

Abstract
Vi antigen, the virulence factor of Salmonella typhi, has been used clinically as a molecular vaccine. TviB and TviC are two enzymes involved in the formation of Vi antigen, a linear polymer consisting of α-1,4-linked N-acetylgalactosaminuronate. Protein sequence analysis suggests that TviB is a dehydrogenase and TviC is an epimerase. Both enzymes are expected to be NAD+ dependent. In order to verify their functions, TviB and TviC were cloned, expressed in Escherichia coli, and characterized. The C-terminal His6-tagged TviB protein, purified from soluble cell fractions in the presence of 10 mM DTT, shows UDP-N-acetylglucosamine 6-dehydrogenase activity, and is capable of catalyzing the conversion of UDP-N-acetylglucosamine (UDP-GlcNAc) to UDP-N-acetylglucosaminuronic acid (UDP-GlcNAcA) with a kcat value of 15.5 ± 1.0 min−1. The Km values of TviB for UDP-GlcNAc and NAD+ are 77 ± 9 μM and 276 ± 52 μM, respectively. TviC, purified as C-terminal hexahistidine-tagged protein, shows UDP-GlcNAcA 4-epimerase and UDP-N-acetylgalactosamine (UDP-GalNAc) 4-epimerase activities. The Km values of TviC for UDP-GlcNAcA and UDP-N-acetylgalactosaminuronic acid (UDP-GalNAcA) are 20 ± 1 μM and 42 ± 2 μM, respectively. The kcat value for the conversion of UDP-GlcNAcA to UDP-GalNAcA is 56.8 ± 0.5 min−1, while that for the reverse reaction is 39.1 ± 0.6 min−1. These results show that the biosynthesis of Vi antigen is initiated by the TviB-catalyzed oxidation of UDP-GlcNAc to UDP-GalNAc, followed by the TviC-catalyzed epimerization at C-4 to form UDP-GalNAcA, which serves as the building block for the formation of Vi polymer. These results set the stage for future in vitro biosynthesis of Vi antigen. These enzymes may also be drug targets to inhibit Vi antigen production.
Typhoid fever (TF), caused by Salmonella typhi and Salmonella paratyphi C, is a serious public health problem in many developing countries.1 These Salmonella strains, similar to many pathogenic bacteria implicated in severe invasive infections of humans, such as Neisseria meningitides, which causes meningitis, Streptococcus pneumoniae, which causes pneumonia, and Haemophilus influenzae type b, which causes septicaemia, are encapsulated by polysaccharides. The capsules in S. typhi and S. praratyhi are linear homopolymers made up of α-1,4-linked N-acetylgalactosaminuronate (GalNAcA), with 60–70% of the monomeric units O-acetylated at the C-3 position.2,3 This capsular polysaccharide, commonly referred as Vi antigen (1), has a molecular mass typically over 200 kDa. It is a virulence antigen which enhances S. typhi virulence in mice and induces immune response in rabbits.3,4 Purified Vi polysaccharide is also licensed as a typhoid vaccine. Patients immunized with purified Vi polysaccharide show good protection against typhoid fever.5
The expression of Vi antigen is associated with three loci, viaA, viaB and ompB on the chromosomes of S. typhi.6,7,8 The viaB region (Figure 1), located at 92 min on the chromosome of S. typhi, consists of 11 open reading frames (ORFs),8,9,10 within which tviB, tviC, tviD and tviE are assigned as the structural genes for Vi antigen. When those genes are coexpressed in E. coli, Vi antigen accumulates inside the E. coli cells.9,11 Since acquisition of the entire viaB region by E. coli results in a Vi-positive phenotype, the remaining ORFs in the viaB locus are believed to be export genes.
FIGURE 1.
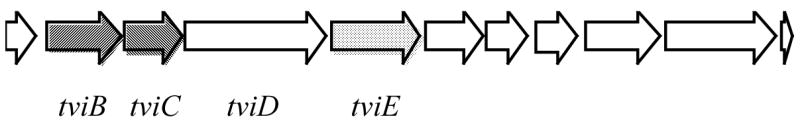
Organization of the viaB locus. The structural genes encoding enzymes involved in Vi antigen biosynthesis are highlighted. On the basis of sequence comparison, tviB likely encodes a dehydrogenase, tviC encodes an epimerase, tviD encodes a cytochrome P450-like enzyme, and tviE encodes a glycosyltransferase.
Sequence analysis showed that protein (TviB) encoded by the tviB gene belongs to the nucleotidyldiphosphohexose dehydrogenase superfamily.12,13,14 TviB exhibits high amino acid sequence identity (65% identity and 83% similarity) with WbpO, which is an UDP-GalNAc 6-dehydrogenase from Pseudomonas aeruginosa PAO1.15 TviB also shows modest sequence identity with WbpA (31% identity and 52% similarity), a well-characterized UDP-GlcNAc 6-dehydrogenase from the same P. aeruginosa strain.16 In view of the sequence similarities and the structure of Vi antigen, TviB is likely an UDP-N-acetylglucosamine (UDP-GlcNAc, 2) or an UDP-N-acetylgalactosamine (UDP-GalNAc, 3) 6-dehydrogenase, catalyzing the conversion of 2 or 3 to the corresponding N-acetylglycosaminuronic acid.
TviC belongs to the short-chain dehydrogenase/reductase (SDR) protein family. Members of this family possess two common features: a SYK catalytic triad and a GXXGXXG motif within a Rossmann fold at the N-terminus for NAD(P)+ binding.17 Both features are conserved in TviC. This enzyme is likely an NAD+-dependent C-4 epimerase, which catalyzes the conversion of UDP-GlcNAc (2) and UDP-GalNAc (3) or that of UDP-N-acetylglucosaminuronic acid (UDP-GlcNAcA, 4) and UDP-N-acetylgalactosaminuronic acid (UDP-GalNAcA, 5). To date, no enzyme catalyzing a C-4 epimerization between 4 and 5 has been reported, and only two UDP-GlcNAc 4-epimerases have been fully characterized, WbgU and WbpP. WbgU is involved in the formation of 2-acetamino-2-deoxy-L-altruronic acid found in the O-antigen repeating unit of Plesiomonas shigelloides O17,18 and WbpP is involved in the O-antigen formation in P. aeruginosa serogroup O6.19 The amino acid sequence of TviC is 62% identical to that of WbgU and 63% identical to that of WbpP.
On the basis of the organization of viaB locus and the proposed functions for TviB and TviC, two pathways are conceivable for the biosynthesis of UDP-GalNAcA (5), the precursor of Vi antigen (1). As depicted in Scheme 1, the reaction may be initiated by C-6 oxidation of UDP-GlcNAc (2), followed by epimerization at C-4 of the resulting UDP-GlcNAcA (4) to give 5 (route A). Alternatively, the reaction may proceed with epimerization at C-4 of 2 to give UDP-GalNAc (3), followed by oxidation of 3 at C-6 to give 5 (route B). Clearly, the substrate specificity of TviB and TviC will determine whether the biosynthesis of Vi antigen proceeds by route A or B. To distinguish between these two pathways, the genes for TviB and TviC were cloned, the proteins expressed in Escherichia coli, purified, and characterized. These studies confirmed that TviB is an UDP-GlcNAc 6-dehydrogenase and TviC is an UDP-GlcNAcA 4-epimerase. Our results also established that the precursor of S. typhi Vi antigen (1), UDP-GalNAcA (5), is derived from UDP-GlcNAc (2) via route A as shown in Scheme 1.
Scheme 1.

EXPERIMENTAL PROCEDURES
General
All chemical reagents used were purchased from Sigma-Aldrich (St. Louis, MO), unless mentioned otherwise. All enzymes used for DNA manipulations were obtained from Invitrogen (Carlsband, CA). Escherichia coli BL21(DE3) and plasmids pET24b(+) and pET28b(+) were purchased from Novagen (Madison, WI), and E. coli DH5α was acquired from New England Biolabs (Beverly, MA). Salmonella choleraesuis subsp. typhi genomic DNA (ATCC 700931) was obtained from the American Type Culture Collection (Manassas, VA). DEAE Sepharose fast flow resin, MonoQ HR 16/10 and Superdex 200 HR 10/30 FPLC columns were purchased from Amersham Pharmacia Biotech (Piscataway, NJ). Ni-NTA Agarose was a product of Qiagen Inc. (Valencia, CA). The oligonucleotide primers for the polymerase chain reaction (PCR) were ordered from IDT DNA Technologies (Coralville, IA). DNA sequencing and N-terminal sequencing of purified enzymes were performed by the Core Facilities in the Institute of Molecular and Cellular Biology at the University of Texas at Austin. Protein concentrations were determined according to Bradford20 using bovine serum albumin as the standard. The general methods and protocols for recombinant DNA manipulations were as described by Sambrook et al.21 The kinetic data were analyzed by non-linear fit using Grafit5 (Erithacus Software Ltd. UK).
Gene Amplification and Cloning of tviB and tviC
The sequences of both tviB and tviC genes in S. choleraesuis subsp. typhi have been reported,9 allowing the design of oligonucleotide primers to amplify these genes from the genomic DNA by PCR. The sequences of the primers used for the amplification of tviB gene were 5′-GGATCCACATATGTTCGGTATAGACGAG-3′ (start primer) and 5′-GGAATTCTTACAATCTCACATCTGAC-3′ (stop primer), and those for tviC gene were 5′-CATGCCATGGCGGCTTACGAAGAACTAC-3′ (start primer) and 5′-GGAATTCACCGAGGAATACAAAGTAG-3′ (stop primer). The incorporated restriction sites of endonuclease NdeI and NcoI in the start primers and EcoRI in the stop primers are shown in italics. The PCR-amplified tviB gene was purified, digested with NdeI and EcoRI restriction enzymes, and ligated into the NdeI/EcoRI restriction sites of the vector, pET24b(+), to give the recombinant plasmid pET-tviB. Likewise, the tviC gene was cloned into the NcoI and EcoRI sites of pET28b(+) vector to give pET-tviC. Both constructs were verified by restriction enzyme digestion and DNA sequencing. These plasmids were then used to transform the expression host strain E. coli BL21(DE3).
Growth of Escherichia coli BL21(DE3)/pET-tviB Cells
An overnight culture of E. coli BL21(DE3)/pET-tviB, grown in Luria-Bertani (LB) medium supplemented with kanamycin (50 μg/mL) at 37 °C, was used (2 mL each) to inoculate six 1 L cultures containing the same medium and antibiotic. These cultures were incubated at 37 °C until the OD600 reached 0.6, induced with 0.5 mM isopropyl β-D-thiogalactoside (IPTG), and followed by a 2-h incubation at 30 °C. The cells were harvested by centrifugation (6000g, 10 min). All of the subsequent manipulations were carried out at 4 °C.
Purification of TviB Protein
The cells collected from the 6 L cultures were re-suspended in buffer A (20 mM Tris·HCl, pH 7.5, 10 mM DTT). The cells were disrupted by sonication on ice and the cell debris was removed by centrifugation at 12,000g for 30 min. The supernatant was applied to a column packed with 200-mL DEAE fast flow resin which was pre-equilibrated with buffer A. The column was washed with buffer A, followed by elution with a linear gradient of 0–500 mM NaCl in buffer A. The purification was monitored by analyzing the collected fractions by SDS-PAGE. The desired fractions were pooled and concentrated by ultrafiltration (Amicon YM-10 membrane). The concentrated proteins were further purified using a FPLC MonoQ HR 16/10 column. The TviB protein, which contains a His6-tag at the C-terminus, was eluted from column with a linear gradient of 0–500 mM NaCl in buffer A over 80 mL. After dialysis against buffer A, the partially purified TviB was applied to a column packed with 100-mL of Reaction Dye Green 19 resin pre-equilibrated with buffer A. A linear gradient of 1–4 M NaCl in buffer A (total volume 1 L) was used as the eluent. Fractions were analyzed by SDS-PAGE and the TviB-containing fractions were pooled, dialyzed against buffer A, and concentrated by Amicon ultrafiltration cell (YM-10 membrane). The resulting TviB were subjected to FPLC Superdex 200 HR 10/30 chromatography using buffer B (20 mM Tris·HCl, pH 7.5, 150 mM NaCl, 10 mM DTT). The purified TviB protein was collected, dialyzed against buffer A in the presence of 15% glycerol, concentrated by ultrafiltration (Amicon YM-10 membrane), and stored at −80 °C.
Growth of Escherichia coli BL21(DE3)/pET-tviC Cells
An overnight culture of E. coli BL21(DE3)/pET-tviC, grown in Luria-Bertani (LB) medium supplemented with kanamycin (50 μg/mL) at 37 °C, was used (1 mL each) to inoculate six 1 L cultures containing the same medium and antibiotic. These cultures were incubated at 37 °C overnight. The cells were harvested by centrifugation (2000g, 10 min).
Purification of TviC Protein
The cells collected from the 6 L cultures were resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, pH 8.0). The cells were disrupted by sonication on ice and the cell debris was removed by centrifugation at 12,000g for 30 min. The supernatant was mixed by slow agitation with 2 mL of Ni-NTA agarose resin for 2 h at 4 °C. The resin was collected by centrifugation (2000g, 3 min) and resuspended in 10 mL of lysis buffer containing 10 mM imidazole. The slurry was poured into a small column, followed by washing with lysis buffer containing 20 mM imidazole to remove non-binding proteins. The TviC protein, which contains a His6-tag at the C-terminus, was eluted with lysis buffer containing 250 mM imidazole. The desired fractions, detected by Bradford reagents and confirmed by SDS-PAGE, were pooled and dialyzed against 20 mM Tris·HCl (pH 7.5) containing 15% glycerol. The purified enzyme was stored at −80 °C.
Molecular Mass Determination
The native molecular masses of TviB and TviC were determined by gel filtration performed on a Pharmacia FPLC equipped with a FPLC Superdex 200 HR 10/30 column. The system was calibrated with protein standards purchased from Sigma-Aldrich (MW-GF-200). The standard and sample protein were individually loaded and eluted from the column with 50 mM sodium phosphate (pH 7.4) containing 150 mM NaCl. The data were analyzed by the method of Andrews.22 The subunit molecular mass was estimated by SDS-PAGE as described by Laemmli.23
Extraction of NAD(H) from Purified TviC
NAD(H) extraction was performed according to a reported procedure19 with minor modification. Briefly, 400 μg TviC was treated with 25 μg of proteinase K in 200 μL of 50 mM Tris·HCl, pH 7.5, for 1 h at 37 °C. After the digestion was completed, the released NAD(H) in the incubation solution was subjected to chemical reduction directly. This was achieved by successive additions of 2 μL aliquot of 10 mg/mL sodium borohydride aqueous solution every 30 min to the above digested solution until no increase in absorption at 340 nm was observed. The stoichiometric ratio between NAD+ and TviC was determined on the basis of the concentrations of NADH (ε340 = 6220 M−1 cm−1) and TviC.
Enzyme Activity Assay for TviB
A typical assay mixture to determine the TviB activity contained 0.25 mM UDP-GlcNAc (2), 1 mM NAD+, 5 mM DTT and 2 mM MgCl2 in 50 μL of 20 mM Tris·HCl buffer, pH 7.5. The reaction was initiated by the addition of TviB (2 μM final concentration), and absorbance at 340 nm was measured during 1 min immediately after the addition of enzyme. An HPLC assay was also developed to determine the TviB activity. This assay is especially useful for testing a series of UDP-sugar derivatives, including UDP-D-glucose (UDP-Glc), UDP-D-galactose (UDP-Gal), and UDP-GalNAc (3), which were used to individually replace 2 in the assay to determine the substrate specificity of TviB. The reaction mixture was incubated at 30 °C for 1 h, and the protein was removed by a Microcon YM-10 centrifugation unit. The filtrate was analyzed by HPLC equipped with a Dionex CarboPac PA-1 (4 × 250 mm) column. The product was eluted by a gradient of water as solvent A and 1 M NH4OAc as solvent B where the gradient ran from 20 to 25% B over 2 min, 25 to 35% B over 35 min, 35 to 100% B over 5 min, followed by 20 min wash by 100% B. The flow rate was 0.6 mL/min, and the detector was set at 267 nm. The retention times were 15.2 min for UDP-GalNAc (3), 15.8 min for UDP-GlcNAc (2), 18.8 min for UDP-Gal, and 19.8 min for UDP-Glc.
Enzyme Activity Assay for TviC
The assay mixture to determine the TviC activity contained 0.25 mM UDP-GlcNAcA (4) and 2 mM MgCl2 in 50 μL of 20 mM Tris·HCl buffer, pH 7.5. To each reaction mixture, 0.1 μM of TviC was added to initiate the reaction. UDP-GlcNAc (2), UDP-GalNAcA (5), and UDP-GalNAc (3), were also tested as possible substrates to determine the specificity of TviC. After incubation at 30 °C for 1 h, the protein in each assay sample was removed by a Microcon YM-10 centrifugation unit, and the filtrate was analyzed by HPLC equipped with a Dionex CarboPac PA-1 (4 × 250 mm) column as described above for TviB assay. The retention times for UDP-GalNAcA (5) and UDP-GlcNAcA (4) were 49.1 and 51.1 min, respectively.
Determination of the Optimal pH for TviB and TviC Activities
To determine the optimal conditions for TviB assay, each 50 μL reaction mixture contained 2 μM TviB, 0.4 mM UDP-GlcNAc (2), 1 mM NAD+, 5 mM DTT and 2 mM MgCl2 in buffers with varied pHs. Activity was evaluated by monitoring the change of absorbance at 340 nm. For the TviC assay, each 50 μL reaction mixture contained 1.7 μg enzyme, 0.75 mM UDP-GlcNAcA (4) and 2 mM MgCl2 in buffers with varied pHs. The reactions were quenched after 5 min, by boiling the reaction mixture for 5 min, and analyzed by HPLC. The substrate and product ratios were calculated based on the integration of the corresponding peaks from the HPLC chromatogram. The reactions were performed at 25 °C in 50 mM potassium phosphate buffer for the pH range between 6.0 and 7.6 in increments of 0.4, in 20 mM Tris·HCl buffer for pH range between 8.0 and 8.8 in increments of 0.4, and in 50 mM glycine-NaOH solution for the pH range between 9.6 and 10.0.
Determination of the Kinetic Parameters for TviB
The rate of the TviB-catalyzed reaction was determined spectrophotometrically by following the formation of NADH (ε340 = 6220 M−1 cm−1) at 25 °C. A series of samples containing 1.6 mM NAD+, 2 mM MgCl2, 5 mM DTT, and varying amounts of UDP-GlcNAc (2, 0.05 to 0.8 mM) in 100 μL of 50 mM potassium phosphate buffer, pH 8.0, was prepared. The reaction was initiated by the addition of TviB (2 μM final concentration), and absorbance at 340 nm was measured during 1 min immediately after the addition of enzyme. The rates were fitted to the Michaelis-Menten equation. To determine the kinetic parameters of NAD+, the assay was repeated with 1 mM UDP-GlcNAc (2) and varying concentrations of NAD+ (0.05 to 1.6 mM) in 100 μL of 50 mM potassium phosphate buffer, pH 8.0, containing 2 mM MgCl2 and 5 mM DTT.
Determination of the Kinetic Parameters of TviC for UDP-GlcNAcA and UDP-GalNAcA
The kinetic parameters of TviC for UDP-GlcNAcA (4) and UDP-GalNAcA (5) were determined by the discontinuous HPLC assay as described above. The substrate and product ratios were calculated from the integration of the corresponding peaks from the HPLC chromatogram. A typical reaction contained 0.01–1mM of UDP-GlcNAcA (4) or UDP-GalNAcA (5) in 40 μL of 50 mM potassium phosphate buffer containing 2 mM MgCl2, pH 8.0. The reaction was initiated by the addition of appropriate amount of TviC (27 pmol for 4 or 53 pmol for 5) and carried out at room temperature. Aliquots were taken every 10 s from samples containing 0.01–0.075 mM substrate, or every 30 s from samples containing 0.1–1 mM substrate, and quenched by boiling in the microcentrifuge tubes for 5 min. The standard Michaelis-Menten equation was used for calculating Km and Vmax.
Determination of the Kinetic Parameters of TviC for UDP-GlcNAc and UDP-GalNAc
The kinetic parameters of TviC for UDP-GlcNAc (2) and UDP-GalNAc (3) were determined by the capillary electrophoresis (CE) analysis. Each assay mixture contained varying concentrations of 0.04–1.2 mM of UDP-GlcNAc or UDP-GalNAc and 2 mM MgCl2 in 40 μL of 50 mM potassium phosphate buffer, pH 8.0. The reaction was initiated by the addition of TviC (267 pmol for 2 or 133 pmol for 3). Aliquots were taken every 30 s from the incubation mixture containing 0.04–0.1 mM UDP-GalNAc (3), every 1 min for those containing 0.2–0.3 mM substrate, and every 2 min from samples containing 0.5–1.2 mM substrate, and quenched by boiling in microcentrifuge tube for 5 min. For UDP-GlcNAc (2), aliquots of reactions were taken every 1 min from samples having substrate concentration of 0.04–0.2 mM, and every 2 min from those containing 0.3–1.2 mM substrate. The reaction was quenched by boiling for 5 min.
Capillary electrophoresis analysis was performed at room temperature using a P/ACE 5000 system (Beckman Instruments, Fullerton, CA) equipped with a diode array detector. The running buffer was 100 mM sodium tetraborate, pH 9.4. The capillary was bare silica 75 × 57 cm with the detector set at 50 cm. The capillary was conditioned before each run by washing with 0.1 N NaOH for 2 min, distilled water for 2 min, and running buffer for 2 min. Samples were introduced by pressure injection for 4 s, and the separation was performed at 22 kV. The retention time of UDP-GlcNAc (2) and UDP-GalNAc (3) were 12.4 and 13.3 min, respectively. Peak integration was done using the Beckman P/ACE Station software. The standard Michaelis-Menten equation was used to calculate Km and Vmax.
Enzymatic Synthesis of UDP-GlcNAcA (4)
A 2-mL reaction mixture containing 0.1 mg/mL of purified TviB, 1 mM NAD+, 2 mM MgCl2, 5 mM DTT, and 0.4 mM UDP-GlcNAc (2) in 20 mM Tris·HCl, pH 7.5, was incubated at 30 °C for 16 h. Protein was precipitated by the addition of ethanol (1 mL) and removed by centrifugation. The supernatant was loaded onto a FPLC MonoQ HR 16/10 column, washed with deionized water, and eluted with a linear gradient from 0 to 250 mM NH4HCO3 over 16 min, 250–500 mM NH4HCO3 over 4 min, followed by a 5-min wash with 500 mM NH4HCO3. UDP-GlcNAcA (4) was eluted during isocratic elution with 500 mM NH4HCO3. The flow rate was 5 mL/min and the detector was set at 280 nm. The conversion of 2 to 4 was nearly quantitative. The fractions containing the desired product were combined and lyophilized. 1H NMR (500 MHz, D2O) δ 7.82 (1 H, d, J6″,5″ 8.0 Hz, 6″-H), 5.86 (1 H, d, J1′,2′ = 4.5 Hz, 1′-H), 5.84 (1 H, d, J5″,6″ = 8.0 Hz, 5″-H), 5.40 (1 H, dd, 1-H, J1,2 = 3.0 Hz), 4.25-4.22 (2 H, m, 2′-H, 3′-H), 4.16 (1 H, m, 4′-H), 4.07 (2 H, m, 5′-H), 4.04 (1 H, d, J5,4 = 10.0 Hz, 5-H), 3.90 (1H, dd, J2,1 = 3.0 Hz, J2,3 = 10.5 Hz, 2-H), 3.69 (1 H, dd, J3,2 = 10.5 Hz, J3,4 = 9.5 Hz, 3-H), 3.46 (1 H, dd, J4,3 =9.5 Hz, J4,5 = 10.0 Hz, 4-H), 1.95 (3 H, s, CH3CO). 13C NMR (125 MHz, D2O) δ 176.0 (C-6), 175.0 (CH3CO), 166.5 (C-4″), 152.6 (C-2″), 141.8 (C-6″), 102.9 (C-5″), 94.4 (C-1), 88.6 (C-1′), 83.5 (C-4′), 74.0 (C-2′), 73.2 (C-5), 72.3 (C-4), 70.9 (C-3), 69.9 (C-3′), 65.2 (C-5′), 53.7 (C-2), 22.3 (CH3CO). 31P NMR (202 MHz, D2O) δ −10.2 (Pα, d, J = 20.8 Hz), −11.9 (Pβ, d, J = 18.9 Hz).
Enzymatic Synthesis of UDP-GalNAcA (5)
A 2-mL reaction mixture containing 0.1 mg/mL of purified TviC, 2 mM MgCl2, and 0.4 mM UDP-GlcNAcA (4), in 20 mM Tris·HCl, pH 7.5, was incubated at 30 °C for 16 h. Protein was precipitated by the addition of ethanol (1 mL) and removed by centrifugation. The desired product was purified by FPLC using the identical conditions for the isolation of UDP-GlcNAcA (4) as described above. The conversion of 4 to 5 was approximately 70%. The purified UDP-GalNAcA (5) was lyophilized, and the identity was confirmed by NMR spectroscopy. 1H NMR (500 MHz, D2O) δ l7.53 (1 H, d, J6″,5″ = 8.5 Hz, 6″-H), 5.60 (1 H, d, J1′,2′= 5.0 Hz, 1′-H), 5.58 (1 H, d, J5″,6″ = 8.5 Hz, 5″-H), 5.23 (1 H, dd, J1,2 = 3.5 Hz, 1-H), 4.13 (1 H, d, J5,4 = 1.5 Hz, 5-H), 3.99 (1 H, dd, J2′,1′ = 5.0 Hz, J2′,3′ = 5.5 Hz, 2′-H), 3.97 (1 H, dd, J4,3 = 3.5 Hz, J4,5 = 1.5 Hz, 4-H), 3.95 (1 H, dd, J3′,2′ = 5.5 Hz, J3′,4′ = 4.5 Hz, 3′-H), 3.91-3.89 (1 H, m, 4′-H), 3.88–3.83 (2 H, m, 2-H, 5′-H), 3.78–3.74 (1 H, m, 5′-H), 3.66 (1 H, dd, J3,4 = 3.5 Hz, J3,2 = 11.0 Hz, 3-H), 1.70 (3 H, s, CH3CO). 13C NMR (125 MHz, D2O) δ 175.4 (C-6), 174.9 (CH3CO), 166.6 (C-4″), 152.1 (C-2″), 141.6 (C-6″), 102.6 (C-5″), 94.4 (C-1), 88.8 (C-1′), 83.1 (C-4′), 73.7 (C-2′), 73.0 (C-5), 69.8 (C-3′), 69.6 (C-4), 67.5 (C-3), 65.1 (C-5′), 59.4 (C-2), 22.2 (CH3CO). 31P NMR (202 MHz, D2O) δ −10.2 (Pα;, d, J = 19.3 Hz), −11.8 (Pβ, d, J = 18.9 Hz).
RESULTS
Expression and Purification of TviB and TviC
Production of recombinant TviB and TviC was made possible by the cloning and expression of the corresponding genes from S. choleraesuis subsp. typhi genomic DNA.9 The tviB gene was cloned in a pET24b(+) vector and the gene product was produced efficiently as a soluble protein in E. coli at 30 °C. This enzyme, which contains a His6-tag at the C-terminus, was purified to near homogeneity by a protocol consisting of DEAE Sepharose, FPLC MonoQ, Reaction Dye, and FPLC Superdex 200 chromatographic steps (Figure 2A). The yield was about 20 mg of TviB per 1 L of culture. DTT (10 mM) was included in all buffers throughout the purification to prevent deactivation via aberrant disulfide bond formation. Chromatography on a FPLC Superdex 200 column, the last purification step, removed the inactive oligomeric forms of TviB. TviB is stable when stored in buffer containing 10 mM DTT. No precipitation was observed even after multiple freeze-thaw from the −80 °C stocks. The Mr of 120.5 kDa, estimated by gel filtration, and the calculated mass of 47,675 Da, based on the translated sequence for each subunit, suggest that TviB likely exists as a homotrimer.
FIGURE 2.

SDS-PAGE of purified TviB and TviC. The molecular weight marks are: 175, 83, 62, 47.5, 32.5, 25 and 16.5 kDa (top to bottom).
The tviC gene was cloned into a pET28b(+) vector and the expression in E. coli was carried out at 37 °C without induction by IPTG. The TviC protein carrying a His6-tag at the C-terminus was purified to near homogeneity by a Ni-NTA column (Figure 2B). The yield was 10 mg per 1 L of culture. The subunit molecular mass of 37.5 kDa, as approximated by SDS-PAGE, correlates well to the predicted value of 40,325 Da calculated from the translated sequence plus the His6-tag. A molecular mass of 73.0 kDa, estimated by the size exclusion chromatography, indicated that the native TviC exists as a homodimer.
NAD(H) Extraction
TviC contains tightly bound NAD(H) coenzyme which could be released by complete digestion of TviC with proteinase K. The free coenzyme was then reduced to NADH by sodium borohydrate. A stoichiometric ratio of 0.7–0.8 mol of NAD(H) per mol of protein was calculated from the absorbance at 340 nm.
Determination of Enzyme Activity of TviB
The activity of TviB as a sugar dehydrogenase was determined using various UDP-sugar derivatives as substrates. Although the progress of the reaction using the natural substrate, UDP-GlcNAc (2), could be followed by the formation of NADH at 340 nm, the slow conversion of other substrate analogues under the incubation conditions rendered this method impractical. Instead, the reaction mixture was analyzed by a HPLC Dionex CarboPAC PA-1 column, which directly detects product formation. It was found that TviB efficiently converts UDP-GlcNAc (2) to UDP-GlcNAcA (4) (Figure 3, trace 4), and the reaction, as expected, requires the addition of NAD+ (Figure 3, traces 4 and 5). All reactions were performed in buffer at pH 7.5 because an analysis of TviB activity over a range of pH from 6.0 to 9.6 revealed the pH optimum at 7.2, with a large decrease in activity near pH 8.4 (Figure 4A). Our results also show that UDP-GalNAc (3), UDP-Glc and UDP-Gal are not substrates for TviB (Figure 3, traces 1, 2 and 3). Thus, TviB is clearly an UDP-GlcNAc 6-dehydrogenase.
FIGURE 3.

The HPLC assay to determine TviB activity. Reaction mixture contained 0.5 μM TviB, 5 mM DTT, 2 mM MgCl2, 0.5 mM NAD+, and 0.25 mM of either UDP-Glc (trace 1), UDP-Gal (trace 2), UDP-GalNAc (3, trace 3), or UDP-GlcNAc (2, trace 4) in 20 mM Tris•HCl buffer (pH 7.5). Incubation with UDP-GlcNAc (2) in the absence of NAD+ was also performed (trace 5). Product formation was detected only in the sample using UDP-GlcNAc (2) as substrate and in the presence of NAD+ (trace 4). The product was confirmed to be UDP-GlcNAcA (4) by NMR. See Experimental Procedures for details. Peaks marked * are the contaminants from NAD+.
FIGURE 4.
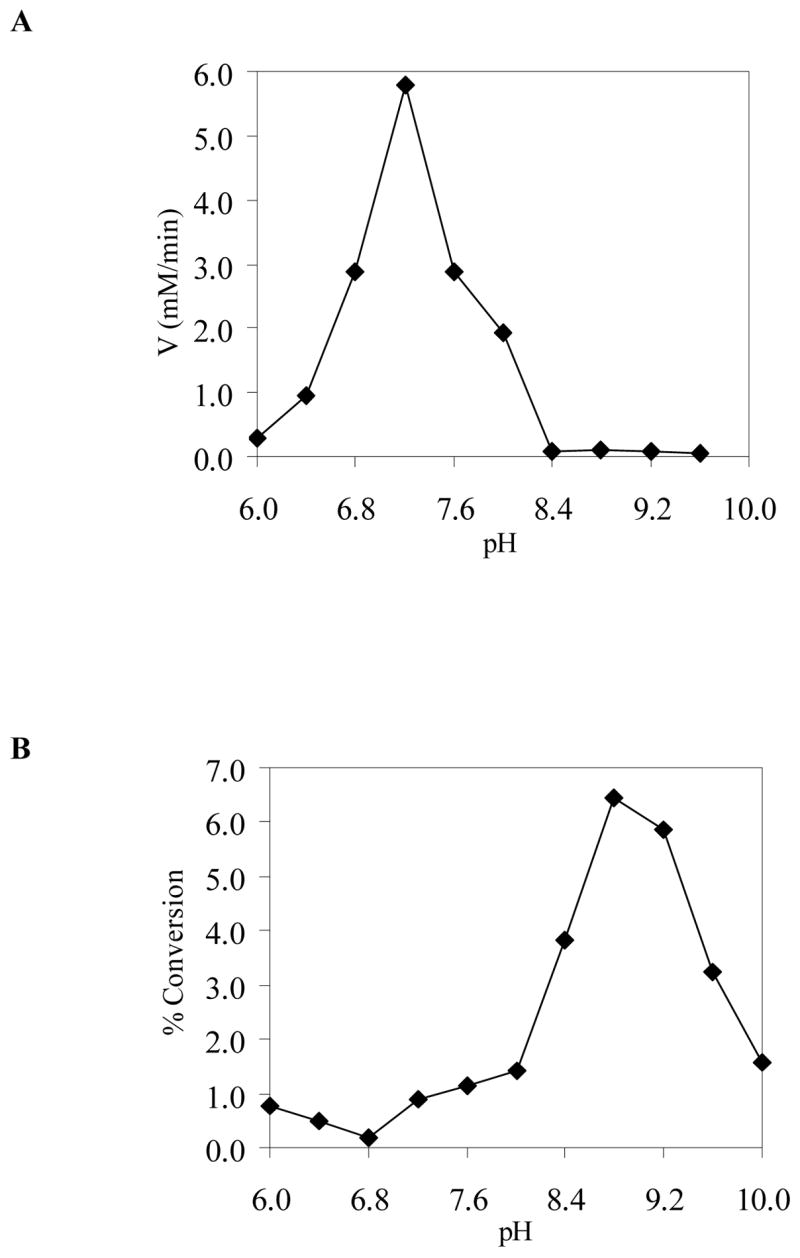
The pH dependence of enzyme activity. All reactions (50 μL total volume) were carried out in 50 mM potassium phosphate buffer for pH 6.0–7.6, in 20 mM Tris·HCl buffer for pH 8.0–8.8, and in 50 mM glycine-NaOH solution for pH 9.6–10.0. (A) Reaction included UDP-GlcNAc (2), NAD+, and TviB. (B) Reaction included UDP-GlcNAcA (4) and TviC. See the Experimental Procedures for details.
Determination of Enzyme Activity of TviC
The C-4 epimerase activity of TviC was investigated by HPLC analysis of the incubation mixtures. As shown in Figure 5, TviC is active using not only UDP-GlcNAcA (4) and UDP-GalNAcA (5) (Figure 5, traces 2 and 1), but also UDP-GlcNAc (2) and UDP-GalNAc (3) (Figure 5, traces 4 and 3). In contrast, neither UDP-Glc nor UDP-Gal is a substrate for TviC (Figure 5, traces 6 and 5). Upon reaching equilibrium, the ratio of UDP-GlcNAcA (4) and UDP-GalNAcA (5) in the incubation mixture is 30:70. For the UDP-GlcNAc (2) and UDP-GalNAc (3) pair, the ratio at equilibrium is approximately 70:30. Although TviC is active over a broad range of pH (from 6 to 10), it has the highest activity at pH 8.8 when using 4 or 5 as substrate (Figure 4B).
FIGURE 5.

The HPLC assay to determine TviC activity. Reactions contained 0.1 μM TviC, 2 mM MgCl2, and 0.25 μM of either UDP-GalNAcA (5, trace 1), UDP-GlcNAcA (4, trace 2), UDP-GalNAc (3, trace 3), UDP-GlcNAc (2, trace 4), UDP-Gal (trace 5), or UDP-Glc (trace 6). Interconversion of UDP-GalNAc and UDP-GlcNAc was observed when either substrate was used (traces 3 and 4). Epimerization between UDP-GalNAcA and UDP-GlcNAcA (traces 1 and 2) was also noted. See the Experimental Procedures for details.
Identification of Reaction Products of TviB and TviC
The products of TviB and TviC reactions can be easily separated from other components of the incubation mixture using FPLC equipped with a MonoQ ion exchange column (Figure 6). When 2 was incubated with TviB, TviC, and the necessary components in a one-pot reaction, product analysis showed the formation of UDP-GalNAc (3) and UDP-GalNAcA (5), in addition to the unreacted starting material UDP-GlcNAc (2) and the TviB product UDP-GlcNAcA (4). While 2 and 3 were co-eluted under the FPLC conditions, they were well resolved by capillary electrophoresis (see Figure 9). Formation of 3 and 5 is clearly due to the action of TviC. Products 4 and 5 were identified by 1H (COSY and HSQC), 13C and 31P NMR spectral analyses. The configuration at C-4 of UDP-GlcNAcA (4) was assigned based on the J3,4 and J4,5 values of 9.5 and 10.0 Hz, respectively, indicating the equatorial orientation for the 4-OH group. The coupling constants for UDP-GalNAcA (5) are 3.5 and 1.5 Hz, respectively, indicating the axial orientation for the 4-OH group.
FIGURE 6.

Separation of products generated in TviB and TviC reactions by FPLC equipped with a MonoQ HR 16/10 column. Fractions containing UDP-GlcNAcA (4) UDP-GalNAcA (5) were collected, lyophilized, and analyzed by NMR.
FIGURE 9.
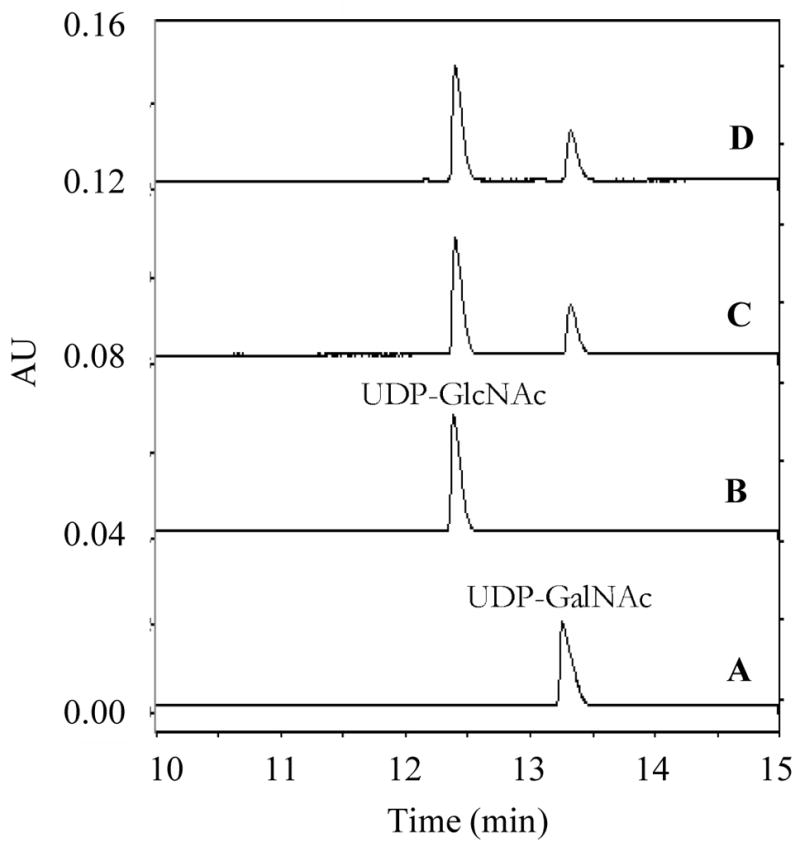
Capillary electrophoresis separation of UDP-GlcNAc and UDP-GalNAc. The capillary was bare silica 75 × 57 cm with the detector set at 50 cm. The running buffer was 100 mM sodium tetraborate, pH 9.4. The separation was performed at 22 kV. Trace A, UDP-GalNAc (3) alone; Trace B, UDP-GlcNAc (2) alone; Trace C, UDP-GalNAc+TviC; Trace D, UDP-GlcNAc+TviC. The retention time of UDP-GalNAc (3) (trace A) and UDP-GlcNAc (2) (trace B) were 13.3 and 12.4 min, respectively.
Determination of the Kinetic Parameters for TviB and TviC
For the sake of comparison, the kinetic studies of TviB and TviC were carried out under the same conditions at room temperature and pH 7.5, at which both enzymes have sufficiently high activities. The kinetic parameters for TviB were determined by varying the concentration of NAD+ and UDP-GlcNAc (2) separately, and monitoring the change of absorbance at 340 nm using a spectrophotometric assay. The Km values for UDP-GlcNAc and NAD+ were determined to be 77 ± 9 μM and 276 ± 52 μM, respectively, and the kcat value was determined to be 15.5 ± 1.0 min−1 (see Figure 7 and Table 1).
FIGURE 7.
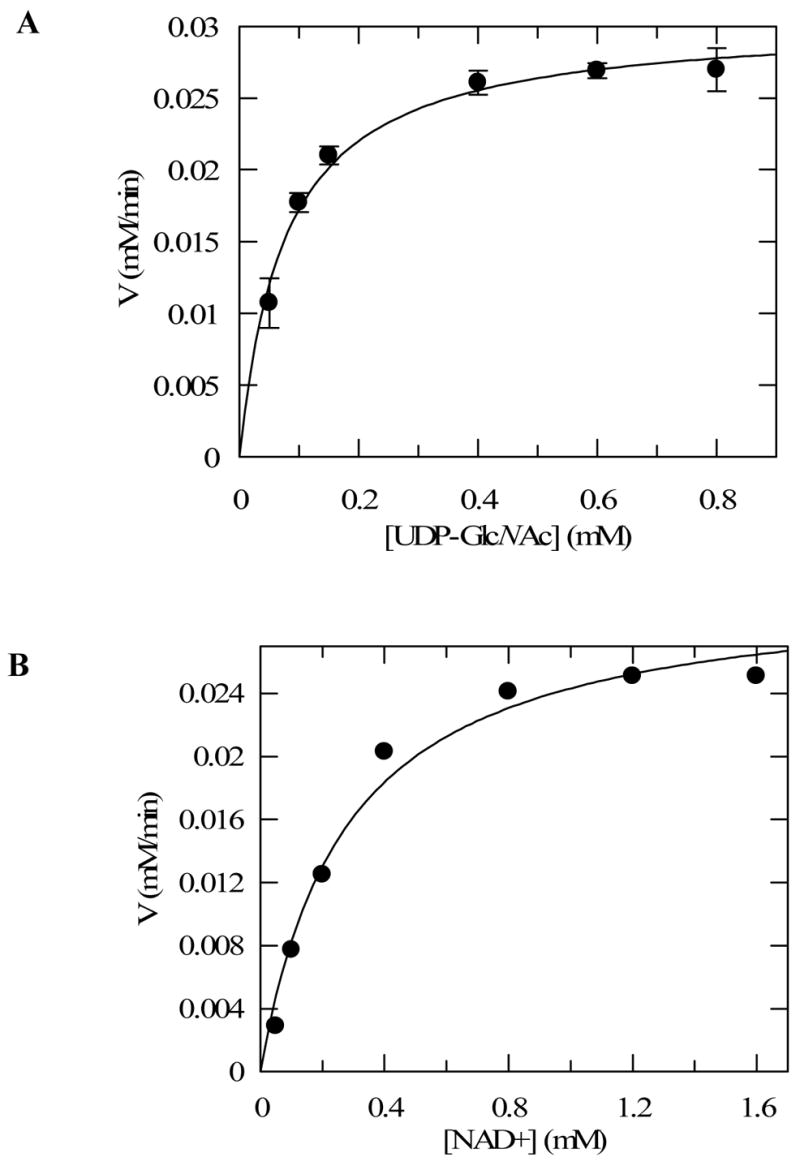
Kinetics analysis of TviB catalyzed reaction. Initial rate versus substrate concentration were plotted, and the data were fitted to Michaelis-Menten equation by nonlinear regression using Grafit 5. (A) Reactions were performed with UDP-GlcNAc (2) as the variable substrate (0.05 – 0.8 mM) and NAD+ at a fixed concentration of 1.6 mM. (B) Reactions were performed with NAD+ as the variable substrate (0.05 – 1.6 mM) and UDP-GlcNAc (2) at a fixed concentration of 1 mM. All reactions contained 2 μM TviB in 100 μL of 50 mM potassium phosphate buffer, pH 8.0, and were conducted at room temperature. See Experimental Procedures for details.
Table 1.
Kinetic Parameters for Reaction Catalyzed by TviB Determined by the Spectrophotometric Assaya
| Substrate |
Km (mM) |
Vmax (mM/min) |
TviB
mM |
kcat (min−1) |
kcat/Km (mM−1min−1) |
|---|---|---|---|---|---|
| UDP-GlcNAc (2) | 0.077±0.009 | 0.031±0.002 | 2.0×10−3 | 15.5±1.0 | 198.8±19.1 |
| NAD+ | 0.276±0.052 | 0.031±0.002 | 2.0×10−3 | 15.5±1.0 | 56.2±8.0 |
See Experimental Procedures for details
Due to the reversible nature of the TviC reaction, the kinetic parameters were determined in both directions. To ensure initial rate measurements, the amount of TviC used allowed only 10% or less substrate conversion after a certain incubation period. As shown in Figure 8 and Table 2, the Km for UDP-GlcNAcA (4) is 20 ± 1 μM and the kcat is 56.8 ± 0.5 min−1 in the forward direction (4 → 5) using the HPLC assay. In the reverse direction, the Km for UDP-GalNAcA (5) and the kcat for the catalysis (5 → 4) were determined to be 42 ± 2 μM and 39.1 ± 0.6 min−1, respectively. Although UDP-GlcNAc (2) and UDP-GalNAc (3) cannot be cleanly separated by HPLC, baseline resolution of these two compounds was achieved by capillary electrophoresis (Figure 9). On the basis of the integration of the elution peaks, the Km values were determined to be 51 ± 3 μM and 71 ± 5 μM for UDP-GlcNAc and UDP-GalNAc, respectively. The kcat value for the forward reaction (2 → 3) is 14.2 ± 0.2 min−1 and that for the reverse direction (3 → 2) is 52.6 ± 0.9 min−1 (see Figure 10 and Table 2).
FIGURE 8.
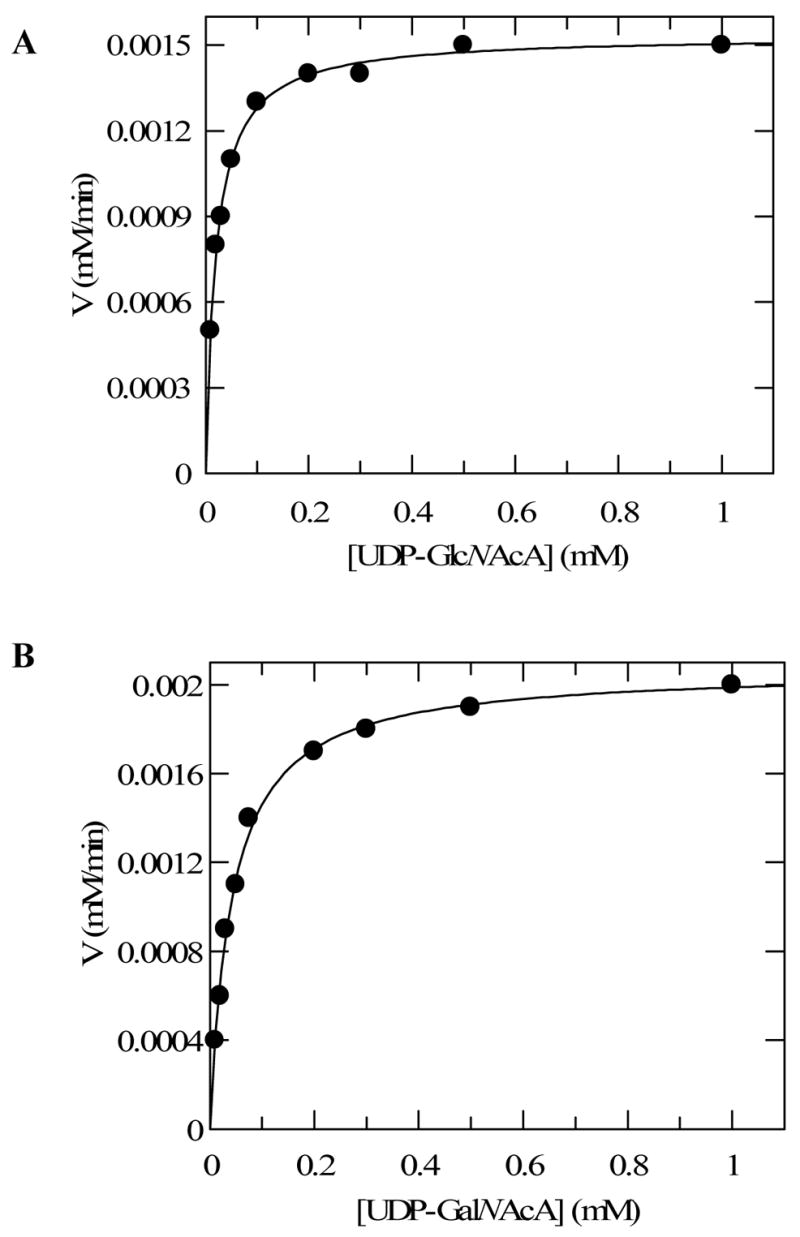
Kinetics analyses of the epimerization of UDP-GlcNAcA and UDP-GalNAcA by TviC using FPLC. The initial rates of epimerization were determined by measuring the slope of the plot of amount of product formation versus time. The data were fitted to Michaelis-Menten equation by nonlinear regression using Grafit 5. (A) Reactions were performed with UDP-GlcNAcA (4) as the substrate using 27 pmol of TviC. (B) Reactions were performed with UDP-GalNAcA (5) as substrate using 53 pmol of TviC. All reactions were conducted at room temperature in 40 μL of 50 mM potassium phosphate buffer containing 2 mM MgCl2, pH 8.0. See Experimental Procedures for details.
Table 2.
Kinetic Parameters for Reactions Catalyzed by TviC Determined by the HPLC Assay and Capillary Electrophoresis Assaya
| Substrate |
Km (mM) |
Vmax (mM/min) |
TviC
mM |
kcat (min−1) |
kcat/Km (mM−1min−1) |
|---|---|---|---|---|---|
| UDP-GlcNAcA (4)b | 0.020±0.001 | 0.0015±0.00001 | 2.7×10−5 | 56.8±0.5 | 2871.6±110.8 |
| UDP-GalNAcA (5)b | 0.042±0.002 | 0.0021±0.00003 | 5.3×10−5 | 39.1±0.6 | 931.3±43.3 |
| UDP-GlcNAc (2)c | 0.051±0.003 | 0.0038±0.00005 | 2.7×10−4 | 14.2±0.2 | 281.6±12.6 |
| UDP-GalNAc (3)c | 0.071±0.005 | 0.0068±0.0001 | 1.3×10−4 | 52.6±0.9 | 741.7±38 |
See Experimental Procedures for details.
Values listed were determined by HPLC assays.
Values listed were determined by the capillary electrophoresis assays.
FIGURE 10.
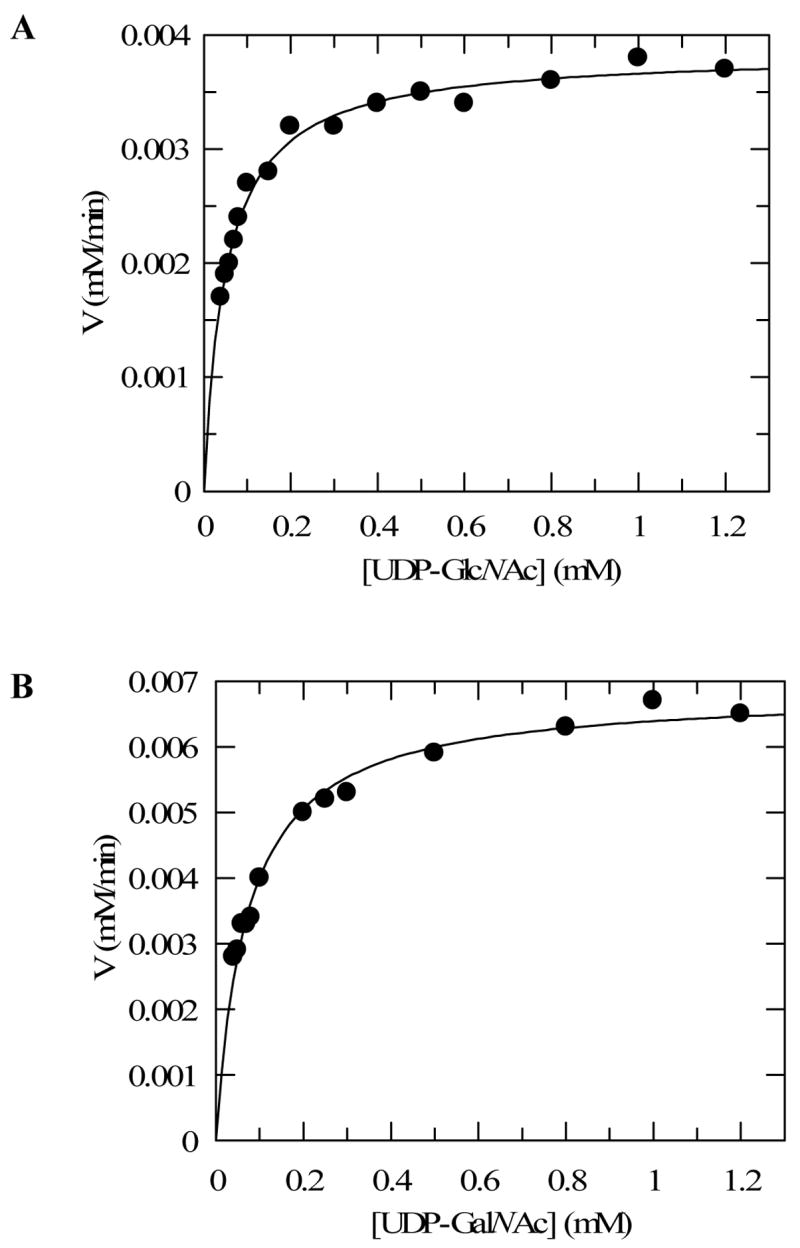
Kinetics analyses of the epimerization of UDP-GlcNAc and UDP-GalNAc by TviC using capillary electrophoresis. The initial rates of epimerization were determined by measuring the slope of the plot of amount of product formation versus time. The data were fitted to Michaelis-Menten equation by nonlinear regression using Grafit 5. (A) Reactions were performed with UDP-GlcNAc (2) as the substrate using 27 pmol of TviC. (B) Reactions were performed with UDP-GalNAc (3) as substrate using 13 pmol of TviC. All reactions were conducted at room temperature in 40 μL of 50 mM potassium phosphate buffer containing 2 mM MgCl2, pH 8.0. See Experimental Procedures for details.
DISCUSSION
Capsular polysaccharides are immunogenic in mammals and, when injected, can induce the production of antibodies capable of neutralizing the corresponding capsulated bacteria. The fact that they are non-toxic and free of the deleterious effects associated with the whole organism has prompted considerable interest in developing capsular polysaccharides as molecular vaccines. The ready availability of the necessary capsular polysaccharides to be used as antigen is essential for the development of such a vaccine. While the producing bacteria are obvious sources for the desired polysaccharides, handling those pathogenic strains can be challenging. Recent advances in synthetic chemistry have made it possible to prepare the targeted polysaccharides in a de novo fashion. However, such a chemical approach is tedious and time consuming, and in many cases, the final product is obtained in low yield. The above problems can be avoided if the desired polysaccharides are prepared enzymatically. Hence, we initiated a study of the biosynthesis of Vi antigen to explore the feasibility of reconstituting its biosynthetic machinery in vitro. Utilization of this machinery may allow direct access to the desired polysaccharides in sufficient quantity.
The genes believed to be directly involved in the biosynthesis of Vi antigen include tviB, tviC, tviD and tviE.6,7,11 Sequence analysis suggests tentative assignments of functions: TviB may be a dehydrogenase, TviC may function as an epimerase, TviD is homologous to cytochrome P450 enzymes, and TviE belongs to the family of glycosyltransferases. Since TviB and TviC are likely involved in the early steps of the biosynthesis of Vi antigen, biochemical characterization of these two proteins to verify their functions will provide the necessary evidence to establish the biosynthetic pathway.
TviB was produced in E. coli as a C-terminus histidine tagged protein. The purified TviB catalyzes the conversion of UDP-GlcNAc (2) to UDP-GlcNAcA (4) in the presence of NAD+. Similar to the well known UDP-glucose dehydrogenase, the mechanism of TviB is expected to be initiated by a NAD+-dependent oxidation at C-6, followed by the addition of a cysteine residue to the nascent C-6 aldehyde group to form a thioester intermediate.12,14 The thioester intermediate is then hydrolyzed to give the glucuronic acid product. Sequence alignments of TviB, WbpA, WbpO, and UDP-glucose dehydrogenase from group A Streptococci (Figure 11) reveals a single conserved cysteine residue at position 261 (using the TviB sequence numbering). This is most likely the catalytically important cysteine in the reaction. In fact, its equivalent in Streptococcal UDP-glucose dehydrogenase (Cys260) has been shown to play such a catalytic role.12 There also exist five other cysteine residues in TviB. The failure to detect any dehydrogenase activity of TviB purified in the absence of DTT suggests an aberrant disulfide linkage or sulfoxide formation among these cysteine residues causing conformational change of the active site, or modification of the essential cysteine residue. It should be noted that Ni-NTA chromotography was not used to purify the heterologously expressed His6-tagged TviB, since 1 mM is the maximum concentration of DTT allowed for Ni-NTA resin purification,24 but 1 mM DTT is not sufficient to prevent oxidation of the cysteine residues.
FIGURE 11.
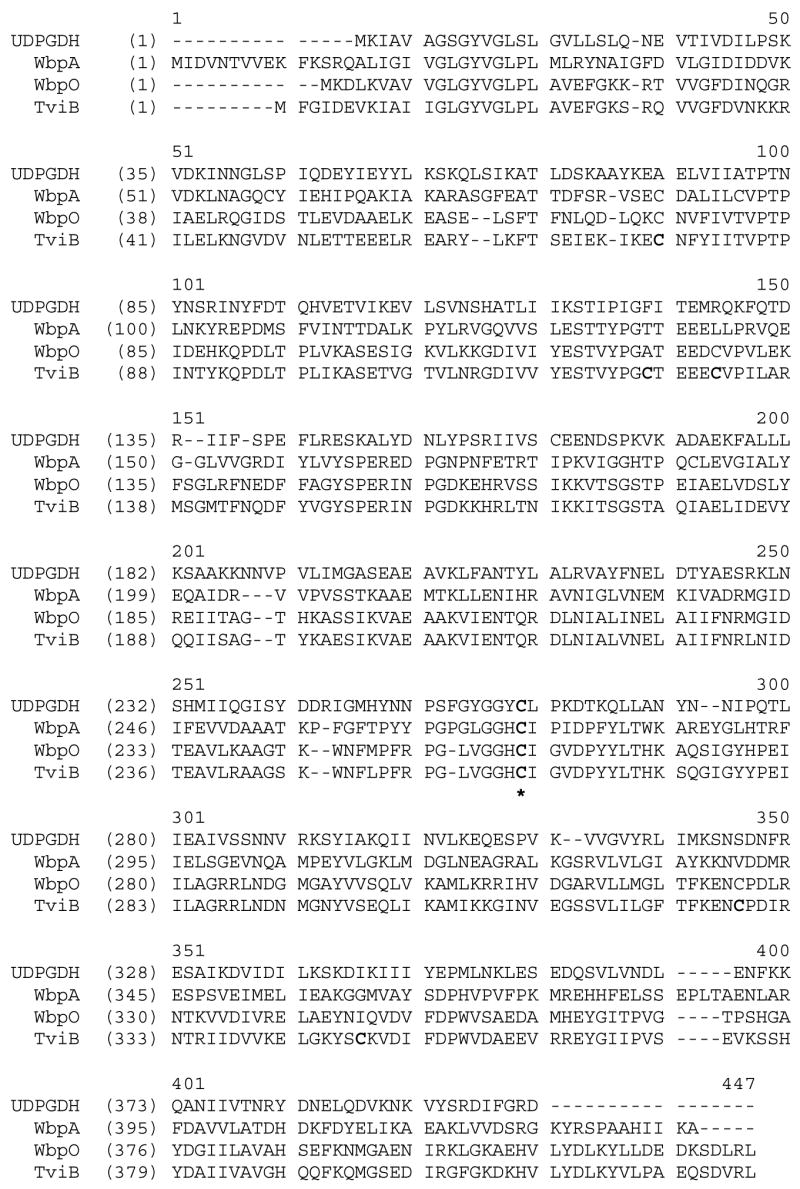
Sequence alignment of TviB and its homologues: WbpA, an UDP-GlcNAc 6-dehydrogenase from Pseudomonas aeruginosa PAO1 (31% identity, 52% similarity),16 WbpO, an UDP-GalNAc 6-dehydrogenase from Pseudomonas aeruginosa PAO1 (65% identity, 83% similarity).15 Also included is the sequence of UDP-Glucose dehydrogenease from Group A Streptococci.12 The six cysteine residues in TviB are highlighted in bold phase. Cys261 (TviB sequence number), which is conserved among all four sequences, is marked with an asterisk.
TviB shares higher sequence identity with WbpO (65%), which is an UDP-GalNAc dehydrogenase, than with WbpA (32%), which is an UDP-GlcNAc dehydrogenase. However, TviB functions as an UDP-GlcNAc dehydrogenase and has no UDP-GalNAc dehydrogenase activity. NMR spectral data confirmed that the product of TviB upon incubation with UDP-GlcNAc (2) is indeed UDP-GlcNAcA (4). The Km values determined for UDP-GlcNAc (2) and NAD+, respectively, are comparable to those reported for WbpA (Km of 94 μM for 2 and 220 μM for NAD+).16 In contrast, the kcat value for TviB reaction was found to be nearly 6-fold lower than that determined for WbpA (15.5 versus 86 min−1). It is possible that TviB-catalyzed dehydrogenation is the rate-limiting step in the Vi antigen biosynthesis. Since capsular polysaccharides are not essential for the bacterial cell structure but enhance virulence and induce immune response in the hosts, slow formation of Vi antigen may allow the invading bacteria to elude the host cell defense system.
The purified TviC, carrying a His6-tag at the C-terminus, is a homodimeric protein. The epimerization catalyzed by TviC is interesting because it occurs at a stereogenic center (C-4) that is not activated by an adjacent keto or other electron withdrawing groups. Examination of the translated sequence of the tviC gene led to the identification of a NAD+-binding motif near the N-terminus,17 suggesting that TviC is likely a pyridine nucleotide-dependent catalyst. Further characterization showed that TviC contains a tightly bound NAD+. UDP-galactose 4-epimerase25–28 is the best studied example of an epimerase whose catalysis is NAD+ dependent. The NAD+ cofactor in this enzyme plays a key role in the formation of a 4-ketosugar intermediate. Subsequent NADH reduction allows the return of the hydride to either face of the pyranose ring, resulting in epimerization at C-4.
An analogous mechanism could also be operative with TviC, which functions as an UDP-hexose 4-epimerase catalyzing the epimerization at C-4 not only of UDP-GlcNAc (2) and UDP-GalNAc (3), but also of UDP-GlcNAcA (4) and UDP-GalNAcA (5). Relaxed substrate specificity is not uncommon among sugar epimerases. For example, GalE,27,28 which is an UDP-Gal 4-epimerase, can accept UDP-GalNAc/UDP-GlcNAc pair as substrates, and WbgU18 and WbgP,19,29 both of which are UDP-GlcNAc 4-epimerases, can also process the UDP-Glc/UDP-Gal pair. However, TviC is unusual because it is aminosugar specific (does not accept UDP-Glc/UDP-Gal) and is the only known epimerase that carries out interconversion between UDP-GlcNAcA and UDP-GalNAcA. The UDP-GalNAcA product (5) obtained from the incubation of UDP-GlcNAcA (4) with TviC was confirmed by NMR analysis.
Kinetic analyses of TviC revealed that Km for UDP-GlcNAcA (4) is 2-fold lower than that of UDP-GalNAcA (5), and the kcat/Km for the forward reaction (4 → 5) is 3-fold greater than that of the reverse reaction. As a result, the equilibrium shifts toward UDP-GalNAcA (5) upon incubation of 4/5 with TviC (4:5 = 30:70). Interestingly, an equilibrium ratio of 70:30 between UDP-GlcNAc (2) and UDP-GalNAc (3) was observed when 2/3 was used in the incubation mixture. This is an interesting example where the stereo-preference of an epimerase for a specific epimer in a pair of related epimers (4 versus 5 in the 4/5 pair, and 2 versus 3 in the 2/3 pair) is reversed and appears to be governed by a functional group (the 5-carboxylate group in TviC case) located adjacent to the reaction center. While both UDP-GlcNAc (2) and UDP-GalNAc (3) are good substrates for TviC, as revealed by the kinetic parameters listed in Table 2, UDP-GlcNAcA (4) and UDP-GalNAcA (5) are the preferred substrate pair. Thus, TviC is a specific UDP-GlcNAcA C-4 epimerase.
Taken together, the biosynthesis of the monomeric precursor for Vi antigen in Salmonella typhi is now established to proceed first by the TviB-catalyzed oxidization of UDP-GlcNAc (2) to UDP-GlcNAcA (4), followed by the TviC-catalyzed epimerization of UDP-GlcNAcA (4) to UDP-GalNAcA (5) (Scheme 1, route A). The favorable equilibrium toward 5 agrees well with the direction of the reaction flux. A supplementary route involving the conversion of cellular UDP-GalNAc (3) to UDP-GlcNAc (2) by TviC may also exist to provide additional 2 for the TviB reaction. The product of the TviB/TviC reactions, UDP-GalNAcA (5), serves as the substrate for TviE, which is the glycosyltranferase catalyzing the formation of the Vi polymer. Interestingly, the order of the same two reactions is reversed in the biosynthesis of O-antigen in P. aeruginosa O6 in which UDP-GalNAcA (5) is the substrate for the corresponding glycosyltransferases, WbpU and WbpT.18,30 In this pathway, UDP-GlcNAc (2) is first epimerized by WbpP to give UDP-GalNAc (3), which is then oxidized by WbpO to form UDP-GalNAcA (5) (analogous to route B in Scheme 1). Apparently, a different path has been evolved in Salmonella typhi to make Vi antigen.
In conclusion, we elucidated the pathway for the biosynthesis of UDP-GalNAcA (5), the precursor of Vi antigen. TviB is only the second enzyme catalyzing the formation of UDP-GlcNAcA (4), which has been biochemically characterized. Moreover, TviC is the first UDP-GlcNAcA C-4 epimerase reported to date, which can perform epimerization reactions between UDP-GlcNAcA (4) and UDP-GalNAcA (5), as well as UDP-GalNAc (3) and UDP-GlcNAc (2). The significance of this work is two-fold: it provides the platform for future in vitro biosynthesis of Vi antigen and it is potentially useful for developing inhibitors that end Vi antigen production.
Acknowledgments
We thank Dr. Klaus D Linse for the help with capillary electrophoresis and Professor Yuan-chun Lee for helpful discussion.
Abbreviations
- COSY
2D correlated spectroscopy
- DEAE
diethylaminoethyl
- DTT
dithiothreitol
- FPLC
fast protein liquid chromatography
- HPLC
high performance liquid chromatography
- HSQC
heteronuclear single quantum coherence
- IPTG
isopropyl β-D-thiogalactoside
- LB
Luria-Bertani
- NAD+
β-nicotinamide adenine dinucleotide
- NADH
β-nicotinamide adenine dinucleotide, reduced form
- ORF
open reading frames
- PAGE
polyacrylamide gel electrophoresis
- PCR
polymerase chain reaction
- SDS
sodium dodecyl sulfate
- TviB
an UDP-N-acetylglucosamine 6-dehydrogenase
- TviC
an UDP-N-acetylglucosaminuronic acid 4-epimerase
- UDP
uridine 5′-diphosphate
- UDP-Glc
UDP-glucose
- UDP-GlcNAc
UDP-N-acetylglucosamine
- UDP-GlcNAcA
UDP-N-acetylglucosaminuronic acid
- UDP-Gal
UDP-galactose
- UDP-GalNAc
UDP-N-acetylgalactosamine
- UDP-GalNAcA
UDP-N-acetylgalactosaminuronic acid
Footnotes
This work was supported in part by a National Institutes of Health Grant (GM35906). H.-w.L. also thanks the National Institute of General Medical Sciences for a MERIT Award.
References
- 1.Baker EE, Whiteside RE, Basch R, Derow MA. The Vi antigen of the Enterobacteriaceae- II. Immunologic and biologic properties. J Immunol. 1959;83:680–686. [PubMed] [Google Scholar]
- 2.Su SC, Bystrichy S. Physical, chemical, antigenic, and immunologic characterization of polygalacturonan, its derivatives, and Vi antigen from Salmonella typhi. Method Enzymol. 2003;363:552–567. doi: 10.1016/S0076-6879(03)01079-6. [DOI] [PubMed] [Google Scholar]
- 3.Virloguex-Payant I, Popoff MY. The Vi antigen of Salmonella typhi. Bull Inst Pasteur. 1996;94:237–250. [Google Scholar]
- 4.Felix A, Pitt R. Virulence of B. typhosus and resistance to O antibody. J Pathol Bacteriol. 1934;38:409–420. [Google Scholar]
- 5.Klugman KP, Koornhof HJ, Robbins JB, Le Cam NN. Immunogenicity, efficacy and serological correlate of protection of Salmonella typhi Vi capsular polysaccharide vaccine three years after immunization. Vaccine. 1996;14:435–438. doi: 10.1016/0264-410x(95)00186-5. [DOI] [PubMed] [Google Scholar]
- 6.Johnson EM, Baron LS. Genetic transfer of the Vi antigen from Salmonella typhosa to Escherichia coli. J Bacteriol. 1969;99:355–359. doi: 10.1128/jb.99.1.358-359.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pickard D, Li J, Roberts M, Maskell D, Hone D, Levine M, Dougan G, Chatfield S. Characterization of defined ompR mutants of Salmonella typhi: ompR is involved in the regulation of Vi polysaccharide expression. Infect Immun. 1994;62:3984–3993. doi: 10.1128/iai.62.9.3984-3993.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson EM, Krauskopf B, Baron LS. Genetic mapping of Vi and somatic antigenic determinants in Salmonella. J Bacteriol. 1965;90:302–308. doi: 10.1128/jb.90.2.302-308.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hashimoto Y, Li N, Yokoyama H, Ezaki T. Complete nucleotide sequence and molecular characterization of viaB region encoding Vi antigen in Salmonella typhi. J Bacteriol. 1993;175:4456–4465. doi: 10.1128/jb.175.14.4456-4465.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waxin H, Virlogeux I, Kolyva S, Popoff MY. Indentification of six open reading fames in the Salmonella enterica subsp. Enterica ser, Typhi viaB locus involved in Vi antigen production. Res Microbiol. 1993;144:363–371. doi: 10.1016/0923-2508(93)90193-6. [DOI] [PubMed] [Google Scholar]
- 11.Virlogeux I, Waxin H, Ecobichon C, Popoff MY. Role of the viaB locus in synthesis, transport and expression of Salmonella typhi Vi antigen. Microbiology. 1995;141:3039–3047. doi: 10.1099/13500872-141-12-3039. [DOI] [PubMed] [Google Scholar]
- 12.Campbell RE, Sala RF, van de Rijn I, Tanner ME. Properties and kinetic analysis of UDP-glucose dehydrogenase from group A Streptococci. J Biol Chem. 1997;272:3416–3422. doi: 10.1074/jbc.272.6.3416. [DOI] [PubMed] [Google Scholar]
- 13.Campbell RE, Mosimann SC, Rijin I, Tanner ME, Strynadka NCJ. The first structure of UDP-glucose dehydrogenase reveals the catalytic residues necessary for the two-fold oxidation. Biochemistry. 2000;39:7012–7023. [PubMed] [Google Scholar]
- 14.Ge X, Penney LC, van de Rijn I, Tanner ME. Active site residues and mechanism of UDP-glucose dehydrogenase. Eur J Biochem. 2004;271:14–22. doi: 10.1046/j.1432-1033.2003.03876.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhao X, Creuzenet C, Belanger M, Egbosimba E, Li J, Lam JS. WbpO, a UDP-N-acetyl-D-galactosamine dehydrogenase from Pseudomonas aeruginosa Serotype O6. J Biol Chem. 2000;275:33252–33259. doi: 10.1074/jbc.M004191200. [DOI] [PubMed] [Google Scholar]
- 16.Miller WL, Wenzel CQ, Daniels C, Larocque S, Brisson J, Lam JS. Biochemical characterization of WbpA, a UDP-N-acetyl-D-glucosamine 6-dehydrogenase involved in O-antigen biosynthesis in Pseudomonas aeruginosa PAO1. J Biol Chem. 2004;279:37551–37558. doi: 10.1074/jbc.M404749200. [DOI] [PubMed] [Google Scholar]
- 17.Reid MF, Fewson CA. Molecular characterization of microbial alcohol dehydrogenases. Crit Rev Microbiol. 1994;20:13–56. doi: 10.3109/10408419409113545. [DOI] [PubMed] [Google Scholar]
- 18.Kowal P, Wang PG. New UDP-GlcNAc C4 epimerase involved in the biosynthesis of 2-acetamino-2-deoxy-L-altruronic acid in the O-antigen repeating units of Plesiomonas shigelloides O17. Biochemistry. 2002;41:15410–15414. doi: 10.1021/bi026384i. [DOI] [PubMed] [Google Scholar]
- 19.Creuzenet C, Belanger M, Wakarchuk WW, Lam JS. Expression, purification and biochemical characterization of WbpP, a new UDP-GlcNAc C4 epimerase from Pseudomonas aeruginosa Serotype O6. J Biol Chem. 2000;275:19060–19067. doi: 10.1074/jbc.M001171200. [DOI] [PubMed] [Google Scholar]
- 20.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Spring Harbor; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 22.Andrews P. Estimation of the molecular weights of proteins by sephadex gel-filtration. Biochem J. 1964;91:222–233. doi: 10.1042/bj0910222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 24.A handbook for high-level expression and purification of 6×His-tagged proteins. 5. QIAGEN Inc; 2001. The QiAexpressionist. [Google Scholar]
- 25.Thoden JB, Frey PA, Holden HM. Crystal structures of the oxidized and reduced forms of UDP-galactose 4-epimerase isolated from Escherichia coli. Biochemistry. 1996;35:2557–2566. doi: 10.1021/bi952715y. [DOI] [PubMed] [Google Scholar]
- 26.Thoden JB, Frey PA, Holden HM. Molecular structure of the NADH/UDP-glucose abortive complex of UDP-galactose 4-epimerase from Escherichia coli: implications for the catalytic mechanism. Biochemistry. 1996;35:5137–5144. doi: 10.1021/bi9601114. [DOI] [PubMed] [Google Scholar]
- 27.Thoden JB, Wohlers TM, Fridovich-Keil JL, Holden HM. Human UDP-galactose 4-epimerase. Accommodation of UDP-N-acetylglucosamine within the active site. J Biol Chem. 2001;276:15131–15136. doi: 10.1074/jbc.M100220200. [DOI] [PubMed] [Google Scholar]
- 28.Schulz JM, Watson AL, Sanders R, Ross KL, Thoden JB, Holden HM, Fridovich-Keil JL. Determinants of function and substrate specificity in human UDP-galactose 4′-epimerase. J Biol Chem. 2004;279:32796–32803. doi: 10.1074/jbc.M405005200. [DOI] [PubMed] [Google Scholar]
- 29.Demendi M, Ishiyama N, Lam JS, Berghuis AM, Creuzenet C. Towards a better understanding of the substrate specificity of the UDP-N-acetylglucosamine C4 epimerase WbpP. Biochem J. 2005;389:173–180. doi: 10.1042/BJ20050263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belanger M, Burrows LL, Lam JS. Functional analysis of genes responsible for the synthesis of the B-band O antigen of Pseudomonas aeruginosa serotype O6 lipopolysaccharide. Microbiology. 1999;145:3505–3521. doi: 10.1099/00221287-145-12-3505. [DOI] [PubMed] [Google Scholar]