1H, t, J = 5.7 Hz, -CONH-).
N-(Olean-3β-hydroxy-18-en-28-oyl)-(1-hydroxymethyl-3-methyl)-butylamide (32)
Yield of 60% from compound 26; mp 253 − 254 °C; MS (ESI-) m/z: 554.7 (M−-1) for C36H61NO3; 1H NMR (300 MHz) δ 0.76, 0.77, 0.86, 0.97, 0.98, 0.99, 1.00 (3H each, s, CH3-23, 24, 25, 26, 27, 29 and 30), 0.91, 0.93 (3H each, d, J = 6.6 Hz, -CH(CH3)2-28 side chain), 3.20 (1H, dd, J = 10.8, 5.1 Hz, H-3), 3.51 (1H, dd, J = 10.5, 6.3 Hz, -CH2OH-28 side chain), 3.66 (1H, dd, J = 10.5, 3.5 Hz, -CH2OH-28 side chain), 4.02 (1H, m, -CH(CH3)2-28 side chain), 5.33 (1H, s, H-19), 5.87 (1H, d, J = 8.1 Hz, -CONH-).
N-[3β-O-(3′,3′-Dimethylsuccinyl)-olean-18-en-28-oyl]-(1-hydroxymethyl-3-methyl)-butylamide (19)
Compound 32 (130 mg, 0.23 mmol) was heated with 2,2-dimethylsuccinic anhydride (180 mg, 1.38 mmol) and DMAP (56 mg, 0.46 mmol) in pyridine (6 mL) to reflux overnight. After an addition of EtOAc (50 mL), the mixture was washed with 2N HCl (5 mL) twice and H2O. Evaporation of EtOAc gave crude disuccinates 33 as an oil. The oil was treated with 2N KOH (7 mL) in a mixture of MeOH (15 mL) and THF (2 mL) under ice-cooling and then kept at rt overnight. The reaction mixture was acidified with 2N HCl and the organic solvent was evaporated. The residue was extracted by CH2Cl2 to give an oil, which was subjected to silica gel column chromatography. Using a mixture of CH2Cl2 and MeOH (100:1), compound 19 was obtained as an oil and was crystallized from EtOAc to give 105 mg of 19 as a white amorphous powder in a yield of 70.0 %; mp 237 − 238 °C; MS (ESI-) m/z: 682.8 (M−-1) for C42H69NO6. 1H NMR (300 MHz) δ 0.76, 0.80, 0.83, 0.87, 0.97, 0.99 (6H) (3H each, s, CH3-23, 24, 25, 26, 27, 29 and 30), 0.90, 0.92 (3H each, d, J = 4.4 Hz, -CH(CH3)2-28 side chain), 1.27, 1.29 (3H each, s, 2×CH3-3′), 3.49 (1H, dd, J = 10.8, 3.9 Hz, -CH2OH-28 side chain), 3.64 (1H, dd, J = 10.8, 6.3 Hz, -CH2OH-28 side chain), 4.03 (1H, m, - NHCH-), 4.49 (1H, dd, J = 9.9, 5.4 Hz, H-3), 5.34 (1H, s, H-19), 5.88 (1H, d, J = 8.1 Hz, -CONH-); Anal. (C42H69NO6), C 73.59, H 10.20, N 1.99.
N[3β-O-(3′,3′-Dimethylsuccinyl)-olean-18-en-28-oyl]-L-leucine (20)
Yield 30% from 30; white amorphous powder; mp 223 − 224 °C; MS (ESI-) m/z: 696.8 (M−-1) for C42H67NO7. 1H NMR (300 MHz, CDCl3) δ 0.76, 0.77, 0.83, 0.85, 0.93 (6H), 1.03 (3H each, s, CH3-23, 24, 25, 26, 27, 29 and 30), 0.99 (6H, d, J = 7.2 Hz, -CH(CH3)2-28 side chain), 1.28, 1.31 (3H each, s, 2×CH3-3′), 4.52 (1H, dd, J = 8.1, 4.8 Hz, H-3), 4.68 (1H, m, -NHCH-), 5.37 (1H, s, H-19), 6.32 (1H, d, J = 8.1 Hz, -CONH-); Anal. (C42H67NO7), C 71.98, H 9.72, N 2.01.
N-[3β-O-(3′,3′-Dimethylsuccinyl) olean-18-en-28-oyl]-aminoundecanoic acid (21)
Yield of 48% from compound 31; white amorphous powder; mp 131 − 132 °C; MS (ESI+) m/z: 790.6 (M++Na) for C47H77NO7. 1H NMR (500 MHz) δ 0.76, 0.80, 0.85, 0.86, 0.96, 0.98, 1.00 (3H each, s, CH3-23, 24, 25, 26, 27, 29 and 30), 1.26, 1.29 (3H each, s, 2×CH3-3′), 4.54 (1H, dd, J = 11.5, 5.0 Hz, H-3), 5.32 (1H, s, H-19), 5.87 (1H, dd, J = 8.5, 3.5 Hz, -CONH-); Anal. (C47H77NO7), C 73.12, H 9.95, N 1.76.
N-[3β-O-2′,2′-Dimethylsuccinyl)-olean-18-en-28-oyl]-aminoundecanoic acid (22)
Yield of 10% from compound 31 as dia-isosteric isomer of 21, white amorphous powder; mp 109 − 111 °C; MS (ESI+) m/z: 790.6 (M++Na) for C47H77NO7. 1H NMR (500 MHz) δ 0.76, 0.76, 0.86, 0.95, 0.96, 0.98, 1.00 (3H each, s, CH3-23, 24, 25, 26, 27, 29 and 30), 1.27, 1.31 (3H each, s, 2×CH3-2′), 4.40 (1H, dd, J = 12.0, 4.5 Hz, H-3), 5.32 (1H, s, H-19), 5.85 (1H, t, J = 7.5 Hz, -CONH-); Anal. (C47H77NO7), C 73.36, H 10.10, N 1.78.
HIV growth inhibition assay in H9 lymphocytes
The evaluation of HIV-1 inhibition was carried out as follows using H9 lymphocytes. The human T-cell line, H9, was maintained in continuous culture with complete medium (RPMI 1640 with 10% fetal calf serum supplemented with L-glutamine at 5% CO2 and 37 °C. Test samples were prepared as described previously6 and to each sample well was added 90 μl of media containing H9 cells at 3 × 105 cells/mL and 45 μL of virus inoculum (HIV-1 IIIIB isolate) containing 125 TCID50. Control wells containing virus and cells only (no drug) and cells only (no virus or drug) were also prepared. A second set of samples were prepared identical to the first and were added to cells under identical conditions without virus (mock infection) for toxicity determinations (IC50 defined below). In addition, AZT was also assayed during each experiment as a positive drug control. On days 1 and 4 post-infection (PI), spent media was removed from each well and replaced with fresh media. On day 6 PI, the assay was terminated and culture supernatants were harvested for analysis of virus replication by p24 antigen capture. The compound toxicity was determined by XTT using the mock-infected sample wells. If a test sample inhibited virus replication and was not toxic, its effects were reported in the following terms: IC50, the concentration of test sample that was toxic to 50% of the mock-infected cells; EC50, the concentration of the test sample that was able to suppress HIV replication by 50%; and the Therapeutic index (TI), the ratio of the IC50 to EC50.
Anti-HIV assay in MT4 cells
An HIV-1 infectivity assay previously described was used in the experiments.13 A 96-well microtiter plate was used to set up the HIV-1 replication assay. HIV-1 at a multiplicity of infection (MOI) of 0.01 was used to infect MT4 cells. Culture supernatants were collected on day 4 post infection for p24 assay using an ELISA kit from ZeptoMetrix Corporation (Buffalo, New York).
Cell fusion assay
A protocol modified from a previously described fusion assay was used in this study.9 TZM cells that expressed luciferase upon fusion with envelope-expressing COS cells were used as fusion partners. The fusion assays were performed by transfecting monkey kidney cells (COS) with an expression vector pSRHS that contained the HIV-1 NL4−3 envelope genes. Electroporation was performed to express the HIV-1 envelope on COS cells. Briefly, COS cells (106) in culture medium were incubated with 2 μg of the envelope expression vector on ice for 10 min. Electroporation was performed using a gene pulser (BioRad, Hercules, CA) with capacitance set at 950 μF and voltage at 150 V. The transfected COS cells were cultured for one day before mixing with TZM cells. TZM cells (10 × 104) were incubated with COS cells (104) in 96-well flat-bottomed plates (Costar) in 100 μl culture medium. Compounds to be tested at various concentrations in 10 μl of culture medium were incubated with the cell mixtures at 37 °C for 24 h. Luciferase activity was quantified using a Biotek luminometer.
Supplementary Material
Figure 1.

Related Triterpene Natural Products.
Figure 2.

Previously Modified Anti-HIV Triterpene Derivatives.
Figure 3.

Newly Synthesized Triterpene Derivatives.
Table 3.
Inhibition of HIV-1 Envelope-mediated Membrane Fusion
| compound | EC50 (μM) | IC50 (μM) |
|---|---|---|
| 8 | >17.1 | >17.1 |
| 9 | 0.0086 | >13.2 |
| 19 | 5.85 | >14.6 |
| 20 | 3.58 | >14.3 |
| 21 | 5.21 | >13.0 |
EC50 is the drug concentration that inhibits HIV envelope-mediated membrane fusion by 50%; IC50 (cytotoxicity) for both cell types were determined in an one-day assay since the fusion assay is also an one-day assay. The highest concentration tested: 10 μg/mL.
Acknowledgment
This investigation was supported by Grant AI-33066 from the National Institute of Allergy and Infectious Diseases (NIAID) awarded to K. H. Lee. Thanks are also due to Dr. Takashi Tatsuzaki, President of Tokiwa Phytochemical Co., Ltd., Chiba, Japan, for supplying 7.
Abbreviations
- BA
Betulinic acid
- MA
moronic acid
- OA
oleanolic acid
- UA
ursolic acid
- GLA
glycyrrhetinic acid
Footnotes
Due to changes in the screening system after 2000, our prior publications reported different activities for 12 − 15 in H9 lymphocytes.12, 16
Supporting Information Available: Elemental analysis data for compounds 16−22. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nakagawa-Goto K, Lee KH. Anti-AIDS Agents 68. The first total synthesis of a Unique Potent Anti-HIV Chalcone from Genus Desmos. Org. Lett. Submitted. [Google Scholar]
- 2.UNAIDS. WHO “AIDS epidemic update. 12−2005”. http://www.unaids.org.
- 3.FDA “Antiretroviral drugs approved by FDA for HIV,”. http://www.fda.gov/oashi/aids/virals.html.
- 4.Kashiwada Y, Hashimoto F, Cosentino LM, Chen CH, Garrett PE, Lee KH. Betulinic acid and dihydrobetulinic acid derivatives as potent anti-HIV agents. J. Med. Chem. 1996;39:1016–1017. doi: 10.1021/jm950922q. [DOI] [PubMed] [Google Scholar]
- 5.Hashimoto F, Kashiwada Y, Cosentino LM, Chen CH, Garrett PE, Lee KH. Anti-AIDS agents 27. Synthesis and anti-HIV activity of betulinic acid and dihydrobetulinic acid derivatives. Bioorg. Med. Chem. 1997;5:2133–2143. doi: 10.1016/s0968-0896(97)00158-2. [DOI] [PubMed] [Google Scholar]
- 6.Li F, Goila-gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, Castillo A, Zoumplis D, Martin DE, Orenstein JM, Allaway GP, Freed EO, Wild CT. PA-457: A potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13555–13560. doi: 10.1073/pnas.2234683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou J, Yuan X, Dismuke D, Forshey BM, Lundquist C, Lee K-H, Aiken C, Chen CH. Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J. Virol. 2004;78:922–929. doi: 10.1128/JVI.78.2.922-929.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun IC, Chen CH, Kashiwada Y, Wu JH, Wang HK, Lee KH. Anti-AIDS agents 49. Synthesis, anti-HIV, and anti-Fusion activities of IC9564 analogues based on betulinic acid. J. Med. Chem. 2002;45:4271–4275. doi: 10.1021/jm020069c. [DOI] [PubMed] [Google Scholar]
- 9.Holz-Smith SL, Sun IC, Jin L, Matthews TJ, Lee KH, Chen CH. Role of human immunodeficiency virus (HIV) type 1 envelope in the anti-HIV activity of the betulinic acid derivative IC9564. Antimicrob. Agents Chemother. 2001;45:60–66. doi: 10.1128/AAC.45.1.60-66.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beatty G, Lalezari J, Eron J, Pollard R, Saag M, Doto J, Salzwedel K, Wild C, Allaway G, Jacobson J, Martin D. Safety and antiviral activity of PA-457, the first-in-class maturation inhibitor, in a 10-day monotherapy study in HIV-1 infected patients.. 45th Conference on Antimicrobial Agents and Chemotherapy; Washington, DC. Dec 16−19.2005. [Google Scholar]
- 11.Sun IC, Wang HK, Kashiwada Y, Shen JK, Cosentino LM, Chen CH, Yang LM, Lee KH. Anti-AIDS agents. 34. Synthesis and structure-activity relationships of betulin derivatives as anti-HIV agents. J. Med. Chem. 1998;41:4648–4657. doi: 10.1021/jm980391g. [DOI] [PubMed] [Google Scholar]
- 12.Kashiwada Y, Wang HK, Nagao T, Kitanaka S, Yasuda I, Fujioka T, Yamagishi T, Cosentino LM, Kozuka M, Okabe H, Ikeshiro Y, Hu CQ, Yeh E, Lee KH. Anti-AIDS agents 30. Anti-HIV activity of oleanolic acid, pomolic acid, and structurally related triterpenoids. J. Nat. Prod. 1998;61:1090–1095. doi: 10.1021/np9800710. [DOI] [PubMed] [Google Scholar]
- 13.Huang L, Yuan X, Aiken C, Chen CH. Bi-functional anti-HIV-1 small molecules with two novel mechanisms of action. Antimicrob. Agents Chemother. 2004;48:663–665. doi: 10.1128/AAC.48.2.663-665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ito J, Chang FR, Wang HK, Park YK, Ikegaki M, Kilgore N, Lee KH. Anti-AIDS agents. 48. Anti-HIV activity of moronic acid derivatives and the new melliferone-related triterpenoid isolated from Brazilian propolis. J. Nat. Prod. 2001;64:1278–1281. doi: 10.1021/np010211x. [DOI] [PubMed] [Google Scholar]
- 15.Baltina LA. Chemical modification of glycyrrhizic acid as a route to new bioactive compounds for medicine. Curr. Med. Chem. 2003;10:155–171. doi: 10.2174/0929867033368538. [DOI] [PubMed] [Google Scholar]
- 16.Kashiwada Y, Nagao T, Hashimoto A, Ikeshiro Y, Okabe H, Cosentino LM, Lee KH. Anti-AIDS agents 38. Anti-HIV activity of 3-O-acyl ursolic acid derivatives. J. Nat. Prod. 2001;63:1619–1622. doi: 10.1021/np990633v. [DOI] [PubMed] [Google Scholar]
- 17.Condra JH, Schleif WA, Blahy OM, Gabryelski LJ, Graham DJ, Quintero JC, Rhodes A, Robbins HL, Roth E, Shivaprakash M, Titus D, Yang T, Teppler H, Squires KE, Deutsch PJ, Emini EA. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature. 1995;374:569–571. doi: 10.1038/374569a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.