

Abstract
Designed, synthetic heterocyclic diamidines have excellent activity against eukaryotic parasites that cause diseases such as sleeping sickness and leishmania and adversely affect millions of people each year. The most active compounds bind specifically and strongly in the DNA minor groove at AT sequences. The compounds enter parasite cells rapidly and appear first in the kinetoplast that contains the mitochondrial DNA of the parasite. With time the compounds are also generally seen in the cell nucleus but are not significantly observed in the cytoplasm. The kinetoplast decays over time and disappears from the mitochondria of treated cells. At this point the compounds begin to be observed in other regions of the cell, such as the acidocalcisomes. The cells typically die in 24–48 hours after treatment. Active compounds appear to selectively target extended AT sequences and induce changes in kinetoplast DNA minicircles that cause a synergistic destruction of the catenated kinetoplast DNA network and cell death.
Keywords: Antiparasitic drugs, Heterocyclic diamidines, Kinetoplast DNA, DNA topology, DNA binding
1. DNA as a biological target for antiparasitic drugs
1.1. The problem of eukaryotic parasites
Diseases caused by eukaryotic parasites, which include malaria, leishmania, and African and America trypanosomiasis, threaten approximately 40% of the human population [1]. There is currently an epidemic spread of these diseases and millions of people are infected with the deadly microorganisms that cause them [1, 2]. The parasites pose a danger to travelers in the affected regions and returning travelers can cause spread of the diseases. The sleeping sickness parasite, Trypanosoma brucei, the Chagas disease parasite Trypanosoma cruzi, and Leishmania limit the health and economic well being of millions of people from the American continents across Africa to the Far East [1].
Decades of attempts to create a vaccine against these eukaryotic parasitic diseases have not been successful in overcoming centuries of evolutionary development of defense mechanisms by the parasites against our immune systems. Because of the variable surface glycoproteins of trypanosomes, for example, the only current hope of escape from severe sickness and eventual death from this disease is chemotherapy [3–5]. Unfortunately, the current agents are overly expensive for most of the infected populations, they typically require multiple doses by injection, and they have unacceptably high toxicity. There is also evidence that drug-resistant strains of the parasites have developed and are spreading [6–8]. It is clearly essential to find new, more effective and less toxic compounds against these diseases.
1.2. Pentamidine is active against eukaryotic parasites
The bisbenzamidine derivative, pentamidine (Figure 1), has been one of the most successful agents against the eukaryotic parasite induced diseases and it has been used clinically against trypanosomiasis, leishmaniasis, and P. carinii pneumonia for over 70 years [7, 9, 10]. Pentamidine is not effective when given orally and it can cause severe toxicity [10,11]. A number of toxic side effects have been reported for pentamidine including abscesses at the injection site, abnormal liver function, hypotension, nephrotoxicity, cardiotoxicity, and hypoglycemia, with possible progression to insulin-dependent diabetes mellitus [12–15]. Some of the toxicity has been shown to arise from toxic metabolites [16,17].
Figure 1.

Pentamidine, furamidine and analogs along with and -AATT- DNA hairpin are shown. All compounds are dications.
Its continued use illustrates the essential need to develop new drugs, especially orally available agents, to replace pentamidine, melarsoprol, an arsenic derivative, and other similar drugs which exhibit significant toxicity. A related diamidine compound, berenil, is not currently used in humans but it is important for control of animal trypanosomal diseases. Given these successes, diamidines represent a promising template for design and development of new drugs against the eukaryotic parasite diseases.
1.3. Initial evidence for DNA as a biological target for diamidines
If a less toxic and orally effective replacement for pentamidine is to be designed, it is useful to start with the compound structural features of that compound that are essential for interactions with the parasite bioreceptor. Pentamidine and berenil have a number of common structural features: a curved shape or the ability to adopt such a shape, terminal diamidines with positive charges and H-bond donating ability, and a relatively flat conformation that has some flexibility in adjustment of dihedral angles. Analysis of these features in comparison to similar agents, suggested that the diamidines should bind to DNA. Initial studies with diamidines indicated that they did bind to DNA and that they did not intercalate [10, 18–23]. The compounds typically exhibited a preference for binding to AT sequences of at least four consecutive AT base pairs in the binding site. In an important series of studies, Tidwell and coworkers prepared a small library of compounds related to pentamidine and evaluated their DNA interactions by thermal melting experiments [24]. They found that compounds with an even number of methylene groups in the linking chain bound more weakly to DNA than compounds with an odd number of methylenes. They correlated the affinity differences with changes in curvature of the compounds from amidine to amidine. Compounds that bound well to AT sequences had an appropriate curvature to match the convex shape and helical twist of the DNA minor groove. The amidines provide H-bond donating -NH groups to interact with H-bond acceptors, N3 of A and O2 of T at the floor of the minor groove in AT sequences.
1.4. Diamidine-DNA complexes
Neidle and coworkers [20, 25] extended the analysis to high resolution when they obtained an x-ray crystallographic structure of pentamidine bound to a four base pair -AATT-sequence (Figure 2A) in a self-complementary duplex, d(CGCGAATTCGCG)2. The close match of the compound curvature to the minor groove shape can easily be seen in Figure 2. The weak binding to the minor groove of related compounds without the critical structural features to complement DNA, such as pentamidine analogs with an even number of methylenes [24], can be understood from the DNA complex structure. Interactions in the DNA-pentamidine crystal structure also illustrate that phenylamidines provide an excellent minor groove recognition unit in AT sequences. They form H-bonds with acceptors on the base pair edges at the floor of the groove, they effectively stack with the walls of the narrow minor groove in AT sequences and they have a positive charge to offset the phosphate charges and negative electrostatic potential of the groove. The steric bulk of the third H-bond in GC base pair containing sequences as well as the generally larger width of the minor groove in such sequences [20, 26, 27] prevents similar molecular recognition between the compounds in Figure 1 and the minor groove in GC sequences.
Figure 2.
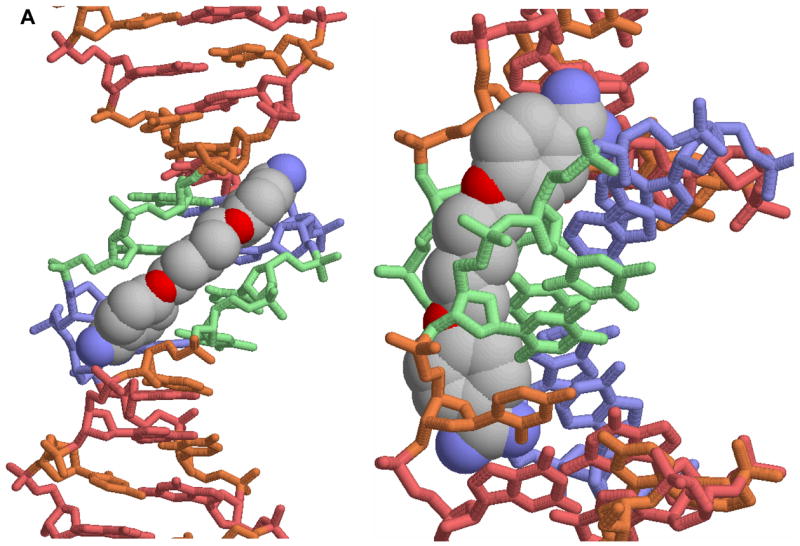

A. A view of the crystal structure of pentamidine bound to the -AATT- site of d(CGCGAATTCGCG)2 is shown [25]. The view on the left is into the minor groove and the view on the right shows how the compound fits into the groove. In this conformation the amidines are H-bonded to TO2 groups and the compound makes contacts with the groove floor and walls. B. A view into the minor groove of the DB75 crystal structure with the same binding site and DNA sequence is shown at the left [53]. The DNA contacts are similar to those with pentamidine but the DB75 structure is preorganized to bind in this orientation. The view on the right is from the major groove through the base pairs to DB75. The red dots identify the amidine to TO2 H-bond interactions. The compound curvature allows DB75 to slide deeply into the groove in AT sequences and make excellent contacts with the bases and groove walls.
As with pentamidine, berenil and other related heterocyclic dications have also been found to bind specifically to AT minor groove sequences [10,18–30]. This similarity and strength of interaction with DNA for these compounds provided initial evidence that DNA binding could be involved in the mechanism of action of the dications. For these reasons emphasis has been placed on design and study of compounds that could target AT DNA sequences but with better properties and lower toxicity than pentamidine [11–17]. A successful example of this design concept is furamidine, DB75 in Figure 1, and a prodrug of that compound is currently in Phase III clinical trials against the parasite that causes sleeping sickness. The prodrug of furamidine is a neutral derivative that is orally effective and it is described in more detail below. The structure of DB75 in the d(CGCGAATTCGCG)2 DNA sequence is very similar to that of pentamidine and is shown in Figure 2B. There is a clear pattern for binding of these compounds which will be described in detail below.
1.5 A strategy for drug development
Our strategy to capitalize on these intrinsic features of the heterocyclic dications, described in this review, involves design and development of new compounds targeted to the DNA minor groove in a variety of complexes that are metabolically robust with limited toxicity, and good cell uptake properties in parasites. We have also developed and describe below prodrug approaches to provide oral bioavailability for the dication class [10, 31]. The prodrug of furamidine has been found to have low toxicity in human trials in African, Asian, Caucasian and Hispanic populations. The availability of a safe and orally effective drug promises to revolutionize the treatment of parasitic diseases. Since there is no effective drug for Chagas’ disease and resistance is emerging for drugs currently available for parasitic diseases, it is urgent that the rational design of new, low-cost, orally-effective dications be intensely pursued.
2. Heterocyclic dications that target the DNA of eukaryotic parasites
2.1 The kinetoplast and its DNA network
Since most of the recent cell uptake studies on diamidines have been conducted with the Leishmania and trypanosome parasites, this section and the remainder of this review will focus on those organisms. Given the observations described above, an understanding of the DNA interactions of the heterocyclic dications should provide an insight into possible mechanisms for the antiparasitic action of diamidines. In addition to their nuclear DNA genome, trypanosomes, leishmania and related organisms have a single mitochondrion with a kinetoplast that contains the mitochondrial DNA [32, 33]. The kinetoplast organelle consists of an unusual DNA network that is made up of thousands of DNA circles topologically linked in a disk-shaped, planar array. The DNA network typically consists of two general types of circular DNAs: a small number of maxicircles that code for rRNAs and mitochondrial proteins that function in energy conversion and thousands of quite small DNA minicircles that are about 1 kb in T. brucei [32–34]. The mRNA from maxicircles must be edited at specific sites to provide a correct reading frame. The reaction is very unusual and involves specific insertion and/or deletion of specific U bases. The DNA minicircles code for guide RNAs that control the editing reaction to modify the mRNAs [32–36]. The kinetoplast DNA (kDNA) minicircles have extensive, closely-spaced, phased AT sequences that yield curved double-helical structures [37, 38]. These AT sequences in the thousands of kinetoplast DNA minicircles provide a compact array of potential cellular targets for diamidines that have AT sequence binding specificity.
2.2 Cellular distribution of synthetic diamidines
Unlike pentamidine, which absorbs below 300 nm, compounds similar to furamidine typically have absorption bands near 360 nm and they emit in the blue region with peaks around 460 nm in solution [18]. The fluorescence of these compounds makes it possible to follow their distribution after cell uptake. Results for heterocyclic diamidines by this method were recently published with DB75 and DB820, a pyridine analog of DB75 (Figure 1), by Mathis et al. [39]. Although dications would be expected to enter most cells slowly by diffusion mechanisms, there are several potential transporters that can effectively carry diamidines into parasite cells [6, 10, 40 – 42]. The P2 adenosine/aminopurine transporter has been identified as the primary diamidine transporter but other transporters can participate in a more limited capacity [6, 40–42]. Both DB75 and DB820 are taken up rapidly through the P2 transporter and are initially distributed to the kinetoplast and nucleus [39, 43]. The kinetoplast has very strong fluorescence with less intensity in the nucleus while very little fluorescence is observed in any other region of the cell in the initial stages. Over time the kinetoplast begins to change shape, break down and can actually disappear. During this period DB75 is also accumulated into the acidocalcisomes, a dication sequestering compartment of the trypanosomes [39, 43, 44] and shows strong acidocalcisome fluorescence. Less intense fluorescence is observed in acidocalcisomes with DB820 at early time points (less than four hours of exposure to the compound) but the acidocalcisome fluorescence becomes stronger at longer exposures [39, 43]. The cells typically die within 24 – 48 hours after treatment with these compounds. Pretreatment of the trypanosomes with either ammonium chloride or the sodium ionophore, monensin, largely blocks acidocalcisome uptake. The pyridine substitution to give DB820 from DB75 does not strongly affect the DNA binding ability of the compound but it does improve the ability to cross the blood-brain barrier. A prodrug of DB820 is thus an excellent candidate for treatment of late stage sleeping sickness where the trypanosomes have invaded the central nervous system. Additional clarification of compound cellular distribution and therapeutic mechanism correlations comes from some recent experiments by Barrett and coworkers [45]. They were able to construct a leishmania mutant that was resistant to mitochondrial, and thus kinetoplast, uptake of diamidines. The compounds could enter mutant cells through the plasma membrane in a normal manner but then could not easily pass through the mitochondrial membrane. Strong diamidine fluorescence was observed in the kinetoplast of the wild-type, diamidine sensitive, but not the mutant, diamidine resistant, leishmania cells. It is clear that diamidines can selectively accumulate in the mitochondrial kinetoplast of wild type but not mutant cells. When the mitochondrial membranes of resistant cells were treated to render them permeable to cations, diamidine fluorescence was observed in the kinetoplast. Resistance is thus caused by changes in the mitochondrial membrane and not by changes in kinetoplast binding. Accumulation of the drug into the kinetoplast of the wild type cells was able to drive much greater accumulation of diamidines into those cells. Much lower amounts of the compound accumulated in the resistant cells even though their plasma membrane diamidine transporter functions as in wild type cells [45]. It is thus binding to the kinetoplast DNA that drives much of the cell uptake of diamidines and not high cell uptake that leads to significant binding.
2.3 Cellular distribution of DB351
A typical example of cell uptake and distribution of compounds related to DB75 is shown for DB351 with trypanosomes in Figure 3. DB351 has a central thiophene in place of the furan of DB75 (Figure 1) and it binds to AT sequences of four or more base pairs in a similar manner and with similar affinity to DB75 (see below). As can be seen, DB351 is taken up by trypanosomes very rapidly and after one hour of exposure it is distributed in both the parasite kinetoplast and nucleus. Over time, the kinetoplast fluorescence becomes weaker and typically, by 24 hours the kinetoplast is visibly disintegrating or has completely disappeared. The cells generally die in 24–48 hours after treatment. It is clear with this compound that kinetoplast DNA destruction is directly correlated with the time course of drug action. Unlike DB75, DB351 does not accumulate extensively in the acidocalcisomes as the kinetoplast degenerates and the cells die. The role of acidocalcisomes in the overall mechanism of killing trypanosomes is not clear and some compounds, as with DB351, do not significantly accumulate in the acidocalcisomes as the cells die.
Figure 3.
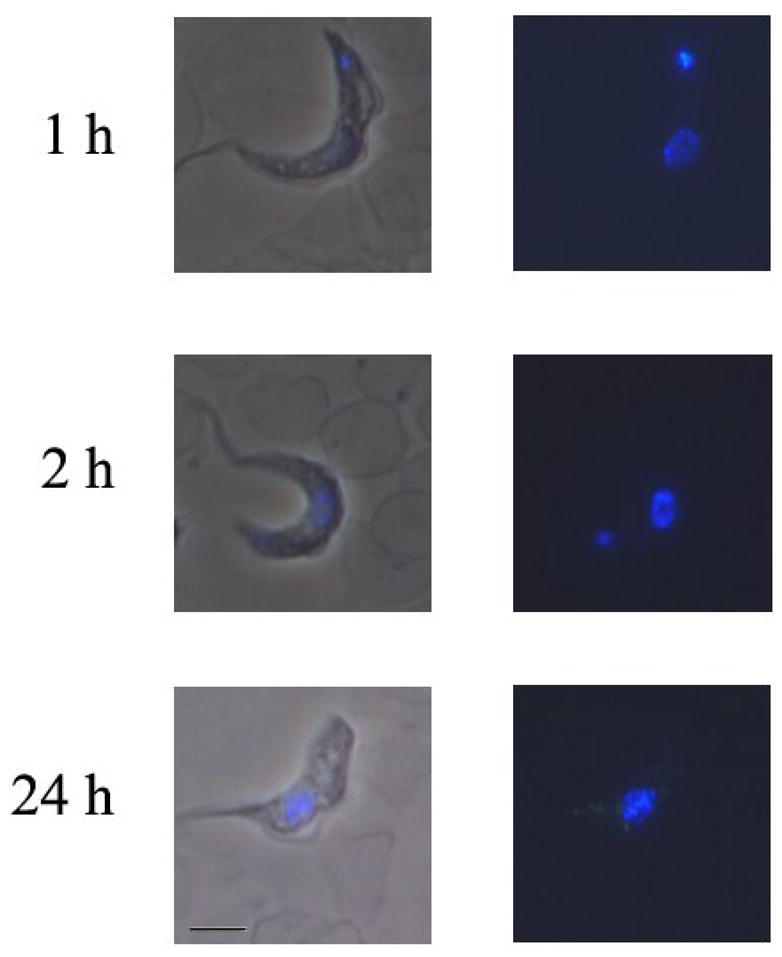
S427 Trypanosoma brucei brucei trypanosomes were incubated in vitro with 500 nM of DB351 for various time points between 1 hour and 24 hours. Trypanosomes were washed and resuspended in freshly isolated mouse blood. Dry smears were prepared and examined using a Nikon Microphot microscope with a UV2A cube. Nuclei are found in the center of trypanosomes, and the kinetoplast is found at the end of trypanosomes.
2.4 The effects of diamidine binding on the kinetoplast DNA network
Diamidine induced destruction of the kinetoplast could occur, for example, by compound interference with the normal topoisomerase II catalyzed opening and rejoining of kDNA minicircles. There are also numerous other DNA replication steps in the complex catenated kinetoplast DNA where compound binding and either induced topological changes or protein inhibition could cause replication errors and DNA degradation. It seems highly probable that exposure of the phased AT sites in kDNA to the diamidines will change a number of DNA topological features as well as modify DNA-protein interactions.
In proposing a mechanism for the parasitic activity and relatively low toxicity of diamidines such as DB75, DB820 and DB351, the intricate structure of the catenated kDNA network must be considered. Since there are thousands of minicircular DNAs in the network, each with multiple closely spaced AT sequences, it would take a very small compound-induced error percentage in the normal biological functions of the kinetoplast to cause destruction of the network, kinetoplast disintegration and cell death. Human cells, which do not contain a similar network, would only display toxicity at higher compound concentrations. In this model, while DB75 and related compounds can bind to nuclear DNA of all cells, the local amounts of bound compound in any DNA sequence at therapeutic concentrations do not reach a high enough amount to cause toxic effects. Minor topological effects of the compounds on the bent, phased A-tract sequences of the kinetoplast DNA network could synergistically sum to cause a biologically catastrophic effect and give the therapeutic result. Some diamidines that bind quite well to DNA and can accumulate in trypanosomes are much less active than DB75 and the reasons for the loss in activity and the steps in cell killing after kinetoplast binding remain to be defined in detail. It should be noted, however, that in treatment of patients up to the Phase III clinical trial level, no significant human toxicity of the diamidines has been observed.
2.5 The design concept
The uncertainty in predicting the compound structural features that are optimal for generating the kDNA effects described above as well as the limited information available on what is optimum for targeting the kinetoplast in a way to generate maximum biological activity present difficulties for the rational design of improved antiparasitic agents. Although the structures in Figure 2 indicate that compound curvature and H-bonding lead to AT specific interactions, the steps after DNA binding that are most important for the mechanism of action -killing of parasite cells - are not clear. Subtle features such as compound induced DNA bending or other topological perturbations of the DNA double helix with amplified effects in catenated kDNA minicircles are probably quite important for biological activity. Compound induced conformational changes could inhibit the ability of topoisomerase II to effectively close the minicircle DNA when it is opened for transcription or replication. Optimization of these additional features, beyond AT sequence specificity, as well as possible effects on DNA-protein interactions could significantly increase compound antiparasitic activity, lead to specific targeting of additional types of parasites, and reduce host toxicity even further. The methods to modify compound structures and properties to improve biological activity, however, is not clear.
In order to expand the base of information on kinetoplast DNA-compound complex features that yield compounds with improved DNA interactions and biological activity, we have taken an empirical approach. The goal is to prepare compounds that have the potential for a variety of DNA interaction features at AT minor groove binding sites that mimic the AT rich region of kinetoplast minicircles. The parent compound for the design studies is DB75, furamidine, whose prodrug is currently in clinical trials. The original syntheses of DB75 [46] and its O-methylamidoxime prodrug DB289 [47] have been described.
Comparison of the stronger AT sequence binding and lower biological toxicity of furamidine relative to pentamidine suggested that the many configurations of the methylene linker of pentamidine reduced the DNA affinity. It also seemed likely that the phenolic linkage of pentamidine could be responsible for a major part of the toxicity of the compound and removal of these linkages was desirable. The goal then is to prepare a library of heterocyclic dications with the furan, phenyl and amidine functions of DB75 modified in ways that would be predicted to affect the DNA binding. Detailed biophysical studies of the DNA complexes of such modified compounds, when correlated to changes in biological activity, should provide a better understanding of the biological mechanism of the dications.
3. The interaction of furamidine and synthetic analogs with kinetoplast DNA model systems
3.1. DB75 binds strongly to AT DNAs that model kinetoplast minicircle sequences
The large number of DNA minicircles with repeated AT rich sequence regions in the kinetoplast provide a large number of binding sites for AT specific diamidines. DB75 and related compounds bind quite strongly to AT sequences that mimic the AT rich regions of kinetoplast minicircles (K> 107 M−1) [18]. As a result, the free drug concentration in equilibrium with cellular AT DNA binding sites is less than 100 nM. This low concentration represents the unbound concentration of compound available for interaction with potential cellular targets other than DNA in parasite cells. As described above, in the first stages of drug action, based on fluorescence microscopy intensity analysis of treated cells, DNA binding accounts for most of the compound in the parasite cells and the cytoplasm shows negligibly small concentrations of the compound. This observation is in complete agreement with the strong binding of the diamidines to AT sequences and explains why they are always observed rapidly and in high concentration in the kinetoplast. The diamidines typically bind 100 or more times more weakly to GC containing sequences than to pure AT sites and this gives them a preference for the AT rich regions in kinetoplast DNA minicircles [48].
A detailed thermodynamic study has been conducted with DB75 and the self-complementary duplex, d(CGCGAATTCGCG)2, shown in Figure 2. We have found that the interactions of diamidines and related compounds with a broad range of DNA sequences and structures can be quantitatively evaluated very effectively with biosensor-surface plasmon resonance (SPR) methods [49,50]. At 25 °C and salt concentrations near physiological SPR and isothermal titration calorimetry studies show that DB75 has a binding constant, K = 1.4 × 107 M−1 (0.1M NaCl, pH = 6.2 buffer), with a small favorable binding enthalpy (−2.2 kcal/mol) and large favorable binding entropy (T.ΔS = 7.5 kcal/mole) for the -AATT- sequence [51, 52]. At higher salt concentration DB75 has a slightly lower binding constant, K = 4.2 ± 0.6 × 106 M−1 (0.2M NaCl, pH =6.2 buffer), as expected for the interaction of a dication with DNA [48, 52]. ITC experiments provide a direct method to obtain the binding enthalpy but the low enthalpy values obtained for diamidines do not allow determination of the K value. The K values obtained from SPR with the enthalpy from calorimetry allow a full thermodynamic characterization of DNA complexes. Such a full thermodynamic picture is essential to complement the structural perspective from crystallography of diamidine-DNA complexes [51–53].
The large entropy for DB75 binding indicates that release of high entropy water from the minor groove in AT sites provides a major part of the binding ΔG°. Recent osmotic stress measurements of DNA–small molecule binding, however, indicate that there is a net water uptake, not release, on complex formation, including diamidine complexes [54]. A possible explanation for these apparently contradictory findings is that exchange of two types of water occurs in the binding process. Highly ordered and, thus, high entropy water is found in the minor groove of AT sequences, such as -AATT-, and is released from the groove in the binding reaction. Release of this type of bound water will dominate the entropy term while general association of a larger amount (per molecule of DNA complex) of relatively unstructured, more mobile and, thus, lower entropy water could dominate the net amount of bound water during complex formation. The net binding entropy would be positive in this situation while the net amount of bound water would increase. The more highly structured the water in the groove, the more dominant the entropy of binding would become to the Gibbs energy of complex formation, and this is what is observed for diamidines binding to sequences such as -AATT- and most of the AT sequences in kinetoplast minicircles.
We thus have a very thorough understanding of the structure, affinity and thermodynamics for the interaction of DB75 with DNA sequences such as those found in kinetoplast minicircles. From this base of reference we can ask how small changes in DB75 structure affect DNA interactions and biological activity.
3.2. The effects of changes in the DB75 phenyls on DNA interactions: pyridyl derivatives
Nitrogen atoms added to the aromatic system of DB75 change the lipophilicity and polarity of the heterocyclic core and thereby could lead to different absorption and distribution profiles (aza analogs, Figure 1B). As described above, the pyridyl derivative DB820, as with DB75, initially localizes in the kinetoplast and nucleus of treated trypanosomes. The most exciting feature of this compound is that, unlike DB75, DB820 can cross the blood brain barrier and may provide a new therapeutic strategy for treatment of late stage sleeping sickness and other diseases [55]. It is thus important to characterize the DNA interactions and biological activities of nitrogen analogs of DB75 in detail. The four compounds DB820, DB867, DB829, and DB944 that closely match the shape and dimensions of furamidine show strong affinity for AT sequences of DNA as reflected by the ΔTm increases (Table 1) for binding to polydA.polydT. Consequently, the use of polydA.polydT is helpful for relative scaling of the affinities of these compounds [31, 56]. The ΔTm values for the aza-analogues are reduced from that of DB75, but are significantly higher than that of pentamidine. The reduction in the ΔTm values of the aza-analogues is consistent with their increased hydrophilic properties as a result of additional nitrogen atoms. Transfer of the pyridyl compounds from aqueous solution to the less polar environment of the minor groove is thus less favorable than with DB75. The aza-analogues were evaluated against T. b. rhodesiense in vitro (Table 1) and DB820, DB867, and DB994 give IC50 values comparable to that of DB75.
Table 1.
ΔTm to polydA.polydT, DNA binding constants to AATT site, and biological activity of derivatives that modified from DB75
More quantitative analysis of the pyridyl interactions was done with SPR methods as described in the Methods section. Sensorgram for DB820 binding to the -AATT- sequence DNA are shown in Figure 4 and a binding isotherm is in Figure 5. Fitting the results, as described in Methods, provided DNA binding constants for DB820, and all other aza-analogues were evaluated in the same manner (Table 1). The compounds bind strongly and specifically to AT DNA models for kinetoplast sequences as with DB75. In summary, aza-analogues of DB75 exhibit high binding affinity for AT sequences and good biological activity against T. b. rhodesiense. The introduction of nitrogen(s) into the phenyl ring(s) of DB75 thus yield compounds with promising antimicrobial activity and good ability to target organisms that have invaded the central nervous system.
Figure 4.
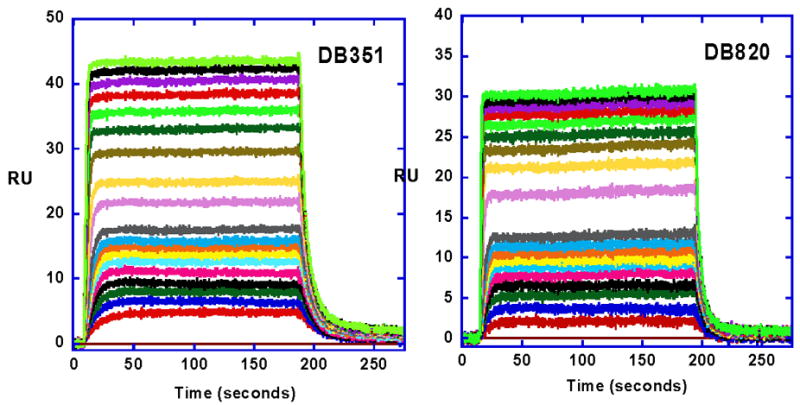
SPR sensorgrams for binding of DB351 and DB820 with an -AATT- site, as in Figure 2, in an immobilized hairpin DNA (Figure 1). The compound concentrations were 10 nM to 1 μM from bottom to top. The experiments were carried out in Tris buffer at 25 °C.
Figure 5.
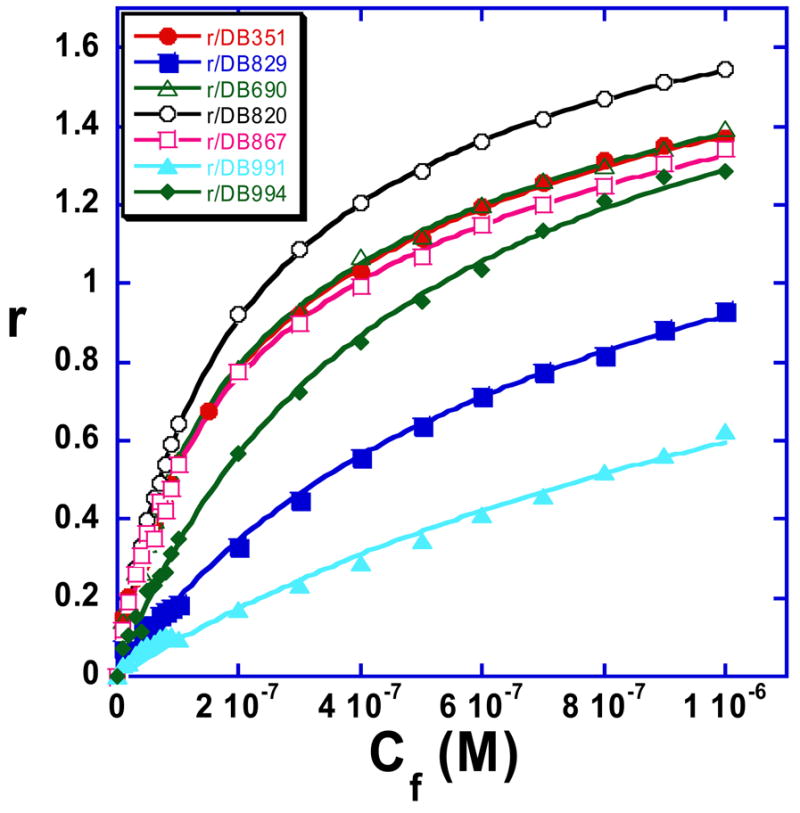
Binding curves from the SPR results in Figure 4 for the DB75 derivatives are shown with r values plotted against the unbound compound concentration, Cf (flow solution). The data were fitted to a two site model using equation 1 and all compounds have one strong binding site in the AATT sequence and some much weaker secondary binding.
3.3. The effects of changes in the DB75 furan on kDNA interactions: DB351
Starting from the parent compound DB75, the furan ring was changed by one atom in the five-member ring to give a thiophene (DB351) in place of furan. Sensorgrams for DB351 are also shown in Figure 4 and binding plots in Figure 5. The compound binds strongly and specifically to AT DNA sequences, comparably to DB75, with K= 9.4×106. The compound also has very good biological activity (Table 1) and clear kinetoplast up take (Figure 3). At 25 °C and salt concentrations near physiological SPR and ITC studies show that DB351 binds to DNA with a small enthalpy (−3.0 kcal/mol) and large entropy (T.ΔS = 6.4 kcal/mole) as with DB75.
An initial evaluation of the topological effects of diamidines on DNA model systems related to kinetoplast DNA was performed with DB351 (Figure 6). DNA ladders were constructed with DNA ligase, such that oligomers with multiple phased A tracts were created (see Methods Section). These ligation ladders provide an excellent opportunity to investigate the topological effects of diamidines in a system that models the AT rich region of kinetoplast minicircle DNA. In this method multiple lengths of DNA differing only by the number of A tracts they contain are prepared and their topology evaluated simultaneously by gel electrophoresis. Two 21 base pair sequences with in phase (cis) and out of phase (trans) A tracts were ligated (Figure 6C). Because A tracts are curved, the cis ladder, which has in phase A tracts, migrates significantly slower than the trans ladder. In a test to see if DB351 has an effect on DNA topology, gel electrophoresis was performed on the cis and trans ladders in the presence of DB351 (Figure 6A). A measurable decrease in migration was detected in the presence of DB351, a result that clearly shows topological changes being induced in the DNA. In the absence of DB351 both the cis and trans fragments migrate anomalously slowly due to the presence of A tracts. When DB351 is bound to the A tracts, their anomalous migration is enhanced. This enhancement is quantitated in Figure 6B where RL values for both ladders in the presence and absence of DB351 are plotted. The increase in RL indicates that the compound causes increased curvature in the DNA. This observation provides a possible explanation for the destruction of the kinetoplast DNA as shown in Figure 3.
Figure 6.

(A) Sequence of trans and cis oligomers used in the gel electrophoresis experiments. (B) Native PAGE of trans (lanes 2 and 4) and cis ligation ladders (lanes 1 and 3) and 20-bp marker (M) in the presence of DB351. Circular products (C) and linear ligation products (L) are marked, where L2 corresponds to the 42-bp dimer, L3 to the 63-bp, and so forth. (C) Plot of relative mobility (where RL= apparent length/actual length) versus actual length (Lact) for trans (red circle) and cis (red triangle) ligation ladders without DB351 and trans (blue circle) and cis (blue triangle) with DB351. Only linear products were used to calculate RL, and missing points correspond to linear ligation products indistinguishable from circular products.
3.4. The effects of changes in the DB75 amidines on DNA interactions: DB244
DB244 (Figure 1) binds strongly and specifically to AT sequence DNA [43]. The compound has a binding constant, K = 2.2 × M−7 with a small enthalpy (−2.3 kcal/mol) and large entropy (T.ΔS = 7.6 kcal/mole) of binding to the -AATT- sequence. Crystallographic structures for DB75 and DB244 complexes with d(CGCGAATTCGCG)2 [51, 53] show similar interactions of the central diphenylfuran ring system of the two compounds with the minor groove. The cyclopentyl groups of DB244 make good contacts with the minor groove and displace additional water from the groove relative to DB75. Interestingly, the biological activity of DB244 is approximately 10 times less than for DB75 in spite of its similar DNA binding affinity, thermodynamics and complex structure. Clearly there are post DNA binding events, such as required topological changes, inhibition of binding of specific control proteins or direct inhibition of key enzymes such as a topoisomerase, that are essential to activity.
3.5. Effects of shape changes in DB75 on DNA complexes: DB555, DB991, DB690, and DB879
As described above, variation in the number of methylene units in the methylene linker of pentamidine analogs changed the compound shape and had a strong effect on the binding of the derivatives to DNA. A similar shape test was performed with DB75 and DB820 by shifting the position of one or both amidine groups from the para to the meta position (Figure 1). This modification significantly changes the amidine to amidine curvature and since the phenyl-amidine groups are critical for DNA complex formation (Figure 2), it also changes the potential for interaction with the DNA minor groove in AT sequences. Based on the curvature model for diamidines, as well as other similar minor groove binding cations, which interact with DNA, the meta derivatives DB555 and DB991 (Figure 1) deviate significantly from the optimum binding geometry. The meta derivatives exhibit much lower DNA affinity than DB75 and DB821 and they also have lower activity against T.b.r. (Table 1).
If the meta amidine of DB555 or DB991 points toward the inner face of the molecule, it will clash with the base pairs at the floor of the minor groove. Rotation about the phenyl-furan bond allows the amidine to point away from the DNA bases but with the loss of the amidine-base interactions. The para amidine in DB555 or DB991 can interact with DNA just as with the amidines of DB75 [31, 57]. In general, a crescent shape remains a critical factor for good DNA minor groove interactions in compounds related to DB75. It is interesting that the antitrypanosomal activity of DB555 is reduced by approximately ten fold relative to DB75, illustrating the linkage (direct or indirect) between a baseline level of required DNA binding affinity for biological activity.
Another question in this series of analogs is what happens if the basic concave shape is maintained but with the oxygen of the furan pointed away from the minor groove, the opposite of DB75, to give DB690. It might be expected that pointing the furan oxygen out of the minor groove where it can interact with water instead of the less polar minor groove would improve binding. What is actually observed, however, is that DB690 binds specifically to AT DNA sequences but slightly weaker than DB75 [9.5×106 vs K> 107 M−1]. This is probably due to slight changes in compound shape that allow DB75 to align the amidine groups more optimally with the groups on bases at the floor of the groove than is possible with DB690.
Clearly, the compound shape and its match with the shape of the DNA minor groove are critical for optimizing compound interactions with the groove as well as contacts with base edges at the floor of the groove. In summary, both furamidine and pentamidine type compounds must match the groove curvature for strong DNA interactions and effective biological activity. This can be through direct contacts or indirect interactions through bound water.
4. Diamidine prodrugs
4.1 The oral bioavailability problem with diamidines
The poor oral bioavailability of aryl diamidines limits their use and represents a challenge for delivery of these highly active compounds. Amidines are strongly basic molecules that typically exhibit pK values greater than 10 [58] and consequently are protonated at physiological pH. Thus the major cause of their low oral bioavailability is attributable to the pK. The promising antiparasitic activity of the diamidines has lead to exploration of various prodrug approaches in efforts to improve bioavailability of this class of compounds. Functional group modification which lowers the pK of the molecule so that it is largely unprotonated at physiological pH is an approach which has received attention by several groups. The use of amidoximes analogues of amidines is a popular specific example of this approach. The potential for this approach has been available in the literature since 1939 [59], however it apparently was not recognized as an amidine prodrug strategy until relatively recently. The preparation of pentamidine dioxime and its conversion to pentamidine in a mouse model has been described [60] and the reductive metabolism of amidoximes has been reviewed [61]. Oral administration of pentamidine dioxime in the immunosupressed rat model for PCP clearly demonstrated the potential of this prodrug approach; however in this example it was only moderately effective [62]. For a prodrug to be effective it must have a good absorption and distribution profile as well as be effectively converted to the active drug in vivo. In the case of prodrugs of diamidines, it is conceivable that bioconversion to mono-amidine- mono-prodrug would yield a compound which if pumped out of the metabolizing cell may not be able to re-enter, due to its cationic character, another cell to complete the conversion to the active diamidine. To fully understand the success or failure of a potential prodrug requires considerable in vitro and in vivo investigations and these are often limited to compounds that have been advanced moderately far along the drug development pathway.
4.2. Prodrugs for furamidine
Furamidine (DB75 in Figure 1) has broad spectrum antimicrobial activity [10, 44, 63, 64]. We have shown that the bis-amidoxime (DB290) and the bis-O-methylamidoxime (DB289) of furamidine are quite effective in an immunosupressed rat model for PCP on both intravenous and oral dosing [47]. Likewise, DB289 was found to be orally active in a mouse model for human African tyrpanosomiasis (HAT) [65]. These results have led to a considerable amount of development work and DB289 has now advanced into two different Phase III trials; one against PCP and the other versus HAT [5].
The metabolism pathway for the conversion of DB289 into furamidine has been elucidated [65, 66]. Sequential O-demethylation and N-dehydroxylation reactions result in the bioconversion of the prodrug into furamidine. The bioconversion has been shown to involve cytochrome P450 catalyzed O-demethylation followed by cytochrome b5/b5 reductase catalyzed reductive dehydroxylation [67]. Despite the moderately complex metabolic pathway for DB289 it is clearly efficient in humans based on results from numerous clinical trials.
In parallel with an in vivo efficacy investigation in the stringent STIB900 mouse model for HAT an in vitro transport and a metabolism study was performed with a series of furamidine amidoximes with varying O-alkyl groups [65]. This study showed that rate of metabolism and efficacy declined with increasing size of the alkyl group [65]. The multiple sites of oxidation on the larger alkyl chains presumably leads to a lower rate of production of the key O- dealkylated product.
4.3 Prodrugs for aza-analogs of furamidine
Aza-analogues of furamidine (Figure 1B), DB820 and DB829, exhibit potent in vitro antitrypanosomal activity; however, they are also limited by poor oral activity due to the positively charged amidine groups [31]. DB844, the methoxyamidine prodrug of DB820, shows potent oral activity in the STIB900 mouse model for early stage African trypanosomiasis, providing 4/4 cures at the remarkably low oral dose of 5 mg/kg. DB868, the methoxyamidine prodrug of DB829, is less effective in the same model, providing only 2/4 cures at 25 mg/kg dosage [31]. However, both prodrugs are quite effective in a late stage mouse model (CNS involvement) for African trypanosomiasis and consequently they are undergoing further evaluation in order to select a candidate drug for possible treatment of late stage HAT [55].
4.4 Successes and failures: prodrugs of other heterocyclic diamidines
All amidoximes or O-methylamidoximes of diamidine systems are not effective prodrugs. For example, amidoximes or O-methylamidoximes of molecules that include benzimidazole moieties have been found to be ineffective [62]. It is not clear if the lack of effectiveness of these molecules is due to poor uptake or if the metabolism products are trapped within cells and not sufficiently released to reach the target organisms. Alternatively, inactive intermediate metabolites may be effluxed from cells which could also reduce quantities of active diamidines formed. In a study of the isomeric 2,8 and 3,7 diamidoximes in the dibenzothiophene series the 2,8 diamidoxime was an effective prodrug whereas the 3,7 isomer was not (see figure 7B) [68]. A similar result was noted for the analogous isomeric set of dibenzofurans [69]. It seems possible that the near linear array is not being recognized by the metabolizing enzymes (vide infra). Further studies are required to develop a full understanding of the lack of effectiveness in these cases. In yet another system, O-methylamidoximes of the highly active linear terphenyl diamidines were found to be ineffective when given orally [70]. In an effort to gain some insight into low effectiveness of these prodrugs the rates of substrate depletion of the linear analogues were compared with that of DB289 using mouse liver microsomes [70]. In this system DB289 gives a t1/2 of less than 15 minutes whereas the t1/2 of all the linear analogues (DB1185, DB1217, DB1257, DB1231) were greater than 90 minutes except DB1203 which gave a t1/2 value of 55 minutes. These results suggest that in mice the poor efficacy of the linear prodrugs is due, at least in part, to slow bioconversion. Studies directly with the cytochrome P450 involved are needed to determine the relative importance of adsorption/distribution and sensitivity of the demethylation step to structure.
Figure 7.

The structures of diamidine prodrugs are shown for a number of different heterocycles.
4.5. Other types of prodrugs
Other prodrugs of amidines which have been found to be effective include O-acyl amidoximes and related compounds. These compounds are double prodrugs which are thought to be first converted by nonspecific esterases to the amidoximes which are then reduced to form the amidine. The O-acetylamidoxime of pentamidine has been prepared and found to be converted to pentamidine in rats after oral administration [71]. Several O-acyl and related analogues of 1,12-bis-(N,N′-acetamidinyl)dodecane have been found to be effective in mouse models for malaria [72]. The bis-O-methylsulfonylamidoxime and the bis-oxadiazolone were the most effective in this model (Figure 7).
Another potential class of diamidine prodrugs which has received attention involves masking the amidine as a carbamate. Carbamates can be converted to the amidine either by chemical or enzymatic (presumably esterases) hydrolysis followed by spontaneous decarboxylation. In this system it is important to attempt to achieve a balance between carbamate chemical stability and its enzymatic bioconversion. For example, labile carbamates are likely to be rapidly hydrolyzed by stomach acid. In such cases oral dosing of labile carbamates would be equivalent to dosing with the diamidine and poor oral availability would be the outcome.
We reported the synthesis and evaluation of a series of alkyl and aryl carbamates of furamidine [73]. The most promising analogues, which seemed to have the needed balance between chemical stability and enzymatic conversion, were the aryl carbonates DB596 and DB539. These compounds gave comparable oral efficacy to DB289 in an immunosuppressed rat model for PCP. A series of N-alkylfuramidine analogues has been reported and several of these compounds showed significant activity in an immunosuppressed rat model for PCP on iv dosing at 1 μmol/kg [74]. In an initial study designed to determine if these compounds could be made orally effective we examined alkyl carbamates of two of the more potent N-alkylfuramdines (DB181 and DB244) [75]. In the systems studied it was found that the 2,2,2-trichloroethyl carbamate of both the N-i-propyl (DB485) and the N-cyc-pentyl (DB517) derivatives of furamidine were quite effective on oral dosing in an immunosuppressed rat model for PCP. Thus it appears that carbamates of amidines and N-alkylamidines on oral dosing can function as effective prodrugs.
It is quite clear that produgs of diamidines can be developed to allow effective oral delivery of the active dications. What is less clear is the most efficient design path to employ. In the development process it is important to gain an understanding of a potential prodrug’s pharmacokinetics properties as well as its metabolism to rationally guide subsequent modifications. Such studies are time consuming, expensive and requires the active collaboration of chemists and pharmacologists.
5. Conclusions
The comparative DNA binding and biological activity results presented here for the compounds of Figure 1 emphasize the importance of the compound shape and its match with the shape of the DNA minor groove for optimizing interactions with the groove as well as contacts with base edges at the floor of the groove. Both furamidine and pentamidine type compounds must match the groove curvature for strong DNA interactions and the best biological activity. An interesting observation from the results in Table 1 is that in this series of DB75 analogs, the activity and DNA binding track closely together (Figure 8) and both log K and Tm are linearly correlated with logIC50. An exception is DB244, which has alkyl amidine groups that slightly increase the DNA affinity relative to DB75 but the biological activity of DB244 is reduced by approximately a factor of 20 relative to DB75. This decreased activity relative to DB75 is not unique to DB244 but is also seen with other derivatives with alkyl amidines.
Figure 8.
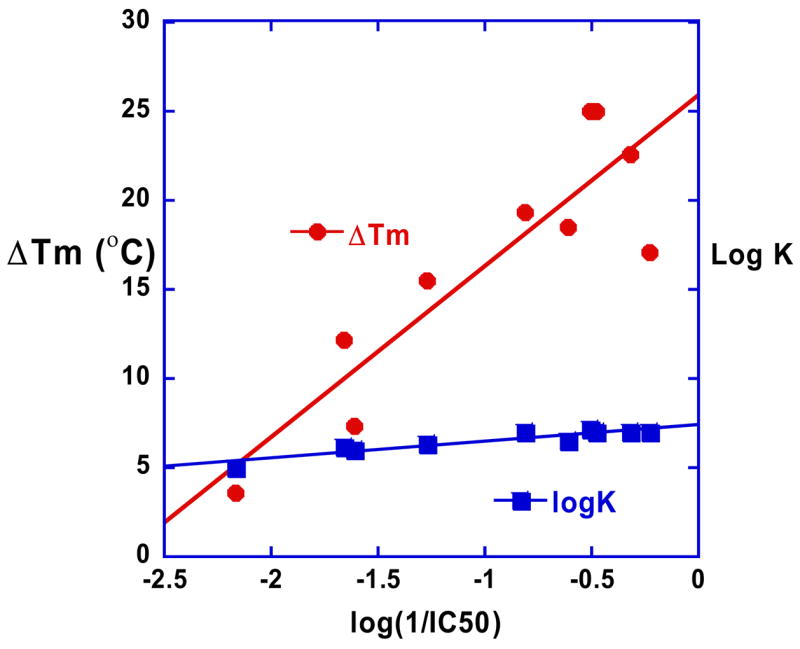
Plot of log (1/IC50) vs ΔTm and log K for the compounds of Figure 1. The correlation between antitrypanosomal activity and binding to AT DNA for this set of compounds can be seen.
The correlation of DNA binding results and biological activity in Figure 8 supports the importance of a DNA interaction step in the mechanism of biological activity of the close analogs of DB75 in Figure 1. It should be emphasized, however, that such linear behavior is expected only for a very closely related series of compounds that have similar uptake, metabolism, and effects at the bioreceptor such that affinity for the receptor is the primary variable among the compounds. Modification of one of the features, such as effects induced by the compound at the bioreceptor will put a discontinuity in any linear plot of affinity versus biological activity and that suggests why DB244 and other alkyl amidines display different IC50 correlation than DB75.
The fluorescence microscopy results shown in Figure 3 strongly support mitochondrial kinetoplast DNA as a key biological target of diamidines. The destruction of the kinetoplast is complete in 24 hours and is correlated with cell death. Results with DNA ladders with A tract models for kinetoplast DNA (Figure 6) provide a mechanism for the DNA destruction. The diamidines induces a topological change that results in increased global bending of the A tract. Such a change in kinetoplast minicircle DNA could topologically disturb the many replication steps involved in removing, copying and reinserting minicircles into the catenated kinetoplast DNA network. This mechanism can explain the correlation of DNA binding with biological activity and the time course of parasite cell death after treatment with diamidines.
On a very exciting final note, it is worth emphasizing that design efforts to modify the structure and sequence recognition of DNA by diamidines has yielded successful results. Diamidines that recognize DNA sequences that contain GC base pairs, including majority GC sequences, have now been described [76–80]. In addition we realize that the curved shape of diamidines is one option for binding but is not a requirement. In fact, essentially linear compounds that incorporate water directly into their DNA recognition have now been synthesized and found to have very strong DNA binding potential [80 – 82]. Clearly the options for therapeutic applications of diamidines are considerably broadened by these new findings. The low toxicity and cell uptake by these compounds makes them very attractive platforms for development against other parasitic diseases and cancer.
6. Methods
6.1. Fluorescence microscopy
S427 trypanosomes (Trypanosoma brucei brucei) were cultured in Complete Baltz Modified Essential Medium (CBMEM) as previously described [39]. DB351 was prepared as 1 mM stock solution in sterile distilled water. Trypanosomes (105 trypanosomes/ml) were incubated with 500 nM of each compound for time periods between 1 hour and 24 hours. At each time point, trypanosomes were washed by centrifugation (16100 g, 20 sec) in a Beckman 5415R microcentrifuge (Beckman Coulter) to remove drugs. Trypanosomes were resuspended in freshly isolated blood from a Swiss Webster mouse. A drop of blood was placed on a microscope slide and smeared with the side of a coverslip. When slides were viewed, a drop of glycerol was added to the slide and covered with a 22 mm square coverslip. Slides were examined using a Nikon Microphot FXA (Garden City, NJ) with a 60 DM × 1.4 NA objective lens, a mercury lamp, and an Optronics DEI 750 CCD camera (Goleta, CA). The microscope was equipped with a Nikon UV2A cube that limits excitation wavelengths to 330–380 nm and emission wavelengths to 420 nm. Phase contrast and fluorescence images were obtained for each time point.
6.2. Biosensor - surface plasmon resonance (SPR)
Biosensor-SPR experiments were conducted with a BIAcore T100 instrument as previously described [49, 50]. 5′-Biotin labeled DNA hairpins (Figure 1) were immobilized on streptavidin-linked sensor chips via non-covalent capture. The tight affinity between biotin and streptavidin yields a highly stable surface over time, allowing for regeneration with relatively harsh conditions. The immobilization wizard in BIAcore T100 supports the immobilization of ligand in any combination of the four flow cells in one run. Immobilization in each flow cell is performed independently in a separate cycle, so that different DNAs can be used in the different flow cells. After washing the chip with 1M NaCl/50 mMNaOH and buffer to remove unlinked streptavidin, a 25 nM DNA solution of a selected sequence was injected over each streptavidin derivatized flow cell until the desired amount of DNA was immobilized. Three flow cells contained DNA and one flow cell was left blank as a reference.
For binding studies the compounds were diluted to different concentrations in degassed and filtered Tris buffer (0.01M Tris HCl and 0.001M EDTA, 0.10 M NaCl adjusted to pH 7.0, and 0.005% surfactant P-20) and the diluted samples were injected over the DNA surface for a selected time. A 10 mM glycine solution at pH 2.0 was used for flow cell surface regeneration. Steady-state binding studies were carried out by averaging the resonance unit values (RU) in the plateau region of the sensorgrams (Figure 4) over a selected time region at different compound concentrations and converting them to r (moles of bound compound per mole of DNA hairpin), r = RUeq/RUmax, where RUeq is the averaged RU at steady state for each concentration and RUmax is the maximum RU value for binding one compound per binding site. The number of binding sites and the equilibrium constant were obtained from fitting plots of RU versus Cfree. Binding results from the SPR experiments were fit with either a single site model (K2 = 0) or with a two site model:
| (1) |
where r represents the moles of bound compound per mole of DNA hairpin duplex, K1 and K2 are macroscopic binding constants, and Cfree is the free compound concentration in equilibrium with the complex.
6.3. Ligation ladders and Gel electrophoresis
Lypholized DNA oligomers were purchased from Integrated DNA Technologies, Inc. (Coraville, IA) with standard desalting and single HPLC purification. These oligomers were ligated using a procedure similar to that described by Maher and coworkers [83–85]. Briefly, complementary strands (100 μM) were annealed in 1X ligation buffer (New England BioLabs, Ipswich, MA) containing 50 mM Tris-HCl, 10 mM MgCl2, 10 mM dithiothreitol, 1 mM ATP, and 25 μg/mL bovine serum albumin. Annealed oligos (3 μM) were incubated for 30 min at 37°C in a reaction (100 μL) containing 20 U of T4 polynucleotide kinase (New England Biolabs) in 1X ligation buffer. These oligomers (0.5 μM) were then assembled into ligation reactions (80 μL) with 400 U of T4 DNA ligase (New England BioLabs) in 1X ligation buffer. Room temperature ligation time was 30 minutes followed by an inactivation time of 10 min at 65°C.
Ligation ladders were separated on 8% native polyacrylamide gels (1.5 mm, 20 cm) prepared from a 40% acrylamide solution (19:1, bisacrylamide:acrylamide) (Bio-Rad, Hercules, CA). Electrophoresis was at 200 V (10 V/cm) at room temperature for 210 min in a BioRad Protean IIxi gel box in 1X TBE buffer (0.89 M Tris, 0.89 M boric acid, 20 mM EDTA, pH 8.3) using a Bio-Rad PowerPac 3000. Samples of ligation ladder (30 μL) and 20-bp molecular marker (Bayou Biolabs, Harahan, LA) were incubated with test compounds (usually 1μL) at a concentration in five-fold molar excess of the free drug concentration in equilibrium with the compound-DNA complex. The 1X TBE running buffer in the lower buffer chamber and the gel itself also contained an excess of the same compound in the ladder samples. In lieu of radiolabeling, gels were stained with SYBR® Gold Nucleic Acid Gel Stain (Molecular Probes, Eugene, OR) at the concentration recommended by the manufacturer for 1 hour. Stained gels were imaged using a UVP BioImaging Systems Epi Chem II Darkroom (UVP, Upland, CA) equipped with a SYBR Gold filter. Data for migration analysis and molecular weight assignment was obtained using ImageQuant TL (Amersham Biosciences, Piscataway, NJ).
Acknowledgments
We wish to thank our outstanding coworkers who have done many of the experiments described in this paper and whose discussions have been critical to our understanding of diamidines and DNA. We also thank an excellent group of scientific collaborators whose contributions over many years have helped to develop the fields of diamidine therapeutics and diamidine-DNA interactions. We would particularly like to emphasize the critical contributions of Drs. Richard R. Tidwell, Stephen Neidle and Christian Bailly. The work in our laboratories has been supported by National Institutes of Health grants AI064200 and GM61587, by the Gates Foundation, and by equipment purchase support from the Georgia Research Alliance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Watkins BM. Drugs for the control of parasitic diseases: current status and development. Trends Parasitol. 2003;19:477–478. doi: 10.1016/j.pt.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization (WHO) Research Results: Nos. 1, 6. and 7; African trypanosomiasis; Fact Sheet Number 259. World Health Organization Publications; Geneva: 2001. [Google Scholar]
- 3.Hutchinson OC, Picozzi K, Jones NG, Mott H, Sharma R, Welburn SC, Carrington M. Variant surface glycoprotein gene repertoires in Trypanosoma brucei have diverged to become strain-specific. BMC Genomics. 2007;8:234–243. doi: 10.1186/1471-2164-8-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vincendeau P, Bouteille B. Immunology and immunopathology of African trypanosomiasis. An Acad Bras Cienc. 2006;78:645–665. doi: 10.1590/s0001-37652006000400004. [DOI] [PubMed] [Google Scholar]
- 5.Barrett MP, Boykin DW, Brun R, Tidwell RR. Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. Br J Pharmacol. 2007;152:1155–1171. doi: 10.1038/sj.bjp.0707354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrett MP, Burchmore RJ, Stich A, Lazzari JO, Frasch AC, Cazzulo JJ, Krishna S. The trypanosomiases. Lancet. 2003;362:1469–1480. doi: 10.1016/S0140-6736(03)14694-6. [DOI] [PubMed] [Google Scholar]
- 7.Bouteille B, Oukem O, Bisser S, Dumas M. Treatment perspectives for human African trypanosomiasis. Fundam Clin Pharmacol. 2003;17:171–181. doi: 10.1046/j.1472-8206.2003.00167.x. [DOI] [PubMed] [Google Scholar]
- 8.Bray PG, Barrett MP, Ward SA, de Koning HP. Pentamidine uptake and resistance in pathogenic protozoa: past, present and future. Trends Parasitol. 2003;19:232–239. doi: 10.1016/s1471-4922(03)00069-2. [DOI] [PubMed] [Google Scholar]
- 9.Bakshi RP, Shapiro TA. DNA topoisomerases as targets for antiprotozoal therapy. Mini Rev Med Chem. 2003;3:597–608. doi: 10.2174/1389557033487863. [DOI] [PubMed] [Google Scholar]
- 10.Tidwell RR, Boykin DW. In: DNA and RNA Binders: From Small Molecules to Drugs. Demeunynck M, Bailly C, Wilson WD, editors. Vol. 2. WILEY-VCH; Weinheim: 2003. pp. 414–460. [Google Scholar]
- 11.Delespaux V, de Koning HP. Drug and drug resistance in African trypanosomiasis. Drug Resist Updat. 2007 Feb-Apr;10(1–2):30–50. doi: 10.1016/j.drup.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Glikson M, Dresner-Feigin R, Pollack A, Wolf D, Galun E, Kaspa RT. Pentamidine-induced cardiotoxicity. Isr J Med Sci. 1990;26:588–589. [PubMed] [Google Scholar]
- 13.Coyle P, Carr AD, Depczynski BB, Chisholm DJ. Diabetes mellitus associated with pentamidine use in HIV-infected patients. Med J Aust. 1996;165:587–588. doi: 10.5694/j.1326-5377.1996.tb138654.x. [DOI] [PubMed] [Google Scholar]
- 14.Kuryshev YA, Ficker E, Wang L, Hawryluk P, Dennis AT, Wible BA, Brown AM, Kang J, Chen XL, Sawamura K, Reynolds W, Rampe D. Pentamidine-induced long QT syndrome and block of hERG trafficking. J Pharm Exp Therap. 2005;312:316–323. doi: 10.1124/jpet.104.073692. [DOI] [PubMed] [Google Scholar]
- 15.Milligan KS, Phillips DL. Perioral numbness associated with intravenous pentamidine administration. Ann Pharmacother. 2007;41:153–156. doi: 10.1345/aph.1H272. [DOI] [PubMed] [Google Scholar]
- 16.Berger BJ, Lombardy RJ, Marbury GD, Bell CA, Dyksytra CC, Hall JE, Tidwell RR. Metaboltic N-hydroxylation of pentamidine in vivo. Antimicrob Agents Chemother. 1990;34:1678–1684. doi: 10.1128/aac.34.9.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger BJ, Naiman NA, Hall JE, Peggins J, Brewer TG, Tidwell RR. Primary and secondary metabolism of pentamidine by rats. Antimicrob Agents Chemother. 1992;36:1825–1831. doi: 10.1128/aac.36.9.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson WD, Tanious FA, Buczak H, Venkatramanan MK, Das BP, Boykin DW. In: Molecular Basis of Specificity in Nucleic Acid-Drug Interactions. Pullman B, Jortner J, editors. Kluwer Academic Publishers; 1990. pp. 331–353. [Google Scholar]
- 19.Waring MJ, Bailly C. The influence of the exocyclic amino group characteristic of GC base pairs on molecular recognition of specific nucleotide sequences in DNA by berenil and DAPI. J Mol Recognit. 1997;10:121–127. doi: 10.1002/(SICI)1099-1352(199705/06)10:3<121::AID-JMR356>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 20.Neidle S. DNA minor-groove recognition by small molecules. Nat Prod Rep. 2001;18:291–309. doi: 10.1039/a705982e. [DOI] [PubMed] [Google Scholar]
- 21.Reddy BS, Sondhi SM, Lown JW. Synthetic DNA minor groove-binding drugs. Pharmacol Ther. 1999;84:1–111. doi: 10.1016/s0163-7258(99)00021-2. [DOI] [PubMed] [Google Scholar]
- 22.Geierstanger BH, Wemmer DE. Complexes of the minor groove of DNA. Annu Rev Biophys Biomol Struct. 1995;24:463–493. doi: 10.1146/annurev.bb.24.060195.002335. [DOI] [PubMed] [Google Scholar]
- 23.Turner PR, Denny WA. The genome as a drug target: sequence specific minor groove binding ligands. Curr Drug Targets. 2000;1:1–14. doi: 10.2174/1389450003349407. [DOI] [PubMed] [Google Scholar]
- 24.Cory M, Tidwell RR, Fairley TA. Structure and DNA binding activity of analogues of 1,5-bis(4- amidinophenoxy)pentane (pentamidine) J Med Chem. 1992;35:431–438. doi: 10.1021/jm00081a003. [DOI] [PubMed] [Google Scholar]
- 25.Edwards KJ, Jenkins TC, Neidle S. Crystal structure of a pentamidine-oligonucleotide complex: implications for DNA-binding properties. Biochemistry. 1992;31:7104–7109. doi: 10.1021/bi00146a011. [DOI] [PubMed] [Google Scholar]
- 26.Hamelberg D, Williams LD, Wilson WD. Influence of the dynamic positions of cations on the structure of the DNA minor groove: sequence-dependent effects. J Am Chem Soc. 2001;123:7745–7755. doi: 10.1021/ja010341s. [DOI] [PubMed] [Google Scholar]
- 27.Bloomfield VA, Crothers DM, Tinoco I., JR . Nucleic Acids: Structures, Properties, and Functions. University Science Books; Sausalito, CA: 2000. [Google Scholar]
- 28.Brown DG, Sanderson MR, Skelly JV, Jenkins TC, Brown T, Garman E, Stuart DI, Neidle S. Crystal structure of a berenil-dodecanucleotide complex: the role of water in sequence-specific ligand binding. EMBO J. 1990;9:1329–1334. doi: 10.1002/j.1460-2075.1990.tb08242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pilch DS, Kirolos MA, Liu X, Plum GE, Breslauer KJ. Berenil [1,3-bis(4′-amidinophenyl)triazene] binding to DNA duplexes and to a RNA duplex: evidence for both intercalative and minor groove binding properties. Biochemistry. 1995;34:9962–9976. doi: 10.1021/bi00031a019. [DOI] [PubMed] [Google Scholar]
- 30.Baraldi PG, Bovero A, Fruttarolo F, Preti D, Tabrizi MA, Pavani MG, Romagnoli R. DNA minor groove binders as potential antitumor and antimicrobial agents. Med Res Rev. 2004;24:475–528. doi: 10.1002/med.20000. [DOI] [PubMed] [Google Scholar]
- 31.Ismail MA, Brun R, Easterbrook JD, Tanious FA, Wilson WD, Boykin DW. Synthesis and antiprotozoal activity of aza-analogues of furamidine. J Med Chem. 2003;46:4761–4769. doi: 10.1021/jm0302602. [DOI] [PubMed] [Google Scholar]
- 32.Shapiro TA, Englund PT. The structure and replication of kinetoplast DNA. Annu Rev Microbiol. 1995;49:117–143. doi: 10.1146/annurev.mi.49.100195.001001. [DOI] [PubMed] [Google Scholar]
- 33.Klingbeil MM, Drew ME, Liu Y, Morris JC, Motyka SA, Saxowsky TT, Wang Z, Englund PT. Unlocking the secrets of trypanosome kinetoplast DNA network replication. Protist. 2001;152:255–262. doi: 10.1078/1434-4610-00066. [DOI] [PubMed] [Google Scholar]
- 34.Simpson L. Kinetoplast DNA in trypanosomid flagellates. Int Rev Cytol. 1986;99:119–179. doi: 10.1016/s0074-7696(08)61426-6. [DOI] [PubMed] [Google Scholar]
- 35.Estevez AM, Simpson L. Uridine insertion/deletion RNA editing in trypanosome mitochondria--a review. Gene. 1999;240:247–260. doi: 10.1016/s0378-1119(99)00437-0. [DOI] [PubMed] [Google Scholar]
- 36.Madison-Antenucci S, Grams J, Hajduk SL. Editing machines: the complexities of trypanosome RNA editing. Cell. 2002;108:435–438. doi: 10.1016/s0092-8674(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 37.Marini JC, Levene SD, Crothers DM, Englund PT. A bent helix in kinetoplast DNA. Cold Spring Harb Symp Quant Biol. 1983;47(Pt 1):279–283. doi: 10.1101/sqb.1983.047.01.033. [DOI] [PubMed] [Google Scholar]
- 38.Marini JC, Effron PN, Goodman TC, Singleton CK, Wells RD, Wartell RM, Englund PT. Physical characterization of a kinetoplast DNA fragment with unusual properties. J Biol Chem. 1984;259:8974–8979. [PubMed] [Google Scholar]
- 39.Mathis AM, Holman JL, Sturk LM, Ismail MA, Boykin DW, Tidwell RR, Hall JE. Accumulation and intracellular distribution of antitrypanosomal diamidine compounds DB75 and DB820 in African trypanosomes. Antimicrob Agents Chemother. 2006;50:2185–2191. doi: 10.1128/AAC.00192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matovu E, Stewart ML, Geiser F, Brun R, Maser P, Wallace LJ, Burchmore RJ, Enyaru JC, Barrett MP, Kaminsky R, Seebeck T, de Koning HP. Mechanisms of arsenical and diamidine uptake and resistance in Trypanosoma brucei. Eukaryot Cell. 2003;2:1003–1008. doi: 10.1128/EC.2.5.1003-1008.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carter NS, Berger BJ, Fairlamb AH. Uptake of diamidine drugs by the P2 nucleoside transporter in melarsen-sensitive and -resistant Trypanosoma brucei brucei. J Biol Chem. 1995;270:28153–28157. doi: 10.1074/jbc.270.47.28153. [DOI] [PubMed] [Google Scholar]
- 42.de Koning HP. Uptake of Pentamidine in Trypanosoma brucie brucei is Mediated by Three Distinct Transporters: Implication for Cross-Resistance with Arsenicals. Mol Pharmacol. 2001;59:586–592. doi: 10.1124/mol.59.3.586. [DOI] [PubMed] [Google Scholar]
- 43.Mathis AM, Bridges AS, Ismail MA, Kumar A, Francesconi I, Anbazhagan M, Hu Q, Tanious FA, Wenzler T, Saulter J, Wilson WD, Brun R, Boykin DW, Tidwell RR, Hall JE. Diphenyl furans and aza analogs: effects of structural modification on in vitro activity, DNA binding, and accumulation and distribution in trypanosomes. Antimicrob Agents Chemother. 2007;51:2801–2810. doi: 10.1128/AAC.00005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson WD, Nguyen B, Tanious FA, Mathis A, Hall JE, Stephens CE, Boykin DW. Dications that target the DNA minor groove: compound design and preparation, DNA interactions, cellular distribution and biological activity. Curr Med Chem Anti-Canc Agents. 2005;5:389–408. doi: 10.2174/1568011054222319. [DOI] [PubMed] [Google Scholar]
- 45.Mukherjee A, Padmanabhan PK, Sahani MH, Barrett MP, Madhubala R. Roles for mitochondria in pentamidine susceptibility and resistance in Leishmania donovani. Mol Biochem Parasitol. 2006;145:1–10. doi: 10.1016/j.molbiopara.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 46.Das BP, Boykin DW. Synthesis and antiprotozoal activity of 2,5-bis(4-guanylphenyl)furans. J Med Chem. 1977;20:531–536. doi: 10.1021/jm00214a014. [DOI] [PubMed] [Google Scholar]
- 47.Boykin DW, Kumar A, Bender BC, Hall JE, Tidwell RR. Anti-Pneumocystis activity of bis-amidoximes and bis-O-Alkylamidoximes. Bioorganic Med Chem Letters. 1996;6:3017–3020. [Google Scholar]
- 48.Wilson WD, Tanious FA, Ding D, Kumar A, Boykin DW, Colson P, Houssier C, Bailly C. Nucleic acid interactions of unfused aromatic cations: Evaluation of proposed minor-groove, major-groove, and intercalation binding modes. J Am Chem Soc. 1998;120:10310–10321. [Google Scholar]
- 49.Nguyen B, Tanious FA, Wilson WD. Biosensor-surface plasmon resonance: quantitative analysis of small molecule-nucleic acid interactions. Methods. 2007;42:150–161. doi: 10.1016/j.ymeth.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 50.Tanious FT, Nguyen B, Wilson WD. Biosensor-surface plasmon resonance methods for quantitative analysis of biomolecular interactions. In: Correia J, Detrich HW III, editors. Methods in cell biology. Chapter 3. Vol. 84. Elsevier Inc; 2008. pp. 53–77. [DOI] [PubMed] [Google Scholar]
- 51.Mazur S, Tanious FA, Ding D, Kumar A, Boykin DW, Simpson IJ, Neidle S, Wilson WD. A thermodynamic and structural analysis of DNA minor-groove complex formation. J Mol Biol. 2000;300:321–337. doi: 10.1006/jmbi.2000.3869. [DOI] [PubMed] [Google Scholar]
- 52.Liu Y, Kumar A, Boykin DW, Wilson WD. Sequence And Length Dependent Thermodynamic Differences In Heterocyclic Diamidine Interactions At AT Base Pairs In The DNA Minor Groove. Biophys Chem. 2007;131:1–14. doi: 10.1016/j.bpc.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laughton CA, Tanious F, Nunn CM, Boykin DW, Wilson WD, Neidle S. A crystallographic and spectroscopic study of the complex between d(CGCGAATTCGCG)2 and 2,5-bis(4-guanylphenyl)furan, an analogue of berenil. Structural origins of enhanced DNA-binding affinity. Biochemistry. 1996;35:5655–5661. doi: 10.1021/bi952162r. [DOI] [PubMed] [Google Scholar]
- 54.Degtyareva NN, Wallace BD, Bryant AR, Loo KM, Petty JT. Hydration changes accompanying the binding of minor groove ligands with DNA. Biophys J. 2007;92:959–965. doi: 10.1529/biophysj.106.097451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ansede JH, Voyksner RD, Ismail MA, Boykin DW, Tidwell RR, Hall JE. In vitro metabolism of an orally active O-methyl amidoxime prodrug for the treatment of CNS trypanosomiasis. Xenobiotica. 2005;35:211–226. doi: 10.1080/00498250500087671. [DOI] [PubMed] [Google Scholar]
- 56.Wilson WD, Tanious FA, Fernandez-Saiz M, Rigl CT. Evaluation of drug-nucleic acid interactions by thermal melting curves. Methods Mol Biol. 1997;90:219–240. doi: 10.1385/0-89603-447-X:219. [DOI] [PubMed] [Google Scholar]
- 57.Nguyen B, Tardy C, Bailly C, Colson P, Houssier C, Kumar A, Boykin DW, Wilson WD. Influence of compound structure on affinity, sequence selectivity, and mode of binding to DNA for unfused aromatic dications related to furamidine. Biopolymers. 2002;63:281–297. doi: 10.1002/bip.10073. [DOI] [PubMed] [Google Scholar]
- 58.Oszczapowicz J. In: The Chemistry of Amidines and Imidates. Patai S, editor. Vol. 2. JohnWiley & Sons; Chichester: 1991. pp. 623–688. [Google Scholar]
- 59.Lamb ID, White AC. Some amidines and amide oximes with trypanocidal activity. J Chem Soc. 1939:1253–1257. [Google Scholar]
- 60.Clement B, Immel M, Terlinden R, Wingen FJ. Reduction of amidoxime derivatives to pentamidine in vivo. Arch Pharm. 1992;325:61–62. doi: 10.1002/ardp.19923250114. [DOI] [PubMed] [Google Scholar]
- 61.Clement B. Reduction of N-Hydroxylated Compounds: Amidoximes (N-Hydroxyamidines) as prodrugs of amidines drug metabolism. Rev. 2002;34:565– 579. doi: 10.1081/dmr-120005643. [DOI] [PubMed] [Google Scholar]
- 62.Hall JE, Kerrigan JE, Ramachandran K, Bender BC, Stanko JP, Jones SK, Patrick DA, Tidwell RR. Anti-Pneumocystis activities of aromatic diamidoxime prodrugs. Antimicro Agents Chemother. 1998;42:666–674. doi: 10.1128/aac.42.3.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Soeiro MNC, de Souza EM, Stephens CE, Boykin DW. Aromatic diamidines as antiparasitic agents. Expert Opin Invest Drugs. 2005;14:957–972. doi: 10.1517/13543784.14.8.957. [DOI] [PubMed] [Google Scholar]
- 64.Werbovetz KA. Diamidines as antitrypanosomal, antileishmanial and antimalarial agents. Curr Opin Invest Drugs. 2006;7:147–157. [PubMed] [Google Scholar]
- 65.Ansede JH, Anbazhagan M, Brun R, Easterbrook J, Hall JE, Boykin DW. O-Alkoxyamidine prodrugs of furamidine: in vitro transport and microsomal metabolism as indicators of in vivo efficacy in a mouse model of Trypanosoma brucei rhodesiense infection. J Med Chem. 2004;47:4335–4338. doi: 10.1021/jm030604o. [DOI] [PubMed] [Google Scholar]
- 66.Zhou L, Voyksner RD, Thakker DR, Stephens CE, Anbazhagan M, Boykin DW, Hall JE, Tidwell RR. Metabolites of an orally active antimicrobial prodrug, 2,5-bis(4-amidinophenyl)furan-bis-O-methylamidoxime, identified by liquid chromatography/tandem mass spectrometry. J Mass Spec. 2004;39:351–360. doi: 10.1002/jms.591. [DOI] [PubMed] [Google Scholar]
- 67.Saulter JY, Kurian JR, Trepanier LA, Tidwell RR, Bridges AS, Boykin DW, Stephens CE, Anbazhagan M, Hall JE. Unusual dehydroxylation of antimicrobial amidoxime prodrugs by cytochrome b5 and NADH cytochrome b5 reductase. Drug Metab Dispos. 2005;33:1886–1893. doi: 10.1124/dmd.105.005017. [DOI] [PubMed] [Google Scholar]
- 68.Patrick DA, Hall JE, Bender BC, McCurdy DR, Wilson WD, Tanious FA, Saha S, Tidwell RR. Synthesis and anti-Pneumocystis carinii pneumonia activity of novel dicaionic dibenzothiophenes and orally active prodrugs. Eur J Med Chem. 1999;34:575–583. doi: 10.1016/s0223-5234(00)80027-6. [DOI] [PubMed] [Google Scholar]
- 69.Wang S, Hall JE, Tanious FA, Wilson WD, Patrick DA, McCurdy DR, Bender BC, Tidwell RR. Dicaionic dibenzofuran derivatives as anti-Pneumocystis carinii pneumonia agents: synthesis, DNA binding affinity, and anti-P. carinii activity in an immunosuppressed rat model. Eur J Med Chem. 1999;34:215–224. [Google Scholar]
- 70.Ismail MA, Arafa RK, Brun R, Wenzler T, Miao Y, Wilson WD, Generaux C, Bridges A, Hall JE, Boykin DW. Synthesis, DNA affinity, and antiprotozoal activity of linear dications: Terphenyl diamidines and analogues. J Med Chem. 2006;49:5324–5332. doi: 10.1021/jm060470p. [DOI] [PubMed] [Google Scholar]
- 71.Clement B, Burenheide A, Rieckert W, Schwarz J. Diacetyldiamidoximeester of pentamidine, a prodrug for treatment of protozoal diseases: synthesis. in vitro and in vivo biotransformation. Chem Med Chem. 2006;1:1260–1267. doi: 10.1002/cmdc.200600079. [DOI] [PubMed] [Google Scholar]
- 72.Ouattara M, Wein S, Calas M, Hoang YV, Vial H, Esacle R. Synthesis and antimalarial activity of new 1,12-bis-(N, N′-acetamidinyl)dodecane derivatives. Bioorg Med Chem Lett. 2007;17:593–596. doi: 10.1016/j.bmcl.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 73.Rahmathullah SM, Hall JE, Bender BC, McCurdy DR, Tidwell RR, Boykin DW. Prodrugs for amidines: synthesis and anti-Pneumocystis carinii activity of carbamates of 2,5-bis(4-amidinophenyl)furan. J Med Chem. 1999;42:3994–4000. doi: 10.1021/jm990237+. [DOI] [PubMed] [Google Scholar]
- 74.Boykin DW, Kumar A, Xiao G, Wilson WD, Bender BC, McCurdy DR, Hall JE, Tidwell RR. 2,5-bis[4-(N-alkylamidino)phenyl]furans as anti-Pneumocystis carinii agents. J Med Chem. 1998;41:124–129. doi: 10.1021/jm970570i. [DOI] [PubMed] [Google Scholar]
- 75.Rahmathullah SM, Tidwell RR, Jones SK, Hall JE, Boykin DW. Carbamate prodrugs of N-alkylfuramidines. Eur J Med Chem. 2008;43 doi: 10.1016/j.ejmech.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 76.Wang L, Bailly C, Kumar A, Ding D, Bajic M, Boykin DW, Wilson WD. Specific molecular recognition of mixed nucleic acid sequences: an aromatic dication that binds in the DNA minor groove as a dimer. Proc Natl Acad Sci USA. 2000;97:12–16. doi: 10.1073/pnas.97.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bailly C, Tardy C, Wang L, Armitage B, Hopkins K, Kumar A, Schuster GB, Boykin DW, Wilson WD. Recognition of ATGA Sequences by the Unfused Aromatic Dication DB293 Forming Stacked Dimers in the DNA Minor Groove. Biochemistry. 2001;40:9770–9779. doi: 10.1021/bi0108453. [DOI] [PubMed] [Google Scholar]
- 78.Tanious F, Wilson WD, Wang L, Kumar A, Boykin DW, Marty C, Baldeyrou B, Bailly C. Cooperative Dimerization of a Heterocyclic Diamidine Determines Sequence-Specific DNA Recognition. Biochemistry. 2003;46:13576–13586. doi: 10.1021/bi034852y. [DOI] [PubMed] [Google Scholar]
- 79.Wang L, Carrasco C, Kumar A, Stephens CE, Bailly C, Boykin DW, Wilson WD. Evaluation of the Influence of Compound Structure on Stacked-Dimer Formation in the DNA Minor Groove. Biochemistry. 2001;40:2511–2521. doi: 10.1021/bi002301r. [DOI] [PubMed] [Google Scholar]
- 80.Munde M, Lee M, Neidle S, Arafa R, Boykin DW, Liu Y, Bailly C, Wilson WD. Induced Fit Conformational Changes of a “Reversed Amidine” Heterocycle: Optimized Interactions in a DNA Minor Groove Complex. J Am Chem Soc. 2007;129:5688–5698. doi: 10.1021/ja069003n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nguyen B, Lee MPH, Hamelberg D, Joubert A, Bailly C, Brun R, Neidle S, Wilson WD. Strong Binding in the DNA Minor Groove by an Aromatic Diamidine with a Shape That Does Not Match the Curvature of the Groove. J Am Chem Soc. 2002;124:13680–13681. doi: 10.1021/ja027953c. [DOI] [PubMed] [Google Scholar]
- 82.Miao Y, Lee MP, Parkinson GN, Batista-Parra A, Ismail MA, Neidle S, Boykin DW, Wilson WD. Out of-shape DNA minor groove binders: Induced fit interactions of heterocyclic dications with the DNA minor groove. Biochemistry. 2005;44:14701–14708. doi: 10.1021/bi051791q. [DOI] [PubMed] [Google Scholar]
- 83.Hardwidge PR, Den RB, Ross ED, Maher LJ., III Relating Independent Measures of DNA Curvature: Electrophoretic Anomaly and Cyclization Efficiency. Journal of Biomolecular Structure & Dynamics. 2000;18:219–230. doi: 10.1080/07391102.2000.10506660. [DOI] [PubMed] [Google Scholar]
- 84.Hardwidge PR, Maher LJ. Experimental evaluation of the Liu-Beveridge dinucleotide step model of DNA structure. Nucleic acids research. 2001;29:2619–2625. doi: 10.1093/nar/29.12.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ross ED, Den RB, Hardwidge PR, Maher LJ., III Improved quantitation of DNA curvature using ligation ladders. Nucleic acids research. 1999;27:4135–4142. doi: 10.1093/nar/27.21.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]