

Abstract
Among the ‘allergic’ conditions involving the lung, asthma is the more frequent and the most extensively investigated, although asthma itself may be caused by different disorders. The triggering event in allergic subjects is the reaction allergen-specific immunoglobulin E (IgE) that activates mast cells and initiates a complex and redundant inflammatory process, where cells, cytokines and adhesion molecules are involved at different stages. In fact, mucosal eosinophilic inflammation is one of the distinctive features of asthma and the particular T helper type 2 (Th2) phenotype of allergic patients favours it. In general, the clinical severity of asthma correlates well with the degree of inflammation. None the less, other phenomena such as non-specific bronchial hyperresponsiveness and remodelling intervene in the pathophysiology of allergic asthma. These phenomena are only partially inflammation-related. In particular, the remodelling of the bronchial wall seems to start very early in life and also seems to be a distinctive histological feature of the asthmatic bronchus. The recent introduction of biological treatments (monoclonal antibodies) has allowed elucidation of some of the pathogenic features of allergic asthma.
Keywords: allergic asthma, bronchial hyperresponsiveness, inflammation, remodelling
General aspects
There are numerous diseases involving the lung, which for historical and cultural reasons are termed ‘allergic’, such as ‘allergic granulomatous’ (Churg–Strauss syndrome) or ‘allergic intrinsic alveolitis’ (hypersensitivity pneumonia) [1]. None the less, in such diseases, immunoglobulins (IgE) are not at all, or are not the only, triggering factors. In the case of ‘allergic bronchopulmonary aspergyllosis’, IgE intervene only in the asthmatic response to Aspergyllus, whereas disease due to the proliferation of fungi in the bronchial tree has different pathogenic mechanisms.
Therefore, we will focus our discussion on allergic asthma (AA), where the initiating condition is the presence of specific IgE towards inhalant allergens, which are bound to the surface of mucosal mast cells. Of note, AA is probably the more extensively studied condition, due to its high prevalence [2] and the well-reproducible pathogenic mechanisms, allowing provocation of the disease in controlled conditions (i.e. allergen-specific bronchial challenge). AA, together with other forms of asthma (aspirin- and exercise-induced), is defined as a chronic inflammatory disorder of the bronchi [3], characterized by attacks of bronchospasm that revert spontaneously or after bronchodilators. Additional features of asthma are the presence of a non-specific bronchial responsiveness (BHR) [4], the remodelling of the bronchial wall [5] and, possibly, the progressive decline of respiratory function [6]. The characteristics (abrupt onset of wheezing, chest tightness, cough, nocturnal awakenings) make the diagnosis of asthma easy to conduct in a clinical setting. The severity of an asthma attack may vary from cough (cough-variant asthma) to life-threatening attacks, with respiratory failure and even respiratory arrest. A pulmonary function test confirms the diagnosis, showing a bronchial obstruction that is reversible after the administration of a short-acting bronchodilator. This aspect differentiates asthma from chronic obstructive pulmonary disease (COPD), where the bronchial obstruction is not fully reversible [7]. Because, between attacks, the pulmonary function may be within the normal range, the presence of BHR can be revealed by means of the bronchial provocation test with histamine, methacholine or adenosine [8]. The methacholine test, in particular, has a good negative predictive value. The management of asthma in the long term is well standardized across guidelines and involves the step-up or step-down aproach with different drugs (preferably inhaled), according to the frequency and severity of symptoms and the level of control [3]. Certainly, inhaled corticosteroids still represent the cornerstone in the long-term treatment of asthma.
Pathophysiological aspects of allergic asthma
As mentioned previously and simplified in Fig. 1, in AA the triggering event is the contact of allergens with the specific IgE that are bound to the mast-cell surface [9]. Of note, the bronchial mucosa is rich in mast cells per se, as happens with the skin and the gut [10]. The term ‘aeroallergens’ (inhalant allergens) encompasses a wide variety of proteins that derive from different sources such as pollens (trees, grasses and weeds), dust mites, urine or saliva from pets, occupational substances and, rarely, foods. The capacity of aeroallergens to reach the bronchial tree depends upon the size of the carrying particles, and increases as the aerodynamic mass decreases. In fact, pollen grains have a large size and are retained efficiently by the nasal filter, so that they usually cause rhinitis, and provoke asthma when their concentration is very high.
Fig. 1.
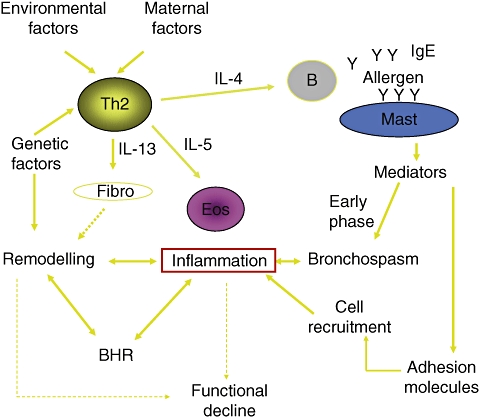
Simplified view of the pathogenic mechanisms of allergic asthma.
Once the allergen is bound to at least two contiguous molecules of specific IgE, the mast cell is activated and immediately releases the substances stored in their granules [11]. Among these mediators histamine is the most important, as via the H1 receptor it causes all the clinical manifestations of the so-called ‘early phase’. These manifestations include increased mucus secretion, vasodilation, stimulation of the nerve ends and bronchospasm. All these phenomena can be reproduced easily by means of the allergen-specific bronchial provocation test. In addition to the release of preformed mediators, activated mast cells also start to synthesize other inflammatory mediators and cytokines [e.g. leukotrienes, interleukin (IL-4) and IL-5]. One of the most prominent effects of the proinflammatory substances is activation of the adhesion machinery [12,13]. This complex system of molecules favours the margination and extravasation of other inflammatory cells, including eosinophils, neutrophils and lymphocytes, so that a mucosal inflammatory infiltration is established. At this point, we should consider that allergic subjects possess a special subset of T lymphocytes, known as T helper type 2 (Th2). These Th2 cells, at variance with the ‘normal’ Th1 cells, secrete high amounts of IL-4, IL-5 and IL-13 [14,15]. These cytokines favour IgE synthesis [16], eosinophil activation and survival [17], and the activation of fibroblasts [18], respectively. The existence of the Th2 subset explains, on one hand, why the allergic subject produces abnormal amounts of specific IgE and, on the other hand, why the mucosal inflammation, once established, can be maintained. In persistent (chronic) asthma, the bronchial eosinophilic inflammation, related to the Th2 microenvironment, is always present and it is, at least in part, independent of the IgE-mediated mechanism. This is confirmed indirectly by the observation that in AA the clinical effect of the specific anti-IgE monoclonal antibody is only partial [19]. Of note, it has been shown consistently that the severity of asthma correlates well with the degree of mucosal inflammation [20], but the direct pathogenic role of eosinophils in asthmatic inflammation has been questioned recently [21], based on the observation that a selective blockage of IL-5 reduces the number of eosinophils in sputum but does not affect the clinical severity of asthma [22]. Indeed, basic and clinical studies have shown that also neutrophils may be involved in asthmatic inflammation [23].
Even more complex is the aspect of non-specific BHR, which is a characteristic feature of asthma. Indeed, it was reasonably believed that BHR was a direct consequence of persistent inflammation of the mucosa [3], but some studies failed to identify a direct correlation between mucosal inflammation and the degree of BHR [24,25]. Conversely, it was shown that the bronchial smooth muscle, if sensitized passively with an IgE-containing serum, increased its rapidity and strength of contraction [26]. In this regard, it was hypothesized that the presence of IgE themselves is partly responsible for the observed BHR. Finally, inhaled corticosteroids control BHR [27], but it is the last to disappear during the treatment, which rapidly controls symptoms and improves pulmonary function [3]. So far, the only effective way to abolish BHR seems to be the physical ablation of the smooth muscle by means of endoscopically applied radiofrequency (bronchial thermoplasty[28]).
Another feature of asthma is the presence of a ‘remodelling’ of the bronchial wall. Remodelling is a series of fine architectural changes of the bronchial wall that involve epithelial disruption, basal membrane thickening, collagen deposition, muscle hyperthrophy and increased vascularization [29]. These features are found specifically in asthma, whereas they are less pronounced in COPD and rhinitis [30,31], which are also chronic inflammatory conditions. Among the mechanisms proposed to explain the remodelling, one of the more interesting is an impairment of the activity of metalloproteinases [32], which in normal conditions control the degradation of subepithelial collagen. Also, in this case it was believed that the remodelling was the result of chronic inflammation, but recent studies with bronchial biopsies have demonstrated clearly that the remodelling process is partially independent of inflammation and may be a constitutive characteristic of the asthmatic bronchus [33–35]. According to more recent epidemiological/pharmacological studies, the remodelling process seems to start early in childhood [36]. For this reason, it has been hypothesized that it may be responsible for the slow but progressive functional decline that is seen also in asthmatic subjects.
Allergic asthma and allergic rhinitis
It is well known that asthma and rhinitis often co-exist, expecially in the case of allergic subjects, so that the term ‘united airways disease’ has been introduced [37]. Several cross-sectional studies have shown clearly the association between rhinitis and asthma: up to 50% of patients with rhinitis have asthma, and rhinitis occurs in up to 80% of patients with asthma [38,39]. This is not surprising, as the respiratory tract is a single morphofunctional entity. It is entirely covered to the smaller bronchi by ciliate epithelium and mucinous glands and is served by an extensive vasculature and innervation. From a pathophysiological viewpoint, the bronchial mucosal inflammation does not differ between asthmatics and rhinitics, as confirmed by segmental bronchial challenge: in pure rhinitics and asthmatics there was no difference in eosinophil count, IL-5 and IL-10 generation in the bronchoalveolar lavage fluid [40]. Moreover, it is known that nasal challenge with allergen may result in increased BHR [41]. What is more surprising is that the segmental bronchial challenge also results in nasal inflammation [42]. Thus it has been hypothesized that the nose bronchi link is bidirectional and also mediated by humoural mechanims. In addition, some observations have suggested that appropriate treatment of rhinitis also has a beneficial effect on concomitant asthma [43–45]. In short, in the case of allergic subjects, asthma and rhinitis can be considered not as different diseases, but as two clinical aspects of a unitary disease of the airways.
Indeed, this hypothesis does not explain some challenging observations. First, the prevalence of asthma is also higher in rhinitis subjects in the absence of allergy [46]. Secondly, the remodelling of asthma is not present or negligible in rhinitis, although the lining mucosa is identical [30,31].
Conclusions
The IgE-mediated reaction that occurs in AA is a well-known pathogenic model. In the allergic subject, who produces an increased amount of specific IgE towards inhalatory proteins, the IgE reaction triggers a complex network of molecular and cellular events which results in mucosal inflammation. The allergic phenotype is characterized by a particular T lymphocyte response that, in turn, favours the inflammatory process. In AA, the inflammation is predominantly eosinophilic in its nature. Once the mucosal inflammation is established, the role of the IgE reaction becomes less important. The inflammatory process is certainly central in asthma, as testified by the functional and clinical efficacy of inhaled corticosteroids, but it does not explain some aspects fully, such as bronchial hyperreactivity and the presence of remodelling since the early years of life.
Finally, it should be remembered that allergy is only one of the possible mechanisms, and other forms of asthma that are clinically undistinguishable from the allergic form exist. This is the reason why, in the last decade, the concept of ‘asthma penotypes’ has emerged [47,48]. This new concept, on one hand, has provided a useful clinical tool and, on the other hand, has highlighted the great heterogeneity of the disease (Fig. 2) and our still incomplete knowledge of the pathogenic mechanisms.
Fig. 2.
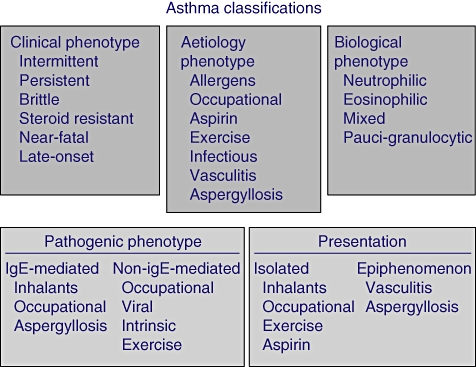
Possible classifications of asthma according to phenotypes.
References
- 1.Mcsharry C, Anderson K, Bourke SJ, Boyd G. Takes your breath away – the immunology of allergic alveolitis. Clin Exp Immunol. 2002;128:3–9. doi: 10.1046/j.1365-2249.2002.01849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;59:469–78. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- 3.Global strategy for the prevention and management of asthma. [February 2nd 2008]. GINA report 2007. Available at: http://www.ginasthma.org.
- 4.Brusasco V, Pellegrino R. Complexity of factors modulating airway narrowing in vivo: relevance to assessment of airway hyperresponsiveness. J Appl Physiol. 2003;95:1305–13. doi: 10.1152/japplphysiol.00001.2003. [DOI] [PubMed] [Google Scholar]
- 5.James A. Airway remodeling in asthma. Curr Opin Pulm Med. 2005;11:1–6. doi: 10.1097/01.mcp.0000146779.26339.d8. [DOI] [PubMed] [Google Scholar]
- 6.Sears MA. Lung function decline in asthma. Eur Respir J. 2007;30:411–13. doi: 10.1183/09031936.00080007. [DOI] [PubMed] [Google Scholar]
- 7.Mauad T, Dolhnikoff M. Pathologic similarities and differences between asthma and chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2008;14:31–8. doi: 10.1097/MCP.0b013e3282f19846. [DOI] [PubMed] [Google Scholar]
- 8.van Schoor J, Joos GF, Pauwels RA. Indirect bronchial hyperresponsiveness in asthma: mechanisms, pharmacology and implications for clinical research. Eur Respir J. 2000;16:514–33. doi: 10.1034/j.1399-3003.2000.016003514.x. [DOI] [PubMed] [Google Scholar]
- 9.Pawankar R, Yamagishi S, Takizawa R, Mast YAGI T. cell-IgE-and mast cell–structural cell interactions in allergic airway disease. Curr Drug Targets Inflamm Allergy. 2003;2:303–12. doi: 10.2174/1568010033484016. [DOI] [PubMed] [Google Scholar]
- 10.Okuda M. Functional heterogeneity of airways mast cells. Allergy. 1999;54(Suppl)(57):50–62. doi: 10.1111/j.1398-9995.1999.tb04406.x. [DOI] [PubMed] [Google Scholar]
- 11.Kambayashi T, Koretzky GA. Proximal signaling events in Fc epsilon RI-mediated mast cell activation. J Allergy Clin Immunol. 2007;119:544–52. doi: 10.1016/j.jaci.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 12.Lampinen M, Carlson M, Hakansson LD, Venge P. Cytokine-regulated accumulation of eosinophils in inflammatory disease. Allergy. 2004;59:793–805. doi: 10.1111/j.1398-9995.2004.00469.x. [DOI] [PubMed] [Google Scholar]
- 13.Kelly M, Hwang JM, Kubes P. Modulating leukocyte recruitment in inflammation. J Allergy Clin Immunol. 2007;120:3–10. doi: 10.1016/j.jaci.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 14.Romagnani S. Immunologic influences on allergy and the TH1/TH2 balance. J Allergy Clin Immunol. 2004;113:395–400. doi: 10.1016/j.jaci.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 15.Romagnani S. Regulation of the T cell response. Clin Exp Allergy. 2006;36:1357–66. doi: 10.1111/j.1365-2222.2006.02606.x. [DOI] [PubMed] [Google Scholar]
- 16.Coffman RL, Ohara J, Bond MW, Carty J, Zlotnik A, Paul WE. B cell stimulatory factor-1 enhances the IgE response of lipopolysaccharide-activated B cells. J Immunol. 1986;136:4538–41. [PubMed] [Google Scholar]
- 17.Wardlaw A. Molecular basis for selective eosinophil trafficking in asthma: a multistep paradigm. J Allergy Clin Immunol. 1999;104:917–26. doi: 10.1016/s0091-6749(99)70069-2. [DOI] [PubMed] [Google Scholar]
- 18.Hershey GK. IL-13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol. 2003;111:677–90. doi: 10.1067/mai.2003.1333. [DOI] [PubMed] [Google Scholar]
- 19.Milgrom H, Fick RB, Jr, Su JQ, et al. Treatment of allergic asthma with monoclonal anti-IgE antibody. rhuMAb-E25 Study Group. N Engl J Med. 1999;341:1966–73. doi: 10.1056/NEJM199912233412603. [DOI] [PubMed] [Google Scholar]
- 20.Louis R, Lau LC, Bron AO, Roldaan AC, Radermecker M, Djukanovic R. The relationship between airways inflammation and asthma severity. Am J Respir Crit Care Med. 2000;161:9–16. doi: 10.1164/ajrccm.161.1.9802048. [DOI] [PubMed] [Google Scholar]
- 21.O'Byrne PM, Inman MD, Parameswaran K. The trials and tribulations of IL-5, eosinophils, and allergic asthma. J Allergy Clin Immunol. 2001;108:503–8. doi: 10.1067/mai.2001.119149. [DOI] [PubMed] [Google Scholar]
- 22.Leckie MJ, ten Brinke A, Khan J, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2144–8. doi: 10.1016/s0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 23.Yoshihara S, Yamada Y, Abe T, Lindén A, Arisaka O. Association of epithelial damage and signs of neutrophil mobilization in the airways during acute exacerbations of paediatric asthma. Clin Exp Immunol. 2006;144:212–16. doi: 10.1111/j.1365-2249.2006.03058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foresi A, Bertorelli G, Pesci A, et al. Inflammatory markers in bronchoalveolar lavage and in bronchial biopsy in asthma during remission. Chest. 1990;98:528–35. doi: 10.1378/chest.98.3.528. [DOI] [PubMed] [Google Scholar]
- 25.Crimi E, Spanevello A, Neri M, et al. Dissociation between airway inflammation and airway hyperresponsiveness in allergic asthma. Am J Respir Crit Care Med. 1998;157:4–9. doi: 10.1164/ajrccm.157.1.9703002. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell RW, Ruhlmann H, Magnussen H, et al. Passive sensitization of human bronchi augments smooth muscle shortening velocity and capacity. Am J Physiol (Lung Cell Mol Physiol) 1994;267:218–22. doi: 10.1152/ajplung.1994.267.2.L218. [DOI] [PubMed] [Google Scholar]
- 27.Cockroft DW. Mechanisms of airway hyperresponsiveness. J Allergy Clin Immunol. 2006;118:551–9. doi: 10.1016/j.jaci.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 28.Cox G, Thomson NC, Rubin AS, et al. AIR Trial Study Group. Asthma control during the year after bronchial thermoplasty. N Engl J Med. 2007;356:1327–37. doi: 10.1056/NEJMoa064707. [DOI] [PubMed] [Google Scholar]
- 29.Chiappara G, Gagliardo R, Siena A, et al. Airway remodelling in the pathogenesis of asthma. Curr Opin Allergy Clin Immunol. 2001;1:85–93. doi: 10.1097/01.all.0000010990.97765.a1. [DOI] [PubMed] [Google Scholar]
- 30.Vignola AM, Paganin F, Capieu L, et al. Airway remodelling assessed by sputum and high-resolution computed tomography in asthma and COPD. Eur Respir J. 2004;24:910–7. doi: 10.1183/09031936.04.00032603. [DOI] [PubMed] [Google Scholar]
- 31.Salib RJ, Howarth PH. Remodelling of the upper airways in allergic rhinitis: is it a feature of the disease? Clin Exp Allergy. 2003;33:1629–33. doi: 10.1111/j.1365-2222.2003.01828.x. [DOI] [PubMed] [Google Scholar]
- 32.Cataldo DD, Gueders MM, Rocks N, et al. Pathogenic role of matrix metalloproteases and their inhibitors in asthma and chronic obstructive pulmonary disease and therapeutic relevance of matrix metalloproteases inhibitors. Cell Mol Biol. 2003;49:875–84. [PubMed] [Google Scholar]
- 33.Payne DN, Rogers AV, Adelroth E, et al. Early thickening of the reticular basement membrane in children with difficult asthma. Am J Respir Crit Care Med. 2003;167:78–82. doi: 10.1164/rccm.200205-414OC. [DOI] [PubMed] [Google Scholar]
- 34.Zagai U, Sköld CM, Trulson A, Venge P, Lundahl J. The effect of eosinophils on collagen gel contraction and implications for tissue remodelling. Clin Exp Immunol. 2004;135:427–33. doi: 10.1111/j.1365-2249.2004.02396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baldwin L, Roche WR. Does remodelling of the airway wall precede asthma? Paediatr Respir Rev. 2002;3:315–20. doi: 10.1016/s1526054202002610. [DOI] [PubMed] [Google Scholar]
- 36.Canonica GW, Rossi GA, Baena Cagnani CE. Airway remodelling in children: when does it start? Curr Opin Allergy Clin Immunol. 2007;7:196–200. doi: 10.1097/ACI.0b013e328082559a. [DOI] [PubMed] [Google Scholar]
- 37.Passalacqua G, Ciprandi G, Canonica GW. United airways disease: therapeutic aspects. Thorax. 2000;55(Suppl)(2):S26–7. doi: 10.1136/thorax.55.suppl_2.S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bousquet J, Vignola AM, Demoly P. Links between rhinitis and asthma. Allergy. 2003;58:691–706. doi: 10.1034/j.1398-9995.2003.00105.x. [DOI] [PubMed] [Google Scholar]
- 39.Annaesi-Maesano I. Epidemiological evidence of the occurrence of rhinitis and sinusitis in asthma. Allergy. 1999;54(Suppl)(57):7–13. doi: 10.1111/j.1398-9995.1999.tb04401.x. [DOI] [PubMed] [Google Scholar]
- 40.Crimi E, Milanese M, Oddera S, et al. Inflammatory and mechanical factors of allergen-induced bronchoconstriction in mild asthma and rhinitis. J Appl Physiol. 2001;91:1029–34. doi: 10.1152/jappl.2001.91.3.1029. [DOI] [PubMed] [Google Scholar]
- 41.Corren J, Adinoff A, Irvin C. Changes in bronchial responsiveness following nasal provocation with allergens. J Allergy Clin Immunol. 1992;89:611–18. doi: 10.1016/0091-6749(92)90329-z. [DOI] [PubMed] [Google Scholar]
- 42.Becky Kelly EA, Busse WW, Jarjour NN. A comparison of the airway response to segmental antigen bronchoprovocation in atopic asthma and allergic rhinitis. J Allergy Clin Immunol. 2003;111:79–86. doi: 10.1067/mai.2003.28. [DOI] [PubMed] [Google Scholar]
- 43.Crystal-Peters J, Neslusan C, Crown WH, Torres A. Treating allergic rhinitis in patients with comorbid asthma: the risk of asthma-related hospitalizations and emergency department visits. J Allergy Clin Immunol. 2002;109:57–62. doi: 10.1067/mai.2002.120554. [DOI] [PubMed] [Google Scholar]
- 44.Ciprandi G, Tosca MA, Passalacqua G, Canonica GW. Long-term cetirizine treatment reduces allergic symptoms and drug prescriptions in children with mite allergy. Ann Allergy Asthma Immunol. 2001;87:222–6. doi: 10.1016/S1081-1206(10)62230-2. [DOI] [PubMed] [Google Scholar]
- 45.Pasquali M, Baiardini I, Rogkakou A, et al. Levocetirizine in persistent allergic rhinitis and asthma: effects on symptoms, qualityof life and inflammatory parameters. Clin Exp Allergy. 2006;36:1161–7. doi: 10.1111/j.1365-2222.2006.02548.x. [DOI] [PubMed] [Google Scholar]
- 46.Leynaert B, Bousquet J, Neukirch C, et al. Perennial rhinitis: an independent risk factor for asthma in nonatopic subjects. J Allergy Clin Immunol. 1999;104:301–4. doi: 10.1016/s0091-6749(99)70370-2. [DOI] [PubMed] [Google Scholar]
- 47.Haldor P, Pavord ID. Noneosinophilic asthma: a distinct clinical and pathologic phenotype. J Allergy Clin Immunol. 2007;119:1043–52. doi: 10.1016/j.jaci.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 48.Green RH, Brightling CE, Bradding P. The reclassification of asthma based on subphenotypes. Curr Opin Allergy Clin Immunol. 2007;7:43–50. doi: 10.1097/ACI.0b013e3280118a32. [DOI] [PubMed] [Google Scholar]