

Abstract
Vaccinia DNA topoisomerase (vTopo) is a prototypic eukaryotic type I topoisomerase that shows high specificity for nucleophilic substitution at a single phosphodiester linkage in the pentapyrimidine recognition sequence 5′-(C/T)+5C+4C+3T+2T+1p↓N-1. This reaction involves reversible transesterification where the active site tyrosine of the enzyme and a 5′ hydroxyl nucleophile of DNA compete for attack at the phosphoryl group. The finite lifetime of the covalent phosphotyrosine adduct allows the enzyme to relax multiple supercoils by rotation of the 5′ OH strand before the DNA backbone is religated. To dissect the nature of the unique sequence specificity, subtle modifications to the major groove of the GGGAA-5′ sequence of the non-scissile strand were introduced and their effects on each step of the catalytic cycle were measured. Although these modifications had no effect on noncovalent DNA binding (KD), or the rate of reversible DNA cleavage (kcl), significant decreases in the cleavage equilibrium (Kcl = kcl/kr) were observed arising from increased rates of 5′ hydroxyl attack (kr) at the phosphotyrosine linkage. These data and other findings support a model where major groove interactions are used to position the phosphotyrosine linkage relative to the mobile 5′ hydroxyl nucleophile. In the absence of native sequence interactions, the phosphotyrosine has a higher probability of encountering the 5′ hydroxyl nucleophile leading to an enhanced rate of ligation and a diminished equilibrium constant for cleavage. By this unusual specificity mechanism the enzyme prevents formation of stable covalent adducts at nonconsensus sites in genomic DNA.
Vaccinia virus topoisomerase I (vTopo) and other eukaryotic type IB enzymes maintain DNA topology by relaxing superhelical tension in DNA. The Topo reaction is comprised of multiple steps as shown in Figure 1: i) non-covalent binding of the enzyme to duplex DNA with a dissociation constant KD, ii) cleavage of a single DNA strand by an active site tyrosine nucleophile leading to formation of a covalent 3′-phosphotyrosyl linkage and a free 5′ -hydroxyl (kcl), iii) release of superhelical tension by a strand rotation mechanism (ksup), and iv) religation of the DNA break by attack of the 5′-hydroxyl nucleophile (kr). Vaccinia topoisomerase is unique among these enzymes in that it exhibits high specificity for cleavage at the consensus pentapyrimidine sequence 5′-(C/T)CCTT↓ (1) (2) (3) (4) (5) (6) (7) (8). However, the remarkable site-specificity of vTopo is not understood at the molecular level because the holoenzyme has proven refractory to crystallization, necessitating the application of other approaches for elucidation of this question.
Fig. 1.

Topoisomerase reaction cycle. A) The three steps in the vTopo reaction involve DNA binding, reversible DNA backbone cleavage described by the rate constants kcl and kr, and for supercoiled DNA substrates, supercoil relaxation while the enzyme is covalently attached to the phosphodiester backbone (ksup). Relaxation occurs by a “free rotation” mechanism in which the noncovalently bound end swivels around the helix axis. The number of supercoils that are removed after a single cleavage event is dependent on the ratio ksup/kr: if kr is rapid there is no opportunity for DNA rotation before the strand is resealed. B) Free energy reaction coordinate profile depicting the thermodynamic and kinetic barriers of the reaction. The rate constants and equilibrium constants refer to the free energy barriers defined by K = exp (-ΔG/RT) and k = (k T/h) exp(-ΔG‡/RT).
One way to define specificity for vTopo is to compare the efficiency of forming the covalent adduct at the consensus site as compared to another site that differs from the consensus sequence. Sequence specific interactions within the consensus sequence are used to kinetically or thermodynamically facilitate the formation of the enzyme-DNA covalent adduct, whereas alternative sequences cannot realize the same benefit, resulting in specificity. This is a useful definition because the lifetime (or concentration) of the covalent phosphotyrosine adduct largely determines how efficiently the enzyme removes supercoils from DNA using a strand rotation mechanism as defined in the legend to Figure 1A (9, 10).
Using this definition, specificity can be achieved at one of three stages of the reaction (Fig. 1B). If strong sequence-specific enzyme-DNA interactions are only used to discriminate at the noncovalent binding step (ES, Fig. 1B), then the relative amount of covalent adduct at the specific sequence would be increased relative to the nonspecific sequence because the specific site has higher occupancy. This follows because the relative amount of covalent adduct is determined by the ratio Kcl/KD. (This analysis makes the limiting assumption that Kcl is the same for the specific and nonspecific substrates.) Alternatively, enzyme interactions with the consensus site could be weak in the noncovalent complex, and then become strong at the transition state for cleavage (ES‡, Fig. 1B). In this case, the activation barrier for cleavage is lowered in the presence of the consensus interactions, thereby increasing the rate of formation of the covalent adduct (E-S, Fig. 1). In this regard, many of the direct enzyme catalytic interactions with the phosphoryl group have been found to act solely by transition state stabilization (11). However, transition state stabilization by itself is unsatisfactory for specificity because it would also give rise to a decrease in the lifetime of the covalent intermediate at the specific site. This follows because stabilization of ES‡ has an equal effect on enhancing the rates of both cleavage and ligation: as the religation rate increases, fewer supercoils are removed each time the covalent complex is formed, resulting in decreased specificity by our definition. Finally, if sequence specific interactions are used to stabilize the covalent complex (E-S, Fig. 1), then its relative concentration would be greater at consensus sites, generating specificity (Fig. 1B). It is clear from this analysis that an understanding of specificity requires measurements of the relative strength of interactions in the ES, ES‡ and E-S complexes as a function of discrete changes in the consensus DNA sequence.
Previous footprinting, chemical protection/interference and UV cross-linking studies have indicated that vTopo makes contacts within the major groove of the GGGAA sequence of the nonscissile strand (4, 7, 12-14). For instance, site-specific modification with benzo-[c]-phenanthrene at N2 and N6 of G can reduce the cleavage rate by a factor of 1000 (6), and site-specific modification of the exocylic amino groups of the +1 and +2 adenines with benzo-[a]-pyrene decreased the rate of cleavage by approximately 103-fold (15). The rate effects on strand cleavage arising from the more subtle major groove modifications of 8-oxoguanine, 8-oxoadenine and 2-aminopurine have also been explored, where deleterious effects as large as 35-fold at the +3G were observed (15). In addition, stereospecific methylphosphonate substitutions of the nonbridging phosphate oxygens in the DNA backbone of this sequence have highlighted the significance of major and minor groove electrostatic interactions towards sequence specific recognition (8). Although these previous findings highlight the significance of major groove interactions, the discrete energetic role of the hydrogen bonding groups that are presented within the major groove of the GGGAA sequence has never been evaluated at each step along the reaction coordinate.
In this paper, we describe a study in which the N7 hydrogen bond acceptor groups of each guanine and adenine within the consensus sequence are replaced with unnatural 7-deaza bases. In separate experiments, the individual guanine O6 and adenine N6 groups are removed to investigate hydrogen bonding at these major groove positions (Fig. 2). Systematic dissection of the energetics of these changes at each step along the reaction pathway shows that major groove hydrogen bonds are in general used to increase the stability of the covalent E-S complex, without having any energetic effect on the noncovalent or transition state complexes (Fig. 1B). A reasonable physical interpretation is that these interactions are used to optimally position the covalent phosphotyrosine linkage relative to the attacking 5′ -hydroxyl nucleophile.
Fig. 2.

DNA substrate used in this study. The normal G/C and A/T base pairs of the consensus sequence were replaced with unnatural bases substitutions in the GGGAA sequence of the consensus site are shown. The arrows indicate potential contact points that are absent with the modified bases. The 7-deaza modification doesn’t affect base pairing whereas the N and P disrupt the hydrogen bonding of the base pair.
MATERIALS AND METHODS
Enzymes
The cloning and purification of wild-type vaccinia topoisomerase has been previously described (16). The enzyme concentration was determined by UV absorption using an extinction coefficient of 41,797 M-1cm-1 in a buffer containing 20 mM sodium phosphate at pH 6.0.
DNA Substrates with Fluorescent Tags
The sequence of the 32 mer DNA substrate containing the consensus cleavage sequence is shown in Figure 2, where FAM is 6-carboxyfluorescein. The synthesis of the non-scissile strand with modified bases incorporated at various sites was accomplished by substituting the modified base during DNA synthesis at these positions. All oligonucleotides were synthesized using an ABI 394 synthesizer using nucleoside phosphoramidites obtained from Glen Research. The oligonucleotides were purified using anion exchange HPLC and then desalted using C-18 reverse phase chromatography. The purity of oligonucleotides was confirmed using electrophoresis through a 20 % polyacrylamide gel containing 7 M urea and MALDI-TOF analysis. The DNA duplexes were prepared in buffer A (20 mM Tris-HCl, 200 mM NaCl, 1mM DTT, pH 8.0) by mixing the two strands in a molar ratio of 1:2 (non-scissile strand was in excess).
Equilibrium Cleavage Measurements
The equilibrium cleavage measurements were performed using Buffer A by titrating FAM32AP/32 mer (60 nM) with increasing concentrations of topo (40-600 nM). The covalent complex was trapped by the addition of 1 volume of 10% SDS after 1h. The fraction covalent complex at each enzyme concentration [counts in covalent complex/(counts in covalent complex + counts in free DNA)] was quantified using ImageQuant software. The fraction covalent complex was plotted against enzyme concentration to obtain the values of the binding constant (KD) and cleavage-religation equilibrium constant (Kcl) using the following equation (eq 1) (16).
| (1) |
where a = 1 + 1/Kcl, b = a[E] + a[S] + KD/Kcl, Kcl = (kcl/kr), [E] and [S] are the total enzyme and substrate concentrations respectively. In this analysis, the counts that migrate as free DNA represent the sum of the counts that were bound non-covalently to the enzyme, and those of the unbound DNA. All measurements were repeated two or three times to estimate errors.
Approach to Equilibrium Kinetics
The rate constant for approach-to-equilibrium in a FAM32AP/32 mer substrate was measured using a KinTek rapid quench instrument. The final enzyme and DNA concentrations were maintained at 1 μM and 100 nM respectively. The enzyme and DNA were mixed and the reactions were quenched using 10% SDS at time intervals ranging between 2.5 and 750 milliseconds. The samples were subjected to electrophoresis on a 10% SDS-PAGE gel. The 6-FAM fluorophore in the free DNA and enzyme-DNA covalent adduct was quantified using Typhoon scanner. Since the dye front ran along with the free DNA band, giving rise to inner filter effects, the samples were loaded on to the gel without the dye component. The fraction of covalent complex formed at each time was fitted to a first order rate equation to obtain kobs which equals the sum of the cleavage and religation rate constants (i.e kobs = kcl + kr) (16). Thus, using the Kcl obtained from equilibrium cleavage measurements the cleavage and religation rate constants (kr) may be calculated using the equations kcl = kobs/(1/Kcl + 1) and kr = kobs/(Kcl + 1). All measurements were repeated two or three times to estimate errors.
RESULTS
Overall Approach
We seek to evaluate the energetic role of vTopo interactions with the DNA major groove by introducing subtle site-specific modifications within the G+5G+4G+3A+2A+1 sequence in the non-scissile strand as shown in Figure 2. In the nomenclature used here, the identity and position of the substituted base is indicated by a letter and number, respectively, where H = 7-deazaguanine, N = 2-aminopurine, B = 7-dezaadenine, and P = purine. Thus, substitution of 7-dezaguanine at position five would be represented as N5, and double N substitutions at positions four and five would be designated N5N4. The 17 single and double DNA substitutions that were synthesized and studied in this work are listed in Table 1. These sequences show H and N substitutions in each of the G positions, B and P substitutions in both of the A sites, and combined H, B and N, B substitutions at both the G and A sites.
Table 1.
Nomenclature, DNA Sequences, and Kinetic and Thermodynamic Parameters for the major groove modified DNAs a
| DNA | Sequence | KD (nM) | Kcl | kcl (s-1) | kr (s-1) |
|---|---|---|---|---|---|
| Unmodified | GGGAA | 118 ± 25 | 1.6 ± 0.3 | 2.2 ± 0.2 | 1.4 ± 0.2 |
| B1 | GGGAB | 94 ± 46 | 0.8 ± 0.2 | 2.8 ± 0.3 | 3.5 ± 0.3 |
| B2 | GGGBA | 74 ± 22 | 0.7 ± 0.1 | 2.2 ± 0.1 | 3.0 ± 0.2 |
| P1 | GGGAP | 64 ± 21 | 1.5 ± 0.3 | 2.5 ± 0.2 | 1.7 ± 0.2 |
| P2 | GGGPA | 72 ± 15 | 2.7 ± 0.6 | 3.1 ± 0.2 | 1.1 ± 0.2 |
| H3 | GGHAA | 77 ± 26 | 0.7 ± 0.1 | 3.2 ± 0.2 | 4.4 ± 0.2 |
| H4 | GHGAA | 59 ± 14 | 0.5 ± 0.03 | 1.6 ± 0.1 | 3.4 ± 0.1 |
| H5 | HGGAA | 75 ± 16 | 0.4 ± 0.03 | 1.5 ± 0.1 | 4.2 ± 0.1 |
| N3 | GGNAA | 55 ± 21 | 1.0 ± 0.2 | 3.2 ± 0.2 | 3.2 ± 0.2 |
| N4 | GNGAA | 171 ± 21 | 0.5 ± 0.03 | 2.4 ± 0.1 | 5.1 ± 0.1 |
| N5 | NGGAA | 148 ± 32 | 0.5 ± 0.05 | 2.3 ± 0.1 | 4.8 ± 0.1 |
| B2B1 | GGGBB | 80 ± 17 | 2.1 ± 0.4 | 4.7 ± 0.2 | 2.2 ± 0.2 |
| H5H4 | HHGAA | 67 ± 21 | 0.5 ± 0.06 | 2.7 ± 0.1 | 5.8 ± 0.1 |
| H5H3 | HGHAA | 110 ± 21 | 0.3 ± 0.02 | 0.9 ± 0.1 | 3.1 ± 0.2 |
| N5N4 | NNGAA | 87 ± 14 | 0.1 ± 0.005 | 0.2 ± 0.1 | 2.5 ± 0.3 |
| N5N3 | NGNAA | 48 ± 18 | 0.2 ± 0.02 | 1.1 ± 0.2 | 4.8 ± 0.2 |
| H3B1 | GGHAB | 72 ± 21 | 1.5 ± 0.3 | 2.2 ± 0.2 | 1.5 ± 0.2 |
| N4B1 | GNGAB | 193 ± 21 | 0.7 ± 0.05 | 5.3 ± 0.1 | 7.7 ± 0.1 |
See Fig. 2 for structures of B, H, P and N.
Because these modifications result in very subtle changes in structure, we anticipated kinetic and thermodynamic consequences of less than 10-fold in the various reaction parameters of vTopo. Accordingly, accurate and precise measurements of KD, Kcl kcl, and kr were required. A new experimental protocol is used here that meets this requirement. This new method has several advantages over previous approaches including i) the use of a non-radioactive 5′ fluorescein probe, ii) measurement of the noncovalent binding constant (KD) using the wild-type enzyme instead of the Y274F surrogate that lacks the covalent nucleophile (16), and iii) measurement of KD, kcl, kr and Kcl using a single approach-to-equilibrium substrate. This new method requires only two experiments to obtain all of these reaction parameters: an equilibrium measurement of both KD and Kcl, and a kinetic measurement of the observed rate constant for approach-to-equilibrium (kobs = kcl + kr). Although only two experiments are required, these parameters are robustly determined by these measurements. In the equilibrium measurement of KD and Kcl, the concentration dependence for formation of the covalent complex assesses the preequilibrium formation of the noncovalent complex (KD = [E][S]/[ES]), and the endpoint provides a direct measurement of the internal equilibrium Kcl = [E-S]/[ES]. For an approach-to-equilibrium involving two species (in this case ES and E-S), only two of the three measurable parameters (kcl, kr or Kcl) are required to characterize the system because once two are known, the other may be calculated using the equation Kcl = kcl/kr. The validity of the two-state approximation, and confirmation of the agreement between approach-to-equilibrium and irreversible reaction kinetic measurements has been confirmed in previous studies (11, 16-18). We show representative data using this approach in the Results and then interpret the findings in the Discussion based on the full results which are summarized in Table 1.
Binding Constant (KD) and Cleavage Equilibrium Constant (Kcl)
Both KD and Kcl were determined from a single experiment by titrating a limiting concentration of DNA with excess topo. As the enzyme concentration is increased, more of the free DNA is bound noncovalently, and by mass action, more covalent complex is formed as well. Thus, upon adding SDS to trap the equilibrium amount of covalent complex at each concentration of enzyme, and resolving the covalent complex from the free DNA by denaturing gel electrophoresis, the binding and internal cleavage equilibria can be extracted from the data using eq 1. As shown in Figure 3A-D for the native DNA, and for the three modified sequences N4, N5 and N5N4, the enzyme-substrate covalent complex (upper band) and the free DNA (lower band) are cleanly resolved. The fraction of the total 5′ FAM-labeled DNA that is covalently bound shows a hyperbolic response to increasing enzyme concentration for each DNA substrate. However, the substituted sequences show large decreases in the amount of covalently bound DNA at saturating concentration of vTopo as compared to the native consensus sequence (Fig. 3E).
Fig. 3.
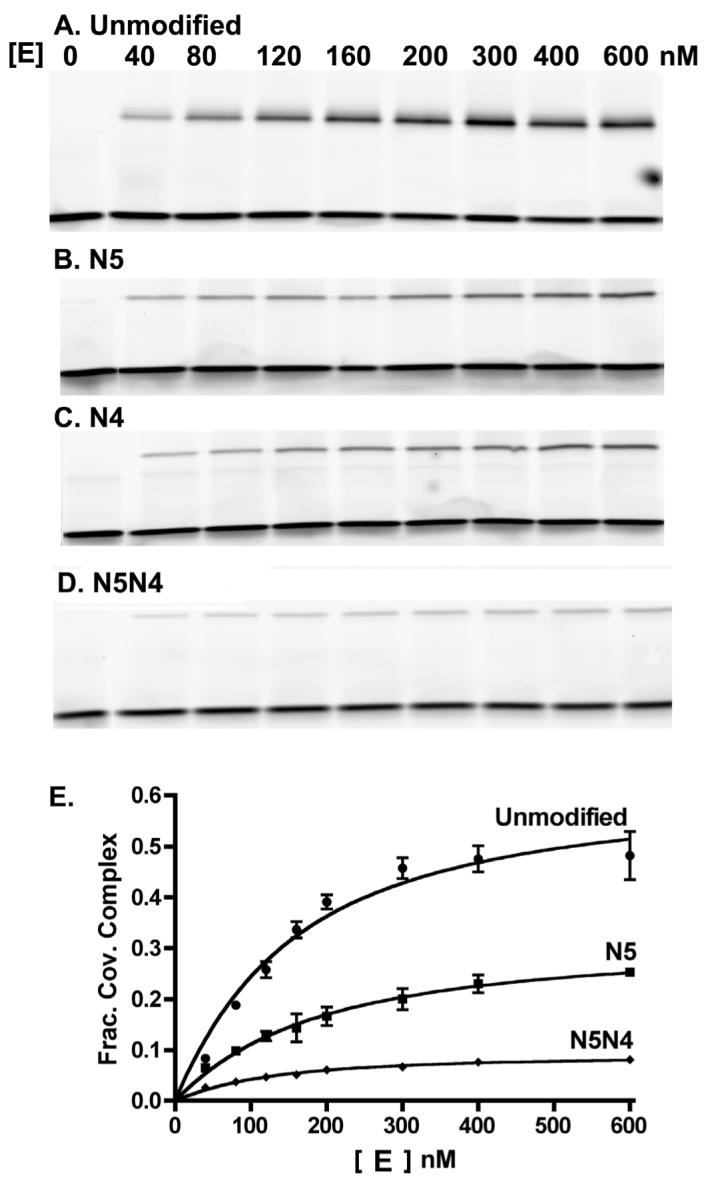
Equilibrium cleavage of the FAM32AP/32 duplex DNA. A-D) SDS-PAGE gels showing the equilibrium reactions of vTopo with unmodified, singly modified (N5, N4) and doubly modified (N5N4) DNA as a function of enzyme concentration. The top band in each panel is the E-S covalent complex, whereas the bottom band is the free DNA. E) Plotting fraction covalent complex against vTopo concentration and fitting to eq 1 provides the binding constant (KD) and the cleavage equilibrium constant (Kcl) for these DNA substrates.
Analysis of the equilibrium cleavage data using eq 1 established that these substitutions have little effect on the binding constant (KD), but that the internal cleavage equilibrium (Kcl) decreased to a varying extent depending on the substitution. The binding constant for the unmodified FAM-32AP/32 mer was 120 ± 50 nM, whereas the substituted DNA sequences differed by less than 2-fold from this value. On the other hand, the cleavage equilibrium for the N5 and N4 substrates was about 3-fold smaller than the native sequence (Kcl (native) = 1.6 ± 0.3). Moreover, the cleavage equilibrium constant for the H5H4, H5H3, N5N4, and N5N3 doubly modified sequences were 3 to 16-fold lower than the consensus sequence (see Discussion and Supplemental Materials). A complete analysis of the effects on KD and Kcl for all the modifications is presented in the Discussion section.
Approach-to-Equilibrium Cleavage Kinetics (kcl)
The cleavage rate constant for each FAM32AP/32 mer equilibrium cleavage substrate was measured using a rapid mix-chemical quench instrument. The enzyme and substrate were rapidly mixed and the reactions were quenched with 10% SDS at various time intervals before separation of the covalently bound DNA product by denaturing polyacrylamide gel electrophoresis (Fig. 4A and 4B). In this analysis, the fraction of covalent complex increases over time until the equilibrium level is reached. The amount of covalent complex at the endpoint for each of these reactions was indistinguishable from that observed in the equilibrium cleavage measurements (compare Fig. 3E with 4C), which establishes that equilibrium was obtained within the time frame of these measurements. The fraction of covalent complex as a function of time was fitted to a first-order rate equation to obtain the observed rate for approach-to-equilibrium kobs = kcl + kr. Despite the large effects of some substitutions on Kcl, the cleavage rates were surprisingly unaffected in nearly all cases (Table 1). The only significant exceptions were the double modifications B2B1 and N4B1, in which kcl was increased 2 and 2.5-fold, and the substrates H5H3 and N5N4, which showed 2.4 and 11-fold smaller kcl values than the consensus sequence (Table 1).
Fig. 4.
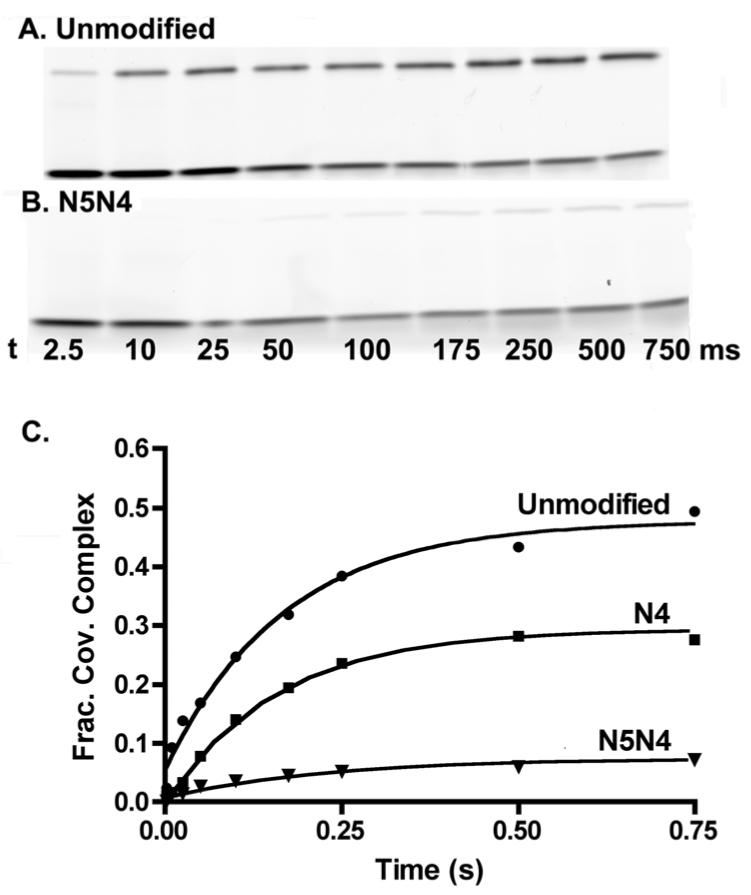
Approach-to-equilibrium cleavage kinetics with the FAM32AP/32 duplex DNA. A, B) SDS-PAGE gels showing the time course for reaction of vTopo with unmodified and doubly modified (N5N4) DNA. These reactions were performed in a rapid mix-chemical quench apparatus. The top band in each gel is the E-S covalent complex, whereas the bottom band is the free DNA. C) The fraction covalent complex at each time point was fitted to a first-order rate expression to obtain the observed cleavage rate kobsd = kcl + kr.
DNA Ligation Rate Constants (kr)
The rates of DNA ligation (kr) were calculated from the cleavage equilibrium constant and the observed rate of cleavage using the equality kr = kobs/(Kcl + 1). For the unmodified FAM32AP/32 mer, kr = 1.1 ± 0.14 s-1. Except for the purine (P) singly modified DNA at the +1 and +2 positions, every other modified DNA was found to increase the religation rate, providing the primary kinetic explanation for the decrease in the cleavage equilibrium constant observed for these modified substrates (see Table 1). The increase in kr was more significant for the G-site substitutions (up to 5-fold) as compared to the A-site alterations, highlighting the primary significance of the G sites.
DISCUSSION
Specific enzymatic recognition of the DNA major groove can take advantage of hydrogen bonding groups that are displayed within this structural feature of B DNA. With respect to the vTopo nonscissile strand consensus sequence, 3′-GGGAA-5′, the G sites present lone pair hydrogen bond acceptors on the N7 and O6 positions, and the A sites present an acceptor site at N7 and a donor site at the six amino group (Fig. 2). These potential interaction sites have been removed using four different unnatural bases. Two of these, 7-deazaguanine (H) and 7-dezaadenine (B), are very subtle modifications that selectively remove the lone pair electrons at the seven position without disrupting any other aspect of the base pair. In contrast, 2-aminopurine (N) and purine (P) substitutions remove the O6 and 6-NH2 groups, respectively, but also disrupt base-pairing at the G and A sites, respectively. Thus, interpretation of the energetic effects of the N and P substitutions must be made more guardedly than for the more conservative H and B substitutions. Finally, introduction of simultaneous modifications at two sites allows one to ask the question whether energetic communication exists between interaction sites. In other words, is the interaction at site one energetically unchanged, enhanced or weakened when the interaction at site two is removed? The strength of these enzyme-DNA contacts can be quantified at each step of the vTopo reaction to reveal important elements of catalytic specificity.
Single G-site Modifications
The effects of single N and H major groove modifications on KD, Kcl, kcl, and kr are summarized in Table 1. The major effect of these G site modifications is to decrease Kcl by as much as 4-fold, with smaller effects on KD. The largest outcomes are seen at the +4 and +5 positions with the 7-deaza (H) substitutions, suggesting that the enzyme donates a hydrogen bond with N7 of G+4 and G+5. Substitution of G+4 and G+5 with N also decreases Kcl by a comparable amount (N lacks the O6 atom of guanine). Since previous studies have found that inosine substitution at these positions had no effect on the cleavage equilibrium (inosine lacks the 2-amino group of guanine) (7), then these findings suggest that vTopo donates hydrogen bonds to both the N7 and O6 acceptor groups of the +4 and +5 guanines, but that the 2 amino group is not an important recognition element (Figure 5A, 5B). Smaller effects are seen when these same substitutions are made at the +3 guanine, a result that is consistent with previous findings where this position was found to be fairly tolerant to both inosine and adenine substitutions (7).
Fig. 5.

Consensus sequence major groove interactions of vTopo and energetic effects of single and double G site modifications. A) Space filling model of duplex DNA containing the consensus sequence. The view is looking into the major groove where the O6 and N7 hydrogen bond accepting groups of the three guanine bases are displayed. B) Hydrogen bond interactions of vTopo with G+5 and G+4 as inferred from H and N substitution at these sites. C) Energetic effect of single and double site G modifications. The single site modification only has an effect on the ground state covalent complex, while the double site modification has an effect on both the ground state covalent complex and the transition state (see text).
Corresponding kinetic studies with this series of singly substituted equilibrium substrates establish that the decreases in Kcl arise almost entirely from increases in the ligation rate (kr) with no large changes in kcl (Table 1). This result indicates that the N7 and O6 single site deletions increase the ligation rate by selectively destabilizing the covalent complex without affecting the stability of the transition state for cleavage and ligation. This conclusion is based on the requirement that energetic perturbations to the transition state for cleavage and ligation must be manifested as equal changes in kcl and kr, which is not observed. These results contrast with the approximately 150-fold decreases in the cleavage rate observed when entire bases were deleted form the +3 and +4 positions (19).2 Possible mechanisms for the selective increases in the ligation rate induced by these modified bases are considered below.
Double G-site Modifications
For the most part, the cleavage equilibrium constants for the double site N and H modifications are reduced from that of the two single modifications by an amount that is expected from independent, multiplicative effects on this equilibrium constant (i.e. additive effects of the two interaction free energies). Surprisingly, the simple multiplicative effects of the double modifications on the cleavage equilibrium mask substantial nonmultiplicative effects on the kinetic constants kcl and kr. The H5H3, N5N4 and N5N3 substrates show large decreases in kcl whereas the calculated multiplicative effects of the two single modifications are small (Table 1). This outcome is the hallmark of energetic cooperativity in the transition state, possibly arising from significant structural changes in the complex brought about by the combined removal of two N7 or O6 hydrogen bond acceptor groups. In contrast to the unexpected deceases in kcl, the combined modifications give rise to much smaller increases in kr than would be expected based on multiplicative effects of the single modifications (Table 1). The different results on the two rate constants can be simply rationalized if the double modifications destabilize the transition state and covalent complex to similar extents, leading to only small changes in the activation barrier for ligation but significant increases in the activation barrier for cleavage.
As summarized in the free energy diagrams in Figure 5C, removal of single N7 or O6 atoms at the +5, +4 and +3 positions selectively destabilizes the covalent complex thereby enhancing the rate of ligation. In contrast, double modifications show equal destabilizing effects in the transition state and covalent complex leading to slower rates of cleavage and lesser damaging effects on ligation.
A-site Modifications
Small changes in KD, Kcl, kcl, and kr are seen upon single and double 7-deaza and purine substitution at A+2 and A+1 (Table 1). Interestingly, purine (P) substitution has little effect at the A+2 and A+1 positions, indicating that the exocyclic amino group of adenine is not an important recognition element. This finding sheds light on previous work in which inosine substitutions at the +1 and +2 positions were found to abrogate cleavage entirely (7). This large effect is likely due to the nonconservative nature of the inosine substitution that replaces the 6-amino group of adenine with a carbonyl group. In contrast, the more subtle P substitution used here simply removes the amino group (Fig. 2). In summary, the N7 and exocyclic amino groups of the A sites are not critical recognition elements.
Combined G and A-Site Modifications
In order to investigate whether there is energetic coupling between the G and A-sites of the consensus sequence, H3B1 and N4B1 doubly modified DNAs were synthesized and their activity with vTopo was measured. As observed for the other substrates, both H3B1 and N4B1 double modifications produced small effects on KD (Table 1). Although the Kcl value for the N4B1 substrate was only modestly smaller than the consensus sequence, and within a factor of two of that anticipated from multiplication of the two single effects, this “null” effect on Kcl was brought about by significant and equal increases in both kcl and kr (Table 1). The H3B1 modification differs in that significant nonmultiplicative effects on Kcl were observed that arise from antagonism of the two H3 and B1 modifications on the religation rate. Minimally these results indicate that the G and A sites can energetically communicate and that combined changes in both sites can give rise to different kinetic outcomes than combined modifications within the G site alone (see above).
Specificity Mechanisms and the Importance of Positioning the Phosphotyrosine Linkage in Topoisomerase Reactions
Ensemble and single molecule measurements have established that vTopo removes multiple DNA supercoils during the lifetime of each covalent intermediate (9, 10). A hallmark of this type of mechanism is that the rate of strand ligation and rotation are competitive kinetic processes. In other words, the lifetime of the covalent complex determines how many supercoils are removed after a single cleavage event. The free rotation mechanism for supercoil release places limits on the mechanisms that vTopo may employ to obtain specificity (i.e. the relative number of supercoils removed from a specific site and a nonspecific site in a given length of time). We consider three possible specificity mechanisms and their outcomes:
Sequence specific interactions could be used by the enzyme to selectively stabilize the transition state for cleavage and ligation. Although such a mechanism would increase the rate of covalent complex formation, it would also be antagonistic with a free rotation mechanism for supercoil release, because the rate of ligation would also increase. We conclude that such interactions cannot in themselves substantially enhance specificity for DNA relaxation because the lifetime of the covalent complex is decreased, and thus, fewer supercoils are removed per cleavage event.
Sequence specific interactions could be used by the enzyme to selectively destabilize (strain) the ground state noncovalent complex thereby lowering the activation barrier for cleavage. Sequence specific contacts such as this could give rise to substantial specificity only if the contacts became stronger in the covalent complex (release of strain). This is required because equal ground state strain in both complexes is energetically equivalent to transition state stabilization which has been discussed in (1).
vTopo could use sequence specific interactions to selectively stabilize the ground state covalent complex thereby increasing its lifetime. In the case of an infinitely stable covalent complex, every rate-limiting cleavage event would lead to complete relaxation of a DNA molecule. This type of mechanism could lead to substantial specificity, especially if balanced with ground state strain in the noncovalent complex and/or transition state stabilization. Of course, an infinitely stable covalent adduct is not compatible with successful replication of DNA, because the enzyme induced strand nicks would be converted into double strand breaks when encountered by a replication fork. Indeed, excessive stabilization of the covalent complex is the poisoning mechanism of the clinically useful topoisomerase drugs(20). Thus, the lifetime of the covalent adduct is limited by this intrinsic biological constraint.
Based on the above considerations, we envision that sequence specific DNA interactions are in part used to optimize the stability of the covalent complex and thereby enhance specificity for supercoil release. Thus, even the subtle major groove perturbations investigated here can have measurable effects on adduct lifetime. What is the physical basis for such sequence specific effects on the stability of the covalent complex? Since most of the effects manifest themselves in the ground state for religation, it is probable that these major groove interactions are used to optimally position the phosphotyrosine target with respect to the 5′-hydroxyl nucleophile. Since the 5′ hydroxyl needs to freely rotate to remove supercoils, it is unlikely that topoisomerase rigidly positions this group. Instead, it seems more likely that the relative position (or mobility) of the phosphotyrosine center determines the efficiency of religation. If this center is too close to the trajectory of the rotating 5′-OH nucleophile, then religation will occur too frequently. Alternatively, if the phosphotyrosine group is rigidly held too far away, religation would never occur. Thus one use of sequence specific interactions is to carefully position the phosphotyrosine moiety relative to the rotating nucleophile such that frequent ligations or overly stable strand nicks are avoided.
Footnotes
This work was supported by National Institutes of Health Research Grant GM 68626 (J.T.S.).
- vTopo
- vaccinia DNA topoisomerase IB
- FAM
- 6-carboxyfluorescein
- N
- 2-aminopurine
- P
- Purine
- B
- Deazaadenine
- H
- Deazaguanine
- buffer A
- 20 mM Tris-HCl 200 mM NaCl, 1mM DTT, pH 8.0.
Although increases in the religation rate, with no significant decreases in the cleavage rate are observed with the +3, +4 and +5 substituted 32/32 mer equilibrium substrates, we do observe 1.5 to 4-fold decreases in the rate of irreversible strand cleavage using similarly substituted 18/24 mer suicide cleavage substrates. The results with the suicide substrates are consistent with the previous base ablation studies of the Shuman group (i.e. cleavage rate decreases) (19). The apparent discrepancy between the effects of major groove alterations using suicide and equilibrium substrates arises from anticooperative interactions between the GGGAA major groove interactions and the downstream DNA regions that are present in the equilibrium substrate and absent in the suicide substrate (Nagarajan and Stivers, unpublished). A comprehensive study of anticooperativity in DNA strand binding, cleavage and religation by vTopo is in progress.
REFERENCES
- 1.Shuman S, Prescott J. Specific DNA cleavage and binding by vaccinia virus DNA topoisomerase I. J Biol Chem. 1990;265:17826–36. [PubMed] [Google Scholar]
- 2.Shuman S. Site-specific interaction of vaccinia virus topoisomerase I with duplex DNA. Minimal DNA substrate for strand cleavage in vitro. J Biol Chem. 1991;266:20576–7. [PubMed] [Google Scholar]
- 3.Shuman S. Site-specific DNA cleavage by vaccinia virus DNA topoisomerase I. Role of nucleotide sequence and DNA secondary structure. J Biol Chem. 1991;266:1796–803. [PubMed] [Google Scholar]
- 4.Sekiguchi J, Shuman S. Vaccinia topoisomerase binds circumferentially to DNA. J Biol Chem. 1994;269:31731–4. [PubMed] [Google Scholar]
- 5.Cheng C, Shuman S. Site-specific DNA transesterification by vaccinia topoisomerase: Role of specific phosphates and nucleosides. Biochemistry. 1999;38:16599–612. doi: 10.1021/bi992001d. [DOI] [PubMed] [Google Scholar]
- 6.Yakovleva L, Handy CJ, Sayer JM, Pirrung M, Jerina DM, Shuman S. Benzo[c]phenanthrene adducts and nogalamycin inhibit DNA transesterification by vaccinia topoisomerase. J Biol Chem. 2004;278:42170–42177. doi: 10.1074/jbc.M401203200. [DOI] [PubMed] [Google Scholar]
- 7.Shuman S, Turner J. Site-specific interaction of vaccinia virus topoisomerase I with base and sugar moieties in duplex DNA. J Biol Chem. 1993;268:18943–50. [PubMed] [Google Scholar]
- 8.Tian L, Claeboe CD, Hecht SM, Shuman S. Remote phosphate contacts trigger assembly of the active site of DNA topoisomerase IB. Structure. 2004;12:31–40. doi: 10.1016/j.str.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 9.Koster DA, Croquette V, Dekker C, Shuman S, Dekker NH. Friction and torque govern the relaxation of DNA supercoils by eukaryotic topoisomerase IB. Nature. 2005;434:671–674. doi: 10.1038/nature03395. [DOI] [PubMed] [Google Scholar]
- 10.Stivers JT, Harris TK, Mildvan AS. Vaccinia DNA topoisomerase I: Evidence supporting a free rotation mechanism for DNA supercoil relaxation. Biochemistry. 1997;36:5212–5222. doi: 10.1021/bi962880t. [DOI] [PubMed] [Google Scholar]
- 11.Nagarajan R, Kwon K, Nawrot B, Stec WJ, Stivers JT. Catalytic phosphoryl interactions of topoisomerase IB. Biochemistry. 2005;44:11476–85. doi: 10.1021/bi050796k. [DOI] [PubMed] [Google Scholar]
- 12.Sekiguchi J, Shuman S. Proteolytic footprinting of vaccinia topoisomerase bound to DNA. J Biol Chem. 1995;270:11636–45. doi: 10.1074/jbc.270.19.11636. [DOI] [PubMed] [Google Scholar]
- 13.Sekiguchi J, Shuman S. Covalent DNA binding by vaccinia topoisomerase results in unpairing of the thymine base 5′ of the scissile bond. J Biol Chem. 1996;271:19436–42. doi: 10.1074/jbc.271.32.19436. [DOI] [PubMed] [Google Scholar]
- 14.Sekiguchi J, Shuman S. Identification of contacts between topoisomerase i and its target DNA by site-specific photocrosslinking. EMBO J. 1996;15:3448–57. [PMC free article] [PubMed] [Google Scholar]
- 15.Yakovleva L, Tian L, Sayer JM, Kalena GP, Kroth H, Jerina DM, Shuman S. Site-specific DNA transesterification by vaccinia topoisomerase: Effects of benzo[alpha]pyrene-dA, 8-oxoguanine, 8-oxoadenine and 2-aminopurine modifications. J Biol Chem. 2003;278:42170–7. doi: 10.1074/jbc.M308079200. [DOI] [PubMed] [Google Scholar]
- 16.Kwon K, Stivers JT. Fluorescence spectroscopy studies of vaccinia type IB DNA topoisomerase. Closing of the enzyme clamp is faster than DNA cleavage. J Biol Chem. 2002;277:345–52. doi: 10.1074/jbc.M109449200. [DOI] [PubMed] [Google Scholar]
- 17.Stivers JT, Jagadeesh GJ, Nawrot B, Stec WJ, Shuman S. Stereochemical outcome and kinetic effects of Rp- and Sp-phosphorothioate substitutions at the cleavage site of vaccinia type I DNA topoisomerase. Biochemistry. 2000;39:5561–72. doi: 10.1021/bi992429c. [DOI] [PubMed] [Google Scholar]
- 18.Kwon K, Jiang YL, Song F, Stivers JT. 19F NMR studies of vaccinia type IB topoisomerase. Conformational dynamics of the bound DNA substrate. J Biol Chem. 2002;277:353–8. doi: 10.1074/jbc.M109450200. [DOI] [PubMed] [Google Scholar]
- 19.Tian L, Sayer JM, Jerina DM, Shuman S. Individual nucleotide bases, not base pairs, are critical for triggering site-specific DNA cleavage by vaccinia topoisomerase. J Biol Chem. 2004;279:39718–26. doi: 10.1074/jbc.M407376200. [DOI] [PubMed] [Google Scholar]
- 20.Liu LF. DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem. 1989;58:351–75. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]