

Abstract
The recently identified type II isopentenyl diphosphate (IPP):dimethylallyl diphosphate (DMAPP) isomerase (IDI-2) is a flavoenzyme that requires FMN and NAD(P)H for activity. IDI-2 is an essential enzyme for the biosynthesis of isoprenoids in several pathogenic bacteria including Staphylococcus aureus, Streptococcus pneumoniae, and Enterococcus faecalis, and thus is considered as a potential new drug target to battle bacterial infections. One notable feature of the IDI-2 reaction is that there is no net change in redox state between the substrate (IPP) and product (DMAPP), indicating that the FMN cofactor must start and finish each catalytic cycle in the same redox state. Here, we report the characterization and initial mechanistic studies of the S. aureus IDI-2. The steady-state kinetic analyses under aerobic and anaerobic conditions show that FMN must be reduced to be catalytically active and the overall IDI-2 reaction is O2 sensitive. Interestingly, our results demonstrate that NADPH is needed only in catalytic amounts to activate the enzyme for multiple turnovers of IPP to DMAPP. The hydride transfer from NAD(P)H to reduce FMN is determined to be pro-S stereospecific. Photoreduction and oxidation-reduction potential studies reveal that the S. aureus IDI-2 can stabilize significant amounts of the neutral FMN semiquinone. In addition, reconstitution of apo-IDI-2 with 5-deazaFMN resulted in a dead enzyme, whereas reconstitution with 1-deazaFMN led to the full recovery of enzyme activity. Taken together, these studies of S. aureus IDI-2 support a catalytic mechanism in which the reduced flavin coenzyme mediates a single electron transfer to and from the IPP substrate during catalysis.
All isoprenoids are derived from two 5-carbon precursors, isopentenyl diphosphate (IPP, 1) and dimethylallyl diphosphate (DMAPP, 2) which are condensed to generate the diverse skeletons of the isoprenoid family. Two pathways for the biosynthesis of these isoprene units have been found, the classical mevalonate (MEV) pathway in animals, fungi, and archaebacteria, and the more recently discovered methylerythritol phosphate (MEP) pathway in green algae, the chloroplasts of higher plants, and most eubacteria.1–3 The end product of the MEV pathway is exclusively IPP (1), which must be isomerized to DMAPP (2), whereas the MEP pathway leads to the formation of both 1 and 2.3–5 The isomerization between 1 and 2 is catalyzed by isopentenyl diphosphate:dimethylallyl diphosphate isomerase (IDI) (Scheme 1). IDI is an essential component of the mevalonate (MEV) pathway because it is the only source of DMAPP.1,2,6 In contrast, the role of IDI in the MEP pathway is uncertain, but it may modulate the concentrations of IPP and DMAPP in the cell.5
Scheme 1.
IDI was first discovered in yeast in the late 1950’s during studies of the MEV pathway.7 Over the years it has been found in a variety of organisms8 and has undergone extensive mechanistic scrutiny. These studies include the stereochemical analysis of the reaction and characterization of a carbocation intermediate during catalysis.9–12 It has been well established that the reaction is initiated by the addition of a proton to the re face of the C3–C4 double bond of IPP (1) followed by removal of the pro-R proton at C2.13–16 The overall transformation is an antarafacial (1,3) proton addition/elimination reaction.
In the late 1990’s a new type of IDI was found in Streptomyces sp. strain CL190.17 This enzyme is a flavoprotein and requires Mg2+ and NAD(P)H for activity. A classification system was subsequently devised with the original coenzyme-free IDI referred to as type I IDI (IDI-1) and the new flavin-containing IDI as type II IDI (IDI-2). Neither form is specific to a given biosynthetic pathway, as IDI-1 and IDI-2 enzymes are found in both MEV and MEP pathways.18,19 As for their distribution in nature, the idi1 gene is found in eubacteria and eukaryotes, while the idi2 gene is found in eubacteria and archaebacteria.19 Interestingly, idi2 has not been found in the genomes of fungi, animals, or plants.18 Several pathogenic gram-positive bacteria including Staphylococcus aureus, Streptococcus pneumoniae, and Enterococcus faecalis, which use the MEV pathway for the biosynthesis of their isoprene compounds, rely exclusively on IDI-2 for the generation of DMAPP, making this enzyme a potential antibiotic target.19
In contrast to the type I enzyme, very little mechanistic data is available for IDI-2. To-date, eight IDI-2 homologues have been purified and characterized to different extents. Five are from eubacteria, including Streptomyces sp. strain CL190,17 Bacillus subtilis,18,20,21 Staphylococcus aureus,17 Synechocystis sp. strain PCC 6803,22 and Thermus thermophilus.23,24 The other three are from archaebacteria, Methanothermobacter thermautotrophicus, 25 Sulfolobus shibatae, 26,27 and Thermococcus kodakaraensis.28 Most reported studies on IDI-2 have focused on the biochemical characterization of the purified proteins. The capability of two IDI-2 enzymes to function as an NADH oxidoreductase was also investigated.18,26
IDI-2 from B. subtilis, S. shibatae, and T. thermophilus are the best-characterized members of the IDI-2 family. A recent crystal structure of the B. subtilis enzyme displayed an octameric structure with one FMN molecule bound per monomer.21 However, attempts to identify binding sites for Mg2+, IPP, and NADP+ were unsuccessful. The enzyme appears to have a flexible active site with no well-defined binding regions for the adenosine moiety of NADPH or the diphosphate moiety of IPP and DMAPP. A recent report on IDI-2 from S. shibatae proposed a flavin radical mechanism based on the lack of activity of apoenzyme reconstituted with 5-deazaFMN, which is incapable of serving as a 1-electron mediator.27 In a study on the IDI-2 enzyme from T. thermophilus, de Ruyck et al. have suggested that the FMN coenzyme may simply play a structural role.23 More recently, Rothman et al. have demonstrated the thermodynamic stabilization of a neutral semiquinone during potentiometric titrations of the T. thermophilus enzyme in the presence of IPP, lending support to a mechanism involving single electron transfer chemistry.24 In addition to electron transfer chemistry, the flavin coenzyme has also been suggested to function in an acid/base chemical mechanism.24,29 Clearly, the actual role of the flavin coenzyme in the IDI-2 reaction remain elusive, and our understanding of the catalytic mechanism of IDI-2 enzymes is still limited.
To better understand this unusual flavin-dependent isomerization reaction, we have initiated a study on the IDI-2 from Staphylococcus aureus and report our initial characterization and mechanistic results herein. On the basis of these data, a radical mechanism in which the enzyme is activated by catalytic amounts of NAD(P)H is supported. The key step likely involves a 1-electron transfer from the reduced FMN to IPP to generate flavin semiquinone and substrate radical intermediates. Deprotonation and electron transfer back to the flavin regenerates the reduced enzyme and yields DMAPP. One-electron flavin chemistry in reactions involving no net change in the redox state of the substrate/product or flavin is rare, but it appears that IDI-2 may now be added to this select group of flavoenzymes.
EXPERIMENTAL PROCEDURES
Materials
Plasmid pQES, which contains the idi2 gene from Staphylococcus aureus, was a generous gift from Professor Haruo Seto of the Tokyo University of Agriculture. The gene included a sequence coding for a His6 tag at the N-terminus of the expressed protein. Escherichia coli M15[pREP4] from Qiagen (Valencia, CA) was used as a host for idi2 gene overexpression. All culture and media components were purchased from BD Diagnostic Systems (Sparks, MD). Kanamycin (Kn) and ampicillin (Ap) were obtained from Shelton Scientific (Shelton, CT) and Fisher Scientific (Fair Lawn, NJ), respectively. Ni-NTA agarose resin for affinity purification of His6-tagged proteins was a product of Qiagen. DEAE Sepharose Fast Flow anion exchange resin was purchased from Amersham Biosciences (Uppsula, Sweden). Polyacrylamide Bio-Gel P-2 was obtained from Bio-Rad (Hercules, CA), and the pre-stained protein marker was purchased from New England BioLabs (Ipswich, MA). IPP (1) was either prepared according to a literature procedure,30 or purchased from Sigma-Aldrich (St. Louis, MO). The radiolabeled [1-14C]IPP (55 mCi/mmol) was obtained from Sigma-Aldrich and was diluted to a specific activity of 11 mCi/mmol for all experiments. The 14C-labeled product generated in radioassays was analyzed with non-aqueous biodegradable counting scintillant (BCS-NA) from Amersham Biosciences (Buckinghamshire, England). 1- and 5-deazariboflavin were synthesized by published methods.31 FAD synthetase, used to prepare 1- and 5-deazaFAD, was cloned from Corynebacterium ammoniagenes genomic DNA, overexpressed in NovaBlue(DE3), and purified by Ni-NTA chromatography. (R)- and (S)-[4-2H]NADH were prepared from NAD+ using yeast alcohol dehydrogenase (YADH)32 and lipoamide dehydrogenase (LDH),33 respectively. Except for anthroquinone-2-sulfonate (AQS), which was obtained from Acros Organics (Geel, Belgium), all other chemicals were obtained from Sigma-Aldrich, VWR International (West Chester, PA), or Fisher Scientific (Pittsburgh, PA).
General
Protein concentrations were determined according to Bradford34 using bovine serum albumin (BSA). The concentration of stock enzyme solutions used in this study was also determined by quantitative amino acid analysis performed by the Protein Chemistry Laboratory at Texas A&M Univeristy, and the IDI-2 concentrations determined by this method were used to scale the IDI-2 concentrations determined by the Bradford assays by a factor of 1.11. All protein concentrations reported herein refer to the concentration of IDI-2 monomer. Protein purity and subunit molecular mass were estimated by SDS-PAGE according to Laemmli.35 Native molecular mass determination was performed using a HPLC equipped with a Superdex 200 HR 10/30 column. The data were analyzed by the method of Andrews.36 A Kodak Carousel Slide projector containing a Sylvania 300W, 82V bulb was used for light irradiation during photoreduction. A Coy Laboratories anaerobic chamber (Grass Lakes, MI) was used to prepare the components of the photoreduction assay and to carry out anaerobic steady-state kinetic assays. Data analyses for dissociation constants and steady-state kinetic parameters were performed using GraFit Data Analysis Software (Version 5.0.1) from Erithacus (Surrey, UK). NMR spectra were acquired on a Varian INOVA 500 MHz spectrometer. Chemical shifts (δ in ppm) were given relative to those of solvents. Coupling constants are provided in hertz (Hz).
Growth of E. coli M15[pREP4]/pQES Cells
A single overnight culture of E. coli M15[pREP4]/pQES was used to inoculate (1 to 20 dilution) six 25-mL samples of LB-Kn-Ap (25 μg/mL Kn, 100 μg/mL Ap). Each culture was grown to an OD600 of 0.6 at 37 °C. Aliquots were then used to inoculate six 1-L samples of the same media. Incubation at 37 °C was continued until an OD600 of 0.6 was reached. After induction with 0.1 mM IPTG, the resulting culture was grown for another 4.5 h at 37 °C. Cells were harvested by centrifugation (4000g, 20 min) and carried forward to the next step.
Purification of IDI-2
All procedures were carried out at 4 °C unless noted otherwise. The cell pellet (typically 20 g from a 6 L culture) was thawed on ice and resuspended in 100 mL of cold lysis buffer (50 mM sodium phosphate, 400 mM NaCl, 10 mM imidazole, 15% glycerol (v/v), pH 7.5). Lysozyme was added to 0.15 mg/mL and the mixture incubated on ice for 30 min. Sonication (36 × 10 s pulses, separated by 20 s cooling periods) was used to disrupt the cell membranes. After centrifugation (10,000g, 30 min), the supernatant was collected and mixed with 19 mL of Ni-NTA agarose resin previously washed two times with cold lysis buffer. The extract/agarose resin mixture was incubated for 1 h on a rotating table at 4 °C and then loaded into a glass column. An initial wash of the column with 20 mL of lysis buffer was followed by washes (20 mL each) of increasing concentrations of imidazole (20, 50, and 100 mM, respectively). A final 40 mL wash, containing 250 mM imidazole, removed all remaining protein from the column. All wash and elution buffers contained 50 mM sodium phosphate (pH 7.5), 400 mM NaCl, and 15% (v/v) glycerol in addition to the imidazole at the concentration noted above. Fractions of 4 mL were collected throughout all washes. Those fractions containing IDI-2, based on SDS-PAGE, were pooled (approximately 90 mL) and dialyzed against four, 4-L volumes of 50 mM sodium phosphate buffer (pH 7.5) containing 15% (v/v) glycerol. After dialysis, the golden yellow protein solution was concentrated to 24 mL, divided into small aliquots, flash frozen by liquid nitrogen, and stored at −80 °C. Typical yield of purified IDI-2 was 144 mg/L of culture.
Identification of Flavin Coenzyme
The method of Light et al was used to identify the flavin coenzyme in IDI-2.37 Accordingly, purified enzyme (150 μL, 78.8 mg/mL) was placed in a foil-covered, 1.5 mL microcentrifuge tube and subjected to a boiling water bath for 10 min. After cooling on ice for 15 min, the sample was centrifuged (16,000g, 20 min) to remove denatured enzyme. The supernatant was transferred to a clean YM-30 centrifugal filter device (Millipore) and centrifuged (16,000g, 30 min) to remove remaining traces of protein. The flow-through was lyophilized and the resulting solid residue (flavin) was redissolved in 20 μL of deionized H2O. This sample was then applied to a C18 reverse-phase HPLC column (250 × 10 mm) and eluted with a linear gradient of 10 to 70% methanol in 5 mM ammonium acetate (pH 6.0). Separate runs were performed using FMN and FAD as standards. A detector set at 360 nm was used to detect the flavins. The retention time for FMN was 20.5 min under the HPLC conditions.
Reconstitution of IDI-2 Apoenzyme
The purified IDI-2 (72 μM) was incubated with 750 μM FMN in 10 mM HEPES buffer (pH 7.5) for 3 h at 4 °C. After incubation, the assay mixture was placed in a YM-30 centrifugal filter device (Millipore) that was previously rinsed with deionized H2O. Unbound flavin was removed by centrifugation (14,000g, 10 min, 4 °C). Cold, 10 mM HEPES buffer (pH 7.5, 100 μL) was added and the device shaken by hand for 30 s. Unbound FMN was again removed by centrifugation (14,000g, 4 min, 4 °C). Cold, 10 mM HEPES buffer (pH 7.5, 100 μL) was then added. The filter was shaken lightly by hand for 30 s, inverted, and centrifuged (4,000g, 4 min, 4 °C) to recover the holoenzyme. To check the extent of reconstitution, the holoenzyme was denatured by SDS (0.2% final concentration), and the released FMN quantified by UV-vis spectroscopy (ε446 = 12,200 M−1 cm−1).38 Control assays without enzyme were carried out to estimate FMN bound to the YM-30 filter. The final holoenzyme spectrum was obtained by subtracting an average background from the spectrum of the reconstituted enzyme solution.
Determination of Dissociation Constants for FMN, 1-deazaFMN, and 5-deazaFMN
The binding affinities of IDI-2 for the oxidized forms of FMN, 1-deazaFMN, and 5-deazaFMN were determined by ultrafiltration or UV-vis difference spectroscopy. The ultrafiltration assays (115 μL) involved the incubation of apo-IDI-2 (35 μM) with increasing amounts (2, 4, 10, 20, 50, and 100 μM) of FMN or 1-deazaFMN in 0.1 M HEPES buffer (pH 7.0) containing 15% glycerol. Following a 15-min incubation at room temperature, each assay was applied to a YM-10 centrifugal filter device (Millipore) and the unbound flavin was removed by centrifugation (14,000g, 30 min). The flow-through was diluted 2-fold and analyzed by UV-vis spectroscopy. Extinction coefficients of 12,200 M−1 cm−1 at 446 nm and 6,800 M−1 cm−1 at 535 nm were used to estimate the concentration of free FMN and 1-deazaFMN, respectively.31–38 Assays were carried out in duplicate and the average ratio of holoenzyme to total enzyme at each flavin concentration was used to calculate the flavin Kd values by fitting to a hyperbolic equation using the GraFit Software (Erithacus) package. Control assays containing the same components but no enzyme were run in parallel.
The UV-vis difference spectroscopy assays were also carried out at room temperature. The reference cell contained 1 mL of 0.1 M HEPES buffer (pH 7.0), 0.15% glycerol (v/v), and 5 mM MgCl2. The sample cell contained the same components (1 mL) along with the purified IDI-2 (12.0 μM). Since glycerol was present in the IDI-2 storage buffer, it was also included in the reference cell to minimize the background absorbance. After blanking the instrument with the above reference and sample cells, an identical aliquot of flavin stock solution was added to each cell and a difference spectrum was immediately recorded. This procedure was continued until the spectrum indicated binding of flavin to the enzyme had reached saturation. Final concentrations of FMN, 1-deazaFMN, and 5-deazaFMN were 200, 100, and 54 μM, respectively. Difference data at 440/493 nm, 333 nm, and 430 nm were fit by GraFit Software (Erithacus) to determine the Kd values for FMN, 1-deazaFMN, and 5-deazaFMN, respectively.
Determination of Steady-State Kinetic Parameters
A steady-state kinetic analysis was performed under aerobic and anaerobic conditions to evaluate the effect of oxygen on activity. The kinetic parameters were determined using the modified Satterwhite activity assay reported by Kaneda et al, which is based on the acid lability of DMAPP.17 Each of the aerobic assay mixtures contained 55.5 nM purified IDI-2, 5 mM MgCl2, 1 mM DTT, 10 μM FMN, 5 mM NADPH and variable concentration of [1-14C]IPP (0.96 to 288 μM) in 0.1 M HEPES buffer (pH 7.0, 37 °C). For the determination of Km,FMN under anaerobic conditions (see below), the concentration of [1-14C]IPP was held constant at 150 μM, and the FMN concentration was varied (0.0039, 0.0157, 0.0786, 0.393, 0.785, and 15.7 μM). For these reactions, IDI-2 (50.3 nM) and FMN (variable) were incubated with 5 mM NADPH for 1 h at 25 °C in 0.1 M HEPES buffer (pH 7.0) containing 5 mM MgCl2 and 1 mM DTT. For each concentration of variable substrate (either IPP or FMN), a 270 μL assay mixture containing all components except substrate was incubated for 10 min at 37 °C. Radiolabeled [1-14C]IPP (30 μL at appropriate concentration) was then added to start the reaction. At different time intervals (0.5, 2.5, 4.5, 6.5, and 8.5 min), a 50 μL aliquot was removed from the reaction mixture, quenched with 200 μL of HCl:MeOH (1:4), and incubated for 10 min at 37 °C. The acid hydrolysis of DMAPP leads to a delocalized allylic cation which then reacts rapidly with water and methanol to give a mixture of methylvinyl carbinol, dimethylallyl alcohol, and their corresponding methyl ethers.39 Petroleum ether (1 mL), with a boiling point of 60 to 95 °C, was used to extract these alcohol products, while unreacted [1-14C]-IPP remained in the aqueous fraction. From the organic layer, 0.3 mL was removed, mixed with 6 mL of non-aqueous scintillation fluid (Amersham Biosciences), and analyzed by a liquid scintillation counter. DPM and specific activity of the [1-14C]IPP substrate were used to calculate the reaction rates. The GraFit Software package was used to fit the data to the Michaelis-Menten equation by non-linear regression analysis.
Initial velocities under anaerobic conditions were measured using procedures identical to those of the aerobic assays except the experiments were carried out in a Coy Laboratories anaerobic chamber (O2 ≤ 5 ppm) containing 95% N2 and 5% H2. Individual components used in the assays were made anaerobic using a Schlenk line and oxygen-free argon. Buffers and substrate were degassed by bubbling argon into solution for times varying from 30 min for microliter volumes to 1 h for volumes of a few milliliters. The purified IDI-2 (1.5 mL, 3.3 μM) was made anaerobic by 40 cycles of vacuum and argon purging (1 s/15 s).
Photoreduction of Purified IDI-2
Photoreduction of enzyme was carried out in an anaerobic cuvette using the method reported by Massey.40,41 The reaction mixture (1 mL) contained 396 μM purified IDI-2, 20 mM EDTA, 1.2 μM 5-deazariboflavin, and 0.1 M potassium phosphate buffer (pH 7.0). The concentration of 5-deazariboflavin was determined using ε396 of 12,000 M−1 cm−1.31 Individual components were made anaerobic prior to mixing using a Schlenk line. Stock solutions (all < 2 mL), were deoxygenated by bubbling oxygen-free argon into solution for 1 h. Glycerol was removed from IDI-2 using a YM-30 centrifugal filter device and enzyme was then deoxygenated using 40 cycles of vacuum/argon (1 s/15 s). Assay components were mixed in a Coy anaerobic chamber. The cuvette was sealed, removed from the chamber, and immediately scanned in a diode array spectrophotometer (Agilent) to serve as time zero. Photoreduction was then carried out using short periods of illumination from a Kodak Carousel Slide Projector. Irradiation of IDI-2 was continued until full reduction was observed as indicated by the bleaching of oxidized FMN absorbance at 452 nm. Throughout the photoreduction experiment, the temperature of the cuvette in the spectrophotometer was maintained at 25 °C by a jacketed cell holder connected to a circulating water bath (Thermo).
Determination of Oxidation-Reduction Potential for E:FMN0x/E:FMNred
The oxidation-reduction potential of the E:FMNox/E:FMNred redox couple was determined using the xanthine oxidase reducing system developed by Massey.42 Accordingly, a 1 mL assay mixture containing 20 μM purified IDI-2, 17 μM FMN, 18 μM AQS, 0.4 mM hypoxanthine, 2 μM methyl viologen, and 0.55 mg of xanthine oxidase (0.084 units/mg) in 0.1 M potassium phosphate buffer (pH 7.0) was prepared under anaerobic conditions. For the reduction performed in the presence of substrate, 150 μM IPP was also included. Since only 15% of the purified IDI-2 used in this experiment contained bound FMN (using ε452 of 9,300 M−1 cm−1, this study), additional FMN (17 μM) was added to bring the total [FMN] up to 20 μM. IDI-2 and xanthine oxidase were degassed separately using a Schlenk line through 40 cycles of vacuum/argon (1 s/15 s). A 2 mL mixture containing the remaining components was degassed by bubbling oxygen-free argon through the solution for 1 h. Individual components were mixed in an anaerobic cuvette inside a Coy anaerobic chamber. The cuvette was sealed, removed from the chamber, and immediately scanned in a diode array spectrophotometer. The temperature of the cuvette was maintained at 25 °C by a jacketed cell holder connected to a circulating water bath. Prior to scanning, 0.1 M potassium phosphate buffer (pH 7.0) was used to blank the instrument. Subsequent scans were recorded at 1 min intervals. The reduction of AQS and IDI-2 was monitored at 332 nm (the isosbestic point for FMNox/FMNred) and 354 nm (the isosbestic point for AQSox/AQSred), respectively. Both isosbestic points were determined in this study. Full reduction of AQS and IDI-2 was achieved after 1 h in the absence of IPP and after ~ 20 min in the presence of IPP.
Data Analysis of Xanthine Oxidase Assay Results
The isosbestic point of free FMNox/FMNred (332 nm), where FMNox and FMNred have the same extinction coefficient, was used to calculate [AQSox] and [AQSred]. Because absorbance from other assay components at this wavelength is negligible, any changes that occur at 332 nm are a result of AQS reduction. On the basis of the total change in A332 and the initial concentration of AQS (18 μM, based on ε256 of 52,095 M−1 cm−1 for AQSox)43, the concentrations of AQSox and AQSred at each time point could be calculated. These concentrations were also calculated using the absorbance change at A327, the isosbestic point for E:FMNox/E:FMNred (Figure 2B). Monitoring AQS reduction at either isosbestic point (327 nm or 332 nm) gave comparable results. Hence, the values from the A332 readings were chosen for subsequent calculations. The concentration of FMNox and FMNred were determined in a similar manner by monitoring the absorption changes at 354 nm, the isosbestic point for AQSox/AQSred. The percent decrease in A354 was used along with the total flavin concentration (20 μM) to calculate [FMNox] and [FMNred] at each time point. Data was plotted according to the method of Minnaert with log([E:FMNox]/[E:FMNred]) as the y-axis and log([AQSox]/[AQSred]) as the x-axis.44 The value of the y-intercept was used in conjunction with Em,dye (−0.225 V for AQS)45 to calculate Em. This is the midpoint oxidation-reduction potential for E:FMNox/E:FMNred.
FIGURE 2.
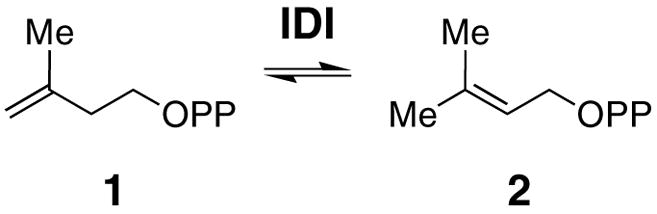
Anaerobic photoreduction of IDI-2. (A) The as-purified IDI-2 (396 μM) in 0.1 M potassium phosphate buffer (pH 7.0) containing 20 mM EDTA and 1.2 μM 5-deazariboflavin was irradiated with a 300-watt slide projector lamp (see Experimental Procedures) at 25 °C under anaerobic conditions. Spectra (top to bottom) were recorded after 0, 9, 13, 24, 57, 167, and 1382 sec of irradiation, respectively. Inset figure shows a plot of absorbance at 583 nm versus absorbance at 452 nm during reduction. The solid lines represent extrapolation of absorbance values to 100% neutral semiquinone. (B) UV-vis absorption spectra for oxidized (solid line) and reduced (dashed line) IDI-2 obtained from the photoreduction data in Figure 2A.
Determination of Em1 and Em2
The separation of Em1 and Em2 the midpoint reduction potentials for the single electron reduction of E:FMNox → E:FMNsem and E:FMNsem → E:FMNred, respectively, was estimated based on percent semiquinone formation observed during photoreduction (Figure 2A, inset). According to Clark, the maximum percent of semiquinone formation at equilibrium is equal to K0.5/(2 + K0.5).46 The semiquinone formation constant, K, can be substituted into (Em1 − Em2) = (2.303RT/F) × (logK) to determine the separation of Em1and Em2.46 Here, R is the gas constant (8.3145 J·K−1·mol−1), T is temperature (K), and F is the Faraday constant (96,485 J·V−1·mol−1). The midpoint potential for the 2-electron reduction obtained from the xanthine oxidase system, Em was used in Em = (Em1 + Em2)/2 to calculate numerical values for Em1 and Em2.
Preparation of 1-deazaFMN
The 1-deazaFMN coenzyme was prepared enzymatically from 1-deazariboflavin using recombinant FAD synthetase from Corynebacterium ammoniagenes and Naja naja snake venom.31 A 250 mL reaction mixture containing 4.6 mg (12 μmol) of 1-deazariboflavin, 9 mg of FAD synthetase, 250 mg (0.45 mmol) of ATP disodium salt, and 225 mg (2.36 mmol) of MgCl2 in 30 mM potassium phosphate buffer (pH 7.5) was incubated in the dark at 37 °C for 24 h. The reaction mixture was applied to a 2.5 × 25 cm DEAE Sepharose anion exchange column (Cl− form) and eluted with three column volumes of H2O, followed by a 1 L, linear gradient of 0 to 0.2 M NaCl in 50 mM potassium phosphate buffer (pH 7.0) to remove the remaining starting material. Fractions containing 1-deazaFAD, based on the purple color, were combined and lyophilized to yield a purple solid. This solid was dissolved in 15 mL of deionized H2O and divided into three portions. Each portion was applied to a 2.5 × 110 cm P2 Biogel column (Bio-Rad) to desalt the final product. The column was eluted with deionized H2O at a flow rate of 10 mL/h. Purple fractions containing 1-deazaFAD were pooled, lyophilized, and carried forward to the second reaction. P2 purified 1-deazaFAD (15 μmol) was incubated with 6 mg of Naja naja snake venom (Sigma-Aldrich) in 150 mL of 20 mM potassium phosphate buffer (pH 7.6) for 1 h at room temperature in the dark. Protein was removed by a Centricon-10 microconcentrator (Millipore) and the filtrate lyophilized to yield a purple solid. This solid was dissolved in minimal water and applied to a P2 Biogel column to separate the products of the cleavage reaction. Fractions containing 1-deazaFMN were combined and lyophilized to yield a purple solid that was stored at −80 °C. 1H NMR (500 MHz, D2O) δ 2.18 (3H, s), 2.30 (3H, s), 3.49 – 4.37 (7H, m), 5.43 (1H, s), 7.33 (2H, s); 13C NMR (125 MHz, D2O) δ 18.5, 20.6, 48.3, 65.3, 72.1, 72.2, 72.3, 85.5, 115.7, 123.5, 131.0, 133.1, 136.5, 138.5, 148.4, 161.2, 161.6, 166.7; 31P NMR (200 MHz, D2O) δ 5.83 (s).
Preparation of 5-deazaFMN
The 5-deazaFMN coenzyme was prepared using the same procedures as for 1-deazaFMN except the starting material was 5-deazariboflavin. The final 5-deazaFMN yellow solid was stored at −80 °C. 1H NMR (500 MHz, D2O) δ 2.19 (3H, s), 2.34 (3H, s), 3.45 – 4.44 (7H, m), 7.30 (1H, s), 7.57 (1H, s), 8.19 (1H, s); 13C NMR (125 MHz, D2O) δ 18.5, 20.9, 47.1, 65.6, 69.9, 72.0, 72.7, 117.3, 120.0, 130.7, 136.7, 139.3, 142.3, 149.8, 156.8, 159.0, 163.3, 168.7; 31P NMR (200 MHz, D2O) δ 4.95 (s).
Stereospecificity of Hydride Transfer from NADH
A 17-mL reaction mixture containing 1 mM (R)- or (S)-[4-2H]NADH, 2 mM IPP (1), 13 μM IDI-2, 10 μM FMN, and 5 mM MgCl2 in 0.1 M potassium phosphate buffer (pH 7.0) was incubated for 14 h at room temperature in the dark. The reaction was terminated by removal of IDI-2 with a Centricon-10 microconcentrator. NAD+ present in the filtrate was purified by FPLC equipped with a MonoQ (10/10) column using a linear gradient of 0.04 to 0.4 M ammonium bicarbonate over a 30 min period with a flow rate of 2 mL/min. The desired fractions, based on an NAD+ standard, were combined, lyophilized, and redissolved in D2O for analysis by 1H NMR. Enzyme activity was confirmed by 1H NMR spectroscopy prior to FPLC purification.
NADPH Consumption and DMAPP Formation During Turnover
Both UV-vis spectroscopy and acid lability assays were carried out over a 30-min period to monitor NADPH consumption and DMAPP (2) formation. Each 53 μL radioassay contained 5.3 μM IDI-2, 0.125 mM NADPH, 5 mM MgCl2, and [1-14C]IPP (at 0.050, 0.500, or 1.200 mM) in 0.1 M HEPES buffer (pH 7.0). The reaction was initiated by adding 3 μL of 94 μM reconstituted IDI-2 to 50 μL of assay mixture. Individual assays were incubated for 2, 5, 10, 15, and 30 min at 37 °C. Each assay was quenched with 0.2 mL of 25% HCl:MeOH and then incubated for an additional 10 min at 37 °C. After extraction with petroleum ether (1 mL), a 0.35 mL sample was removed from the organic layer, mixed with 7 mL of non-aqueous scintillation fluid, and analyzed by liquid scintillation spectrometry.
Complementary UV-vis spectroscopy assays (0.178 mL) were used to monitor the NADPH consumption. The reaction mixture contained 5.3 μM IDI-2, 0.125 mM NADPH, 5 mM MgCl2, and 0.05, 0.5, or 1.2 mM IPP (1) in 0.1 M HEPES buffer (pH 7.0). The assay mixture was incubated for 10 min at 37 °C before adding 9 μL of 105 μM reconstituted IDI-2 to initiate the reaction. After enzyme was added, absorbance at 340 nm was recorded at specific time points during a 30-min period. Duplicate reactions were carried out at each IPP concentration. Average values were used to quantify NADPH consumption. Control assays without IDI-2 were used to determine the background reading of NADPH consumption. The temperature within the quartz cuvette was maintained at 37 °C using a jacketed cell holder connected to a circulating water bath. All UV-vis readings were recorded on a diode array spectrophotometer.
Correlation between NADPH Consumption and IDI-2 Concentration
A second set of experiments with procedures identical to the UV-vis assays used to monitor NADPH consumption (described above) were conducted to determine the relationship between NADPH consumption and IDI-2 concentration. Absorbance at 340 nm was monitored over 30 min for assays (178 μL) containing 0.125 mM NADPH, 1.2 mM IPP (1), and 5 mM MgCl2 in 0.1 M HEPES buffer (pH 7.0). As in the above UV-vis assays, reconstituted IDI-2 was added to the reaction mixture and absorbance at 340 nm was monitored for 30 min. Five different IDI-2 concentrations were investigated (2.0, 4.1, 9.3, 14, and 22 μM).
Preparation of IDI-2 Apoenzyme
IDI-2 (0.3 mL, 13 mg/mL) was dialyzed against 1.5 L of 0.1 M potassium phosphate buffer containing 2 M KBr and 15% glycerol (pH 7.0).47 Dialysis buffer was exchanged at 6, 12, and 18 h. Removal of bound flavin was monitored by the decrease in yellow color of the enzyme solution. At the end of dialysis, the complete removal of bound FMN was confirmed by UV-vis spectroscopy. Potassium bromide buffer was exchanged with 0.1 M potassium phosphate buffer (pH 7.5) containing 15% glycerol using a YM-30 centrifugal filter device. Small aliquots of the apoenzyme (50 μL) were flash frozen by liquid nitrogen and stored at −80 °C.
Activity of IDI-2 Apoenzyme Reconstituted with 1- and 5-deazaFMN
Acid-lability activity assays were carried out with IDI-2 apoenzyme in the presence of FMN, 1-deazaFMN, and 5-deazaFMN to investigate the catalytic role of the flavin coenzyme. Each assay mixture (55 μL) contained approximately 100 μM free flavin, 4.9 μM apo-IDI-2, 5 mM NADPH (or dithionite at 5 and 50 mM), 5 mM MgCl2, and 50 μM [1-14C]IPP in 0.1 M HEPES buffer (pH 7.0). The reaction was initiated by the addition of 5 μL of apo-IDI-2 to 50 μL of assay solution. After a 10-min incubation period at 37 °C, the reaction was quenched with 0.2 mL of 25% HCl:MeOH plus 0.5 mL of water. The quenched reaction was incubated for an additional 10 min at 37 °C, saturated with NaCl, and extracted twice with 0.5 mL of toluene. A 0.3 mL sample was removed from the combined extracts, mixed with 7 mL of nonaqueous scintillation fluid, and analyzed by liquid scintillation counter. A negative control (no enzyme) confirmed that DMAPP (2) formation was enzyme-catalyzed and did not occur from a reaction between NADPH (or dithionite) and IPP (1). Previous assays also demonstrated that IDI-2 apoenzyme was inactive in the absence of FMN.
RESULTS
Overexpression and Purification of IDI-2
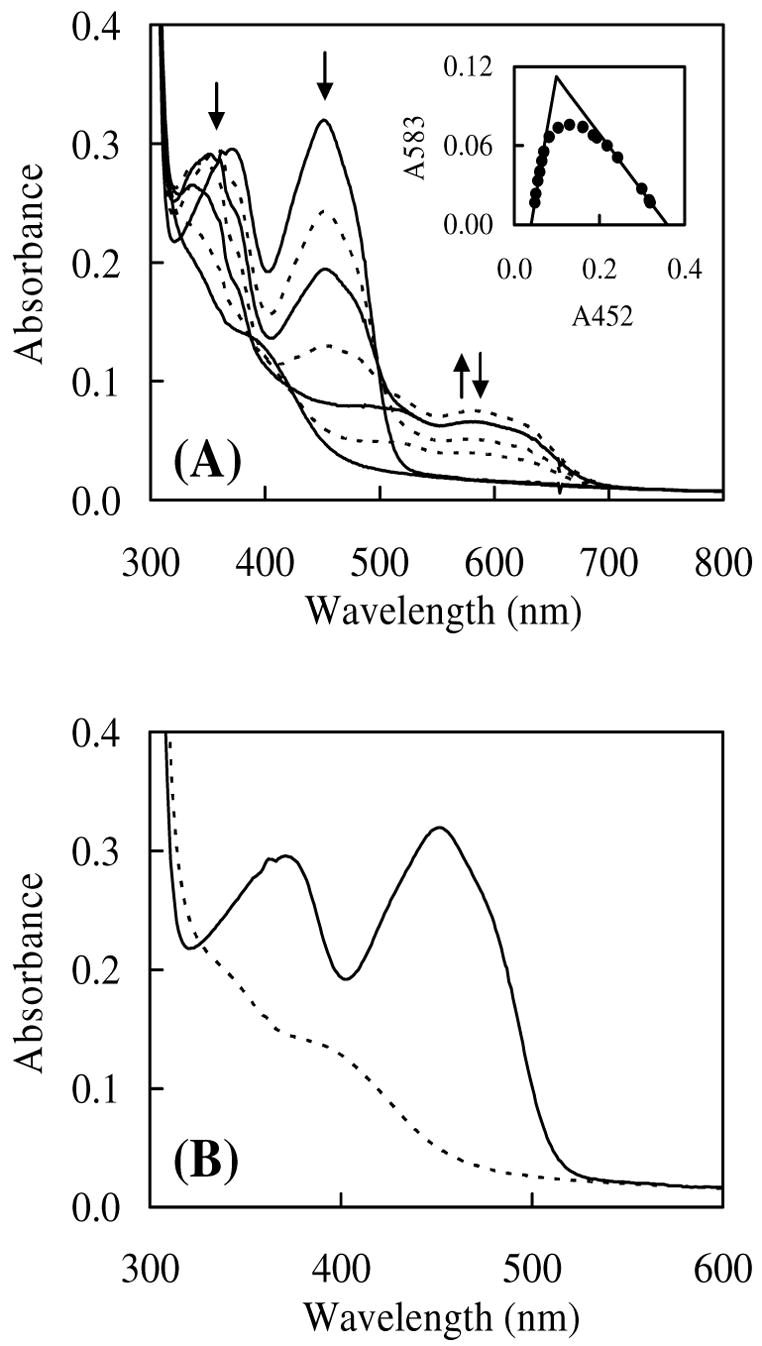
Recombinant IDI-2 was purified as an N-terminal His6-tagged protein to greater than 90% homogeneity as shown by SDS-PAGE. Migration of the protein monomer during SDS-PAGE was consistent with its calculated monomeric molecular weight of 40.1 kDa, which includes the His6-tag. In agreement with a previous report, the native enzyme is a homotetramer with a molecular mass of 154 kDa as determined by gel filtration results.17 The UV-vis spectrum (Figure 1) of the purified IDI-2 is typical for that of a flavoenzyme. HPLC analyses of released flavin confirmed that the enzyme-bound flavin is FMN. However, most of the flavin coenzyme (85 to > 90% depending on the batch) was lost during the purification (Figure 1). Results of the reconstitution experiments established a stoichiometric ratio of 0.85 mol FMN per mol monomer of enzyme. This is consistent with the crystal structure of B. subtilis IDI-2 in which each monomer binds a single molecule of FMN.21
FIGURE 1.
UV-vis absorption spectra of the as-purified IDI-2 (103 μM, dashed line) and the reconstituted IDI-2 (103 μM, solid line) in 10 mM HEPES buffer, pH 7.5.
Binding Affinity of Flavin Coenzyme
Upon binding to the apoenzyme, there is a slight shift of the absorption maxima in the UV-vis spectrum of FMN. The peaks typically observed at 375 and 446 nm for the free FMN are shifted to 373 and 452 nm, respectively. A noticeable reduction of the extinction coefficient of the second major peak also occurs; the free FMN has an ε of 12,200 M−1 cm−1 at 446 nm38 which decreases to 9,300 M−1 cm−1 at 452 nm upon binding to IDI-2. Both ultrafiltration and UV-vis difference spectroscopy were used to quantify the affinity of the apoenzyme for FMN. The latter approach exploited the shift in absorbance peaks mentioned above when FMN binds to the apoenzyme. As shown in Table 1, the results are similar for both methods with ultrafiltration providing a dissociation constant of 34 ± 3 μM, and UV-vis difference spectroscopy yielding an average value of 21 μM. Taken together, these data offer an average Kd for FMNOX of 27.5 μM.
Table 1.
Dissociation Constants Obtained from Ultrafiltration and UV-Vis Difference Spectroscopy Experiments
| Kd (μM) | ||
|---|---|---|
| Flavin Cofactor | Ultrafiltration | UV-vis Difference Spectroscopy |
| FMN | 34 ± 3 | 24 ±5 (λ = 440nm) 18 ±5 (λ = 493nm) |
| 1-deazaFMN | N.D.a | 97 ± 19 (λ = 333 nm) |
| 5-deazaFMN | N.D.a | 16 ± 3 (λ = 430nm) |
Not determined.
Binding affinities for the deazaflavin analogues, 1- and 5-deazaFMN, were also determined. The 5-deazaFMN, which is only capable of 2-electron transfer to and from the isoalloxazine ring system has a similar binding affinity as FMN (Kd = 16 ± 3 μM). The 1-deazaFMN, which is capable of both 1- and 2-electron transfers, is bound less tightly. Its Kd value of 97 μM is four to five-fold higher than that of FMN and 5-deazaFMN, suggesting that the N1 nitrogen of FMN plays an important role in cofactor binding.
Photoreduction of IDI-2
Photoreduction of IDI-2 led to a broad absorption peak centered at 583 nm (Figure 2A), which is characteristic for the neutral form of FMN semiquinone.48,49 Also observed is a clear shift of the peak centered at 373 nm during reduction. Because photoreduction conducted at pH 6, 9, and 10.5 (data not shown) all resulted in the appearance of the 583 nm peak, the FMN semiquinone intermediate (whose pKa ~8.5 in solution)49 apparently remains neutral over this pH range. Under our photoreduction conditions, the maximum semiquinone formation occurred after 24 s of irradiation. Approximately 68% of bound FMN was converted to neutral semiquinone as estimated by the extrapolated lines of the plot of the absorbance at 583 versus that at 452 nm (Figure 2A, inset). The semiquinone intermediate appeared to be relatively stable because a total irradiation time of 23 min was necessary to generate the fully reduced species, E:FMNred. Isosbestic points occurred near 355 and 500 nm for the oxidized FMN to semiquinone transition, 389 and 424 nm for the semiquinone to the reduced transition, and at 327 nm for the oxidized and reduced forms of the enzyme.
Electronic Absorption of IDI-2
The UV-vis spectrum for the oxidized and reduced IDI-2, obtained from the aforementioned photoreduction experiments, are shown in Figure 2B. The concentration of bound FMNOX at time zero was estimated to be 34 μM using ε452 of 9,300 M−1 cm−1 (this study). This concentration, combined with a theoretical maximum semiquinone absorbance of A583 = 0.112 (Figure 2A, inset), provided an estimated ε583 of 3,300 M−1 cm−1. A similar approach was used to calculate ε395 of 3,900 M−1 cm−1 for E:FMNred. The peak at 395 nm is typical for a neutral, reduced flavin49 whose N1 is protonated (Figure 2B), although a second absorption peak at ~ 350 nm (corresponding to the anionic reduced flavin) is also visible in the spectrum of the reduced coenzyme.
Steady-State Kinetics Parameters Determined Under Aerobic Conditions
Steady-state kinetic analyses carried out under aerobic conditions using NADPH as the reducing agent gave the kinetic parameters shown in Table 2 (row 1). A decrease in initial velocity was observed when the concentration of IPP (1) exceeded 75 μM (data not shown). This inhibitory behavior was reproducible, but was not reported in the first published study on this enzyme.17 The reason(s) for this decrease are not fully understood, but no inhibition was observed at high [IPP] when 10 mM sodium dithionite was used as the reducing agent in place of NADPH. Dithionite is a strong reducing agent that not only reduces flavins and flavoproteins, but also acts as an oxygen scavenger, reducing molecular oxygen to hydrogen peroxide. No decrease in initial velocity at high IPP concentrations was observed in the dithionite assays, suggesting that O2 may be the cause of inhibition in the previous experiment. The dramatic increase in Kcat to 0.57 ± 0.02 s−1 in the dithionite-reduced sample (Table 2, row 2, versus 0.065 ± 0.008 s−1 in the NADPH-reduced sample) also supported that E:FMNred is the active form of the enzyme.
Table 2.
Steady-State Kinetic Parameters for IDI-2 from Staphylococcus aureus
| Conditions | kcat (sec−4) | Km for IPP (μM) | kcat/Km (M−1 sec−1) |
|---|---|---|---|
| Aerobic with 5 mM NADPH as reducing agent | 0.065 ± 0.008 | 4.8 ± 10.7 | 1.4 × 104 |
| Aerobic with 10 mM dithionite as reducing agent | 0.57 ± 0.02 | 11.9± 1.8 | 4.8 × 104 |
| Anaerobic with 5 mM NADPH as reducing agent | 0.69 ± 0.03 | 16.8 ±3.0 | 4.1 × 104 |
Steady-State Kinetics Parameters Determined Under Anaerobic Conditions
To firmly establish O2 as the culprit for the inhibition, a third set of assays was carried out under anaerobic conditions using 5 mM NADPH as the reducing agent. All reaction conditions and work-up procedures were identical to those used in the aerobic/NADPH assay. The kinetic parameters (Table 2, row 3: Kcat = 0.69 s−1 and Km = 16.8 ± 3.0 μM for IPP) were similar to those obtained from the aerobic/dithionite experiments. Interestingly, no inhibition was observed at high IPP concentration under the anaerobic conditions. In addition, a Km of 0.2 ±0.1 μM was also determined anaerobically for FMN in the presence of IPP using NADPH as the reducing agent. This is about 70-fold lower than the Kd (14 μM) for reduced FMN binding to apo-IDI-2, and demonstrates that binding of the reduced coenzyme to IDI-2 is much tighter in the presence of IPP (1). The steady-state kinetic parameters determined by the anaerobic/NADPH assay are considered to be more relevant to catalysis under physiological conditions. These results strongly indicate that E:FMNred is the active form of IDI-2, and that E:FMNred can be readily oxidized by O2, which diminishes the enzyme activity.
Stereospecificity of Hydride Transfer from NADH
To establish the stereospecificity of NADH oxidation in IDI-2 activation, several incubations with stereospecifically labeled NADH were performed and the oxidized NAD+ was isolated. Based on high-field NMR analysis, it was found that NAD+ isolated from incubation mixtures of (R)-[4-2H]NADH retained the deuterium on the nicotinamide ring (i.e., no 4-H signal in the δ 8.5 to 9.0 range33). On the contrary, incubation mixtures with (S)-[4-2H]NADH led to the release of all isotopic labeling (i.e., the spectra showed a doublet at δ 8.55). Thus, reduction of the IDI-2 bound FMN by NADH is pro-S stereospecific with regard to the removal of one of the diastereotopic methylene hydrogens at C4 of the dihydronicotinamide ring. The same stereoselectivity was inferred for the NADPH coenzyme. These results show that FMN reduction by NAD(P)H is clearly enzyme-mediated.
NADPH Consumption during Enzymatic Turnover
NADH or NADPH is required for IDI-2 activity as a reducing agent for FMN. Even though a large excess of NADH or NADPH is typically used in the assay17, we anticipated that only catalytic amounts are required to convert the oxidized enzyme into its active, reduced form. Two sets of experiments were carried out to test this hypothesis. First, we determined the total amounts of NADPH consumed and DMAPP (2) formed in the same reaction using complimentary UV-vis and radioassays. As shown in Figure 3A (top curve), multiple turnovers of IPP to DMAPP occurred at the expense of only a small amount of NADPH. The data used to plot the NADPH curve are the average values of the assay results. The range of values for the replicate measurements is small and falls within the symbols shown on the curve. Clearly, NADPH consumption is independent of IPP concentration. The amount of NADPH consumption was also measured using a constant IPP (1) concentration (1.2 mM) and variable enzyme concentrations (2.0 to 22 μM). As shown in Figure 3B, the amount of NADPH consumption is directly proportional to the enzyme concentration. A calculated ratio of 0.91 μmol NADPH consumed per μmol enzyme used was obtained. These results strongly support a catalytic role for NADPH as previously suggested by Yamashita et al26.
FIGURE 3.
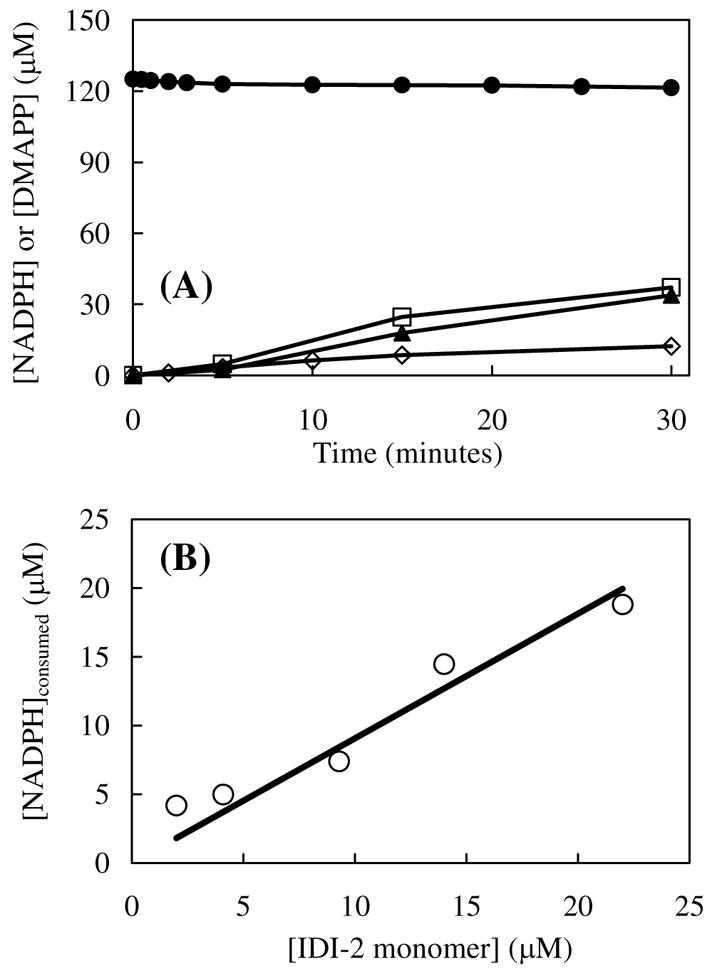
(A) Stoichiometry of NADPH consumption versus DMAPP formation over a 30-min incubation period with IDI-2. The UV-vis assays (0.178 mL) contained 5.3 μM reconstituted IDI-2, 0.125 mM NADPH, 5 mM MgCl2, and varying concentrations of IPP (0.05, 0.50 and 1.20 mM) in 0.1 M HEPES buffer (pH 7.0). Each assay was incubated for 30 min at 37 °C as described in the Experimental Procedures. NADPH concentration (●) was determined based on the absorbance at 340 nm. DMAPP formation was determined by the radioassay using variable amounts of [1-14C]IPP (0.05 (◇), 0.50 (□), or 1.20 (▲) mM). (B) Linear relationship between NADPH consumption and enzyme concentration during turnover. Each assay contained 0.125 mM NADPH, 1.2 mM IPP, and 5 mM MgCl2, and variable amounts of reconstituted IDI-2 (2.0, 4.1, 9.3, 14, or 22 μM) in 0.1 M HEPES buffer (pH 7.0). NADPH consumption over a 30 min period was calculated from the total change in absorbance at 340 nm.
Activity of IDI-2 Apoenzyme Reconstituted with 1- and 5-deazaFMN
The 1- and 5-deazaFMN analogues were used to probe the catalytic role of enzyme-bound FMN. Both analogues are capable of two-electron transfers, but only 1-deazaFMN is able to mediate 1-electron chemistry.31 The results of assays using the reconstituted enzymes are shown in Figure 4. It was found that apoenzyme incubated with either analogue was inactive in the presence of 5 mM NADPH. Ineffective coenzyme binding was ruled out since flavin concentrations were at, or above, the respective Kd values determined in this study. However, because both deazaflavins have redox potentials lower than FMN,31 the resistance to reduction by NADPH may have prevented activation of the enzyme. Thus, sodium dithionite instead of NADPH was used in the incubation mixture to ensure the complete reduction of reconstituted IDI-2. Unfortunately, in the presence of 5 mM dithionite, the activity of deazaflavin-reconstituted enzyme remained negligible, and inspection of the assay mixtures indicated that the flavins were mostly oxidized. When the dithionite concentration was increased to 50 mM, a complete reduction of the reconstituted enzymes was noted. Interestingly, the reduced 5-deazaFMN, which most certainly binds as tightly as the oxidized form in the active site, was catalytically incompetent. In contrast, reconstitution with 1-deazaFMN provided full activity. These results provide support for a 1-electron transfer role of the flavin coenzyme during catalysis.
FIGURE 4.
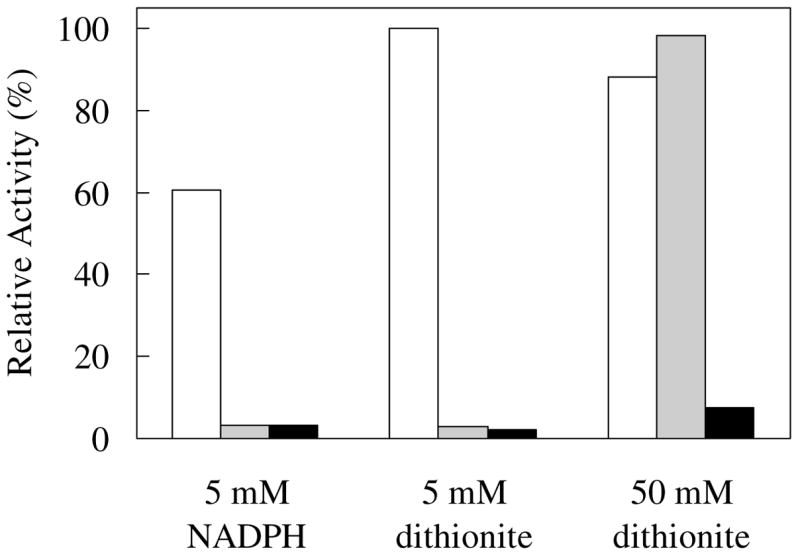
Activity of IDI-2 apoenzyme in the presence of FMN (white), 1-deazaFMN (grey), and 5-deazaFMN (black). The incubation mixture contained 4.9 μM apo-IDI-2, 5 mM NADPH (or 5 or 50 mM dithionite), 50 μM [1-14C]IPP, 5 mM MgCl2, and 100 μM flavin coenzyme in 0.1 M HEPES buffer (pH 7.0). All reactions were incubated for 10 min at 37 °C, quenched with 25% HCl:MeOH, saturated with NaCl, and extracted with toluene or petroleum ether. The radioactivity of a 0.3 mL sample removed from the organic extracts was measured by liquid scintillation counting (see Experimental Procedures for details).
Oxidation-Reduction Potentials of IDI-2
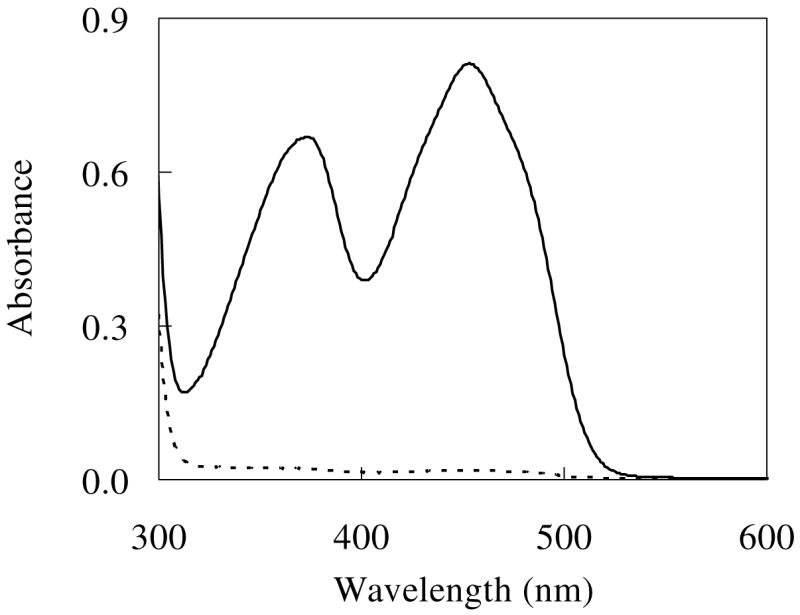
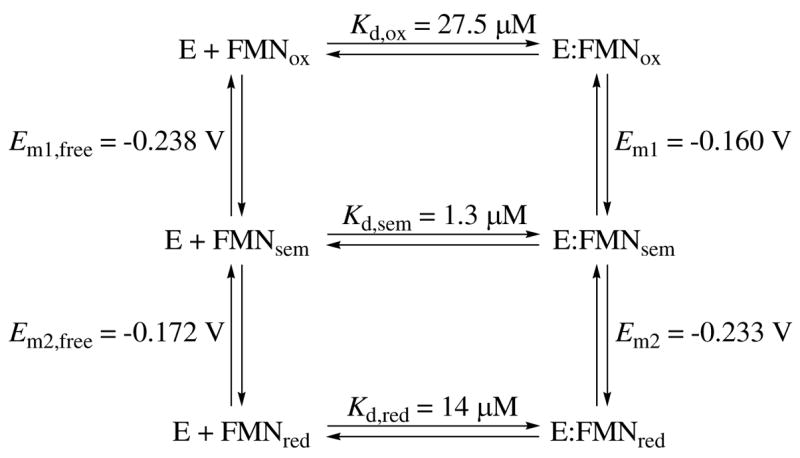
The midpoint oxidation-reduction potential for E:FMNox/E:FMNred was determined using the xanthine oxidase reducing system developed by Massey.42 Select scans are shown from assays in the absence and presence of IPP (Figure 5A and 5B, respectively). In the absence of IPP, the reduced form of the anthroquinone-2-sulfonate reference dye (AQS, Em = −0.225 versus SHE at pH 7.0)45 began to accumulate after ~ 20 min as observed by the increase in A381 The concentration of oxidized and reduced AQS and FMN were determined as described in Experimental Procedures. A plot of log([E:FMNox]/[E:FMNred]) versus log([AQSox]/[AQSred]) was constructed according to the method of Minnaert (Figure 5A, inset).44 The slope of the line was unity as expected for equilibrium between a two-electron donor and two-electron acceptor. Using the y-intercept as described in the Experimental Procedures, a redox potential of −0.197 V was obtained for the FMNox/FMNred couple. This value is nearly equivalent to that of free FMN (−0.205 V, Table 3) indicating that IDI-2 binds FMNox and FMNred with similar affinities. The dissociation constant for FMNred was deduced from the thermodynamic cycles of Scheme 2 and Equation 1. Indeed, the reduced form is only slightly stabilized as reflected by a small reduction in Kd from 27.5 μM for the oxidized FMN (an average value of Kd derived from the ultrafiltration and UV-vis difference spectroscopy assays) to 14 μM for the reduced FMN.
FIGURE 5.
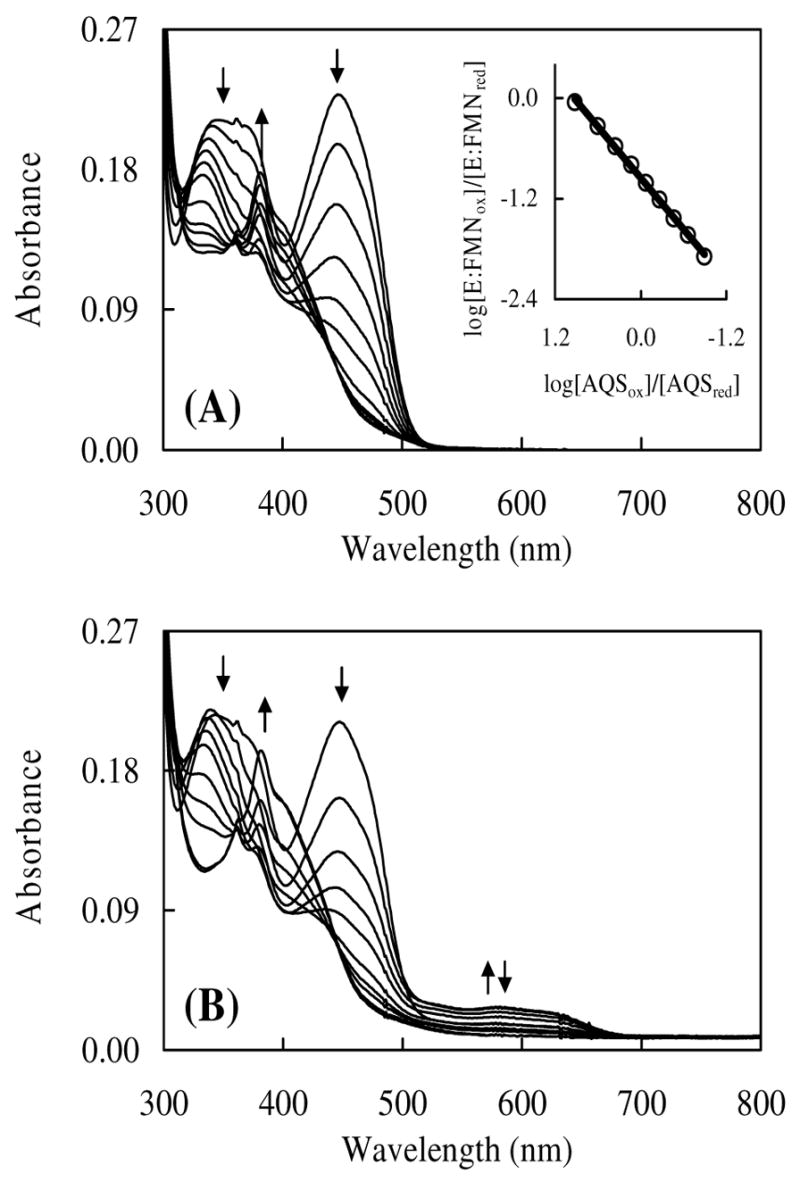
(A) Anaerobic reduction of IDI-2 by the xanthine oxidase reducing system in the absence of IPP. IDI-2 (20 μM) in 0.1 M potassium phosphate buffer (pH 7.0), containing 17 μM FMN, 0.4 mM hypoxanthine, 2 μM methyl viologen, and 18 μM anthroquinone-2-sulfonate (AQS) was incubated under anaerobic conditions at 25 °C with 0.55 mg bovine milk xanthine oxidase (0.084 units/mg). Spectra from top to bottom were recorded at 0, 7.5, 11.5, 15.5, 19.5, 23.5, 31.5, 39.5, 43.5, 51.5 and 59.5 min. Increased absorbance at 381 nm began after 20 min due to the accumulation of reduced AQS. The inset figure shows the Minnaert plot obtained during reduction. Reduction of AQSox was monitored at 332 nm, the isosbestic point for FMNox and FMNred- Reduction of FMN was monitored at 354 nm, the isosbestic point for AQSox and AQSred. (B) Anaerobic reduction of IDI-2 by the xanthine oxidase reducing system in the presence of IPP. Contents and procedures were identical to (A) except for the inclusion of 150μM IPP in the assay. Spectra from top to bottom were recorded at 0.03, 1, 2, 3, 4, 6, 8, 10, 20, and 56 min. The last two spectra are virtually identical. Increased absorbance at 381 nm began after 4 min due to the accumulation of reduced AQS.
Table 3.
Midpoint Reduction Potentials (vs. SHE) for FMN in the Absence and Presence ofIDI-2
| Eml (V)a | Em2 (V)b | Em (V)c | |
|---|---|---|---|
| FMN (pH 7.0)d | −0.238 | −0.172 | −0.205 |
| FMN + DI-2(pH7.0)e | −0.160 | −0.233 | −0.197 |
Midpoint potential (n = 1) of FMNox/FMNsem redox couple.
Midpoint potential (n = 1) of FMNsem/FMNred redox couple.
Midpoint potential (n = 2) of FMNox/FMNred redox couple.
Data from reference 51
Data from this study.
Scheme 2.
| (1) |
The midpoint redox potentials Em1 and Em2 were estimated using results from the photoreduction experiments. Maximum semiquinone formation (approximately 68%) occurred after 24 s of irradiation. At this time point, an A583 value of 0.076, compared to a theoretical maximum of 0.112 was observed (Figure 2A, inset). This provided a semiquinone formation constant, K, of 17.2, which indicates a positive separation of Em1 and Em2.46 From the Nernst equation, (Em1 – Em2) was calculated to be 0.073 V.46 Using this data along with the midpoint potential of −0.197 V for E:FMNox/E:FMNred, values of −0.160 and −0.233 V were calculated for Em1 and Em2, respectively. The more positive Em1 value indicates that IDI-2 apoenzyme stabilizes semiquinone formation by making the first electron transfer more favorable than the second. This is in contrast to the potentials of free FMN (Table 3) which destabilize semiquinone formation. The source of semiquinone stabilization by IDI-2 may be ascribed to the tighter binding of semiquinone (Kd,Sem = 1.3 μM) as determined from the thermodynamic cycles shown in Scheme 2 and Equation 2.
| (2) |
DISCUSSION
IDI-2 from Staphylococcus aureus is an intriguing flavoenzyme. It requires a FMN coenzyme to catalyze an overall nonredox reaction. Interestingly, it was purified essentially as an apoenzyme containing 5–20% of its stoichiometric flavin content. This is unusual for flavoenzymes, and is due to its relatively weak binding affinity (Kd ~27.5 μM) for FMNox. Dissociation constants of 97 and 16 μM were determined for the oxidized 1-deazaFMN and 5-deazaFMN, respectively. The low affinity for 1-deazaFMN suggests hydrogen bonding to N1 of the isoalloxazine ring system may be important for binding of FMN. In fact, amino acid sequence alignments of reported IDI-2 enzymes reveal an absolutely conserved lysine residue (Lys 186 in S. aureus) in the vicinity of FMN that could serve as the hydrogen bond donor to N1. The proposed role of this residue is supported by the recent X-ray crystal structures of the homologous enzymes from Bacillus subtilis 21 and Thermus thermophilus.23 The similarity of the dissociation constants for FMN and 5-deazaFMN is also notable. Because 5-deazaFMN does not reconstitute the isomerase activity, these results strongly suggest that the flavin coenzyme of IDI-2 is not used simply to stabilize the enzyme active site or to maintain the enzyme conformation as previously proposed.23 In contrast, the reduced FMN coenzyme appears to play a direct role in catalysis, perhaps as a redox cofactor that mediates electron transfer to and from IPP (1) during its conversion into DMAPP (2).
Because reduced 5-deazaFMN does not restore activity in either S. aureus or Sulfolobus shibatae enzymes,27 a mechanism involving the transient two-electron reduction of IPP50 can be excluded. Instead, the complete recovery of activity when apoenzyme is reconstituted with reduced 1-deazaFMN suggests that single electron transfer chemistry may be operative in catalysis. Additional evidence for the involvement of single electron chemistry is provided by the photoreduction experiments in which IDI-2 was shown to stabilize the neutral form of the FMN semiquinone. While these observations do not ensure that formation of FMNsem is catalytically relevant, they do show that the IDI-2 active site has the necessary framework in place to bind FMNsem tighter (Kd = 1.3 μM) than both FMNox (Kd = 27.5 μM) and FMNred (14 μM). The result of this tighter binding is the stabilization of a significant amount (~68%) of semiquinone after it is formed by one electron reduction of E:FMNox. Thermodynamically, this creates a 73 mV positive separation between Em1 (−0.160 V) and Em2 (−0.233 V) in the S. aureus enzyme.
To gain more insight into the mechanism of IDI-2, we used two sets of experiments to examine whether binding of the IPP substrate increases the affinity of the apoenzyme for FMNred. First, we carried out anaerobic steady-state kinetic experiments in which the concentration of free FMNred was incrementally increased in the presence of saturating IPP (1). From this data, we calculated a Km value for FMNred of 0.2 μM, which is similar to the value reported by Yamashita for the Sulfolobus shibatae enzyme.26 This represents a significant increase in binding affinity for FMNred, compared to a Kd value of 14 μM for FMNred in the absence of IPP. This may be ascribed to a conformational change upon IPP binding that creates more favorable binding interactions with the FMN coenzyme. A conformational change could also affect the polarity, hydrophobicitiy, and rigidity of the FMN environment, which might modulate the redox properties of IDI-2. Such substrate-induced binding effects are common in flavoenzymes51 and, importantly, the X-ray crystal structures for IDI-2 enzymes also suggest that substantial ordering of active site loops is likely required to generate the catalytically competent, closed ternary complex.21–23
In a second experiment, we used the xanthine oxidase reducing system to investigate the effect of IPP binding on the redox potentials of IDI-2. The reaction contents and procedures were identical to the original assays except 150 μM IPP (~ 9 Km) was included. The results of this experiment (Fig 5B) were dramatically different. In the presence of IPP, FMNox reduction was much faster and progressed through a neutral semiquinone species, whereas reduction by the xanthine oxidase system in the absence of IPP led to the direct formation of FMNred (Figure 5A). Maximum semiquinone formation occurred at ~ 2 min and was estimated to be 42% of the total FMN using ε583 of 3300 M−1 cm−1. Reduction to the fully reduced FMN was complete in 20 min, as compared to 1 h in the absence of IPP, suggesting that Em for the E:FMNox/E:FMNred couple may be higher in the presence of IPP. Although the reaction under our conditions was too fast to calculate the redox potentials, these results clearly suggest that IPP binding shifts Em1 to more positive values relative to Em2 Our results are entirely consistent with a recent report by Poulter and coworkers, where they demonstrated a similar positive separation between Em1 and Em2 in the presence of IPP for the IDI-2 enzyme from Thermus thermophilus using potentiometric titrations.24 Under their titration conditions, however, they were able to determine values for Em1 (−83 mV) and Em2 (−196 mV) in the presence of IPP. The increase in Em1 observed in the presence of IPP leads to a thermodynamic stabilization of the semiquinone in both enzymes, which may help to prevent the undesirable oxidation of FMNsem during turnover.
Taken together, these findings suggest a catalytic mechanism which is similar to that proposed previously by Hemmi27 and Bornemann.50 As shown in Scheme 3, activation of the enzyme occurs through transfer of the 4S hydride from NAD(P)H to enzyme-bound FMNox (3) to generate FMNred (4). Following substrate binding, Rothman et al. have shown that the anionic FMNred becomes protonated to form neutral FMNred.24 We have noted similar observations in our studies on the S. aureus enzyme.29 From this enzyme:substrate complex, protonation of the IPP double bond could be achieved by an active site amino acid (path a) or by the N5 atom of the neutral FMNred (path b). Though the N5-H is not typically very acidic,52 hydrogen bonding with the strictly conserved Thr67 residue could serve to lower its pKa. At present, the identity of the general acid (AH) in path a cannot be discerned from the available crystal structures.21–23 The generation of the 3° carbocation (5), could help to establish a driving force for electron transfer either from C4a (path a) or from N5 (path b) of FMNred. Electron transfer would generate a substrate radical (6) and a neutral FMNsem (either 7 or 8). In path a, electron transfer from C4a would be facilitated by deprotonation of the N1-H of FMNred, which could be achieved by the aforementioned Lys 186 residue. In path b, the electron could be transferred to 5 from the N5 atom. Both of these single electron transfer mechanisms could explain the inactivity of 5-deazaFMN and the full activity observed with 1-deazaFMN. In addition, Eguchi and co-workers have recently shown that an N5-alkyl adduct forms when Methanocaldococcus jannaschii IDI-2 is incubated with a known epoxide inactivatior of IDI-1.53 This suggests that the N5-C4a region of the flavin is in close proximity to the reactive inhibitor intermediates, and that the N5 atom appears to accumulate sufficient electron density to form a covalent adduct with the inactivator.
Scheme 3.

The pro-R stereospecificity of proton abstraction at C2 of IPP (1) during turnover was separately determined.54 This deprotonation would likely be effected by a general base, with concomitant electron transfer back to FMNsem to complete the catalytic cycle. Once again, the available crystal structures provide no indication of what this general base could be. Interestingly, recent crystallographic and biochemical studies on chorismate synthase (another enzyme that uses a reduced FMN cofactor to catalyze a reaction with no net redox change) have suggested that deprotonation of the substrate radical intermediate is carried out by the N5 atom of an anionic semiquinone.55–57 We have noticed that IDI-2 and chorismate synthase share many apparent mechanistic similarities.29 Future structural data will be helpful in determining whether the N5 atom of FMNred in IDI-2 is positioned properly to perform a similar role as an active site base.
If the cryptic redox cycles depicted in Scheme 3 are operative, the redox potential of the FMNsem/FMNred couple will be critical in determining the energetic barriers for electron transfer to and from IPP during turnover. As noted by Rothman et al., the formation of a stable flavin smeiquinone/IPP radical pair seems feasible in the IDI-2 reaction based on Em2 (the reduction potential for the E:FMNred/E:FMNsem couple in the presence of IPP) and the reduction potential for the t-butyl cation (which would be similar to the carbocation generated upon IPP protonation as in Scheme 3).24 Substrate binding effects on flavoenzyme redox potentials, such as those observed with both S. aureus and T. thermophilus IDI-2, are common and are one of the key tools used by flavoenzymes to catalyze the variety of reactions seen in nature. These effects could include changes in the hydrophobicity and polarity of the active site, as well as changes in hydrogen bonding interactions between active site amino acids and flavin functional groups when substrate binds. Clearly, more structural studies, coupled with site-directed mutagenesis and biochemical characterization of the mutant enzymes are necessary to characterize the IDI-2 active site.
Overall, the results presented in this work have provided much needed characterization and mechanistic information for this unusual flavoenzyme. Although the photoreduction experiments, oxidation-reduction studies, and activity assays with FMN and its deazaFMN analogues support a mechanism involving single electron transfer reactions and the obligate intermediacy of a flavin semiquinone in the S. aureus enzyme, the catalytic competence of the neutral semiquinone has not yet been established and other mechanistic possibilities can not be rigorously excluded at this time.24–29 Experiments geared towards further dissecting the mechanism of this enzyme are currently in progress. Because IDI-2 is essential for the survival of many pathogenic bacteria, and because it has a distinct catalytic mechanism from the IDI-1 employed by humans, this enzyme represents a promising new antiomicrobial target. Further mechanistic investigation of this unusual enzyme could therefore form the basis for the development of new antibiotics.
Abbreviations
- Ap
ampicillin
- AQS
anthroquinone-2-sulfonate
- DEAE
diethylaminoethyl
- DMAPP
dimethylallyl diphosphate
- DTT
dithiothreitol
- DPM
disintegrations per minute
- EDTA
ethylenediaminetetraacetic acid
- Em
midpoint reduction potential
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- FPLC
fast protein liquid chromatography
- HEPES
(N-[2-hydroxyethyl]piperazine-N′-[2-ethanesulfonic acid])
- HPLC
high performance liquid chromatography
- IDI, isopentenyl diphosphate
dimethylallyl diphosphate isomerase
- IPP
isopentenyl diphosphate
- IPTG
isopropyl β-D-thiogalactoside
- Kn
kanamycin
- LDH
lipoamide dehydrogenase
- LB
Luria-Bertani
- MEP
methylerythritol phosphate
- MEV
mevalonate
- NAD+
β-nicotinamide adenine dinucleotide
- NADH
β-nicotinamide adenine dinucleotide, reduced form
- NADP+
β-nicotinamide adenine dinucleotide phosphate
- NADPH
β-nicotinamide adenine dinucleotide phosphate, reduced form
- NTA
nitrilotriacetic acid
- PAGE
polyacrylamide gel electrophoresis
- SDS
sodium dodecyl sulfate
- YADH
yeast alcohol dehydrogenase
Footnotes
This work was supported in part by a Welch Foundation Grant (F-1511) and a National Institutes of Health Grant (GM40541)
References
- 1.Koyama T, Ogura K. Isopentenyl diphosphate isomerase and prenyltransferases. In: Cane DE, editor. Comprehensive Natural Products Chemistry. Vol. 2. Elsevier; Amsterdam: 1999. pp. 69–96. [Google Scholar]
- 2.Kellogg BA, Poulter CD. Chain elongation in the isoprenoid biosynthetic pathway. Curr Opin Chem Biol. 1997;1:570–578. doi: 10.1016/s1367-5931(97)80054-3. [DOI] [PubMed] [Google Scholar]
- 3.Rohmer M. Mevalonate-independent methylerythritol phosphate pathway for isoprenoid biosynthesis. Elucidation and distribution. Pure Appl Chem. 2003;75:375–387. [Google Scholar]
- 4.Adam P, Hecht S, Eisenreich W, Kaiser J, Grawert T, Arigoni D, Bacher A, Rohdich F. Biosynthesis of terpenes: Studies on l-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase. Proc Natl Acad Sci USA. 2002;99:12108–12113. doi: 10.1073/pnas.182412599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rohdich F, Hecht S, Bacher A, Eisenreich W. Deoxyxylulose phosphate pathway of isoprenoid biosynthesis. Discovery and function of ispDEFGH genes and their cognate enzymes. Pure Appl Chem. 2003;75:393–405. [Google Scholar]
- 6.Qureshi N, Porter JW. In: Biosynthesis of Isoprenoid Compounds. Porter JW, Spurgeon SL, editors. Vol. 1. Wiley; New York: 1981. pp. 47–94. [Google Scholar]
- 7.Agranoff BW, Eggerer H, Henning U, Lynen F. Isopentenol pyrophosphate isomerase. J Am Chem Soc. 1958;81:1254–1255. [PubMed] [Google Scholar]
- 8.Ramos-Valdivia AC, van der Heijden R, Verpoorte R. Isopentenyl diphosphate isomerase: A core enzyme in isoprenoid biosynthesis. A review of its biochemistry and function. Nat Prod Rep. 1997;14:591–603. doi: 10.1039/np9971400591. [DOI] [PubMed] [Google Scholar]
- 9.Street IP, Christensen DJ, Poulter CD. Hydrogen exchange during the enzyme-catalyzed isomerization of isopentenyl diphosphate and dimethylallyl diphosphate. J Am Chem Soc. 1990;112:8577–8578. [Google Scholar]
- 10.Reardon JE, Abeles RH. Mechanism of action of isopentenyl pyrophosphate isomerase: Evidence for a carbonium ion intermediate. Biochemistry. 1986;25:5609–5616. doi: 10.1021/bi00367a040. [DOI] [PubMed] [Google Scholar]
- 11.Reardon JE, Abeles RH. Time-dependent inhibition of isopentenyl pyrophosphate isomerase by 2-(dimethylamino)ethyl pyrophosphate. J Am Chem Soc. 1985;107:4078–4079. [Google Scholar]
- 12.Muehlbacher M, Poulter CD. Isopentenyl-diphosphate isomerase: Inactivation of the enzyme with active-site-directed irreversible inhibitors and transition-state analogues. Biochemistry. 1988;27:7315–7328. doi: 10.1021/bi00419a021. [DOI] [PubMed] [Google Scholar]
- 13.Cornforth JW, Cornforth RH, Popjak G, Yengoyan L. XX Steric course of decarboxylation of 5-pyrophosphomevalonate and of the carbon to carbon bond formation in the biosynthesis of farnesyl pyrophosphate. J Biol Chem. 1966;241:3970–3987. [PubMed] [Google Scholar]
- 14.Clifford K, Cornforth JW, Mallaby R, Phillips GT. Stereochemistry of isopentenyl pyrophosphate isomerase. J Chem Soc D No. 1971;24:1599–1600. [Google Scholar]
- 15.Popjak G, Cornforth JW. Substrate stereochemistry in squalene biosynthesis. Biochem J. 1966;101:553–568. [PMC free article] [PubMed] [Google Scholar]
- 16.Poulter CD, Rilling HC. Prenyltransferases and isomerase. In: Porter JW, Spurgeon SL, editors. Biosynthesis of Isoprenoid Compounds. Vol. 1. Wiley; New York: 1981. pp. 162–209. [Google Scholar]
- 17.Kaneda K, Kuzuyama T, Takagi M, Hayakawa Y, Seto H. An unusual isopentenyl diphosphate isomerase found in the mevalonate pathway gene cluster from Streptomyces sp. strain CL190. Proc Natl Acad Sci USA. 2001;98:932–937. doi: 10.1073/pnas.020472198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laupitz R, Hecht S, Amslinger S, Zepeck F, Kaiser J, Richter G, Schramek N, Steinbacher S, Huber R, Arigoni D, Bacher A, Eisenreich W, Rohdich F. Biochemical characterization of Bacillus subtilis type II isopentenyl diphosphate isomerase, and phylogenetic distribution of isoprenoid biosynthesis pathways. Eur J Biochem. 2004;271:2658–2669. doi: 10.1111/j.1432-1033.2004.04194.x. [DOI] [PubMed] [Google Scholar]
- 19.Kuzuyama T, Seto H. Diversity of the biosynthesis of the isoprene units. Nat Prod Rep. 2003;20:171–183. doi: 10.1039/b109860h. [DOI] [PubMed] [Google Scholar]
- 20.Takagi M, Kaneda K, Shimizu T, Hayakawa Y, Seto H, Kuzuyama T. Bacillus subtilis ypgA gene is fni, a nonessential gene encoding type 2 isopentenyl diphosphate isomerase. Biosci Biotechnol Biochem. 2004;68:132–137. doi: 10.1271/bbb.68.132. [DOI] [PubMed] [Google Scholar]
- 21.Steinbacher S, Kaiser J, Gerhardt S, Eisenreich W, Huber R, Bacher A, Rohdich F. Crystal structure of the type II isopentenyl diphosphate:dimethylallyl diphosphate isomerase from Bacillus subtilis. J Mol Biol. 2003;329:973–982. doi: 10.1016/s0022-2836(03)00527-8. [DOI] [PubMed] [Google Scholar]
- 22.Barkley SJ, Desai SB, Poulter CD. Type II isopentenyl diphosphate isomerase from Synechocystis sp. strain PCC 6803. J Bacteriol. 2004;186:8156–8158. doi: 10.1128/JB.186.23.8156-8158.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Ruyck J, Rothman S, Poulter CD, Wouters J. Structure of Thermus thermophilus type 2 isopentenyl diphosphate isomerase inferred from crystallography and molecular dynamics. Biochem Biophys Res Comm. 2005;338:1515–1518. doi: 10.1016/j.bbrc.2005.10.114. [DOI] [PubMed] [Google Scholar]
- 24.Rothman SC, Helm TR, Poulter CD. Kinetic and spectroscopic characterization of Type II isopentenyl diphosphate isomerase from Thermus thermophilus: Evidence for formation of substrate-induced flavin species. Biochemistry. 2007;46 doi: 10.1021/bi0616347. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barkley SJ, Cornish RM, Poulter CD. Identification of an archael type II isopentenyl diphosphate isomerase in Methanothermobacter thermautotrophicus. J Bacteriol. 2004;186:1811–1817. doi: 10.1128/JB.186.6.1811-1817.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamashita S, Hemmi H, Ikeda Y, Nakayama T, Nishino T. Type 2 isopentenyl diphosphate isomerase from a thermoacidophilic archaeon Sulfolobus shibatae. Eur J Biochem. 2004;271:1087–1093. doi: 10.1111/j.1432-1033.2004.04010.x. [DOI] [PubMed] [Google Scholar]
- 27.Hemmi H, Ikeda Y, Yamashita S, Nakayama T, Nishino T. Catalytic mechanism of type 2 isopentenyl diphosphate:dimethylallyl diphosphate isomerase: Verification of a redox role of the flavin cofactor in a reaction with no net redox change. Biochem Biophys Res Comm. 2004;322:905–910. doi: 10.1016/j.bbrc.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 28.Siddiqui MA, Yamanaka A, Hirooka K, Bamaba T, Kobayashi A, Imanaka T, Fukusaki E-i, Fujiwara S. Enzymatic and structural characterization of type II isopentenyl diphosphate isomerase from hyperthermophilic archaeon Thermococcus kodakaraensis. Biochem Biophys Res Comm. 2005;331:1127–1136. doi: 10.1016/j.bbrc.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 29.Mansoorabadi SO, Thibodeaux CJ, Liu H-w. The diverse roles of flavin coenzymes – Nature’s most versatile thespians. J Org Chem. 2007;72 doi: 10.1021/jo0703092. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davisson VJ, Woodside AB, Poulter CD. Synthesis of allylic and homoallylic isoprenoid pyrophosphates. Methods Enzymol. 1985;110:130–144. doi: 10.1016/s0076-6879(85)10068-6. [DOI] [PubMed] [Google Scholar]
- 31.Hersh LB, Walsh C. Preparation, characterization, and coenzymic properties of 5-carba-5-deaza and 1-carba-l-deaza analogs of riboflavin, FMN, and FAD. Methods Enzymol. 1980;66:277–287. doi: 10.1016/0076-6879(80)66469-6. [DOI] [PubMed] [Google Scholar]
- 32.Gassner G, Wang L, Bade C, Ballou DP. Reaction of phthalate dioxygenase reductase with NADH and NAD: Kinetic and spectral characterization of intermediates. Biochemistry. 1994;33:12184–12193. doi: 10.1021/bi00206a022. [DOI] [PubMed] [Google Scholar]
- 33.Arnold LJ, You K-s. The hydride transfer stereospecificity of nicotinamide adenine dinucleotide linked enzymes: A proton magnetic resonance technique. Methods Enzymol. 1978;54:223–232. doi: 10.1016/s0076-6879(78)54017-2. [DOI] [PubMed] [Google Scholar]
- 34.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 35.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 36.Andrews P. Estimation of the molecular weights of proteins by Sephadex gel-filtration. Biochem J. 1964;91:222–233. doi: 10.1042/bj0910222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Light DR, Walsh C, Marietta MA. Analytical and preparative high-performance liquid chromatography separation of flavin and flavin analog coenzymes. Anal Biochem. 1980;109:87–93. doi: 10.1016/0003-2697(80)90014-7. [DOI] [PubMed] [Google Scholar]
- 38.Aliverti A, Curti B, Vanoni MA. Methods in Molecular Biology. In: Chapman SK, Reid GA, editors. Flavoprotein Protocols. Vol. 131. Humana Press; Totowa, New Jersey: 1999. pp. 9–23. [Google Scholar]
- 39.Barnard GF. Prenyltransferase from Human Liver. Methods Enzymol. 1985;110:155–167. doi: 10.1016/s0076-6879(85)10071-6. [DOI] [PubMed] [Google Scholar]
- 40.Massey V, Hemmerich P. A photochemical procedure for reduction of oxidation-reduction proteins employing deazariboflavin as catalyst. J Biol Chem. 1977;252:5612–5614. [PubMed] [Google Scholar]
- 41.Massey V, Hemmerich P. Photoreduction of flavoproteins and other biological compounds catalyzed by deazaflavins. Biochemistry. 1978;17:9–17. doi: 10.1021/bi00594a002. [DOI] [PubMed] [Google Scholar]
- 42.Massey V. In: Flavins and Flavoproteins. Curti B, Ronchi S, Zanetti G, editors. Walter de Gruyter & Co.; Berlin: 1990. pp. 59–66. [Google Scholar]
- 43.Personal communication with Albert Vanneste, Quality Control Manager, ACROS Organics, Belgium.
- 44.Minnaert K. Measurement of the equilibrium constant of the reaction between cytochrome c and cytochrome a. Biochim Biophys Acta. 1965;110:42–56. doi: 10.1016/s0926-6593(65)80093-5. [DOI] [PubMed] [Google Scholar]
- 45.Fultz MF, Durst RA. Mediator compounds for the electrochemical study of biological redox systems: A compilation. Anal Chim Acta. 1982;140:1–18. [Google Scholar]
- 46.Clark WM. Oxidation-Reduction Potentials of Organic Systems. William & Wilkins; Baltimore: 1960. pp. 184–190. [Google Scholar]
- 47.Husain M, Massey V. Reversible resolution of flavoproteins into apoproteins and free flavins. Methods Enzymol. 1978;53:429–437. doi: 10.1016/s0076-6879(78)53047-4. [DOI] [PubMed] [Google Scholar]
- 48.Massey V, Palmer G. On the existence of spectrally distinct classes of flavoprotein semiquinones. A new method for the quantitative production of flavoprotein semiquinones. Biochemistry. 1966;5:3181–3189. doi: 10.1021/bi00874a016. [DOI] [PubMed] [Google Scholar]
- 49.Müller F. In: Chemistry and Biochemistry of Flavoenzymes. Müller F, editor. Vol. 1. CRC Press; Boca Raton, Florida: 1991. pp. 1–71. [Google Scholar]
- 50.Bornemann S. Flavoenzymes that catalyse reactions with no net redox change. Nat Prod Rep. 2002;19:761–772. doi: 10.1039/b108916c. [DOI] [PubMed] [Google Scholar]
- 51.Stankovich MT. In: Chemistry and Biochemistry of Flavoenzymes. Müller F, editor. Vol. 1. CRC Press; Boca Raton, Florida: 1991. pp. 401–425. [Google Scholar]
- 52.Macheroux P, Ghisla S, Sanner C, Ruterjans H, Muller F. Reduced flavin: NMR investigation of N(5)-H exchange mechanism, estimation of ionization constants and assessment of properties as biological catalyst. BMC Biochem. 2005;6:1–11. doi: 10.1186/1471-2091-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoshino T, Tamegai H, Kakinuma K, Eguchi T. Inhibition of type 2 isopentenyl diphosphate isomerase from Methanocaldococcus jannaschii by a mechanism-based inhibitor of type 1 isopentenyl diphosphate isomerase. Bioorg Med Chem. 2006;14:6555–6559. doi: 10.1016/j.bmc.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 54.Kao C-l, Kittleman W, Zhang H, Seto H, Liu H-w. Stereochemical analysis of isopentenyl diphosphate isomerase type II from Staphylococcus aureus using chemically synthesized (S)- and (R)-[2-2H]isopentenyl diphosphates. Org Lett. 2005;7:5677–5680. doi: 10.1021/ol0524050. [DOI] [PubMed] [Google Scholar]
- 55.Maclean J, Ali S. The structure of chorismate synthase reveals a novel flavin binding site fundamental to a unique chemical reaction. Structure. 2003;11:1499–1511. doi: 10.1016/j.str.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 56.Kitzing K, Auweter S, Amrhein N, Macheroux P. Mechanism of chorismate synthase: Role of the two invariant histidine residues in the active site. J Biol Chem. 2004;279:9451–9461. doi: 10.1074/jbc.M312471200. [DOI] [PubMed] [Google Scholar]
- 57.Rauch G, Ehammer H, Bornemann S, Macheroux P. Mutagenic analysis of an invariant aspartate residue in chorismate synthase supports its role as an active site base. Biochemistry. 2007;46:3768–3774. doi: 10.1021/bi602420u. [DOI] [PubMed] [Google Scholar]