

Abstract
Chromosome abnormalities are frequently associated with cancer development. The 8;21(q22;q22) chromosomal translocation is one of the most common chromosome abnormalities identified in leukemia. It generates fusion proteins between AML1 and ETO. Since AML1 is a well-defined DNA-binding protein, AML1-ETO fusion proteins have been recognized as DNA-binding proteins interacting with the same consensus DNA-binding site as AML1. The alteration of AML1 target gene expression due to the presence of AML1-ETO is related to the development of leukemia. Here, using a 25-bp random double-stranded oligonucleotide library and a polymerase chain reaction (PCR)-based DNA-binding site screen, we show that compared with native AML1, AML1-ETO fusion proteins preferentially bind to DNA sequences with duplicated AML1 consensus sites. This finding is further confirmed by both in vitro and in vivo DNA-protein interaction assays. These results suggest that AML1-ETO fusion proteins have a selective preference for certain AML1 target genes that contain multimerized AML1 consensus sites in their regulatory elements. Such selected regulation provides an important molecular mechanism for the dysregulation of gene expression during cancer development.
Introduction
The t(8;21)(q22;q22) translocation is associated with nearly 40% of cases of the French-American-British (FAB) M2 subtype of acute myeloid leukemia (AML) and 8% to 20% of all cases of AML.1–4 The AML1-ETO fusion protein generated due to this translocation contains the N-terminus of AML1 (also known as RUNX1, CBFα2, PEBP2αB) and almost full-length ETO (also known as MTG8).5,6 The AML1 portion of AML1-ETO contains the Drosophila melanogaster protein Runt homology domain, which is essential for binding to DNA with its heterodimerization partner CBFβ, and lacks the C-terminal region required for transcriptional regulation.7–13 AML1 modulates both growth and differentiation, and the analysis of Aml1 knockout mice demonstrates that AML1 is a crucial factor for definitive hematopoiesis.14,15 In vitro studies showed that AML1 regulates the expression of growth factor receptors, cytokines, and enzymes involved in hematopoiesis.16–21 More recently, AML1 was shown to have a role in the self-renewal potential and expansion of hematopoietic stem cells.22–24 ETO contains 4 Nervy homology regions (NHR1-4) (reviewed by Peterson et al25). NHR1 has sequence homology to TAF110 (TATA box binding protein–associated factor 110) and has been shown to associate with E proteins and Gfi-1. NHR2 contains a hydrophobic amino acid heptad repeat (HHR) and is critical for interactions with ETO family members (MTG8, MTG16, and MTGR1), oligomerization of AML1-ETO, and protein-protein interaction with proteins such as mSin3A, Gfi-1, BCL6, HDAC1, and HDAC3. NHR3 includes a predicted coiled-coil structure with a potential PKA-anchoring protein (AKAP) function. NHR4 also known as the myeloid-Nervy-DEAF1 (MYND) homology domain contains 2 zinc chelating centers and mediates protein-protein interactions with the corepressor complexes of nuclear receptor corepressor (NCoR) and silencing mediator for retinoid and thyroid hormone receptors (SMRT). Targeted disruption of Eto revealed an important role in the gastrointestinal system.26 The ETO portion of AML1-ETO was shown to recruit NCoR/SMRT-Sin3–histone deacetylase (HDAC) complexes, decreasing histone acetylation and chromatin accessibility at AML1 targets.27–29
Various AML1 consensus DNA-binding sequences have been reported as PuACCPuCA (TGT/CGGTT/C),30 TTTGCGGTTA/T,31 and PyGPyGGTPy (T/CGC/TGGTT/C).32 Furthermore, PCR-based random sequence screening with purified bacterially expressed GST-tagged AML1a (aa's 1-250) determined the AML1 consensus DNA-binding sequence as TGT/CGGT.7 Since AML1-ETO contains the AML1 DNA-binding domain, it is generally believed that AML1-ETO binds to the same DNA sequence as AML1.
Previously, we reported that although full-length AML1-ETO is not leukemogenic in mice, a C-terminal truncated version of this fusion protein (AML1-ETOtr) is highly leukemogenic.33 Moreover, we identified an alternatively spliced variant of AML1-ETO that includes ETO exon 9a and generates the fusion protein AML1-ETO9a that is almost identical to AML1-ETOtr and is also strongly leukemogenic.34 The major difference between AML1-ETO9a and the full-length AML1-ETO is that AML1-ETO9a is missing 178 amino acids from the C-terminal end of ETO that contain the NHR3 and NHR4 domains.25 Additional studies revealed that disruption of the NHR4 domain by deletion or point mutation of a cysteine residue in the zinc center motif transforms AML1-ETO into a leukemogenic protein (Ahn et al, manuscript in preparation). These findings, in addition to the general understanding that zinc finger domains are involved in protein-DNA interaction besides protein-protein interaction, led us to address whether full-length AML1-ETO binds to other DNA sequences besides the AML1 consensus binding site. Therefore, to characterize DNA-binding affinities for full-length AML1, AML1-ETO, or leukemogenic AML1-ETOtr/AML1-ETO9a, we screened DNA-binding sequences in a 25-bp random double-stranded oligonucleotide library. Although we did not identify any new AML1-ETO consensus binding sites, our results interestingly demonstrate that compared with AML1, the AML1-ETO fusion proteins preferentially bind DNA sequences containing duplicated AML1 consensus sites, suggesting that AML1-ETO fusion proteins have a selective preference for AML1 target genes with such multimerized AML1 consensus sites and this is likely a part of the mechanism by which they disrupt normal hematopoiesis and promote leukemogenesis.
Methods
Cell culture
HEK293T, Kasumi-1, K562, U937T, and U937T-AML1-ETO cells were cultured as previously described.34,35 3xFLAG-AML1-ETO9a stably transfected K562 cells were maintained with 4 μg/mL puromycin (Calbiochem/EMD, San Diego, CA).
Plasmid DNA construction
The cDNAs of human AML1b, AML1-ETO, AML1-ETO9a, AML1-ETOtr, or the NHR2 domain–deleted AML1-ETO were subcloned into pcDNA6-V5–6xHis expression vector (Invitrogen, Carlsbad, CA) with an N-terminal HA-tag. AML1-binding site containing basal TK promoter-luciferase reporter (pBAAE#35-TK-luc) and another luciferase reporter including a 2-kb fragment of the 5′ upstream region of the human separase gene (phSeparase-luc) were prepared as described in Document S1 (available on the Blood website; see the Supplemental Materials link at the top of the online article).
Screening of DNA-binding sequences
DNA-binding site selection was performed as described previously7 with some modifications. Please see Document S1 for details.
Nuclear protein preparation
Nuclear protein was prepared from vector-transfected 293T cells or normal cultured Kasumi-1 cells as described previously36 with some modifications. Please see Document S1 for details.
Electrophoretic mobility shift assays
N-terminal HA and C-terminal V5-6xHis-tagged AML1b, AML1-ETO, or AML1-ETO9a protein were prepared as nuclear extracts from 293T cells transfected with each expression construct. Electrophoretic mobility shift assays (EMSAs) were performed as described previously36,37 with some modifications. Please see Document S1 for details.
DNA affinity purification assay
Oligonucleotide-binding assay was performed as described previously38,39 with some modifications. Please see Document S1 for details.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was performed based on a previous publication40 with some modifications. Please see Document S1 for details.
Establishment of phSeparase-luc stable U937T-AML1-ETO or U937T transformants
phSeparase-luc stable U937T-AML1-ETO or U937T cells were generated by nucleofection using Cell Line Nucleofector Kit V (Amaxa, Gaithersburg, MD). Two selected polyclonal transfected pools were prepared. Please see Document S1 for details.
Luciferase reporter assay
Luciferase reporter assay was performed as described previously.38,41,42 Please see Document S1 for details.
Northern blot and reverse-transcription–PCR
Examination of mRNA level was carried out as described previously38,42,43 with some modifications. Please see Document S1 for details.
Western blotting analysis
Immunoblotting was performed as described previously38,41 with some modifications. Please see Document S1 for details.
Results
Screening DNA-binding sequences of AML1 and AML1-ETO
To analyze whether AML1-ETO and the leukemogenic mutant of AML1-ETOtr recognize other DNA-binding sequences in addition to the consensus AML1-binding site, we performed a site selection assay using a 25N random double-stranded oligonucleotide library and purified HA-tagged AML1, AML1-ETO, and AML1-ETOtr proteins (Figure 1A,B). After incubation with the oligonucleotide library and following extensive washes, the bound DNA sequences were amplified by PCR (Figure 1C) and this PCR product was used for repeated selection. After 4 rounds of selection, the bound DNA fragments were subcloned into a plasmid vector and their sequences were determined (Table S1). Most of our selected clones contained the AML1 consensus DNA-binding site, TGT/CGGT (underlined in Table S1), which was reported by Meyers et al.7 Further, no new AML1-ETO or AML1-ETOtr consensus DNA-binding sites were identified in our assay. Interestingly, compared with the DNA-binding sequences isolated with AML1, AML1-ETO and AML1-ETOtr preferentially bound DNA that contained 2 or 3 repeated AML1 DNA-binding motifs as shown in Table 1. The spaces between 2 AML1-binding sites varied from 0 to 14 bp among these clones (Table S1). Interestingly, as shown in Table S1, we also observed that AML1-ETOtr has a higher preference for the TGCGGT sequence (detected 85 times) compared with TGTGGT (detected 17 times). In addition, a binding selection procedure using GST–C-terminus–ETO, which is the portion included in AML1-ETO but lacking in AML1-ETOtr/9a, was performed. This protein was not able to bind specific DNA sequences, as multiple cycles of PCR amplification using the random 25-mer library gave a few clones without any specific pattern in their sequences. Thus there is no evidence to support the idea that the C-terminal portion of AML1-ETO directly binds to DNA.
Figure 1.

Strategy for screening of DNA-binding sequence for AML1, AML1-ETO, and AML1-ETOtr. (A) Schematic representation of HA-tagged proteins used for the screening of DNA-binding sequence. The Runt homology domain is indicated with a black box and 4 Nervy homology regions (NHRs) are shown as gray boxes. The predicted zinc finger motif in the C-terminal region of AML1-ETO is indicated with ▴. (B) The expression of AML1 (A), AML1-ETO (AE), and AML1-ETOtr (tr) in cell lysates was examined by immunoblotting with anti-HA antibody. Control (C) is the lysates from vector-transfected cells. (C) Target DNA sequence including 25N random sequence.  indicate the position of forward or reverse primers for PCR amplification, and boxes show the position of restriction enzyme digestion sites for subcloning.
indicate the position of forward or reverse primers for PCR amplification, and boxes show the position of restriction enzyme digestion sites for subcloning.
Table 1.
Consensus AML1 DNA-binding sites, TGCGGT (1) and TGTGGT (2), in the selected DNA sequences
| AML1 (A) | AML1-ETO (AE) | AML1-ETOtr (AEtr) | N-terminal AML1* | |
|---|---|---|---|---|
| (1) × 1 time | 20 | 9 | 7 | 13 |
| (2) × 1 time | 7 | 3 | 0 | 13 |
| (1) × 2-3 times | 14 | 25 | 33 | 0 |
| (2) × 2-3 times | 0 | 0 | 1 | 5 |
| (1) and (2) mixed | 14 | 17 | 9 | 1 |
| No site | 9 | 3 | 0 | 6 |
| No. of total clones | 64 | 57 | 50 | 38 |
| 1 site, % | 49 | 22 | 14 | 81 |
| 2-3 sites,† % | 51 | 78 | 86 | 19 |
The result of N-terminal AML1 is from Meyers et al.7
By Fisher exact test, the difference between AML1/N-terminal AML1 and AML1-ETO/ AML1-ETOtr is statistically significant (P = .001).
In vitro comparison of DNA-binding affinities of AML1 and AML1-ETO
To analyze the observed difference in DNA-binding affinity between AML1 and AML1-ETO, we prepared nuclear extracts from 293T cells transfected with expression constructs for HA-tagged AML1, AML1-ETO, or AML1-ETO9a (Figure 2A). EMSA analysis was performed with the human macrophage colony-stimulating factor (M-CSF) receptor FMS/CSF1R probe, containing one TGTGGT AML1-binding site. As reported previously,20 AML1 strongly bound the c-FMS probe (Figure 2B lane 2). On the other hand, the binding affinities of both AML1-ETO and AML1-ETO9a were weak (Figure 2B lanes 3,4). The specific bands detected in each sample were super shifted with the anti-HA antibody (Figure 2B lanes 5-7).
Figure 2.

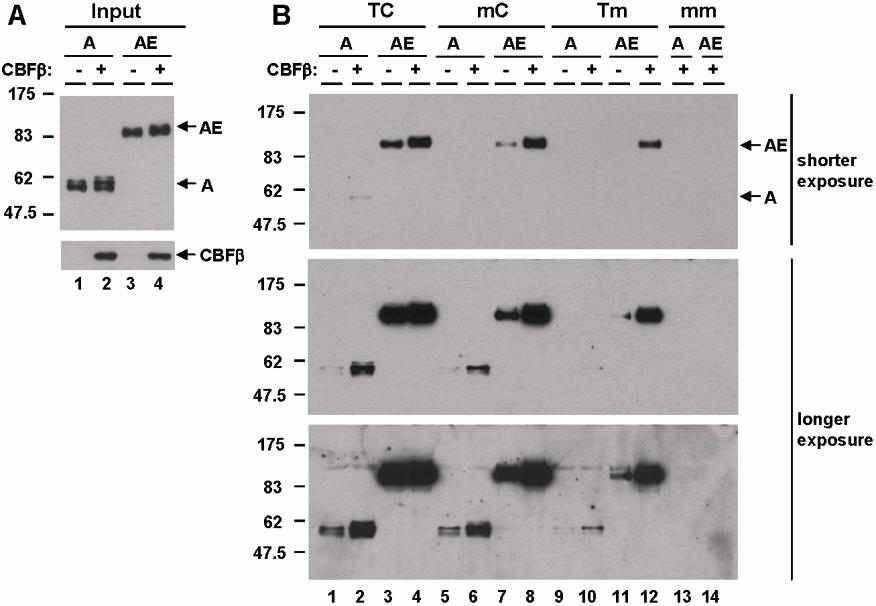
EMSA for comparison of DNA-binding activity. (A) Relative levels of AML1 (A), AML1-ETO (AE), and AML1-ETO9a (AE9a) protein expression in nuclear extracts are shown. Control (C) was prepared from 293T cells transfected with vector. show the position of AML1-, AML1-ETO (AE)–, and AML1-ETO9a (AE9a)–specific bands. (B) EMSA with human M-CSF receptor probe containing the TGTGGT consensus and nuclear extracts shown in panel A was performed. Anti-HA antibody was added to lanes 5 through 7 and supershifts were detected (◀). (C) EMSA with wild-type human separase (TC) probe and nuclear extracts shown in panel A was performed. Anti-HA antibody was added to lanes 5-8 and supershifts were detected (◀). (D) Schematic diagram of the human separase 5′ upstream region, exon 1, and exon 2. Two AML1 consensus sites are present with a space of 2 nucleotides (TGTGGTagTGCGGT) in the 5′-upstream region. Mutants of AML1 consensus sites used in this study are also listed. The −2 kb of 5′-upstream region (bold line) is fused with luciferase for promoter analysis in Figure 4. EMSA with wild-type (TC) and mutant (mC and Tm) human separase probes and nuclear extracts shown in panel A was performed.
Recently, we reported a function of AML1-ETOtr in the development of aneuploidy through the alteration of spindle checkpoint components.44 Specifically, in the presence of mitotic poisons, cells expressing AML1-ETOtr contain active/cleaved separase (extra spindle poles like 1 [ESPL1]) resulting in the cleavage of cohesin that is required for keeping the centrosome of sister chromatids intact. Analysis of the promoter region of the human separase gene identified a duplicated AML1 consensus TGTGGTagTGCGGT sequence in its 5′-upstream region (Figure 2D). Both AML1-ETO and AML1-ETO9a bound strongly to the human separase wild-type TC probe (5′-GAGTGGTGTGGTAGTGCGGTCGGGG-3′) compared with AML1 (Figure 2C lanes 2-4). These binding complexes were super shifted by the anti-HA antibody (Figure 2C lanes 6-8). To further study the importance of double binding sites for AML1-ETO binding, we designed mutant probes, as previously reported,7 that disrupt either one or both AML1-binding sites in the TC probe (Figure 2D). AML1-ETO has a higher binding affinity for the TC wild-type probe than for the mC probe, and we could not detect a clear band with the Tm probe on this gel (Figure 2D lanes 3, 6, and 9). AML1 showed a much weaker binding affinity than AML1-ETO with the TC probe. However, it has almost equal binding affinity to the TC wild-type and mC probes (Figure 2D lanes 2 and 5). The binding of AML1 to the Tm probe was also undetectable (Figure 2D lane 8), but we could see a faint band when we used 3-fold more AML1 protein (data not shown). On the other hand, even with 3-fold more AML1-ETO protein we could not detect any specific band for either AML1-ETO or AML1-ETO9a (data not shown). The lower binding affinity of AML1-ETO compared with AML1 to the sequence with one T site was also observed with c-fms probe in Figure 2B. Overall, EMSA analysis determined that the binding affinity of AML1-ETO for the TC double site was the highest; it became weaker with mC, and undetectable with Tm. On the other hand, AML1 showed a weaker binding affinity than that of AML1-ETO to the TC probes, with an equal affinity to both TC and mC probes and a lower affinity to the Tm probe.
Higher DNA-binding affinity of AML1-ETO for double AML1 sites compared with AML1
EMSA analysis is a one-dimensional assay to detect the binding of transcription factors to DNA. Since AML1-ETO is hypothesized to bind DNA as dimer/tetramer based on its structure data, we further examined its DNA-binding affinity in a 3-dimensional manner by performing DNA affinity purification assays using a biotinylated human separase probe, which can be isolated from a DNA-protein–binding reaction by streptavidin magnetic beads. We prepared nuclear extracts from 293T cells transfected with HA-tagged AML1 or AML1-ETO expression constructs, which showed a similar level of expression of these proteins (Figure 3A). The nuclear extracts were used for DNA affinity purification assays with wild-type and mutant human separase probes as illustrated in Figure 2D. AML1-ETO displayed a higher binding affinity than AML1 to the TC probe (Figure 3B lanes 5 and 6). The mutant CC probe with 2 C sites (Figure 2D) was also bound more strongly by AML1-ETO than AML1 (Figure 3B lanes 2 and 3). On the other hand, with the mC probe, the binding affinity of AML1-ETO became weak and almost equal to that of AML1 (Figure 3B lanes 8 and 9). The Tm probe again displayed no detectable binding to AML1-ETO and binding by AML1 was also weak, but still detectable after longer exposure (Figure 3B lanes 11 and 12). The double mutant mm probe did not bind to AML1 or AML1-ETO in the protein-DNA reaction (Figure 3B lanes 13 and 14). The overall binding affinity of AML1 among probes showed it to be almost equal among CC, TC, and mC probes, but low with the Tm probe (Figure 3C lanes 1-4). The binding affinity of AML1-ETO among these probes showed that it was high with the CC and TC probes, significantly lower with the mC probe, and undetectable with the Tm probe (Figure 3C lanes 6-9). These results are highly reproducible with independent sets of nuclear extracts. Furthermore, we also showed that the AML1 heterodimer partner CBFβ enhanced DNA binding of both AML1 and AML1-ETO and that the preference of AML1-ETO for double sites remained the same in the presence or absence of CBFβ (Figure S1).
Figure 3.

DNA-binding affinity assay. (A) Expression level of AML1 (A) and AML1-ETO (AE) was examined by immunoblotting with anti-HA antibody in nuclear extracts. Control (C) was prepared from 293T cells transfected with vector. (B) DNA-binding affinity assay using wild-type and mutant separase probes with nuclear extracts shown in panel A was performed. The protein binding was detected by immunoblotting with anti-HA antibody. show the position of both AML1 (A) and AML1-ETO (AE). (C) DNA affinity purification assay using wild-type and mutant separase probes with nuclear extracts shown in panel A was performed. AML1 reaction (10 μL) and AML1-ETO reaction (5 μL) were loaded. (D) DNA affinity purification assay with Kasumi-1 cells was performed. Input lane shows the relative expression of endogenous AML1 and AML1-ETO examined by immunoblotting with anti-AML1 antibody in nuclear extracts. The protein-binding activity of wild-type (TC) or mutant (mm) separase probes was examined by immunoblotting with anti-AML1 antibody and shown in IP lanes. (E) DNA affinity purification assay with Kasumi-1 cells using wild-type or mutant SPARC and PU.1 probes was performed. Input lane shows the relative expression of endogenous AML1 and AML1-ETO examined by immunoblotting with anti-AML1 antibody in nuclear extracts. The protein-binding activity of wild-type (AC for SPARC and CT for PU.1) or mutant (mm) probes was examined by immunoblotting with anti-AML1 antibody and shown in IP lanes. (F) Expression level of full-length (AE) and NHR2-deleted (AEΔ) AML1-ETO in nuclear extracts was examined by immunoblotting with anti-HA antibody (input, lanes 1 and 2). The results of DNA affinity purification assay using wild-type (TC) or mutant (mC) separase probes with full-length (lane 3) and NHR2-deleted (lanes 4-6) AML1-ETO are shown. show the position of both full-length (AE) and NHR2-deleted (AEΔ) AML1-ETO.
To analyze the binding affinity of endogenous AML1-ETO and AML1, we prepared nuclear extracts from Kasumi-1 cells, which are derived from a t(8;21) AML patient.45 Expression levels of AML1 and AML1-ETO protein were almost equal in the input sample (Figure 3D lane 1). After collection of biotinylated TC probe by streptavidin magnetic beads, more AML1-ETO was associated with the TC probe than AML1 (Figure 3D lane 2) and the control mm probe did not associate with the proteins (Figure 3D lane 3). Furthermore, oligonucleotides with a single AML1-binding site showed weaker interaction with AML1-ETO (Figure S2).
Thus, both the DNA affinity assay and EMSA analysis determined that the binding affinity of AML1-ETO to the TC or CC probes was the highest, it decreased with the mC probe, and it was almost undetectable with the Tm probe. On the other hand, compared with AML1-ETO, AML1 showed a weaker but equal binding affinity to the TC, CC, and mC probes, and an even weaker but detectable affinity for the Tm probe. Importantly, the higher binding affinity of AML1-ETO compared with AML1 was reproducible with endogenous proteins from the t(8;21)-positive Kasumi-1 cells.
To further test the difference in binding affinity, we used an oligonucleotide with double AML1-binding sites derived from our recently identified AML1-ETO target gene SPARC (secreted protein acidic and rich in cysteine/osteonectin/BM40).46 SPARC is a secreted calcium-binding matricellular glycoprotein and has roles in the regulation of cell adhesion, proliferation, tumorigenesis, metastasis, and efficient platelet production.47–49 The human SPARC gene contains an ACCACA13nTGCGGT sequence in its upstream regulatory promoter region. In addition, we also used an oligonucleotide with double AML1-binding sites from human PU.1 gene upstream enhancer that has the TGCGGT3nTGTGGT sequence.50 In DNA-binding affinity assays, endogenous AML1-ETO from Kasumi-1 cells showed a significantly higher binding affinity for both SPARC and PU.1 probes compared with AML1 (Figure 3E lanes 1 and 4). DNA-binding affinity of AML1-ETO for mutant probes of SPARC and PU.1 showed again that AML1-ETO–binding affinity was higher with wild-type double site probes (AC for SPARC and CT for PU.1) than mutant single-site probes (mC and Am for SPARC and Cm and mT for PU.1; data not shown). Furthermore, these results also showed that the spacing of 13 nucleotides in the SPARC oligonucleotide compared with 3 nucleotides in the PU.1 oligonucleotide between the 2 AML1-binding sites did not obviously affect the binding affinity of AML1-ETO for these 2 probes. In addition, the orientation of the AML1 consensus sites did not affect the protein-DNA complex of AML1-ETO with the SPARC probe, although most of our screening clones have double AML1-binding sites in the same direction (Table S1). In summary, endogenous AML1-ETO also has a stronger binding affinity for human SPARC and PU.1 regulatory sequences with duplicate AML1 consensus sites than AML1.
Recently the structure of the NHR2 domain of ETO was resolved and suggested to form tetramerized molecules of AML1-ETO.51 We therefore tested whether the NHR2 domain-deletion mutant of AML1-ETO influences its DNA-binding affinity. The DNA-binding affinity assay with human separase TC probe revealed that wild-type AML1-ETO has a much higher binding affinity than the NHR2-deleted AML1-ETO (Figure 3F lanes 3 and 4). More importantly, NHR2-deleted AML1-ETO did not show a significant difference of binding affinity between TC and mC (Figure 3F lanes 5 and 6). Thus these observations suggest that the NHR2 domain is instrumental in stabilizing the protein-DNA complex of AML1-ETO and in determining the preference of AML1-ETO for double AML1-binding sites.
AML1-ETO affects promoter activity in a double site–dependent manner
Next, to examine the effect of different binding sites on the transcriptional activity of AML1-ETO, we performed luciferase reporter assays. We used an oligonucleotide sequence containing 2 AML1-binding sites identified from the initial binding assay with AML1-ETO (clone no. 35) and cloned it upstream of a minimal TK promoter in a luciferase reporter construct to form pBAAE#35-TK-luc. The promoter activity was suppressed by both AML1-ETO and AML1-ETO9a, but not by AML1 (Figure 4A). The suppression was stronger by full-length AML1-ETO than by AML1-ETO9a. We then compared the effect of AML1-ETO on the TC wild-type and its mutant mC and Tm sequences, and observed that the suppression of promoter activity was more effective on the TC compared with the mC or Tm mutants (Figure 4B). Equal expression level of each protein was confirmed by immunoblotting (data not shown). Thus AML1-ETO demonstrated a strong effect on a promoter with double AML1-binding sites that was not induced by AML1. Furthermore, the effect of AML1-ETO was greater on the promoter containing double AML1-binding sites (TC) than the ones with a single AML1-binding site (mC and Tm).
Figure 4.

Promoter activity assay. (A) Luciferase reporter activity was examined with various amounts (nanograms) of pcDNA6-HA-AML1–, pcDNA6-HA-AML1-ETO–, or pcDNA6-HA-AML1-ETO9a–transfected 293T cells. pBAAE35-TK-luc (TC, 50 ng, wild type), pCMV5-CBFb 21.5 (150 ng), and pNull-renilla luciferase (5 ng) were cotransfected. The value of y-axis is a fold change of luciferase activity compared with vector alone. The results are presented as the mean value of 3 independent experiments; error bars represent standard deviation. The almost equal level of protein expression induced from each amount of plasmid DNA was confirmed by immunoblotting with anti-HA antibody (data not shown). (B) Luciferase reporter assay was performed with 293T cells transfected with 50 ng wild-type (TC) or mutant (mC and Tm) of pBAAE35-TK-luc, 150 ng pCMV5-CBFb 21.5, and 5 ng pNull-renilla luciferase and increasing amount of pcDNA6-HA-AML1-ETO expression plasmid. (C) U937T-AML1-ETO cells were stably transfected with wild-type phSeparase-luc (TC) or the constructs including mutated AML1-binding sites (mC, Tm, mm). Two independent cell pools stably transfected with each construct were used in this study. The value of y-axis is a fold change of luciferase activity compared with day 0 of tetracycline (tet) withdrawal. The results are presented as the mean value of 3 independent experiments; error bars represent standard deviation. At day 1 of the tet withdrawal, statistically significant P values compared with mm control (*t test, P < .001 for TC, mC, and Tm) are indicated. At day 2 of the tet withdrawal, statistically significant P value compared with mm control (**t test, P = .005 for TC) is indicated. The expression of AML1-ETO was controlled by tet withdrawal, and protein expression was confirmed by Western blotting using anti-AML1 antibody (◀). Ponceau staining shows the relative amount of protein loading. As a negative control, all of these phSeparase-luc constructs were also stably transfected into parental U937T cells and no change of luciferase activity was detected after tet withdrawal (data not shown).
As shown in Figure 2D, the promoter of the human separase gene contains a duplicated AML1 consensus sequence TGTGGTagTGCGGT. We made human separase promoter-luciferase constructs with either wild-type promoter (TC) or AML1-binding site mutated promoter (mC, Tm, and mm). These reporter constructs were stably transfected into U937T-AE cells, which express AML1-ETO upon withdrawing tetracycline from the culture medium.35 Pools of transfected cells were used to examine the promoter activity in the presence and absence of AML1-ETO (Figure 4C). Following tetracycline removal for 1 day, AML1-ETO expression was clearly detected by Western blotting, and TC, mC, and Tm, but not mm, cells showed a decrease in promoter activity. At 2 days of tetracycline withdrawal, cell pools with all 4 reporter constructs showed a decrease in promoter activity. As we reported previously,35 AML1-ETO induction causes cell growth arrest leading to apoptosis in this cell line. The decrease in promoter activity of the mm construct at day 2 of AML1-ETO expression indicated that there was an AML1-binding site–independent effect of AML1-ETO on the separase promoter. The significantly stronger suppressive activity of AML1-ETO on the wild-type TC promoter than on mutant promoters seemed to be the result of a combinatory effect of both double AML1-binding sites and other regions of the promoter. On the other hand, the promoters with a single AML1 site (mC and Tm) did not show such a significant difference from the promoter without AML1-binding sites (mm).
As controls, we also generated stable transfectants with these 4 promoter-luciferase constructs using the U937T parental cell line, which has no AML1-ETO in the absence of tetracycline. The pools of these stable transfectants did not show any change in luciferase activity following tetracycline removal (data not shown).
AML1-ETO binds to the human separase promoter region in vivo
Next, we examined the in vivo binding of AML1-ETO to the promoter region of human separase by ChIP assay. To study whether the AML1-ETO or AML1-ETO9a fusion proteins bind to the endogenous separase promoter, we used U937T-AE cells inducibly expressing AML1-ETO and K562 cells stably expressing FLAG-tagged AML1-ETO9a. Interestingly, following AML1-ETO expression, the protein expression of AML1 was dramatically decreased (Figure 5A top). Real-time quantitative PCR with ChIP DNA samples normalized with input DNA confirmed AML1-ETO binding to the endogenous promoter region of the human separase gene (Figure 5A bottom). Furthermore, ChIP assays with AML1-ETO9a K562 cells also showed the binding of AML1-ETO9a to the endogenous separase promoter (Figure 5B).
Figure 5.

ChIP assay for human separase promoter region. (A) U937T-AML1-ETO cells cultured in the presence or absence of tetracycline (Tet) for 24 hours were used in ChIP assay. The induction of AML1-ETO expression and a decrease of endogenous AML1 were detected by immunoblotting using anti-AML1 antibody. ChIP assay was performed with anti-AML1 antibody and bound DNA was examined by real-time quantitative PCR to show the binding of AML1-ETO to the upstream region of the separase gene. The relative binding activity was calculated by Ct value differences of ChIP DNA normalized with input DNA and that of cells without AML1-ETO was set to 1. The results are presented as the mean value of 3 independent experiments + standard deviation. (B) ChIP assay was performed with parental (P) and 3xFLAG-AML1-ETO9a (AE9a) stably transfected K562 cells using anti-FLAG M2 agarose. Immunoprecipitation of 3xFLAG-AML1-ETO9a was confirmed by immunoblotting with anti-FLAG antibody. The results of qPCR with ChIP DNA are shown as the mean value of 3 independent experiments + standard deviation. The relative binding activity was calculated by Ct value differences of ChIP DNA normalized with input DNA and that of parental cells was set to 1. Statistically significant P value compared with control (t test, P = .03) is indicated. (C) ChIP assay was performed with anti-FLAG M2 agarose using 293T cells transfected with wild-type (TC) phSeparase-luc or vectors with mutated AML1-binding sites (mC or Tm). pFLAG (C) or pFLAG-AML1-ETO (AE) vector was cotransfected. The expression (input) and immunoprecipitation (IP) of FLAG-AML1-ETO was confirmed by immunoblotting using anti-FLAG antibody (top). The relative binding of AML1-ETO to phSeparase-luc was examined by qPCR and it was calculated by Ct value differences of ChIP DNA normalized with input DNA. That of AML1-ETO–free sample (C) for each phSeparase-luc construct was set to 1 (bottom). The results are presented as the mean value of 3 independent experiments + standard deviation.
To confirm the dependence of AML1-ETO binding on 2 AML1 consensus binding sites by ChIP assay, we cotransfected wild-type and mutant human separase reporter vectors with or without FLAG-AML1-ETO into 293T cells. Equivalent expression and amount of immunoprecipitation of FLAG-AML1-ETO protein was confirmed by immunoblotting (Figure 5C bottom). Quantitative real-time PCR with ChIP DNA samples normalized with input DNA showed that AML1-ETO binds strongly to the wild-type TC sequence in the human separase promoter compared with the mutants, mC or Tm (Figure 5C bottom). PCR amplification with input DNA showed almost equal amount of product from preimmunoprecipitation nuclear extracts (data not shown).
AML1-ETO suppresses endogenous separase expression
Next, we examined whether AML1-ETO affects the expression level of separase. Separase mRNA levels were decreased after AML1-ETO expression at day 2 following tetracycline removal in U937T-AE cells whether measured by Northern blotting (Figure 6A left) or by real-time quantitative reverse-transcription–PCR (RT-qPCR) (Figure 6B left). On the other hand, the level of separase mRNA was not decreased in parental U937T cells 2 days after tetracycline withdrawal (Figure 6A,B right). There is a minor increase of separase mRNA in both U937T and U937T-AE cells on day 1 after tetracycline removal (Figure 6A,B). Further, we examined separase protein expression (Figure 6C). Correlated to the change in mRNA level, both full-length and cleaved forms of separase protein52,53 were also decreased after AML1-ETO expression at day 2 after tetracycline removal in U937T-AE cells. The tetracycline removal itself did not effect separase protein expression in the control U937T cells (Figure 6C right).
Figure 6.

AML1-ETO suppressed separase and PU.1 expression. (A) Northern blotting for separase (top) with days 0, 1, and 2 of tetracycline-removed (−Tet) U937T-AML1-ETO (left) and parental U937T (right) cells was performed. The ethidium bromide staining of the 18S rRNA shows the relative loading of RNA. (B) Separase expression was examined by RT-qPCR with days 0, 1, and 2 of tetracycline-removed U937T-AML1-ETO (left) and U937T (right) cells. The relative separase mRNA level compared with day 0, which is set to 1, is shown. The results are presented as the mean value of 3 independent experiments + standard deviation. Statistically significant P value (t test) is indicated only for U923T-AML1-ETO. (C) Immunoblotting with protein extracts from U937T-AML1-ETO (left) or U937T cells (right) was performed using anti-separase antibody. Both full-length (220 kDa) and cleaved (150 kDa) forms of separase were detected (). Tubulin blots are shown as loading controls. (D) Northern blotting for PU.1 (top) with days 0, 1, and 2 of tetracycline-removed (−Tet) U937T-AML1-ETO (left) and parental U937T (right) cells was performed. The ethidium bromide stainings of the 18S rRNA are also shown (bottom).
Last, we examined PU.1 expression since it is a well-known AML1 target with a duplicated AML1 consensus (TGCGGT3nTGTGGT) in its upstream regulatory element (URE).54 In agreement with the results of the DNA affinity purification assay (Figure 3E), the PU.1 mRNA level was dramatically decreased at both day 1 and day 2 of AML1-ETO expression in U937T-AML1-ETO cells as measured by Northern blotting (Figure 6D left). The tetracycline removal itself had no effect on the PU.1 mRNA expression level in U937T cells (Figure 6D right).
Discussion
The C-terminal truncated AML1-ETO proteins, either the natural t(8;21) fusion protein AML1-ETO9a or a mutant AML1-ETOtr, are strongly leukemogenic in contrast to the traditionally recognized full-length AML1-ETO protein.25 Since the C-terminal truncated region of AML1-ETO contains 2 zinc centers, we questioned whether they provide an additional DNA-binding domain for full-length AML1-ETO compared with AML1. To clarify this possibility, we performed a PCR-based consensus site screen with full-length AML1 and AML1-ETO as well as AML1-ETOtr or the C-terminus of ETO alone. Our assay did not identify any DNA-binding site for the C-terminal region of ETO. However, interestingly, we determined that both full-length and a C-terminal truncated form of AML1-ETO have a preference for multimerized AML1 consensus DNA-binding motifs compared with full-length AML1 (78%-86% compared with 51%). We verified that AML1-ETO had a higher DNA-binding affinity for duplicated AML1 consensus sites than single sites in both in vitro and in vivo assays. In contrast, AML1 did not show such a predominant affinity for double binding sites. Furthermore, compared with AML1, AML1-ETO displayed a substantially stronger effect on promoter-luciferase reporter constructs with 2 AML1-binding sites than on those with single sites. These results support the idea that t(8;21) fusion proteins preferentially regulate selected AML1 target genes with duplicated or multimerized AML1-binding sites within certain regions of their control elements. Such selected regulation may directly affect hematopoietic cell differentiation, proliferation, and survival during leukemogenesis. Furthermore, when a simple basal promoter-luc reporter construct with a double AML1-binding site (pBAAE#35-TK-luc) was used in the promoter activity analysis (Figure 4A), AML1-ETO produced stronger repression than AML1-ETO9a. However, besides preferentially binding to duplicated AML1 sites, the ability of AML1-ETO and AML1-ETO9a to activate or repress any specific target genes also depends on the availability of their cofactors and other DNA-binding transcription factors adjacent to their binding sites. Examples might include their role in regulating MDR1 promoter activity and Notch target gene expression.55,56
It is worth noting that among our cloned multimerized AML1-binding sites for AML1, AML1-ETO, and its C-terminal truncated form (Table S1) and upstream regions of potential AML1-ETO target genes46 (and data not shown), a putative new variation of the consensus-binding site TGC/TGGTtN(0-13)TGCGGT is present in 50% to 86% of the double site sequences. The orientation of these duplicated sites was predominantly the same. Interestingly, through a preliminary frequency survey of the AML1-binding sites in the genome, we found that the TGCGGT consensus, which gave a higher binding affinity for both AML1 and AML1-ETO in this study, was much less common than TGTGGT in both the human and mouse genomes (data not shown).
AML1-ETO and ETO family members form a tetramer through an α-helix structure of the NHR2 domain in ETO.57 Mutation of the NHR2 domain affects transcriptional regulation of some target genes and its clonogenic potential in primary murine bone marrow.57–59 Furthermore, this domain is critical for leukemogenesis by AML1-ETO9a (Yan and D.-E.Z., manuscript submitted, June 2008). Here, we also show that the preference for duplicated AML1 DNA-binding sites by AML1-ETO depends on the NHR2 oligomerization domain. Thus oligomerized AML1-ETO may have a higher binding affinity for target genes with multiple AML1-binding motifs in their promoter regions mediated by each runt domain, leading to stronger competitive binding for certain AML1 target genes (Figure 7). Through the flexibility of DNA in 3 dimensions, 2 AML1 consensus sites separated by an extensive distance in the real genome might also function as a duplicate site. Further studies of AML1-ETO target gene sequences will be required to determine the contribution of spacing and orientation of multimerized AML1 consensus sites to AML1-ETO–binding preferences. However, our current analysis with the SPARC and PU.1 duplicated AML1 consensus sequences suggests that the interspacing of 10 extra nucleotides in the SPARC sequence compared with the PU.1 sequence does not affect the efficiency of AML1-ETO binding to the duplicated AML1 consensus sites.
Figure 7.

A model of a high DNA-binding activity of tetrameric AML1-ETO to duplicated AML1-binding sites. Runt homology DNA-binding domain (Runt), 4 Nervy homology regions (NHR1-4), and N-terminus (N) of AML1-ETO are marked. The oligomerization provides an advantage to AML1-ETO to duplicated binding sites (TGT/CGGTspaceTGT/CGGT) compared with AML1.
Our findings are reminiscent of tetrameric and dimeric transcription factors, such as p53,60 Stat5α,61 and the developmental POU proteins.62 These proteins are known to bind bipartite or palindromic DNA consensus sites. In their target promoters, mutation of one part of the bipartite/palindromic sites dramatically affects their binding to DNA. In the case of the runt domain of AML1, binding to DNA at the consensus site leads to bending of DNA at the site of interaction, which is further stabilized by the heterodimer partner CBFβ.63–65 Further, as DNA bending by transcription factors also influences their transcriptional activity on their targets, the spacing between 2 or multiple AML1 consensus sites may also determine and/or enhance the level of regulation by AML1-ETO of its targets. In fact, the ability to form a dimer is quite essential for the function of several fusion proteins in leukemogenesis.66
In addition to revealing the preference of AML1-ETO for duplicated AML1 sites, we also reported separase as a novel AML1-ETO target gene and used it as a model to demonstrate the regulation of AML1-ETO by multiple sites. Separase is essential for proper chromosome segregation,67,68 and an inactivating mutation causes genomic instability and increased frequency of epithelial cancer in a zebrafish model.69 Since separase expression is modulated in AML1-ETO–expressing U937T cells, t(8;21) patients might have deregulated separase expression that may contribute to the deregulation of the cell cycle and the associated gain and loss of chromosomes observed in these patients.25
The finding in this study that AML1-ETO and its C-terminal truncated mutants preferentially bind multimerized AML1 consensus sites provides a novel insight into the molecular mechanisms of how t(8;21)-related fusion proteins may selectively regulate AML1 target genes or even additional new target genes that promote the onset of acute myeloid leukemia. With the rapid development of new tools to study the human genome and to perform bioinformatic analyses, we expect that additional important AML1-ETO target genes will be discovered.
Supplementary Material
Acknowledgments
We thank Dr Joseph Biggs for helpful suggestions and critical reading of the paper, all other members of D.-E.Z.'s laboratory for valuable discussions, Dr Prasad V. Jallepalli for separase antibody, and Dr Katherine A. Fitzgerald for a detailed protocol of in vitro DNA-protein binding assay.
This work was supported by National Institutes of Health (Bethesda, MD) grant CA104509. The Stein Endowment Fund (La Jolla, CA) has partially supported the departmental molecular biology service laboratory for DNA sequencing and oligonucleotide synthesis. A.J.O. was initially supported by the Japan Society for Promotion of Science (Tokyo, Japan) and the Mochida Memorial Foundation (Tokyo, Japan). F.O. was initially supported by the Uehara Memorial Foundation (Tokyo, Japan) postdoctoral fellowship.
This is paper 19186 from The Scripps Research Institute.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.J.O. designed and performed the research and wrote the paper; L.F.P. was involved in experimental design, data analysis, and paper preparation; F.O. provided experimental advice and assisted with paper preparation; A.B. provided technical advice and performed some of the immunoprecipitation experiments; and D.-E.Z. supervised experimental design, data analysis, and paper preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Dong-Er Zhang, Mail Drop 0815, Room 5328, Moores Cancer Center, University of California, San Diego, 3855 Health Sciences Drive, La Jolla, CA 92093; e-mail: d7zhang@ucsd.edu.
References
- 1.Downing JR. The AML1-ETO chimaeric transcription factor in acute myeloid leukaemia: biology and clinical significance. Br J Haematol. 1999;106:296–308. doi: 10.1046/j.1365-2141.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 2.Friedman AD. Leukemogenesis by CBF oncoproteins. Leukemia. 1999;13:1932–1942. doi: 10.1038/sj.leu.2401590. [DOI] [PubMed] [Google Scholar]
- 3.Lutterbach B, Hiebert SW. Role of the transcription factor AML-1 in acute leukemia and hematopoietic differentiation. Gene. 2000;245:223–235. doi: 10.1016/s0378-1119(00)00014-7. [DOI] [PubMed] [Google Scholar]
- 4.Nucifora G, Rowley JD. AML1 and the 8;21 and 3;21 translocations in acute and chronic myeloid leukemia. Blood. 1995;86:1–14. [PubMed] [Google Scholar]
- 5.Miyoshi H, Shimizu K, Kozu T, et al. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci U S A. 1991;88:10431–10434. doi: 10.1073/pnas.88.23.10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyoshi H, Kozu T, Shimizu K, et al. The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J. 1993;12:2715–2721. doi: 10.1002/j.1460-2075.1993.tb05933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyers S, Downing JR, Hiebert SW. Identification of AML-1 and the (8;21) translocation protein (AML-1/ETO) as sequence-specific DNA-binding proteins: the runt homology domain is required for DNA binding and protein-protein interactions. Mol Cell Biol. 1993;13:6336–6345. doi: 10.1128/mcb.13.10.6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogawa E, Inuzuka M, Maruyama M, et al. Molecular cloning and characterization of PEBP2 beta, the heterodimeric partner of a novel Drosophila runt-related DNA binding protein PEBP2 alpha. Virology. 1993;194:314–331. doi: 10.1006/viro.1993.1262. [DOI] [PubMed] [Google Scholar]
- 9.Speck NA, Stacy T, Wang Q, et al. Core-binding factor: a central player in hematopoiesis and leukemia. Cancer Res. 1999;59:1789s–1793s. [PubMed] [Google Scholar]
- 10.Wang SW, Speck NA. Purification of core-binding factor, a protein that binds the conserved core site in murine leukemia virus enhancers. Mol Cell Biol. 1992;12:89–102. doi: 10.1128/mcb.12.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westendorf JJ, Hiebert SW. Mammalian runt-domain proteins and their roles in hematopoiesis, osteogenesis, and leukemia. J Cell Biochem. 1999;(suppl 32–33):51–58. doi: 10.1002/(sici)1097-4644(1999)75:32+<51::aid-jcb7>3.3.co;2-j. [DOI] [PubMed] [Google Scholar]
- 12.Petrovick MS, Hiebert SW, Friedman AD, et al. Multiple functional domains of AML1: PU. 1 and C/EBPalpha synergize with different regions of AML1. Mol Cell Biol. 1998;18:3915–3925. doi: 10.1128/mcb.18.7.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanno T, Kanno Y, Chen LF, et al. Intrinsic transcriptional activation-inhibition domains of the polyomavirus enhancer binding protein 2/core binding factor alpha subunit revealed in the presence of the beta subunit. Mol Cell Biol. 1998;18:2444–2454. doi: 10.1128/mcb.18.5.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/s0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Stacy T, Binder M, et al. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci U S A. 1996;93:3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank R, Zhang J, Uchida H, et al. The AML1/ETO fusion protein blocks transactivation of the GM-CSF promoter by AML1B. Oncogene. 1995;11:2667–2674. [PubMed] [Google Scholar]
- 17.Hohaus S, Petrovick MS, Voso MT, et al. PU. 1 (Spi-1) and C/EBP alpha regulate expression of the granulocyte-macrophage colony-stimulating factor receptor alpha gene. Mol Cell Biol. 1995;15:5830–5845. doi: 10.1128/mcb.15.10.5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nuchprayoon I, Meyers S, Scott LM, et al. PEBP2/CBF, the murine homolog of the human myeloid AML1 and PEBP2 beta/CBF beta proto-oncoproteins, regulates the murine myeloperoxidase and neutrophil elastase genes in immature myeloid cells. Mol Cell Biol. 1994;14:5558–5568. doi: 10.1128/mcb.14.8.5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uchida H, Zhang J, Nimer SD. AML1A and AML1B can transactivate the human IL-3 promoter. J Immunol. 1997;158:2251–2258. [PubMed] [Google Scholar]
- 20.Zhang DE, Fujioka K, Hetherington CJ, et al. Identification of a region which directs the monocytic activity of the colony-stimulating factor 1 (macrophage colony-stimulating factor) receptor promoter and binds PEBP2/CBF (AML1). Mol Cell Biol. 1994;14:8085–8095. doi: 10.1128/mcb.14.12.8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang DE, Hetherington CJ, Meyers S, et al. CCAAT enhancer-binding protein (C/EBP) and AML1 (CBF alpha2) synergistically activate the macrophage colony-stimulating factor receptor promoter. Mol Cell Biol. 1996;16:1231–1240. doi: 10.1128/mcb.16.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun W, Downing JR. Haploinsufficiency of AML1 results in a decrease in the number of LTR-HSCs while simultaneously inducing an increase in more mature progenitors. Blood. 2004;104:3565–3572. doi: 10.1182/blood-2003-12-4349. [DOI] [PubMed] [Google Scholar]
- 23.Taniuchi I, Osato M, Egawa T, et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–633. doi: 10.1016/s0092-8674(02)01111-x. [DOI] [PubMed] [Google Scholar]
- 24.Tsuzuki S, Hong D, Gupta R, et al. Isoform-specific potentiation of stem and progenitor cell engraftment by AML1/RUNX1. PLoS Med. 2007;4:e172, 880–896. doi: 10.1371/journal.pmed.0040172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peterson LF, Boyapati A, Ahn EY, et al. Acute myeloid leukemia with the 8q22;21q22 translocation: secondary mutational events and alternative t(8;21) transcripts. Blood. 2007;110:799–805. doi: 10.1182/blood-2006-11-019265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calabi F, Pannell R, Pavloska G. Gene targeting reveals a crucial role for MTG8 in the gut. Mol Cell Biol. 2001;21:5658–5666. doi: 10.1128/MCB.21.16.5658-5666.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gelmetti V, Zhang J, Fanelli M, et al. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol. 1998;18:7185–7191. doi: 10.1128/mcb.18.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lutterbach B, Westendorf JJ, Linggi B, et al. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol Cell Biol. 1998;18:7176–7184. doi: 10.1128/mcb.18.12.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Hoshino T, Redner RL, Kajigaya S, Liu JM. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc Natl Acad Sci U S A. 1998;95:10860–10865. doi: 10.1073/pnas.95.18.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamachi Y, Ogawa E, Asano M, et al. Purification of a mouse nuclear factor that binds to both the A and B cores of the polyomavirus enhancer. J Virol. 1990;64:4808–4819. doi: 10.1128/jvi.64.10.4808-4819.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thornell A, Hallberg B, Grundstrom T. Binding of SL3-3 enhancer factor 1 transcriptional activators to viral and chromosomal enhancer sequences. J Virol. 1991;65:42–50. doi: 10.1128/jvi.65.1.42-50.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melnikova IN, Crute BE, Wang S, Speck NA. Sequence specificity of the core-binding factor. J Virol. 1993;67:2408–2411. doi: 10.1128/jvi.67.4.2408-2411.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan M, Burel SA, Peterson LF, et al. Deletion of an AML1-ETO C-terminal NcoR/SMRT-interacting region strongly induces leukemia development. Proc Natl Acad Sci U S A. 2004;101:17186–17191. doi: 10.1073/pnas.0406702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan M, Kanbe E, Peterson LF, et al. A previously unidentified alternatively spliced isoform of t(8;21) transcript promotes leukemogenesis. Nat Med. 2006;12:945–949. doi: 10.1038/nm1443. [DOI] [PubMed] [Google Scholar]
- 35.Burel SA, Harakawa N, Zhou L, et al. Dichotomy of AML1-ETO functions: growth arrest versus block of differentiation. Mol Cell Biol. 2001;21:5577–5590. doi: 10.1128/MCB.21.16.5577-5590.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang DE, Hetherington CJ, Chen HM, Tenen DG. The macrophage transcription factor PU. 1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol Cell Biol. 1994;14:373–381. doi: 10.1128/mcb.14.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peterson LF, Boyapati A, Ranganathan V, et al. The hematopoietic transcription factor AML1 (RUNX1) is negatively regulated by the cell cycle protein cyclin D3. Mol Cell Biol. 2005;25:10205–10219. doi: 10.1128/MCB.25.23.10205-10219.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joo A, Aburatani H, Morii E, Iba H, Yoshimura A. STAT3 and MITF cooperatively induce cellular transformation through upregulation of c-fos expression. Oncogene. 2004;23:726–734. doi: 10.1038/sj.onc.1207174. [DOI] [PubMed] [Google Scholar]
- 39.Severa M, Coccia EM, Fitzgerald KA. Toll-like receptor-dependent and -independent viperin gene expression and counter-regulation by PRDI-binding factor-1/BLIMP1. J Biol Chem. 2006;281:26188–26195. doi: 10.1074/jbc.M604516200. [DOI] [PubMed] [Google Scholar]
- 40.Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14:804–816. [PMC free article] [PubMed] [Google Scholar]
- 41.Okumura F, Zou W, Zhang DE. ISG15 modification of the eIF4E cognate 4EHP enhances cap structure-binding activity of 4EHP. Genes Dev. 2007;21:255–260. doi: 10.1101/gad.1521607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peterson LF, Boyapati A, Ranganathan V, et al. The hematopoietic transcription factor AML1 (RUNX1) is negatively regulated by the cell cycle protein cyclin D3. Mol Cell Biol. 2005;25:10205–10219. doi: 10.1128/MCB.25.23.10205-10219.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okumura AJ, Peterson LF, Lo MC, Zhang DE. Expression of AML/Runx and ETO/MTG family members during hematopoietic differentiation of embryonic stem cells. Exp Hematol. 2007;35:978–988. doi: 10.1016/j.exphem.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Boyapati A, Yan M, Peterson LF, et al. A leukemia fusion protein attenuates the spindle checkpoint and promotes aneuploidy. Blood. 2007;109:3963–3971. doi: 10.1182/blood-2006-09-045583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asou H, Tashiro S, Hamamoto K, et al. Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8;21 chromosome translocation. Blood. 1991;77:2031–2036. [PubMed] [Google Scholar]
- 46.Peterson LF, Wang Y, Lo MC, et al. The multi-functional cellular adhesion molecule CD44 is regulated by the 8;21 chromosomal translocation. Leukemia. 2007;21:2010–2019. doi: 10.1038/sj.leu.2404849. [DOI] [PubMed] [Google Scholar]
- 47.Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 2001;19:816–827. doi: 10.1016/s0945-053x(00)00133-5. [DOI] [PubMed] [Google Scholar]
- 48.Framson PE, Sage EH. SPARC and tumor growth: where the seed meets the soil? J Cell Biochem. 2004;92:679–690. doi: 10.1002/jcb.20091. [DOI] [PubMed] [Google Scholar]
- 49.Lehmann S, O'Kelly J, Raynaud S, et al. Common deleted genes in the 5q- syndrome: thrombocytopenia and reduced erythroid colony formation in SPARC null mice. Leukemia. 2007;21:1931–1936. doi: 10.1038/sj.leu.2404852. [DOI] [PubMed] [Google Scholar]
- 50.Rosenbauer F, Wagner K, Kutok JL, et al. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU. 1. Nat Genet. 2004;36:624–630. doi: 10.1038/ng1361. [DOI] [PubMed] [Google Scholar]
- 51.Liu Y, Chen W, Gaudet J, et al. Structural basis for recognition of SMRT/N-CoR by the MYND domain and its contribution to AML1/ETO's activity. Cancer Cell. 2007;11:483–497. doi: 10.1016/j.ccr.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zou H, Stemman O, Anderson JS, Mann M, Kirschner MW. Anaphase specific auto-cleavage of separase. FEBS Lett. 2002;528:246–250. doi: 10.1016/s0014-5793(02)03238-6. [DOI] [PubMed] [Google Scholar]
- 53.Chestukhin A, Pfeffer C, Milligan S, DeCaprio JA, Pellman D. Processing, localization, and requirement of human separase for normal anaphase progression. Proc Natl Acad Sci U S A. 2003;100:4574–4579. doi: 10.1073/pnas.0730733100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Y, Okuno Y, Zhang P, et al. Regulation of the PU. 1 gene by distal elements. Blood. 2001;98:2958–2965. doi: 10.1182/blood.v98.10.2958. [DOI] [PubMed] [Google Scholar]
- 55.Hines R, Boyapati A, Zhang DE. Cell type dependent regulation of multidrug resistance-1 gene expression by AML1-ETO. Blood Cells Mol Dis. 2007;39:297–306. doi: 10.1016/j.bcmd.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salat D, Liefke R, Wiedenmann J, Borggrefe T, Oswald F. ETO, but not leukemogenic fusion protein AML1/ETO, augments RBP-J{kappa}/SHARP-mediated repression of Notch target genes. Mol Cell Biol. 2008;28:3502–3512. doi: 10.1128/MCB.01966-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y, Cheney MD, Gaudet JJ, et al. The tetramer structure of the Nervy homology two domain, NHR2, is critical for AML1/ETO's activity. Cancer Cell. 2006;9:249–260. doi: 10.1016/j.ccr.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 58.Minucci S, Maccarana M, Cioce M, et al. Oligomerization of RAR and AML1 transcription factors as a novel mechanism of oncogenic activation. Mol Cell. 2000;5:811–820. doi: 10.1016/s1097-2765(00)80321-4. [DOI] [PubMed] [Google Scholar]
- 59.Hug BA, Lee SY, Kinsler EL, Zhang J, Lazar MA. Cooperative function of Aml1-ETO corepressor recruitment domains in the expansion of primary bone marrow cells. Cancer Res. 2002;62:2906–2912. [PubMed] [Google Scholar]
- 60.Kitayner M, Rozenberg H, Kessler N, et al. Structural basis of DNA recognition by p53 tetramers. Mol Cell. 2006;22:741–753. doi: 10.1016/j.molcel.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 61.Soldaini E, John S, Moro S, et al. DNA binding site selection of dimeric and tetrameric Stat5 proteins reveals a large repertoire of divergent tetrameric Stat5a binding sites. Mol Cell Biol. 2000;20:389–401. doi: 10.1128/mcb.20.1.389-401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacobson EM, Li P, Leon-del-Rio A, Rosenfeld MG, Aggarwal AK. Structure of Pit-1 POU domain bound to DNA as a dimer: unexpected arrangement and flexibility. Genes Dev. 1997;11:198–212. doi: 10.1101/gad.11.2.198. [DOI] [PubMed] [Google Scholar]
- 63.Bravo J, Li Z, Speck NA, Warren AJ. The leukemia-associated AML1 (Runx1)–CBF beta complex functions as a DNA-induced molecular clamp. Nat Struct Biol. 2001;8:371–378. doi: 10.1038/86264. [DOI] [PubMed] [Google Scholar]
- 64.Bartfeld D, Shimon L, Couture GC, et al. DNA recognition by the RUNX1 transcription factor is mediated by an allosteric transition in the RUNT domain and by DNA bending. Structure. 2002;10:1395–1407. doi: 10.1016/s0969-2126(02)00853-5. [DOI] [PubMed] [Google Scholar]
- 65.Li Z, Yan J, Matheny CJ, et al. Energetic contribution of residues in the Runx1 Runt domain to DNA binding. J Biol Chem. 2003;278:33088–33096. doi: 10.1074/jbc.M303973200. [DOI] [PubMed] [Google Scholar]
- 66.So CW, Lin M, Ayton PM, Chen EH, Cleary ML. Dimerization contributes to oncogenic activation of MLL chimeras in acute leukemias. Cancer Cell. 2003;4:99–110. doi: 10.1016/s1535-6108(03)00188-0. [DOI] [PubMed] [Google Scholar]
- 67.Kumada K, Yao R, Kawaguchi T, et al. The selective continued linkage of centromeres from mitosis to interphase in the absence of mammalian separase. J Cell Biol. 2006;172:835–846. doi: 10.1083/jcb.200511126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wirth KG, Wutz G, Kudo NR, et al. Separase: a universal trigger for sister chromatid disjunction but not chromosome cycle progression. J Cell Biol. 2006;172:847–860. doi: 10.1083/jcb.200506119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shepard JL, Amatruda JF, Finkelstein D, et al. A mutation in separase causes genome instability and increased susceptibility to epithelial cancer. Genes Dev. 2007;21:55–59. doi: 10.1101/gad.1470407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}