

Abstract
This work investigated the functional role of nuclear factor–κB (NF-κB) in respiratory burst activity and in expression of the human phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase genes CYBB, CYBA, NCF1, and NCF2. U937 cells with a stably transfected repressor of NF-κB (IκBα-S32A/S36A) demonstrated significantly lower superoxide release and lower CYBB and NCF1 gene expression compared with control U937 cells. We further tested Epstein-Barr virus (EBV)-transformed B cells from patients with anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID), an inherited disorder of NF-κB function. Superoxide release and CYBB gene expression by EDA-ID cells were significantly decreased compared with healthy cells and similar to cells from patients with X-linked chronic granulomatous disease (X910 CGD). NCF1 gene expression in EDA-ID S32I cells was decreased compared with healthy control cells and similar to that in autosomal recessive (A470) CGD cells. Gel shift assays demonstrated loss of recombinant human p50 binding to a NF-κB site 5′ to the CYBB gene in U937 cells treated with NF-κB inhibitors, repressor-transfected U937 cells, and EDA-ID patients' cells. Zymosan phagocytosis was not affected by transfection of U937 cells with the NF-κB repressor. These studies show that NF-κB is necessary for CYBB and NCF1 gene expression and activation of the phagocyte NADPH oxidase in this model system.
Introduction
The phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is a multiprotein complex with both membrane-bound and cytosolic components. Flavocytochrome b, the membrane-bound component of the oxidase complex, is a heterodimer composed of a 22-kDa polypeptide subunit (termed p22phox) and a 91-kDa glycoprotein subunit (gp91phox).1 NADPH oxidase activation requires assembly of the flavocytochrome b with several cytosolic proteins, including p47phox, p67phox, p40phox, and a GTP-binding protein, either rac1 in macrophages or rac2 in neutrophils.2 The activated complex generates large quantities of superoxide and other reactive oxygen species essential for the microbicidal function of phagocytes.
Molecular genetic defects affecting components of the NADPH system lead to chronic granulomatous disease (CGD), a primary immunodeficiency characterized by early onset of severe recurrent infections.1 The genes CYBB and NCF1 are the most frequent sites of mutations (∼60% and 30% of the cases) leading, respectively, to the X-linked and one of the autosomal forms of CGD.3,4 The CYBB gene (MIM 306400), encoding gp91phox, is localized at chromosome Xp21.1 and contains 13 exons spanning 30 kilobases (kb).5 The CYBB promoter region includes several cis-elements involved in gene regulation by interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α).6 Several trans-activators and repressors have also been characterized. Removal of repressor elements permits interactions of both widely expressed and IFN-γ–responsive transcriptional activators with cognate binding sites in the CYBB proximal promoter.7,8
The NCF1 gene (MIM 608512) is localized at chromosome 7q11.23 and encodes p47phox, a cytosolic component of the oxidase.3 Li et al9 characterized the NCF1 promoter and identified a functional binding site for the transcriptional factor PU.1. Further experiments confirmed that differentiation-dependent up-regulation of NCF1 gene transcription is associated with changes in PU.1 phosphorylation and increased binding affinity.10
The transcription factor nuclear factor–κB (NF-κB) is a heterodimer formed from members of the mammalian rel gene family, which includes p105/p50, p100/p52, p65 (RelA), RelB, and c-Rel.11,12 The general mechanism of activation of the conventional and most common NF-κB complex (p50/RelA) starts with its sequestration in the cytoplasm by interaction with a family of inhibitory proteins, termed IκBs, including IκBα, IκBβ, IκBγ, IκBε, and the proto-oncogene Bcl-3. Activation by extracellular signals induces phosphorylation of IκB by specific IκB kinases (IκKα and IκKβ) on critical serine residues, Ser32 and Ser36, within the N-terminal signal response domain.13 IκB phosphorylation leads rapidly to its ubiquitinization and rapid proteolytic degradation, thus releasing the NF-κB heterodimer to move into the cell nucleus. There it interacts with κB-responsive promoter elements to modulate transcription of genes involved in myriad functions, including proinflammatory cytokines, chemokines, adhesion molecules, and inducible enzymes that regulate both the innate and adaptive immune responses.14 The inhibition of this system can be accomplished by varied pharmacological tools, such as nonsteroidal anti-inflammatory drugs, anti-inflammatory steroid hormones, and toxins.15 We have previously demonstrated that dexamethasone inhibits the NADPH oxidase system of differentiated THP-1 monocyte/macrophage cells by inhibiting CYBB and NCF1 gene expression.16
Gauss et al17 recently proposed that TNF-α–dependent activation of NF-κB up-regulates the human phagocyte NADPH oxidase activity, leading to enhanced superoxide production, increased CYBB, NCF1, and NCF2 gene expression, and further NF-κB activation, potentially contributing to sustained oxidant production in chronic inflammation. Anrather et al18 have identified 2 putative NF-κB binding sites in the murine gp91phox gene promoter and demonstrated suppression of gp91phox expression by IκBα in murine monocytic and microglial cell lines. In addition, we have recently identified (C.P., A.C.-N., and P.E.N., manuscript in preparation) a putative NF-κB binding site within a DNase I hypersensitive region of the distant 5′ flanking region,19 approximately 15 kb upstream of the human CYBB transcription start site. To date, no NF-κB binding sites have been identified in the human CYBA, NCF1, or NCF2 genes.
The present work investigates the role of NF-κB on human CYBB (gp91phox), CYBA (p22phox), NCF1 (p47phox), and NCF2 (p67phox) gene expression with the use of pharmacological inhibitors, a human myelomonocytic cell line stably transfected with an IκB “super-inhibitor,” and Epstein-Barr virus (EBV)–transformed B cells from patients with anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) due to naturally occurring mutations in the NF-κB pathway.
Methods
Full institutional review board approval for these studies was obtained at all of the participating institutions: State University of Campinas Medical School, Campinas SP, Brazil; University of Paris René Descartes, Necker Medical School; Institute of Biomedical Sciences, University of São Paulo (São Paulo, Brazil); and University of Massachusetts Medical School (Worcester, MA).
Cells and culture conditions
The human monocytic U937 cell line derived from a diffuse histiocytic lymphoma was obtained from the ATCC (Rockville, MD). U937 cells transfected with an empty pCMV3 plasmid vector or a pCMV3 vector containing a FLAG-tagged construct of an IκBα with mutations of serines 32 and 36 to alanine were kindly provided by Dr Carlos V. Paya.20 The IκB-S32A/S36A mutant impedes phosphorylation of IκB, leading to sequestration of NF-κB in the cytoplasm.
U937 cells were cultured (0.5-1 × 106 cells/mL) in RPMI 1640 complete medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mmol/L l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified 5% CO2 atmosphere. To induce differentiation, wild-type or mutated U937 cells were cultured with IFN-γ (100 U/ml) and TNF-α (1000 U/ml) for up to 72 hours, in the presence or absence of NF-κB inhibitor dexamethasone (1 μmol/L) or gliotoxin (10 μmol/L).16,21
EBV-transformed B cells prepared from peripheral blood mononuclear cells express the phagocyte flavocytochrome b and possess NADPH oxidase activity.22,23 Patient-derived cell lines accurately reproduce the biochemical and molecular defects of CGD.22–24 EBV-transformed B cells were prepared, according to procedures published previously,22 from healthy donors, patients with X-linked or autosomal recessive CGD, and 2 patients with EDA-ID. The X-linked CGD patient had a CYBB mutation characterized previously as an A>G substitution at the −2 position in the splice acceptor/donor site of intron 9, leading to exon 10 deletion, lack of gp91-phox expression, and an impairment of the respiratory burst.25 The autosomal recessive CGD patients have the following gene mutations: CYBA, deletion of exons 2 to 5; NCF1, gt deletion (common to most A47 CGD kindreds26); and NCF2, splice site mutation in exon4.
EBV-transformed B-cell lines were prepared from peripheral blood of previously described patients27–30 with EDA-ID and severe recurrent infections. One patient had X-linked EDA-ID caused by a hemizygous hypomorphic mutation in the gene encoding NEMO/IKKγ (W420X), the regulatory subunit of the IκB kinase (IKK) complex,27 leading to impairment of NF-κB activation in this patient's cells.28,29 The second patient had an autosomal-dominant form of EDA-ID associated with a heterozygous hypermorphic mutation (S32I) at serine 32 of IκBα.30 This gain-of-function mutation enhanced the inhibitory capacity of IκBα by preventing its phosphorylation and degradation, resulting in impaired NF-κB activation.30
All reagents were free of endotoxin (< 10 pg/mL as tested by Limulus amebocyte lysate assay). Informed consent was obtained from all patients in accordance with the Declaration of Helsinki.
Superoxide release
Superoxide release was assessed by a superoxide dismutase-inhibitable cytochrome c reduction assay, modified as described previously.31 In brief, U-937 or EBV-transformed B cells were cultured in 6-well polystyrene plates (106 cells/well) for up to 72 hours. On the day of the experiment, the plates were centrifuged, the supernatant was removed, and the cells were incubated in Hanks balanced salt solution (without phenol red) containing cytochrome c (625 μg/mL) for 1 hour at 37°C in a humidified 5% CO2 atmosphere. Half of the wells received superoxide dismutase (75 U/mL) at the beginning of the incubation. In another set of identical plates, phorbol 12-myristate 13-acetate (PMA; 30 nM) was used during the incubation period as an activator of superoxide release. After incubation, all plates were placed on ice, and the remaining wells received superoxide dismutase. The plates were centrifuged again, and the optical absorbance of the supernatants measured at 550 nm. The amount of superoxide released was calculated using an extinction coefficient of 0.021 nM−1 cm−1.
Real-time PCR
Total RNA was isolated from U-937 or EBV-transformed B cells (107 cells) by TRIzol (Invitrogen, Carlsbad, CA), according to manufacturer's instructions. Reverse transcription of 3 μg total RNA was performed with SuperScript II RT and random hexamers. The cDNA was amplified for real-time polymerase chain reaction (PCR) using the SYBR Green Real Time PCR System (Applied Biosystems, Foster City, CA). Each 25-μL PCR mix contained 12 ng/μL cDNA, 8 μL Sybr Green Master Mix (1× Sybr Green Buffer, 3 mmol/L MgCl2, 200 nmol/L dNTP blend, 0.63 U Amplitaq Gold, 0.25 U AmpErase), and the following specific primers: gp91phox (GenBank NM_00397): forward. 5′-TTG TGG AAA CCC TCC TAT GA-3′; reverse, 5′-AAA ACC GCA CCA ACC TCT CA-3′; p47phox (GenBank NM_000256): forward, 5′-CCT CTT TCC AGT GCA TTT AAG G-3′); reverse, 5′-GAT GTG ACG GAT GAA GGT GTC-3′; p67phox (GenBank NM_000433): forward, 5′-CGG ACA AGA AGG ACT GGA AG-3′; reverse, 5′-GGA AGT AAG CCA CTG CCA AG-3′; p22phox (GenBank NM_000101): forward, 5′-ATG TGG GCC AAC GAA CAG-3′; reverse, 5′-GTA CTC CAG CAG GCA CAC AA-3′; and β actin (GenBank NM_001101): forward, 5′-TCA CCG AGC GCG GCT-3′; reverse, 5′-TAA TGT CAC GCA CGA TTT CCC-3′. Cycle conditions for the ABI 5700 thermal cycler were as follows for gp91phox: 50°C for 2 minutes, 95°C for 10 minutes, and then 35 cycles of 95°C for 15 seconds and 60°C for 1 minute. Cycle conditions for the ABI 7500 thermal cycler were as follows for p22phox, p47phox, and p67phox: 50°C for 2 minutes, 95°C for 10 minutes, and then 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. After 35 cycles, a dissociation curve (melting curve) was generated in the range of 60°C to 95°C. Parallel amplification of β-actin (GenBank NM_001101) was used to normalize the amount of template. The reactions were run in triplicate, and the results were represented as means plus or minus SD. Standard curves for each amplification product were generated from 10-fold dilutions of pooled cDNA to determine primer efficiency.
Electromobility gel shift assays
Nuclear extracts from 107 U-937 or EBV-transformed B cells were prepared by a modification of the technique described by Dignam.32 After an initial 10-minute incubation with 0.1 mmol/L diisopropylfluorophosphate, cells were washed twice in ice-cold PBS and collected by centrifugation. Cells were resuspended in buffer A (50 mmol/L NaCl, 10 mmol/L HEPES, pH 8.0, 500 mmol/L sucrose, 1 mmol/L EDTA, 0.5 mmol/L spermidine, 0.15 mmol/L spermine, and 0.2% Triton X-100), and nuclei were collected by centrifugation. The nuclear pellet was washed in buffer B (50 mmol/L NaCl, 10 mmol/L HEPES pH 8.0, 25% glycerol, 0.1 mmol/L EDTA, 0.5 mmol/L spermidine, 0.15 mmol/L spermine), before being resuspended in buffer C (350 mmol/L NaCl, 10 mmol/L HEPES, pH 8.0, 25% glycerol, 0.1 mmol/L EDTA, 0.5 mmol/L spermidine, and 0.15 mmol/L spermine). After a 30-minute incubation at 4°C, the nuclear debris was removed by centrifugation and the nuclear extracts were divided into aliquots and stored at −80°C. Protein concentration was determined by Bradford assay. Protease inhibitors and β-mercaptoethanol were added to all buffers immediately before use.
Approximately 0.5 ng of [γ-32P]ATP 5′-end labeled probe was mixed with 0.012 gel shift units of recombinant human p50 or 10 μg of nuclear extract in a total volume of 25 μL containing 25 mmol/L HEPES, pH 8.0, 1 mmol/L EDTA, 3.5 mmol/L spermidine, 6 mmol/L MgCl2, 100 mmol/L NaCl, 0.15% Nonidet P40, 10% glycerol, 10 mmol/L dithiothreitol, 1 mg/mL bovine serum albumin (Sigma), and 0.5 μg of poly(dIdC). After incubation at 20°C for 15 minutes, anti-p50 antibody (Santa Cruz Biotechnology), was added to specified samples as described in the text and after a further 10-minute incubation, the protein-bound and unbound probe were separated on a 6% native polyacrylamide gel in 0.5× TBE. Oligonucleotide probe names and sequences were: NF-κB cons (NFκB consensus site; obtained from Promega, Madison, WI), AGTTGAGGGGACTTTCCCAGGC; NF-κB cybb (CYBB distant 5′ NF-κB site), CGATAAG-GGGCTTTCCTGTTCA.
Band densitometry was analyzed by Imagemaker Total Lab V1.1 software (Pharmacia, Uppsala, Sweden), to provide relative quantification of NF-κB binding activity.
Phagocytosis of zymosan particles
U937 cells (2 × 106 cells/mL)—either wild-type, transfected with SSFV empty vector, or transfected with the IκB-S32A/S36A NF-κB inhibitor—were treated or not with IFN-γ (100U/mL) for 48 hours before incubation with Zymozan particles (2 × 107 particles/mL) at 37°C for 2 hours. Zymosan particles were opsonized or not with 10% human fresh serum for 30 minutes at 37°C before phagocytosis assays. The number of cells phagocytizing zymosan particles was determined by counting cells containing 2 or more zymosan particles in at least 5 fields (minimum of 200 cells) and was expressed as a percentage of the total number of U937 cells.
Statistics
Results are expressed as means plus or minus SD. Descriptive statistics were performed and comparisons among groups were established by the Mann-Whitney test. A P value less than .05 was considered significant. Phagocytosis assays were analyzed by 1-way ANOVA; P value less than .05 was considered significant.
Results
To test the functional effect of NF-κB inhibition on human myelomonocytic cells, we first examined superoxide release (a measure of phagocyte NADPH oxidase activity) by the U937 cell line, in the presence of NF-κB inhibitors, and by U937 subclones transfected with an IκB S32A/S36A “super-repressor” construct or its empty vector. As shown in Figure 1A, the NF-κB inhibitors dexamethasone (1 μmol/L) and gliotoxin (10 μmol/L) inhibited superoxide release by U937 cells induced to differentiate by IFN-γ and TNF-α (P < .05 in all situations; n = 8, Mann-Whitney test). Panel B shows similar results in cells stably transfected with the empty vector pCMV3. Panel C demonstrates complete suppression of superoxide generation in a U937 subclone transfected with IκB S32A/S36A, including cells not exposed to any exogenous inhibitor (lane2). U937/IκB (S32A/S36A) cells were not further inhibited by dexamethasone or gliotoxin, because their level of superoxide generation was already at a background level similar to that of CGD cell lines.
Figure 1.

PMA-stimulated superoxide release by U937 cells. Wild-type U937 cells (A) and U937 cells transfected with empty vector (B) show that the NF-κB inhibitors dexamethasone and gliotoxin inhibited superoxide release by myelomonocytic cells induced to differentiate by IFN-γ and TNF-α (*P < .05 in all situations; n = 8, Mann-Whitney test). (C) Complete suppression of superoxide generation in a U937 subclone transfected with IκB S32A/S36A, including cells not exposed to any exogenous inhibitor (lane 2). In all panels: lanes 1, 3, and 5, undifferentiated U937 cells; lanes 2, 4, and 6, U937 cells induced to differentiate by IFN-γ and TNF-α; lanes 1 and 2, no inhibitor; lanes 3 and 4, dexamethasone (1 μmol/L); lanes 5 and 6, gliotoxin (10 μmol/L).
To determine whether the inhibitory effect occurred at the gene expression level, we measured transcript levels by real-time PCR amplification of CYBB, CYBA, NCF1, and NCF2 cDNA, compared with serially diluted standards. The patterns paralleled those observed for superoxide generation. As shown in Figure 2A,B, treatment with the NF-κB inhibitors dexamethasone and gliotoxin decreased the levels of CYBB mRNA in U937 cells induced to differentiate by IFN-γ and TNF-α (P < .05 in all situations; n = 8, Mann-Whitney test). This finding indicates that gliotoxin, which could also inhibit superoxide generation at the level of NADPH oxidase activity,33 functioned in these experiments primarily at the level of gene expression. Panel C shows the marked down-regulation of CYBB transcript steady-state levels in the U937 subclone transfected with IκB S32A/S36A, with or without further treatment with exogenous inhibitors.
Figure 2.

CYBB gene expression in U937 cells. Wild-type U937 cells (A) and U937 cells transfected with empty vector (B) show that the NF-κB inhibitors dexamethasone and gliotoxin decreased the levels of CYBB gene expression in myelomonocytic cells induced to differentiate by IFN-γ and TNF-α (P < .05 in all situations; n = 8, Mann-Whitney test). (C) Marked down-regulation of CYBB transcript levels in a U937 subclone transfected with IκB S32A/S36A, not affected by treatment with exogenous inhibitors. In all panels: lanes 1, 3, and 5, undifferentiated U937 cells; lanes 2, 4, and 6, U937 cells induced to differentiate by IFN-γ and TNF-α; lanes 1 and 2, no inhibitor; lanes 3 and 4, dexamethasone (1 μmol/L); lanes 5 and 6, gliotoxin (10 μmol/L).
We also tested the effects of more specific suppression of NF-κB function by an IκB “super-repressor” that has been mutated to prevent phosphorylation at the critical serines. Figure 3A,C shows the down-regulation of, respectively, CYBB and NCF1 gene expression in a U937 subclone transfected with the IκB S32A/S36A inhibitor compared with U937 wild-type cells or to cells transfected with empty vector (P < .05 in all situations; n = 4, Mann-Whitney test). Panel B shows that CYBA gene expression was not affected by transfection of U937 cells with empty vector or IκB S32A/S36A. Panel D shows that NCF2 gene expression was only partially inhibited in U937 cells by transfected with IκB S32A/S36A (P > .05 in all situations; n = 4, Mann-Whitney test).
Figure 3.

CYBB, CYBA, NCF-1, and NCF-2 gene expression in U937 cells induced to differentiate by IFN-γ and TNF-α. (A and C) respective down-regulation of CYBB and NCF-1 transcript levels in the U937 subclone transfected with IκB S32A/S36A, compared with the wild-type and U937 cells transfected with empty vector (*P < .05 in all situations; n = 4, Mann-Whitney test). (B) CYBA gene expression was not affected. (D) NCF-2 gene expression was not significantly inhibited by transfection with IκB S32A/S36A (P > .05 in all situations; n = 4, Mann-Whitney test).
To test whether similar suppression of the NADPH oxidase system would take place with a naturally occurring mutation of NEMO or IKBA, we examined superoxide generation and CYBB, CYBA, NCF1, and NCF2 transcript levels in EBV-transformed B cells from 2 patients with EDA-ID compared with cell lines from healthy donors and from patients with X-linked and autosomal recessive CGD. As shown in Figure 4, superoxide generation by EDA-ID cells was significantly lower than that in a normal B-cell line (P < .05, n = 6, Mann-Whitney test) and equivalent to that in the X-linked CGD cell line. Likewise, Figure 5 shows diminished levels of CYBB transcripts (P < .05, n = 6, Mann-Whitney test) in the EDA-ID cell lines, equivalent to that in the X-linked CGD cell line.
Figure 4.

Superoxide release by EBV-transformed B cell lines. Cell lines were derived as follows: lane 1, healthy donor; lane 2, a patient with X-linked CGD; and lanes 3 and 4, 2 patients with EDA-ID. Superoxide generation in EDA-ID cells was significantly lower than that in the normal cell line (P < .05, n = 6, Mann-Whitney test) and equivalent to that in the X-linked CGD cell line.
Figure 5.

CYBB gene expression in EBV-transformed B-cell lines. Cell lines were derived as follows: lane 1, healthy donor; lane 2, a patient with X-linked with CGD; and lanes 3 and 4, the designated patients with EDA-ID. CYBB gene expression by EDA-ID cells was significantly lower than that in the normal cell line (P < .05, n = 6, Mann-Whitney test) and equivalent to that in the X-linked CGD cell line.
Figure 6 shows CYBB, CYBA, NCF1, and NCF2 gene expression in EDA-cell lines compared with healthy control subjects and CGD cell lines from patients with X-linked CGD or with autosomal recessive CGD due to defects in p22phox, p47phox, and p67phox. Panel A confirms that CYBB gene expression in EDA-ID cells was significantly lower than that in the normal cell line or other CGD cell lines (P < .05, n = 4, Mann-Whitney test) and equivalent to that in the X-linked CGD cell line. Panel B shows that CYBA gene expression was affected only in the p22phox-deficient CGD cell line (P < 0.05, n = 4, Mann-Whitney test). Panel C shows that NCF1 gene expression by EDA-ID IκB S32I was significantly lower than that in the normal cell line or other CGD cell lines (P < .05, n = 4, Mann-Whitney test) and equivalent to that in the p47phox-deficient CGD cell line. Panel D shows that NCF2 gene expression was only partially affected in the EDA-ID IκB S32I cell line (P > 0.05, n = 4, Mann-Whitney test) but almost abolished in the p67phox-deficient CGD cell line (P < 0.05, n = 4, Mann-Whitney test). Thus, EDA-ID B cell lines with 2 different mutations affecting NF-κB function show impairments of superoxide generation and CYBB expression equivalent to that of a cell line from a patient with X-linked CGD. In addition, the EDA-ID IκB S32I cell line also shows an impairment of NCF1 gene expression, similarly to that observed in the U937 cell transfected with IκB S32A/S36A, suggesting that this gene is also sensitive to NF-κB impairment.
Figure 6.

CYBB, CYBA, NCF-1, and NCF-2 gene expression in EBV-transformed B-cell lines. Cell lines were derived as follows: lane 1, healthy donor; lane 2, X-linked CGD; lane 3, A22-CGD; lane 4, A47-CGD; lane 5, A67-CGD; lane 6, EDA-ID IκB S32I; lane 7, EDA-ID NEMO/IKKγ W420X. (A) CYBB gene expression in EDA-ID cells was significantly lower than that in the normal cell line or other CGD cell lines (P < .05, n = 4, Mann-Whitney test) and equivalent to that in the X-linked CGD cell line. (B) CYBA gene expression was only affected in the A22 CGD cell line (P < .05, n = 4, Mann-Whitney test). (C) NCF-1 gene expression by EDA-ID IκB S32I was significantly lower than that in the normal cell line or other CGD cell lines (P < .05, n = 4, Mann-Whitney test) and equivalent to that in the A47 CGD cell line. (D) NCF-2 gene expression was only partially affected in the EDA-ID IκB S32I cell line (P > .05, n = 4, Mann-Whitney test) but almost abolished in the A67 CGD cell line (P < .05, n = 4, Mann-Whitney test).
To investigate the binding activity of NF-κB to CYBB gene, we performed electrophoretic mobility shift assays (EMSA) with wild-type or transfected U937 cells and EBV-transformed B-cell lines from a healthy donor, an X-linked CGD patient, and the patients with 2 EDA-ID. Figure 7A shows that NF-κB, immunoprecipitated by antibody to p50, specifically bound to a radiolabeled 22-bp oligonucleotide representing the distant upstream NF-κB CYBB site. The left and middle groups of lanes show that nuclear extract from U937 and U937 pMCV3 cells, differentiated with IFN-γ and TNFα, bound significantly more probe than U937 IκB S32A/S36A cells (50-fold mean increase, n = 6, P < .05, Mann Whitney test). Dexamethasone and gliotoxin inhibited NF-κB binding to the CYBB site in U937 and U937 pMCV3 cells differentiated with INF-γ and TNFα (5-fold mean inhibition for dexamethasone and 10-fold mean inhibition for gliotoxin, n = 6, P < .05, Mann-Whitney test), confirming the inhibitory effect of these drugs on NF-κB regulation of CYBB expression.
Figure 7.

Electrophoretic mobility shift assay of NF-κB binding activity in U937 cells and EBV-transformed B-cell lines. (A) In wild-type (left panels) and pMCV3-transfected (middle panels) U937 cells, induction by IFN-γ and TNF-α caused an increase in binding activity of p50/Rel to an oligonucleotide representing the distant upstream NF-κB site in the CYBB gene. Binding activity was inhibited by dexamethasone (DEXA) or gliotoxin (GTX). No NF-κB binding activity was observed in U937 transfected with the super-inhibitor IκB S32A/S36A, regardless of pharmacological treatment. Representative image, n = 6. (B) Cell lines were derived as follows: lane 1, healthy donor; lane 2, a patient with CGD; and lanes 3 and 4, 2 patients with EDA-ID. Normal B-cell nuclei showed considerable binding activity of p50/Rel to an oligonucleotide representing the distant upstream NF-κB site in the CYBB gene, as also observed in the B-cell line from a patient with X-linked CGD. Reduced formation of the NF-κB complex was observed in B-cell lines derived from the patients with EDA-ID. Representative image, n = 4.
The specificity of this binding was demonstrated by competition by a 100-fold excess of unlabeled CYBB NF-κB oligonucleotide, and of NF-κB consensus oligonucleotide (n = 6; data not shown). The recognition sequence of the NF-κB consensus site differs by only 1 base pair from the CYBB site (see sequences in “Methods”). In addition, an unrelated transcription factor binding site (AP-1) fails to compete for protein binding (data not shown). Figure 7B shows that p50 binds to the upstream NF-κB site of the CYBB gene in EBV-transformed B cells from healthy donors and the CGD patient (lanes 1 and 2) but not from either of the EDA-ID patient cell lines, both of which show severe impairment of the NF-κB pathway (n = 4).
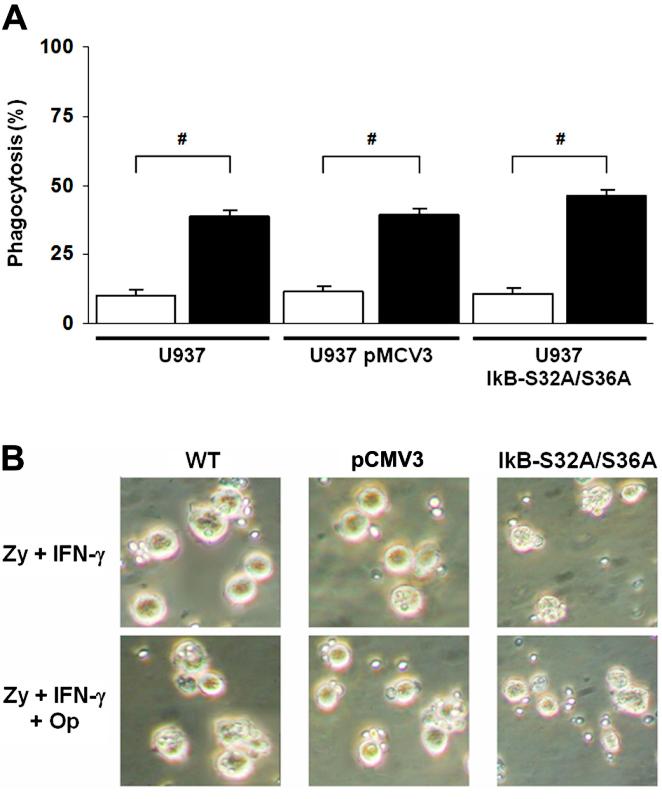
To test whether inhibition of the NF-κB pathway (or the transfection procedure) caused a general impairment of other cell functions, we examined the ability of U937 cells to ingest zymosan particles. Wild-type U937 cells and cells transfected with empty vector or with the IκB-S32A/S36A NF-κB inhibitor all showed similar phagocytic activity of zymosan particles (Figure S1; P < .01, 1-way analysis of variance [ANOVA], n = 3). Opsonizing the zymosan with human fresh serum caused a significant increase in the phagocytic activity of U937 cells with maintenance of normal function in the inhibitor-transfected cells (Figure S2; P < .05, 1-way ANOVA, n = 3).
Discussion
NF-κB is an essential transcription factor for multiple genes related to the immune response and development.12,14 Our previous studies of dexamethasone, a multifunctional steroid hormone that inhibits NF-κB function among many other effects, demonstrated inhibition of the phagocyte NADPH oxidase system at the transcriptional level (CYBB and NCF1 genes) in THP-1 myelomonocytic cells.16 We hypothesized that NF-κB could be a key element in this inhibitory effect. The current study investigated the role of NF-κB in superoxide release and the expression of the CYBB gene, encoding the gp91phox component of NADPH oxidase, in U937 myelomonocytic cells.
Our results show that the NF-κB inhibitors dexamethasone and gliotoxin diminished both superoxide release and CYBB gene expression, confirming our previous studies in a different cell line.16 In addition, dexamethasone and gliotoxin prevented NF-κB binding to the consensus region as demonstrated by EMSA assays (NF-κB/p50/DNA consensus binding sequence).
After the results using pharmacologic inhibitors on U937 cells, we further analyzed the interaction between NF-κB and the CYBB, CYBA, NCF1, and NCF2 genes, using the model of U937 cells transfected with a mutated IκB S32A/S36A gene, which cannot be phosphorylated at the 2 target serines and hence avoids proteosome degradation. This modified gene acts as a super-inhibitor of NF-κB activity.20 The same parameters were evaluated in cells with an empty pCMV3 vector as a negative control. These experiments showed that cells transfected with the super-inhibitor demonstrated diminished respiratory burst activity, CYBB and NCF1 gene expression, comparable with results with dexamethasone.
To confirm the suppression of NF-κB by IκB S32A/S36A, we performed gel shift assays using U937 cells with or without the mutated gene. In cells stimulated with IFN-γ and TNF-α and transfected with the super-inhibitor, there was no detectable binding activity of NF-κB to an oligonucleotide probe representing its consensus binding site, confirming that the transcription factor was not translocated to the nucleus. This binding activity was also inhibited by dexamethasone and gliotoxin, coincident with the inhibitory effect of these drugs on superoxide release and CYBB expression. These findings provide a functional basis for the addition of NF-κB as a new transcriptional element in the cast of proteins that take part on the regulation of CYBB gene expression.
Our findings in human myelomonocytic cells confirm and extend the previous report by Anrather et al18 in a murine model, in which they observed a lack of NAPDH oxidase activity in leukocytes, fibroblasts, and neural cells after overexpression of IκBα or knockout of p65/RelA. We also wished to test the relevance of the impairment of the NAPDH oxidase activity caused by natural defects in the NF-κB pathway, particularly whether NF-κB dysfunction would lead to a defect in the ability of the immune system to prevent infections.
Primary defects in the NADPH oxidase system interfere with the microbicidal activity of phagocytic cells, leading to the phenotype of CGD. On the other hand, defects in components of NF-κB activation can result in a heterogeneous group of immunodeficiencies.34–37 One form of defective NF-κB activation derives from hypomorphic mutations in genes encoding protein kinases responsible for the phosphorylation of IκB in the IκB kinase complex by the regulatory unit NEMO (NF-κB essential modulator). The resultant clinical syndrome of EDA-ID may be inherited either as an X-linked Mendelian recessive trait or by autosomal recessive or dominant inheritance.34–37 We included in this study a case of autosomal dominant EDA-ID derived from a mutation affecting the IκBα molecule at serine 32, similar to the in vitro construct used in the present study.30
EBV-transformed B cells mimic genetic disorders of the NADPH oxidase,22,23 and provide a useful model system for testing its molecular defects. The current study used B-cell lines from 2 patients with EDA-ID to examine the effects of inherited defects in NF-κB activation on the function of the NADPH oxidase. Fresh blood cells were not available from these kindreds, because the X-linked EDA-ID patient carrying the X420W NEMO mutation is deceased, and the patient with autosomal dominant EDA-ID carrying the S32I IκBα mutation has received a hematopoietic stem cell transplant.29 We compared superoxide release in 2 EDA-ID cell lines, compared with EBV-transformed B cells from a healthy control subject and from patients with X-linked, A22, A47, and A67 CGD. One patient with EDA-ID has X-linked disease caused by a hemizygous hypomorphic mutation in the gene encoding NEMO/IKKγ (W420X), the regulatory subunit of the IκB kinase (IKK) complex,27 leading to impairment of NF-κB activation in this patient's cells. The other patient has an autosomal-dominant form of EDA-ID associated with a heterozygous hypermorphic missense mutation (S32I) at serine 32 of IκBα.30 This gain-of-function mutation enhances the inhibitory capacity of IκBα by preventing its phosphorylation and degradation, resulting in impaired NF-κB activation.
Experiments performed with EBV-transformed B cells from the patients with EDA-ID showed decreased expression of the CYBB gene and consequently impaired respiratory burst, similar to the biochemical features of cells from patients with X-linked CGD.1,25 In addition the EDA-ID IκB S32I cell line showed diminished expression of NCF1, similar to a p47phox-deficient CGD cell line, confirmed the findings in the IκB S32A/S36A transfected U937 cell line. These results suggest an additional mechanism for the susceptibility to infections in EDA-ID patients,27,30 because the molecular defect in NF-κB activation also appears to affect cells of the myeloid lineage.
Pneumococcus is by far the most common infecting agent in EDA-ID, found in more than 80% of patients,27,30,38 whereas pneumococcus and other catalase-positive organisms are not associated with CGD.38 On the other hand, staphylococcus is both the second most common causative agent in patients with EDA-ID and the most frequent pathogen in patients with CGD. Patients with EDA-ID also show increased susceptibility to infection by mycobacteria.27,28,39 X-linked EDA-ID due to a hypomorphic mutation in NEMO was first reported in a boy who died of miliary tuberculosis,40 but patients with EDA-ID present more often with less virulent mycobacteria, such as Mycobacterium avium complex.39 Patients with CGD are susceptible to Mycobacterium tuberculosis41–45 and, in regions that use immunizations with bacille Calmette-Guérin (BCG), frequently present with adverse reactions to the organism.41–43,46
The current studies suggest that common defects in NADPH oxidase activity may provide a basis for the clinical overlap between CGD and EDA-ID. The differences in phenotype probably occur because NF-κB is a ubiquitous transcription factor in the immune system, involved in the expression of genes related not only to innate immunity but also to the acquired immune system. However, because the current medical literature includes descriptions of about 3000 patients with CGD, but only approximately 40 patients with EDA-ID, the comparison of clinical phenotypes can only be speculative at this time. It is not yet known whether key pathogens in CGD threaten most patients with EDA-ID or only a subset with specific mutations. In particular, staphylococcal disease, which is common in both patient groups, may in part reflect impaired respiratory burst function. This hypothesis will be tested in future studies of phagocytic function in leukocytes from patients with NEMO mutations of diverse severity.
NF-κB was not detected in the nuclei of EBV-transformed B cells from patients with EDA-ID but was present in nuclei of EBV-transformed B cells from patients with X-CGD. This result was expected, because the 2 entities derive from different molecular mechanisms. In X-CGD, an intrinsic defect in the gp91phox protein directly compromises NADPH oxidase activity. The absence of NF-κB in the nuclei of EDA-ID B cells, together with the decreased expression of CYBB, confirms the hypothesis that NF-κB plays an important role in the control of CYBB gene transcription, not only in human myelomonocytic cells but also in a primary immunodeficiency disorder. Furthermore, NF-κB dysfunction also affects NCF1 gene expression in this model system, providing a second possible reason for defective respiratory burst activity in patients with EDA-ID.
We conclude that proper binding of NF-κB is necessary for human CYBB and NCF1 gene expression and NADPH oxidase activity. Defects in this pathway found in certain forms of ectodermal dysplasia caused by mutations affecting the NF-κB pathway may lead to a CGD-like functional defect in myeloid cells, in addition to the known myeloid and lymphoid immune defects and may contribute to the severe immunodeficiency. Future studies examining primary phagocytes from EDA-ID patients for respiratory burst and bactericidal activity will help to correlate specific NF-κB pathway mutations with biochemical defects, as well as with agents causing infections.
Supplementary Material
Acknowledgments
This work was supported by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico), FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo) grant 06/52483-1, and US National Institutes of Health grant DK54369.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.L.-B. designed research, performed research, collected data, analyzed and interpreted data, and performed statistical analysis. C. Prando performed research, collected data, analyzed and interpreted data, and performed statistical analysis. J.B. contributed vital new reagents and collected data. W.C.A.-F. performed research, collected, analyzed, and interpreted data, and performed statistical analysis. P.V.S.P. collected, analyzed, and interpreted data. J.R. performed research, collected data, and analyzed and interpreted data. C. Padden performed research and collected, analyzed and interpreted data. J.-L.C. contributed vital new reagents and analytical tools, and collected, analyzed and interpreted data. P.E.N. designed research, contributed vital new reagents and analytical tools, analyzed and interpreted data, and revised the manuscript. A.C.-N. designed research, analyzed and interpreted data, performed statistical analysis, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Peter E. Newburger, MD, University of Massachusetts Medical School, LRB 404, 364 Plantation Street, Worcester MA 01605; e-mail: peter.newburger@umassmed.edu.
References
- 1.Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol. 2004;122:277–291. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- 3.Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Curr Opin Immunol. 2003;15:578–584. doi: 10.1016/s0952-7915(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 4.Rae J, Newburger PE, Dinauer MC, et al. X-Linked chronic granulomatous disease: Mutations in the CYBB gene encoding the gp91-phox component of respiratory-burst oxidase. Am J Hum Genet. 1998;62:1320–1331. doi: 10.1086/301874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skalnik DG, Dorfman DM, Perkins AS, et al. Targeting of transgene expression to monocyte/macrophages by the gp91-phox promoter and consequent histiocytic malignancies. Proc Natl Acad Sci U S A. 1991;88:8505–8509. doi: 10.1073/pnas.88.19.8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindsey S, Huang W, Wang H, et al. Activation of SHP2 protein-tyrosine phosphatase increases HoxA10-induced repression of the genes encoding gp91(PHOX) and p67(PHOX). J Biol Chem. 2007;282:2237–2249. doi: 10.1074/jbc.M608642200. [DOI] [PubMed] [Google Scholar]
- 7.Bei L, Lu Y, Eklund EA. HOXA9 activates transcription of the gene encoding gp91Phox during myeloid differentiation. J Biol Chem. 2005;280:12359–12370. doi: 10.1074/jbc.M408138200. [DOI] [PubMed] [Google Scholar]
- 8.Skalnik DG. Transcriptional mechanisms regulating myeloid-specific genes. Gene. 2002;284:1–21. doi: 10.1016/s0378-1119(02)00387-6. [DOI] [PubMed] [Google Scholar]
- 9.Li SL, Valente AJ, Zhao SJ, Clark RA. PU. 1 is essential for p47(phox) promoter activity in myeloid cells. J Biol Chem. 1997;272:17802–17809. doi: 10.1074/jbc.272.28.17802. [DOI] [PubMed] [Google Scholar]
- 10.Marden CM, Stefanidis D, Cunninghame-Graham DS, Casimir CM. Differentiation-dependent up-regulation of p47(phox) gene transcription is associated with changes in PU. 1 phosphorylation and increased binding affinity. Biochem Biophys Res Commun. 2003;305:193–202. doi: 10.1016/s0006-291x(03)00727-7. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. 2006;25:6706–6716. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 13.Pennington KN, Taylor JA, Bren GD, Paya CV. IkappaB kinase-dependent chronic activation of NF-kappaB is necessary for p21(WAF1/Cip1) inhibition of differentiation-induced apoptosis of monocytes. Mol Cell Biol. 2001;21:1930–1941. doi: 10.1128/MCB.21.6.1930-1941.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moynagh PN. The NF-kappaB pathway. J Cell Sci. 2005;118:4589–4592. doi: 10.1242/jcs.02579. [DOI] [PubMed] [Google Scholar]
- 15.D'Acquisto F, May MJ, Ghosh S. Inhibition of nuclear factor kappa B (NF-B): an emerging theme in anti-inflammatory therapies. Mol Interv. 2002;2:22–35. doi: 10.1124/mi.2.1.22. [DOI] [PubMed] [Google Scholar]
- 16.Condino-Neto A, Whitney C, Newburger PE. Dexamethasone but not indomethacin inhibits human phagocyte nicotinamide adenine dinucleotide phosphate oxidase activity by down-regulating expression of genes encoding oxidase components. J Immunol. 1998;161:4960–4967. [PubMed] [Google Scholar]
- 17.Gauss KA, Nelson-Overton LK, Siemsen DW, et al. Role of NF-kappaB in transcriptional regulation of the phagocyte NADPH oxidase by tumor necrosis factor-alpha. J Leukoc Biol. 2007;82:729–741. doi: 10.1189/jlb.1206735. [DOI] [PubMed] [Google Scholar]
- 18.Anrather J, Racchumi G, Iadecola C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem. 2006;281:5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- 19.Lien LL, Lee YS, Orkin SH. Regulation of the myeloid-cell-expressed human gp91-phox gene as studied by transfer of yeast artificial chromosome clones into embryonic stem cells: Suppression of a variegated cellular pattern of expression requires a full complement of distant cis elements. Mol Cell Biol. 1997;17:2279–2290. doi: 10.1128/mcb.17.4.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Asin S, Taylor JA, Trushin S, Bren G, Paya CV. Ikappakappa mediates NF-kappaB activation in human immunodeficiency virus-infected cells. J Virol. 1999;73:3893–3903. doi: 10.1128/jvi.73.5.3893-3903.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pahl HL, Krauss B, Schulze-Osthoff K, et al. The immunosuppressive fungal metabolite gliotoxin specifically inhibits transcription factor NF-kappaB. J Exp Med. 1996;183:1829–1840. doi: 10.1084/jem.183.4.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Condino-Neto A, Newburger PE. NADPH oxidase activity and cytochrome b558 content of human Epstein-Barr-virus-transformed B lymphocytes correlate with expression of genes encoding components of the oxidase system. Arch Biochem Biophys. 1998;360:158–164. doi: 10.1006/abbi.1998.0958. [DOI] [PubMed] [Google Scholar]
- 23.Volkman DJ, Buescher ES, Gallin JI, Fauci AS. B cell lines as models for inherited phagocytic diseases: superoxide generation in chronic granulomatous disease and granules in Chediak-Higashi syndrome. J Immunol. 1984;133:3006–3009. [PubMed] [Google Scholar]
- 24.Dusi S, Nadalini KA, Donini M, et al. Nicotinamide-adenine dinucleotide phosphate oxidase assembly and activation in EBV-transformed B lymphoblastoid cell lines of normal and chronic granulomatous disease patients. J Immunol. 1998;161:4968–4974. [PubMed] [Google Scholar]
- 25.Agudelo-Flórez P, Prando-Andrade CC, Lopez JA, et al. Chronic granulomatous disease in Latin American patients: clinical spectrum and molecular genetics. Pediatr Blood Cancer. 2006;46:243–252. doi: 10.1002/pbc.20455. [DOI] [PubMed] [Google Scholar]
- 26.Chanock SJ, Barrett DM, Curnutte JT, Orkin SH. Gene structure of the cytosolic component phox-47 and mutations in autosomal recessive chronic granulomatous disease. Blood. 1991;78:165a. [Google Scholar]
- 27.Döffinger R, Smahi A, Bessia C, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet. 2001;27:277–285. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 28.Dupuis-Girod S, Corradini N, Hadj-Rabia S, et al. Osteopetrosis, lymphedema, anhidrotic ectodermal dysplasia, and immunodeficiency in a boy and incontinentia pigmenti in his mother. Pediatrics. 2002;109:e97. doi: 10.1542/peds.109.6.e97. [DOI] [PubMed] [Google Scholar]
- 29.Dupuis-Girod S, Cancrini C, Le Deist F, et al. Successful allogeneic hemopoietic stem cell transplantation in a child who had anhidrotic ectodermal dysplasia with immunodeficiency. Pediatrics. 2006;118:e205–e211. doi: 10.1542/peds.2005-2661. [DOI] [PubMed] [Google Scholar]
- 30.Courtois G, Smahi A, Reichenbach J, et al. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplalia and T cell immunodeficiency. J Clin Invest. 2003;112:1108–1115. doi: 10.1172/JCI18714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Condino-Neto A, Newburger PE. Interferon-gamma improves splicing efficiency of CYBB gene transcripts in an interferon-responsive variant of chronic granulomatous disease due to a splice site consensus region mutation. Blood. 2000;95:3548–3554. [PubMed] [Google Scholar]
- 32.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishida S, Yoshida LS, Shimoyama T, et al. Fungal metabolite gliotoxin targets flavocytochrome b558 in the activation of the human neutrophil NADPH oxidase. Infect Immun. 2005;73:235–244. doi: 10.1128/IAI.73.1.235-244.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishikomori R, Akutagawa H, Maruyama K, et al. X-linked ectodermal dysplasia and immunodeficiency caused by reversion mosaicism of NEMO reveals a critical role for NEMO in human T-cell development and/or survival. Blood. 2004;103:4565–4572. doi: 10.1182/blood-2003-10-3655. [DOI] [PubMed] [Google Scholar]
- 35.Kim S, La Motte-Mohs RN, Rudolph D, Zuniga-Pflucker JC, Mak TW. The role of nuclear factor-kappaB essential modulator (NEMO) in B cell development and survival. Proc Natl Acad Sci U S A. 2003;100:1203–1208. doi: 10.1073/pnas.0337707100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puel A, Picard C, Ku CL, Smahi A, Casanova JL. Inherited disorders of NF-kappaB-mediated immunity in man. Curr Opin Immunol. 2004;16:34–41. doi: 10.1016/j.coi.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 37.Puel A, Reichenbach J, Bustamante J, et al. The NEMO mutation creating the most-upstream premature stop codon is hypomorphic because of a reinitiation of translation. Am J Hum Genet. 2006;78:691–701. doi: 10.1086/501532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winkelstein JA, Marino MC, Johnston RB, Jr, et al. Chronic granulomatous disease-Report on a national registry of 368 patients. Medicine. 2000;79:155–169. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 39.Filipe-Santos O, Bustamante J, Chapgier A, et al. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol. 2006;18:347–361. doi: 10.1016/j.smim.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 40.Frix CD, III, Bronson DM. Acute miliary tuberculosis in a child with anhidrotic ectodermal dysplasia. Pediatr Dermatol. 1986;3:464–467. doi: 10.1111/j.1525-1470.1986.tb00652.x. [DOI] [PubMed] [Google Scholar]
- 41.Alcaïs A, Fieschi C, Abel L, Casanova JL. Tuberculosis in children and adults: two distinct genetic diseases. J Exp Med. 2005;202:1617–1621. doi: 10.1084/jem.20052302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lau YL, Chan GC, Ha SY, Hui YF, Yuen KY. The role of phagocytic respiratory burst in host defense against Mycobacterium tuberculosis. Clin Infect Dis. 1998;26:226–227. doi: 10.1086/517036. [DOI] [PubMed] [Google Scholar]
- 43.Movahedi M, Aghamohammadi A, Rezaei N, et al. Chronic granulomatous disease: a clinical survey of 41 patients from the Iranian primary immunodeficiency registry. Int Arch Allergy Immunol. 2004;134:253–259. doi: 10.1159/000078774. [DOI] [PubMed] [Google Scholar]
- 44.Bustamante J, Aksu G, Vogt G, et al. BCG-osis and tuberculosis in a child with chronic granulomatous disease. J Allergy Clin Immunol. 2007;120:32–38. doi: 10.1016/j.jaci.2007.04.034. [DOI] [PubMed] [Google Scholar]
- 45.Barese C, Copelli S, Zandomeni R, et al. X-linked chronic granulomatous disease: first report of mutations in patients of Argentina. J Pediatr Hematol Oncol. 2004;26:656–660. doi: 10.1097/01.mph.0000139455.29962.be. [DOI] [PubMed] [Google Scholar]
- 46.Mouy R, Fischer A, Vilmer E, Seger RA, Griscelli C. Incidence, severity, and prevention of infections in chronic granulomatous disease. J Pediatr. 1989;114:555–560. doi: 10.1016/s0022-3476(89)80693-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}