

Abstract
The most dangerous and life-threatening manifestation of allergic diseases is anaphylaxis, a condition in which the cardiovascular system is responsible for the majority of clinical symptoms and for potentially fatal outcome. The heart is both a source and a target of chemical mediators released during allergic reactions. Mast cells are abundant in the human heart, where they are located predominantly around the adventitia of large coronary arteries and in close contact with the small intramural vessels. Cardiac mast cells can be activated by a variety of stimuli including allergens, complement factors, general anesthetics and muscle relaxants. Mediators released from immunologically activated human heart mast cells strongly influence ventricular function, cardiac rhythm and coronary artery tone. Histamine, cysteinyl leukotrienes and platelet-activating factor (PAF) exert negative inotropic effects and induce myocardial depression that contribute significantly to the pathogenesis of anaphylactic shock. Moreover, cardiac mast cells release chymase and renin that activates the angiotensin system locally, which further induces arteriolar vasoconstriction. The number and density of cardiac mast cells is increased in patients with ischaemic heart disease and dilated cardiomyopathies. This observation may help explain why these conditions are major risk factors for fatal anaphylaxis. A better understanding of the mechanisms involved in cardiac mast cell activation may lead to an improvement in prevention and treatment of systemic anaphylaxis.
Keywords: anaphylaxis, heart, mast cell, mediators
Introduction
The prevalence of allergic diseases has increased greatly over recent decades. While recent data suggest that the trend of prevalence, at least for asthma, may be reaching a plateau, the number of patients with other allergic disorders such as rhinitis, urticaria and food and drug allergy is expected to continue to rise [1]. Anaphylaxis is the most dramatic clinical presentation of allergy and is frequently a medical emergency in both paediatric and adult patients [2]. Systemic anaphylaxis is a relatively rare occurrence, with a lifetime prevalence estimated at 0·05–2% [3]. According to recent epidemiological data, the rate of fatal anaphylaxis ranges from 2 to 20% of cases [4].
Systemic anaphylaxis is a typical example of cardiovascular involvement in allergic diseases. Cardiac and peripheral vascular symptoms dominate the clinical picture and are often the leading cause of death [5]. While the skin (urticaria and angioedema) and the respiratory tract (laryngeal oedema and bronchospasm) are the main organs involved in the early stages of anaphylaxis, dysfunction of the central and peripheral cardiovascular systems usually dictates the outcome of anaphylactic events [6]. Cardiovascular manifestations of anaphylaxis include hypotension and shock, cardiac arrhythmias, ventricular dysfunction and cardiac arrest [7]. While it was believed generally that coronary arteries were not involved primarily in the haemodynamic derangement associated with anaphylaxis, recent observations indicate that coronary blood flow can be impaired during anaphylaxis and this may contribute significantly to an unfavourable outcome [8,9]. A pre-existing coronary artery disease is now considered to be a negative prognostic factor of anaphylaxis [10]. In addition, acute ischaemic events, including angina and myocardial infarction, are considered currently as part of the clinical picture of anaphylaxis [11]. Major advances have been made in the last few years to understand the pathophysiology of the anaphylactic reaction and to provide better identification of risk factors. This review will focus upon recent clinical and experimental evidence indicating the heart as a central organ of anaphylaxis.
Pathogenesis of anaphylaxis
Most anaphylactic reactions are due to the acute and massive release of mediators from tissue mast cells and blood basophils. In classic anaphylaxis, activation of mast cells and basophils is mediated by the interaction of an allergen with immunoglobulin E (IgE) bound to the high-affinity receptor (FcεRI) expressed on the cell surface [12]. Allergen-induced clustering of IgE generates multiple intracellular signals leading to the release of preformed and de novo synthesized mediators [13]. Anaphylactoid reactions are clinically identical to anaphylactic reactions, the only difference being the absence of demonstrable IgE against the eliciting allergen. In this case, alternative mechanisms (non-IgE-mediated) of mast cell and basophil activation have been proposed. Alternative immunological mechanisms include involvement of immune complexes, immunoglobulins other than IgE (IgG and IgM), platelets, T cells and macrophages [14,15]. Activation of the complement system or coagulation cascade have also been shown to play a role in anaphylactoid reactions [16].
The release of mast cell- and basophil-derived mediators usually occurs within minutes. Both preformed (histamine, tryptase, chymase, carboxypeptidase A) and de novo synthesized lipid mediators [cysteinyl leukotriene C4, prostaglandin D2, and platelet activating factor (PAF)] are secreted very rapidly from mast cells and basophils activated either immunologically or non-immunologically [13,17]. Cytokines such as tumour necrosis factor (TNF)-α, interleukin (IL)-4, IL-6 and IL-13 are released hours after mast cell activation and they are thought to have a role in biphasic or protracted anaphylaxis [18].
The heart as a source of mediators of anaphylaxis
Mast cells are distributed widely within human organs and tissues. These cells are particularly abundant at surfaces in contact with the external environment such as the skin, the lung and the gastrointestinal tract. However, several other tissues contain mast cells, supporting a physiological role for these cells. In the early 1990s the presence and functional activities of human cardiac mast cells were examined extensively. Mast cells in the human heart are located mainly between myocardial fibres, around blood vessels and in the arterial intima [19]. In particular, mast cells are often detected in close contact with small intramural coronary arteries as well as in the wall of large epicardial vessels. Cardiac mast cells have a full capacity to respond to IgE-mediated stimuli releasing large quantities of mediators [12,19]. In addition, cardiac mast cells differ from mast cells isolated from other tissues, such as the lung and the skin, in their ability to be activated by other stimuli such as anaphylatoxins (C3a and C5a), substance P and eosinophilic cationic proteins [19]. Interestingly, these mast cells can also be activated by certain muscle relaxants and radiocontrast media, which may be particularly relevant to anaphylaxis occurring during general anaesthesia or radiodiagnostic procedures [20]. Activated mast cells in the human heart release the full set of mediators released by mast cells from other tissues [21]. However, they release unusually large quantities of chymase compared to lung or skin mast cells [19]. In addition, recent data demonstrate that cardiac mast cells contain and release, upon immunological activation or during myocardial ischaemia, significant quantities of renin [22]. According to these studies, renin released from cardiac mast cells initiates activation of the local (cardiac) renin–angiotensin system (RAS). Angiotensin I, generated by mast cell-derived renin, is then converted to angiotensin II by the interstitial angiotensin-converting enzyme (ACE). It is interesting to note that chymase has a potent ACE activity and is able to convert angiotensin I into angiotensin II even when ACE is blocked by ACE inhibitors [23]. Angiotensin II activates AT1 receptors expressed on sympathetic nerve endings and promotes local secretion of noradrenaline [23]. These observations suggest that mast cells play an important role in the activation of cardiac RAS with the consequent stimulation of the adrenergic system. Adrenergic hyperactivity may be responsible, at least in part, for cardiac arrhythmias, myocardial infarction and sudden cardiac death associated with anaphylaxis.
Studies performed with human hearts obtained from recipients of transplants have shown clearly that cardiac tissue from patients with ischaemic or dilated cardiomyopathies contains a significantly larger number of mast cells compared to hearts from normal individuals who have died accidentally [24]. Consequently, the cardiac tissue from patients with heart diseases contains and releases larger quantities of mast cell-derived mediators upon immunological and non-immunological activation [24]. These observations may contribute to explaining why cardiac manifestations of anaphylaxis are more severe in patients with ischaemic or degenerative heart diseases.
Cardiovascular effects of histamine
Early studies from Endou and Levi [25] showed that large quantities of histamine were released from the heart during anaphylaxis and that this mediator exerted major cardiovascular effects in the guinea pig. Vigorito et al.[26] first showed that intracoronary injection of low doses of histamine in humans induced a rapid decrease in the mean aortic pressure and an increase in coronary blood flow. These effects are due mainly to the activation of H1 receptors expressed on vascular smooth muscle. Furthermore, in healthy individuals, histamine may induce arrhythmias and atrioventricular conduction blocks. In a subset of patients with coronary artery disease, intravenous injection of histamine caused a decrease in coronary blood flow and, in some cases, severe spasm of large coronary arteries [27]. It has been shown recently that histamine induces tissue factor expression and function in human endothelial cells and vascular smooth muscle cells [28]. Tissue factor activates factor X, which leads in turn to thrombin formation. These data indicate that histamine is mainly responsible for hypotension and tachycardia secondary to the release of catecholamines associated with anaphylaxis. However, these observations suggest that histamine may be involved in arrhythmias and ischaemic changes that can also be a cause of death in anaphylaxis.
Cardiovascular effects of eicosanoids
Lipid mediators derived from arachidonic acid are referred to collectively as eicosanoids and include prostaglandins, thromboxanes and leukotrienes. Human heart mast cells are a major source of cysteinyl leukotriene C4 (LTC4) and prostaglandin D2 (PGD2) [19]. Cardiovascular effects of LTC4 and its metabolite LTD4 in man were studied in the late 1980s. Intravenous or intracoronary infusion of LTD4 causes a rapid and sustained increase in coronary vascular resistance [29,30]. This effect of leukotrienes is due mainly to a marked constriction of small intramural coronary vessels [29]. Coronary smooth muscle cells express both the CysLT1 and CysLT2 receptors and it is not known currently whether coronary constriction is mediated by one or both receptors. The cardiovascular effects of PGD2 in humans are not known; however, studies in experimental animals indicate that this eicosanoid induces coronary constriction and arrhythmias in isolated perfused hearts [31].
Thus, current evidence suggest that eicosanoids may influence the contractile function of the heart profoundly by reducing myocardial perfusion. This negative effect may contribute to myocardial depression and impaired ventricular function, which are the main haemodynamic events of anaphylactic shock.
Cardiovascular effects of PAF
PAF was identified initially as a lipid mediator of anaphylaxis which was able to induce aggregation and release of mediators, particularly thromboxanes and serotonin, from human platelets [32,33]. PAF in the heart can be released not only by mast cells but also by basophils, neutrophils, eosinophils and tissue macrophages [33,34]. Studies in experimental animals have demonstrated convincingly that PAF influences several myocardial functions. In particular, PAF causes a severe reduction of coronary blood flow and a significant depression of myocardial contractility [35]. This mediator has a direct arrhythmogenic effect due to its capacity to interact with ionic channels on myocardiocytes [35]. In addition to these haemodynamic effects, PAF promotes the recruitment and activation of neutrophils and eosinophils within the cardiac tissue [36]. More importantly, PAF, generated by mast cells infiltrating the atherosclerotic plaque, may contribute to its instability and rupture by inducing local platelet aggregation and release of lytic enzymes by macrophages [37]. PAF released within systemic circulation induces peripheral vasodilation with relative hypovolaemia and severe hypotension [36]. In addition, PAF-induced platelet activation is a major cause of disseminated intravascular coagulation (DIC), associated frequently with fatal anaphylaxis [38]. Taken together, these observations suggest that PAF is a mediator capable of reproducing most, if not all, haemodynamic dysfunctions of severe anaphylaxis. It is important to note that the majority of cardiovascular effects of PAF are evident at picomolar concentrations, which indicates that the circulating levels of this potentially harmful mediator need to be controlled tightly. Inactivation of PAF is carried out mainly by acetyl hydrolase, an enzyme present in large quantities in plasma and associated with low-density lipoproteins (LDL) [39,40]. It has been shown that genetic deficiency of acetyl hydrolase is associated with severe asthma, early atherosclerosis and susceptibility to septic shock [39]. Interestingly, patients with systemic anaphylaxis have elevated plasma levels of PAF that correlate significantly with the severity of anaphylaxis [41]. Moreover, patients with food-induced fatal anaphylaxis have reduced plasma acetyl hydrolase activity compared to those with non-fatal anaphylaxis or normal individuals [41]. These observations support strongly a primary role of PAF in the cardiovascular manifestations of anaphylaxis and suggest that a deficiency in the enzymatic system involved in PAF degradation may be a risk factor for severe anaphylaxis.
Concluding remarks
Anaphylaxis is a life-threatening event in which prominent cardiovascular dysfunction is caused by mediators released locally from cardiac mast cells (Fig. 1). Both preformed and de novo synthesized mediators concur to produce deleterious effects on heart function that, when added to those exerted on peripheral circulation, result in a rapid and dramatic deterioration of the clinical picture. In the last few years our understanding of cells and mediators involved in anaphylaxis has advanced greatly. An emerging concept is that anaphylaxis and myocardial ischaemia are much more connected than thought previously. Anaphylaxis is more severe and can be more frequently fatal in patients with coronary artery disease for at least three mechanisms: (i) mast cells are more abundant and produce more mediators in hearts with ischaemic cardiomyopathy; (ii) atherosclerotic lesions make coronary arteries more susceptible to the effects of mast cell- and basophil-derived mediators; and (iii) drugs used frequently by patients with ischaemic heart disease, such as beta-blockers and ACE inhibitors, may aggravate symptoms or limit efficacy of treatment of anaphylaxis. On the other hand, anaphylaxis can be an event precipitating an acute myocardial ischaemia. Anaphylactic reactions presenting as acute angina or myocardial infarction are being reported increasingly [11]. The coincidental occurrence of chest pain and allergic reaction, accompanied by the typical electrocardiographic and laboratory findings of myocardial ischaemia, is referred to today as Kounis syndrome and is attributed to an acute coronary spasm or plaque rupture induced by mediators of allergic inflammation [42]. Further studies are necessary to define the mechanisms of allergy-induced myocardial dysfunction, but they will probably provide major improvements in the assessment of risk factors and in the treatment of anaphylaxis.
Fig. 1.
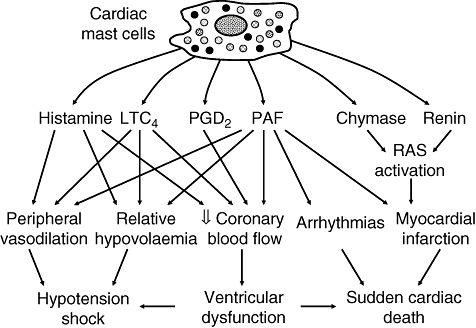
Schematic representation of the cardiovascular effects of mediators released from cardiac mast cells.
Acknowledgments
Part of the work described in this review was supported by grants from the Ministero dell'Istruzione, dell'Università e della Ricerca (M. T., G. M.).
References
- 1.Lawson JA, Senthilselvan A. Asthma epidemiology: has the crisis passed? Curr Opin Pulm Med. 2005;11:79–84. doi: 10.1097/01.mcp.0000147861.60768.24. [DOI] [PubMed] [Google Scholar]
- 2.Simons FE. Anaphylaxis, killer allergy: long-term management in the community. J Allergy Clin Immunol. 2006;117:367–77. doi: 10.1016/j.jaci.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Lieberman P, Camargo CA, Jr, Bohlke K, et al. Epidemiology of anaphylaxis: findings of the American College of Allergy, Asthma and Immunology Epidemiology of Anaphylaxis Working Group. Ann Allergy Asthma Immunol. 2006;97:596–602. doi: 10.1016/S1081-1206(10)61086-1. [DOI] [PubMed] [Google Scholar]
- 4.Pumphrey R. Anaphylaxis: can we tell who is at risk of a fatal reaction? Curr Opin Allergy Clin Immunol. 2004;4:285–90. doi: 10.1097/01.all.0000136762.89313.0b. [DOI] [PubMed] [Google Scholar]
- 5.Low I, Stables S. Anaphylactic deaths in Auckland, New Zealand: a review of coronial autopsies from 1985 to 2005. Path. 2006;38:328–32. doi: 10.1080/00313020600820831. [DOI] [PubMed] [Google Scholar]
- 6.Marone G, Bova M, Detoraki A, Onorati AM, Rossi FW, Spadaro G. The human heart as a shock organ in anaphylaxis. Novartis Found Symp. 2004;257:133–49. discussion 49–60, 276–85. [PubMed] [Google Scholar]
- 7.Golden DB. What is anaphylaxis? Curr Opin Allergy Clin Immunol. 2007;7:331–6. doi: 10.1097/ACI.0b013e3281f8290c. [DOI] [PubMed] [Google Scholar]
- 8.Lombardi A, Vandelli R, Cere E, Di Pasquale G. Silent acute myocardial infarction following a wasp sting. Ital Heart J. 2003;4:638–41. [PubMed] [Google Scholar]
- 9.Wagdi P, Mehan VK, Burgi H, Salzmann C. Acute myocardial infarction after wasp stings in a patient with normal coronary arteries. Am Heart J. 1994;128:820–3. doi: 10.1016/0002-8703(94)90282-8. [DOI] [PubMed] [Google Scholar]
- 10.Simons FE, Frew AJ, Ansotegui IJ, et al. Risk assessment in anaphylaxis: current and future approaches. J Allergy Clin Immunol. 2007;120:S2–24. doi: 10.1016/j.jaci.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Mueller UR. Cardiovascular disease and anaphylaxis. Curr Opin Allergy Clin Immunol. 2007;7:337–41. doi: 10.1097/ACI.0b013e328259c328. [DOI] [PubMed] [Google Scholar]
- 12.Marone G, Patella V, de Crescenzo G, Genovese A, Adt M. Human heart mast cells in anaphylaxis and cardiovascular disease. Int Arch Allergy Immunol. 1995;107:72–5. doi: 10.1159/000236935. [DOI] [PubMed] [Google Scholar]
- 13.Marone G, Triggiani M, de Paulis A. Mast cells and basophils: friends as well as foes in bronchial asthma? Trends Immunol. 2005;26:25–31. doi: 10.1016/j.it.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 14.Kemp SF, Lockey RF. Anaphylaxis: a review of causes and mechanisms. J Allergy Clin Immunol. 2002;110:341–8. doi: 10.1067/mai.2002.126811. [DOI] [PubMed] [Google Scholar]
- 15.Willemse A, Noordzij A, Rutten VP, Bernadina WE. Induction of non-IgE anaphylactic antibodies in dogs. Clin Exp Immunol. 1985;59:351–8. [PMC free article] [PubMed] [Google Scholar]
- 16.Ebo DG, Bosmans JL, Couttenye MM, Stevens WJ. Haemodialysis-associated anaphylactic and anaphylactoid reactions. Allergy. 2006;61:211–20. doi: 10.1111/j.1398-9995.2006.00982.x. [DOI] [PubMed] [Google Scholar]
- 17.Mustafa FB, Ng FS, Nguyen TH, Lim LH. Honeybee venom secretory phospholipase A2 induces leukotriene production but not histamine release from human basophils. Clin Exp Immunol. 2008;151:94–100. doi: 10.1111/j.1365-2249.2007.03542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finkelman FD, Rothenberg ME, Brandt EB, Morris SC, Strait RT. Molecular mechanisms of anaphylaxis: lessons from studies with murine models. J Allergy Clin Immunol. 2005;115:449–57. doi: 10.1016/j.jaci.2004.12.1125. [DOI] [PubMed] [Google Scholar]
- 19.Patella V, Marino I, Lamparter B, Arbustini E, Adt M, Marone G. Human heart mast cells. Isolation, purification, ultrastructure, and immunologic characterization. J Immunol. 1995;154:2855–65. [PubMed] [Google Scholar]
- 20.Stellato C, Casolaro V, Ciccarelli A, Mastronardi P, Mazzarella B, Marone G. General anaesthetics induce only histamine release selectively from human mast cells. Br J Anaesth. 1991;67:751–8. doi: 10.1093/bja/67.6.751. [DOI] [PubMed] [Google Scholar]
- 21.Genovese A, Bouvet JP, Florio G, Lamparter-Schummert B, Bjorck L, Marone G. Bacterial immunoglobulin superantigen proteins A and L activate human heart mast cells by interacting with immunoglobulin E. Infect Immun. 2000;68:5517–24. doi: 10.1128/iai.68.10.5517-5524.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mackins CJ, Kano S, Seyedi N, et al. Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J Clin Invest. 2006;116:1063–70. doi: 10.1172/JCI25713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reid AC, Silver RB, Levi R. Renin: at the heart of the mast cell. Immunol Rev. 2007;217:123–40. doi: 10.1111/j.1600-065X.2007.00514.x. [DOI] [PubMed] [Google Scholar]
- 24.Patella V, Marino I, Arbustini E, et al. Stem cell factor in mast cells and increased mast cell density in idiopathic and ischemic cardiomyopathy. Circulation. 1998;97:971–8. doi: 10.1161/01.cir.97.10.971. [DOI] [PubMed] [Google Scholar]
- 25.Endou M, Levi R. Histamine in the heart. Eur J Clin Invest. 1995;25(Suppl)(1):5–11. [PubMed] [Google Scholar]
- 26.Vigorito C, Giordano A, De Caprio L, et al. Effects of histamine on coronary hemodynamics in humans: role of H1 and H2 receptors. J Am Coll Cardiol. 1987;10:1207–13. doi: 10.1016/s0735-1097(87)80120-1. [DOI] [PubMed] [Google Scholar]
- 27.Vigorito C, Poto S, Picotti GB, Triggiani M, Marone G. Effect of activation of the H1 receptor on coronary hemodynamics in man. Circulation. 1986;73:1175–82. doi: 10.1161/01.cir.73.6.1175. [DOI] [PubMed] [Google Scholar]
- 28.Steffel J, Akhmedov A, Greutert H, Luscher TF, Tanner FC. Histamine induces tissue factor expression: implications for acute coronary syndromes. Circulation. 2005;112:341–9. doi: 10.1161/CIRCULATIONAHA.105.553735. [DOI] [PubMed] [Google Scholar]
- 29.Marone G, Giordano A, Cirillo R, Triggiani M, Vigorito C. Cardiovascular and metabolic effects of peptide leukotrienes in man. Ann NY Acad Sci. 1988;524:321–33. doi: 10.1111/j.1749-6632.1988.tb38555.x. [DOI] [PubMed] [Google Scholar]
- 30.Vigorito C, Giordano A, Cirillo R, Genovese A, Rengo F, Marone G. Metabolic and hemodynamic effects of peptide leukotriene C4 and D4 in man. Int J Clin Lab Res. 1997;27:178–84. doi: 10.1007/BF02912454. [DOI] [PubMed] [Google Scholar]
- 31.Hattori Y, Levi R. Effect of PGD2 on cardiac contractility: a negative inotropism secondary to coronary vasoconstriction conceals a primary positive inotropic action. J Pharmacol Exp Ther. 1986;237:719–24. [PubMed] [Google Scholar]
- 32.Golino P, Ambrosio G, Ragni M, et al. Short-term and long-term role of platelet activating factor as a mediator of in vivo platelet aggregation. Circulation. 1993;88:1205–14. doi: 10.1161/01.cir.88.3.1205. [DOI] [PubMed] [Google Scholar]
- 33.Snyder F. Platelet-activating factor and its analogs: metabolic pathways and related intracellular processes. Biochim Biophys Acta. 1995;1254:231–49. doi: 10.1016/0005-2760(94)00192-2. [DOI] [PubMed] [Google Scholar]
- 34.Triggiani M, Schleimer RP, Warner JA, Chilton FH. Differential synthesis of 1-acyl-2-acetyl-sn-glycero-3-phosphocholine and platelet-activating factor by human inflammatory cells. J Immunol. 1991;147:660–6. [PubMed] [Google Scholar]
- 35.Montrucchio G, Alloatti G, Camussi G. Role of platelet-activating factor in cardiovascular pathophysiology. Physiol Rev. 2000;80:1669–99. doi: 10.1152/physrev.2000.80.4.1669. [DOI] [PubMed] [Google Scholar]
- 36.Braquet P, Touqui L, Shen TY, Vargaftig BB. Perspectives in platelet-activating factor research. Pharmacol Rev. 1987;39:97–145. [PubMed] [Google Scholar]
- 37.Kovanen PT, Kaartinen M, Paavonen T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation. 1995;92:1084–8. doi: 10.1161/01.cir.92.5.1084. [DOI] [PubMed] [Google Scholar]
- 38.Choi IH, Ha TY, Lee DG, et al. Occurrence of disseminated intravascular coagulation (DIC) in active systemic anaphylaxis: role of platelet-activating factor. Clin Exp Immunol. 1995;100:390–4. doi: 10.1111/j.1365-2249.1995.tb03711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karasawa K. Clinical aspects of plasma platelet-activating factor-acetylhydrolase. Biochim Biophys Acta. 2006;1761:1359–72. doi: 10.1016/j.bbalip.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 40.Triggiani M, Chilton FH. Metabolism of platelet-activating factor in the guinea-pig heart. J Mol Cell Cardiol. 1992;24:1101–11. doi: 10.1016/0022-2828(92)93175-j. [DOI] [PubMed] [Google Scholar]
- 41.Vadas P, Gold M, Perelman B, et al. Platelet-activating factor, PAF acetylhydrolase, and severe anaphylaxis. N Engl J Med. 2008;358:28–35. doi: 10.1056/NEJMoa070030. [DOI] [PubMed] [Google Scholar]
- 42.Kounis NG. Kounis syndrome (allergic angina and allergic myocardial infarction): a natural paradigm? Int J Cardiol. 2006;110:7–14. doi: 10.1016/j.ijcard.2005.08.007. [DOI] [PubMed] [Google Scholar]