

Abstract
klotho is an aging suppressor gene and extends life span when overexpressed in mice. Klotho protein was recently demonstrated to function as a hormone that inhibits insulin/insulin-like growth factor-1 (IGF-1) signaling. Here we show that Klotho protein increases resistance to oxidative stress at the cellular and organismal level in mammals. Klotho protein activates the FoxO forkhead transcription factors that are negatively regulated by insulin/IGF-1 signaling, thereby inducing expression of manganese superoxide dismutase. This in turn facilitates removal of reactive oxygen species and confers oxidative stress resistance. Thus, Klotho-induced inhibition of insulin/IGF-1 signaling is associated with increased resistance to oxidative stress, which potentially contributes to the anti-aging properties of klotho.
A defect in klotho gene expression in mice leads to a syndrome closely resembling human aging, including shortened life span, infertility, growth arrest, hypoactivity, skin atrophy, premature thymic involution, arteriosclerosis, osteoporosis, and pulmonary emphysema (1). Conversely, overexpression of the klotho gene extends the life span in the mouse (2), indicating that the klotho gene functions as an aging suppressor gene in mammals that delays aging when overexpressed and accelerates the development of aging-like disorders when disrupted. The klotho gene encodes a single-pass transmembrane protein and is expressed only in limited tissues, notably the distal convoluted tubules in the kidney and the choroid plexus in the brain (1). The extracellular domain of Klotho protein is shed and secreted into the blood. It then binds to a high affinity but as yet unidentified cell-surface Klotho receptor and signals suppression of tyrosine phosphorylation of insulin/IGF-12 receptors and insulin receptor substrates (IRS), association of IRS with phosphatidylinositol 3-kinase (PI3K), and serine phosphorylation of Akt/PKB (2). Thus, Klotho protein is a hormone that inhibits the intracellular insulin/IGF-1 signaling cascade. This activity likely contributes to the suppression of aging by Klotho, because inhibition of insulin-like signaling is an evolutionarily conserved mechanism for extending life span (see Ref. 3 for review). Indeed, we observed alleviation of aging-like phenotypes in Klotho deficient mice (KL−/− mice) by genetic perturbation of insulin/IGF-1 signaling, indicating that the activity of Klotho that inhibits insulin/IGF-1 signaling accounts for its anti-aging properties, at least in part (2). It remains to be determined whether or not the inhibitory effect of Klotho protein on IRS, PI3K, and Akt is entirely dependent on its ability to inhibit insulin/IGF-1 receptors. Notably, single nucleotide polymorphisms in the human KLOTHO gene are associated with longevity (4) and common age-related diseases including coronary artery disease (5), osteoporosis (6-8), and stroke (9), strongly arguing that Klotho regulates aging in humans.
Increased resistance to oxidative stress is associated with increased longevity in various species (see Ref. 10 for review). In the current study, we show that the anti-aging hormone Klotho has an activity to increase resistance to oxidative stress both in vitro and in vivo.
MATERIALS AND METHODS
Production and Purification of Recombinant Klotho Protein
A soluble form of recombinant Klotho protein with the V5 epitope tag was generated using a Drosophila Expression System (Invitrogen) and purified using Sepharose conjugated with anti-V5 antibody (Sigma) as described previously (2).
Measurement of Lipid Oxidation
HeLa cells grown on glass coverslips were incubated with or without 200 pm Klotho protein for 16 h in the presence or absence of 100 μm paraquat. A fluorescence probe for lipid oxidation (C11-BODIPY581/591, Molecular Probes) was added 30 min before the fluorescence microscopy imaging. The fluorescence excitation/emission of the C11-BODIPY581/591 shifts from 581/591 nm to 490/510 nm upon oxidation. Cellular images were captured at excitation/emission = 580/600 nm (the non-oxidized form, red fluorescence) and at excitation/emission = 490/510 nm (the oxidized form, green fluorescence) and then merged to demonstrate the fraction of the oxidized C11-BODIPY581/591.
Flow Cytometry
CHO or HeLa cells were incubated in serum-free DMEM with or without 200 pm Klotho protein for 16 h in the presence of 100 μm paraquat. The cells were harvested by trypsin treatment, stained with both annexin V-FITC and propidium iodide according to the manufacturer's protocol (Pharmingen) and analyzed by flow cytometry (FACStar, BD Biosciences) set for FL1 (annexin V) and FL2 (propidium iodide). A total of 20,000 cells were counted for each sample.
Mice
Generation of KL−/− mice and transgenic mice that overexpress Klotho (EFmKL48) was described previously (1). All animal experiments were approved by the Institutional Animal Care and Research Advisory Committee of the University of Texas Southwestern Medical Center at Dallas.
Insulin Stimulation of Mice
KL−/−, wild-type, and EFmKL48 male mice at 8 weeks of age were administered with insulin (Humulin R, Lilly, 0.2 unit/g of body weight) or saline by intravenous injection via the inferior vena cava. Hind limb muscles were excised 5 min after the injection, frozen immediately in liquid nitrogen, and stored at −80 °C until used for the immunoblot and the protein microarray analysis.
Measurement of Urinary Creatinine and 8-Hydroxydeoxyguanosine (8-OHdG)
Urinary 8-OHdG and creatinine were measured in four 16-week-old male wild-type and EFmKL48 mice using DNA Damage ELISA Kit (Stressgen) and creatinine assay kit (Cayman Chemicals), respectively.
Paraquat Tolerance Test
Twelve 16-week-old male wild-type mice and EFmKL48 mice were administered 75 mg/kg paraquat (methyl viologen, Sigma) by intraperitoneal injection. The mice were checked for survival every 12 h and censored at 10 days.
Plasmids
The plasmid constructs encoding mouse FOXO1, FOXO3a, and FOXO4 were constructed by subcloning PCR-amplified cDNA fragments into the pcDNA3.1/myc-His mammalian expression vector (Invitrogen). The FHRE-Luc reporter (a firefly luciferase reporter construct under the control of a portion of the Fas ligand promoter) was a kind gift from Dr. Michael E. Greenberg (Harvard Medical School). The SOD2-Luc reporter (pSODLUC-3340, a firefly luciferase reporter construct under the control of the human SOD2 promoter) was a kind gift from Dr. Boudewijin M. T. Burgering (University Medical Center Utrechit).
Cell Culture and Transfection
COS cells, HeLa cells, and CHO cells were maintained in DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin. Subconfluent cells were transfected with the plasmids using the FuGENE 6 transfection reagent (Roche Applied Science) according to the manufacturer's protocol.
Immunoblot Analysis
The cells treated as indicated were snap-frozen in liquid nitrogen and lysed in a lysis buffer containing inhibitors for phosphatase and proteinase as previously described (2). The mouse tissue lysates were prepared by homogenizing the frozen muscles in the same lysis buffer. The cell and tissue lysates were subjected to SDS-PAGE and transferred to Hybond C Extra membrane (Amersham Biosciences). The membrane was incubated with anti-Akt antibody (Cell Signaling), anti-phospho-Akt antibody (Cell Signaling), anti-myc antibody (Santa Cruz), anti-FOXO1 antibody (Santa Cruz), anti-phospho-FOXO antibody that recognizes all three FOXOs phosphorylated at the three conserved serine/threonine residues (Cell Signaling), anti-SOD2 antibody (Upstate Biotechnology), or anti-actin antibody (Chemicon) and then with horseradish peroxidase-linked secondary antibodies (Amersham Biosciences). The signals were detected with SuperSignal West Dura system (Pierce).
Preparation of Cytosolic and Nuclear Fractions
HeLa cells treated with Klotho protein as indicated were lysed in low salt lysis buffer (same as the lysis buffer except NaCl was reduced to 10 mm) and centrifuged at 850 × g for 10 min. The supernatants were used as the cytoplasmic fractions. The pellets were resuspended in high salt lysis buffer (same as the lysis buffer except NaCl was increased to 300 mm) and rotated for 1 h at 4 °C and centrifuged at 18,300×g. The supernatants were used as the nuclear fractions.
Immunocytochemistry
HeLa cells transfected with the FOXO1-myc expression vector were seeded on glass coverslips. The coverslips were transferred to serum-free DMEM and then incubated with or without 400 pm Klotho protein for 1 h. The cells were fixed with 4% formaldehyde/phosphate-buffered saline for 15 min at room temperature. The fixed cells were incubated with the anti-myc antibody for 1 h and then with anti-mouse IgG antibody conjugated with FITC for 1 h at room temperature. Subcellular localization of the FOXOs was determined by fluorescence microscopy.
Luciferase Assay
HeLa cells were transfected with FHRE-Luc or SOD2-Luc together with pRL-CMV (a Renilla luciferase reporter construct under the control of the CMV promoter, Promega). The transfected cells were treated with or without 400 pm Klotho protein for 16 h in the serum-free DMEM. The cell lysates were subjected to the luciferase assay using the dual-luciferase reporter assay system (Promega). The firefly luciferase activity was normalized relative to the Renilla luciferase activity.
Northern Blot Analysis
A cDNA fragment encoding mouse SOD2 was PCR-amplified from mouse liver total RNA by using SuperScript II one-step RT-PCR system (Invitrogen) and specific primers (5′-CCGAATTCCAGCGGTCGTGTAAACCTCA-3′ and 5′-CCAAGCTTGTGAGGTTTCACTTCTTGCA-3′). The amplified fragment was subcloned into the EcoRI-HindIII site in pBluscript II SK(−) vector (Strata-gene). The SOD2 cDNA probe was labeled with [α-32P]dCTP with the Megaprime DNA labeling system (Amersham Biosciences). Total RNA was prepared from hind limb muscles of four 8-week-old male KL−/−, wild-type, and EFmKL48 mice by using TRIzol reagent (Invitrogen). Aliquotes of total RNA (5 μg) from the four mice with the identical genotype were pooled (total 20 μg), subjected to 1% formaldehyde-agarose gel electrophoresis, and transferred to Hybond N+ membrane (Amersham Biosciences). The membrane was hybridized with the labeled SOD2 cDNA probe in Rapid-hyb buffer (Amersham Biosciences). The membrane was re-probed with a probe for actin mRNA for a loading control.
Chromatin Immunoprecipitation Assay
HeLa cells were transfected with the FOXO1-myc expression vector and incubated in serum-free DMEM overnight. The cells were then incubated with or without 400 pm Klotho protein for 1 h in the presence or absence of wortmannin (50 nm). The chromatin immunoprecipitation assay was performed essentially according to the method previously reported (11). After cross-linking with 1% formaldehyde/phosphate-buffered saline at 37 °C for 10 min, the cells were lysed in SDS lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris-HCl, pH 8.1, and protease inhibitors), sonicated to shear genomic DNA to an average size of ∼500 bp and then diluted in dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 167 mm NaCl, 16.7 mm Tris-HCl, pH 8.1, protease inhibitors). After removing a part for evaluation of input DNA, the lysates were precleared with sheared salmon sperm DNA, normal mouse IgG, and protein G-Sepharose (Amersham Biosciences) for 2 h at 4°C. The precleared lysates were immunoprecipitated with anti-myc antibody (Covance) overnight and then with protein G-Sepharose for 1 h at 4 °C. The precipitates were washed sequentially with low salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, 150 mm NaCl), high salt wash buffer (same as the low salt wash buffer except NaCl was increased to 500 mm), LiCl wash buffer (0.25 mm LiCl, 1% Nonidet P-40, 1% deoxycholate, 1 mm EDTA, 10 mm Tris-HCl, pH 8.1), and TE buffer (10 mm Tris-HCl, pH 8.0, 1 mm EDTA). The FOXO-DNA complexes were eluted twice with 1% SDS and 100 mm NaHCO3. The eluates and the part of the lysates removed before the immunoprecipitation were heated at 65 °C overnight to dissociate the cross-linking and then treated with Protease K for 1 h at 45°C.Genomic DNA fragments were purified by phenol/chloroform extraction followed by ethanol precipitation and subjected to PCR using primers that amplify the SOD2 promoter region containing a FOXO-binding element at position −1,249 (5′-GAGTATCTATAACCTGGTCCCAGCC-3′ and 5′-GCTGAACCGTTTCCGTTGCTTCTTGC-3′). The amplification condition was 93 °C for 15 s, 64 °C for 30 s, and 72 °C for 30 s for 25–30 cycles. The PCR products (216 bp) were analyzed by 4–20% polyacrylamide gel electrophoresis and stained with ethidium bromide.
Reverse-phase Protein Microarray
The reverse-phase protein microarrays were performed as described previously (12), except that array slides were processed manually or using Mozaic Immunostainer slide racks (Zymed Laboratories Inc.) and a robotic liquid handling system (MultiPorbe II, PerkinElmer Life Sciences). Briefly, cell and tissue lysates were arrayed on nitrocellulose-coated FAST slides (Schleicher & Schuell) using a printing robot (SpotArray microarray printing system, PerkinElmer Life Sciences). Duplicate or quadricate samples were printed of serial dilution curves with a buffer control (lysis buffer only). Each slide was probed with anti-Akt antibody (Akt), anti-myc antibody (FOXO1), antibody specific to phosphorylated Akt (p-Akt) or FOXO (p-FOXO1), anti-SOD2 antibody, or anti-actin antibody followed by an amplification method using a tyramide-based avidin/biotin system (catalyzed signal amplification system, Dako). For background controls, identical slides were incubated without the primary antibody (antibody diluent only). A pegylated, streptavidin-conjugated Quantum Dot 655-Sav (Quantum Dot Corp.) was used as a final fluorescent detector. The slides were visualized using Alpha Innotech's 9900 Fluorometer with a 655-nm narrow bandwidth emission filter (Omega Optical). The spot intensities of SOD2 were normalized relative to those of actin to correct the protein loads between the spots. Measurement of spot intensities and calculations of the relative SOD2 protein amounts based on the duplicate serial dilution curves and replicate experiments were carried out by the MicroVigene software (Vigene Tech.).
RESULTS
To test the hypothesis that Klotho might act as an anti-aging factor by mediating increased resistance to oxidative stress in mammalian cells, we first treated HeLa cells with paraquat, an herbicide that generates superoxide, and compared lipid oxidation in living cells in the presence and absence of a soluble form of recombinant Klotho protein. A fluorescent probe (C11-BODIPY581/591) was used to measure oxidized lipid (13). Klotho protein treatment significantly suppressed paraquat-induced increases in lipid oxidation (Fig. 1A). Next we asked whether Klotho might protect cells from apoptosis induced by oxidative stress, which potentially promotes the survival of irreplaceable cells and contributes to the suppression of aging (14). We treated CHO cells with paraquat and compared the number of apoptotic cells in the presence and absence of Klotho protein. Klotho protein treatment reduced the annexin V-positive, apoptotic cell population but preserved the annexin V-negative, non-apoptotic cell population (Fig. 1, B–D). A similar result was obtained in HeLa cells (data not shown). These observations indicate that Klotho protein confers oxidative stress resistance on mammalian cells.
FIGURE 1. Klotho increases resistance to oxidative stress.
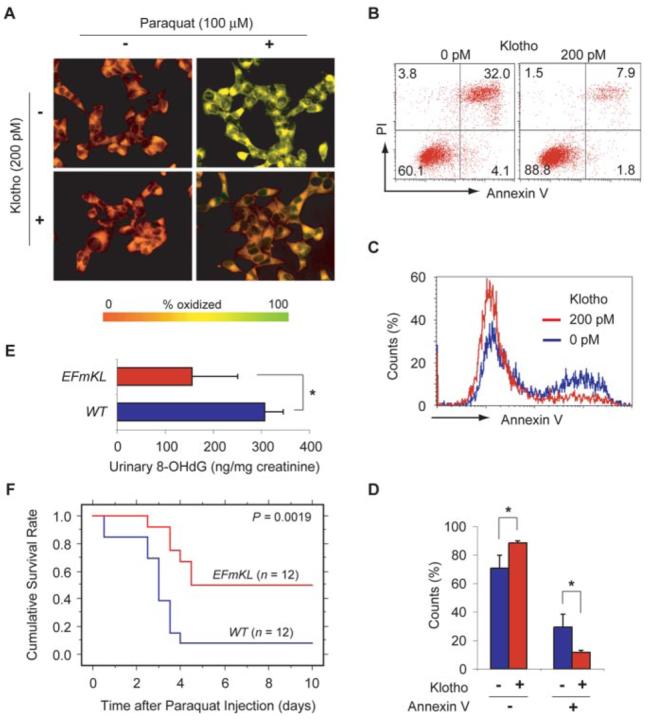
A, the effect of Klotho protein on lipid oxidation in cultured cells. HeLa cells left untreated (−) or treated (+) with 100 μm paraquat in the presence (+) or absence (−) of 200 pm recombinant Klotho protein were loaded with a fluorescent lipid oxidation probe C11-BODIPY581/591. Yellow represents high oxidation and red represents low oxidation. A representative result from four independent experiments is shown. B–D, the effect of Klotho protein on cell death induced by oxidative stress. B, representative dot plots of CHO cells stained with annexin V-FITC and propidium iodide (PI). The cells were treated with 100 μm paraquat in the presence (right panel) or absence (left panel) of 200 pm Klotho protein. The bottom right quadrant represents early apoptotic cells. The top right and left quadrants represent late apoptotic and necrotic cells, respectively. The percentage counts of each quadrant are indicated. C, representative histograms of the CHO cells based on annexin V binding in the presence (red) and absence (blue) of 200 pm Klotho protein under the paraquat-induced oxidative stress. D, mean percentages of annexin V-positive and negative cells in the presence and absence of Klotho protein. The annexin V-negative cells virtually represent live cells under these experimental conditions, because necrotic cell counts were <4% (the top left quadrant in B). *, p < 0.01 by ANOVA (n = 4). E, urinary 8-OHdG levels in wild-type mice (WT) and EFmKL48 mice (EFmKL). *, p = 0.02 by ANOVA (n = 4). F, Kaplan-Meier analysis of survival after the paraquat challenge between age-matched male wild-type mice (WT) and EFmKL48 mice (EFmKL). p = 0.0019 by log-rank test.
Since urinary 8-OHdG is a biological marker of in vivo oxidative DNA damage (15), we compared urinary 8-OHdG levels of long lived Klotho-overexpressing transgenic mice (EFmKL48) (2) with those of wild-type mice. The average urinary 8-OHdG excretion in EFmKL48 mice was approximately one-half of that in wild-type mice (Fig. 1E), indicating that Klotho overexpression reduced overall oxidative DNA damage in mice. Additionally, we observed that EFmKL48 mice survived a challenge with a lethal dose of paraquat significantly longer than wild-type mice (Fig. 1F), strongly suggesting that Klotho confers oxidative stress resistance at the organismal level as well.
To dissect the mechanism by which Klotho protein increases resistance to oxidative stress, we investigated a potential link between the intracellular insulin/IGF-1/PI3K/Akt signaling pathway and the metabolism of reactive oxygen species. Recent studies have identified FoxO forkhead transcription factors (FOXOs) as downstream targets of insulin-like signaling that regulate organismal aging (16-21). FOXOs are negatively regulated by phosphorylation at three conserved serine/threonine residues by Akt (22-24). Once activated by suppression of insulin/IGF-1 signaling, FOXOs up-regulate expression of the manganese superoxide dismutase (MnSOD or SOD2) gene encoding a mitochondrial enzyme that detoxifies superoxide (25). These observations have led to the hypothesis that the anti-aging hormone Klotho, possessing a potent activity that inhibits insulin/IGF-1 signaling, may activate FOXOs and increase SOD2 expression, thereby facilitating removal of reactive oxygen species and increasing resistance to oxidative stress.
To test this hypothesis we asked whether the Klotho protein could reduce Akt and FOXO phosphorylation. We stimulated HeLa cells with Klotho protein and observed that this protein reduced the basal level of Akt and FOXO1 phosphorylation in a dose-dependent manner (Fig. 2A). The effect of Klotho protein on Akt and FOXO1 was evident within 10 min and 15 min, respectively (Fig. 2A). In addition, protein microarray analysis demonstrated that Klotho protein suppressed both basal and insulin-induced phosphorylation of Akt and FOXO1 (Fig. 2B). Klotho protein also suppressed phosphorylation of FOXO3a and FOXO4 (Fig. 2C). Similar results were obtained by using CHO, COS, H4IIE, and 3T3-L6 cells (data not shown). These observations confirm that treatment of cells with Klotho protein reduces phosphorylation of FOXOs. Since the effect of Klotho protein was more pronounced on the FOXO1 than on the FOXO3a or FOXO4 proteins (Fig. 2C), we focused on FOXO1 in subsequent experiments. The precise mechanism behind the difference in Klotho action between the three FOXOs and its physiological significance remain to be determined.
FIGURE 2. Klotho protein reduces FOXO phosphorylation and promotes its nuclear translocation.
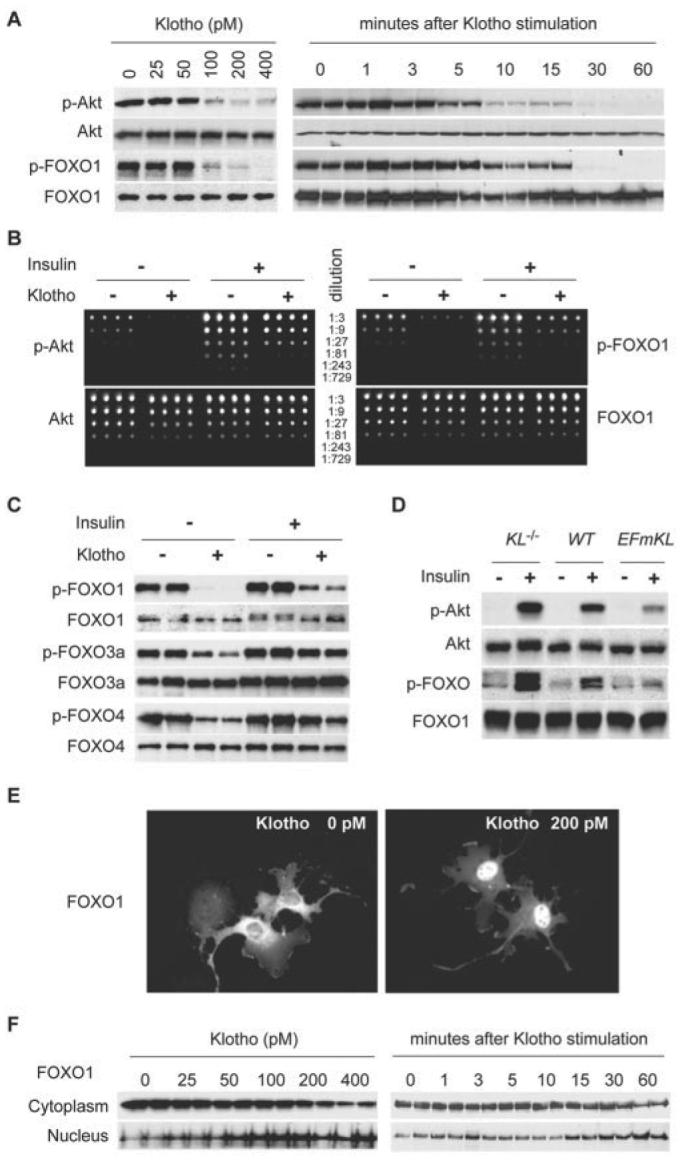
A, the effect of Klotho protein on basal levels of Akt and FOXO phosphorylation. HeLa cells transfected with the FOXO1-myc expression vector were stimulated with various concentrations of Klotho protein as indicated for 30 min (left panels). Cell lysates were immunoblotted with anti-Akt antibody (Akt), anti-myc antibody (FOXO1), or antibody specific to phosphorylated Akt (p-Akt) or FOXO (p-FOXO1). To determine the time course, the cells were stimulated with 200 pm Klotho protein for various time periods as indicated (right panels). B, protein microarray analysis of Akt and FOXO1 phosphorylation. HeLa cells transfected with FOXO1-myc expression vector were incubated in the presence or absence of 200 pm Klotho protein for 30 min and then stimulated with 10 nm insulin or left untreated for 30 min. Cell lysates were arrayed on nitrocellulose-coated slides in quadricate in a serial dilution format as indicated. The slides were probed with anti-Akt antibody (Akt), anti-myc antibody (FOXO1), or antibody specific to phosphorylated Akt (p-Akt) or FOXO (p-FOXO1). Representative blots from four independent experiments are shown. C, Klotho reduces both basal and insulin-induced phosphorylation of all three FOXOs. HeLa cells transfected with the FOXO1-myc, FOXO3a-myc, or FOXO4-myc expression vector were treated as described for B. Cell lysates were analyzed in the same way as described for A. D, Klotho reduces insulin-induced phosphorylation of Akt and FOXO in mice. The lysates of muscles from KL−/− mice (KL−/−), wild-type (WT), and EFmKL48 mice (EFmKL) administered with insulin or saline were immunoblotted with anti-phospho-Akt antibody (p-Akt), anti-Akt antibody (Akt), anti-phosopho-FOXO antibody (p-FOXO), or anti-FOXO1 antibody (p-FOXO1). Representative blots from four independent experiments are shown. E, Klotho-induced nuclear translocation of FOXO1. HeLa cells transfected with the FOXO1-myc expression vector were incubated with or without 200 pm Klotho protein for 30 min and then stained with anti-myc antibody. F, Klotho-induced nuclear translocation of FOXO1 determined by immunoblotting. HeLa cells transfected with the FOXO1-myc expression vector were incubated with the indicated concentrations of Klotho protein for 30 min and then fractionated into cytoplasm and nuclear fractions. Each fraction was immunoblotted with anti-myc antibody (left panels). To determine the time course, the cells were incubated with 200 pm Klotho protein for the indicated time periods (right panels).
In an attempt to determine whether a comparable inhibitory effect of Klotho on Akt and FOXO phosphorylation occurs at the organismal level, we analyzed tissues from KL−/− mice, wild-type mice, and transgenic mice that overexpress Klotho (EFmKL48). EFmKL48 mice express the exogenous klotho gene under the control of the human elongation factor 1α promoter, in addition to the endogenous expression, resulting in ∼2-fold higher blood Klotho concentration and 20–30% longer maximum and average life span than wild-type mice (2). Klotho protein was undetectable in the blood from KL−/− mice (2). Insulin-induced phosphorylation of Akt and FOXO in the muscle was significantly enhanced in KL−/− mice and attenuated in EFmKL48 mice, showing an inverse correlation with the blood Klotho level (Fig. 2D). These observations are consistent with the notion that the circulating Klotho protein has an activity that reduces FOXO phosphorylation in vivo.
To test whether the Klotho-induced decrease in FOXO phosphorylation is associated with its nuclear translocation, we performed immunocytochemical analysis. Klotho protein promoted translocation of FOXO1 from the cytoplasm to the nucleus (Fig. 2E). Furthermore, immunoblot analysis confirmed that Klotho protein resulted in an increase of FOXO1 in the nuclear fraction and a reciprocal decrease in the cytoplasmic fraction in a dose-dependent manner. This effect was evident within 15 min after the addition of Klotho protein (Fig. 2F). The dose response and the time course of nuclear translocation of FOXO1 were inversely correlated with the level of its phosphorylation (Fig. 2A). These observations indicate that Klotho protein signals nuclear trans-location of FOXO.
We next investigated whether the Klotho-induced nuclear translocation of FOXO results in the transcriptional activation of target genes. We transfected HeLa cells with a luciferase reporter construct containing a three tandem array of the forkhead-responsive element derived from the Fas ligand gene promoter (22) and then incubated the cells with Klotho protein to activate endogenous FOXO. Klotho protein greatly stimulated the activity of this promoter (Fig. 3A). Klotho protein also increased the activity of a human SOD2 gene promoter, which is known to contain canonical FOXO binding sites (Fig. 3A) (25). Furthermore, a chromatin immunoprecipitation assay revealed that Klotho protein treatment increased the amount of FOXO1 bound to the native SOD2 gene promoter (Fig. 3B), demonstrating that the Klotho-induced transcriptional activation occurs via direct binding of FOXO1 to the SOD2 promoter. The increase in the promoter activity was associated with an increase in SOD2 protein levels (Fig. 3C). To quantify the Klotho-induced increase in SOD2 protein we performed a quantitative protein microarray analysis (Fig. 3D). Klotho protein increased the SOD2 protein levels 1.5-, 2.3-, and 5.8-fold within 16 h in COS, HeLa, and CHO cells, respectively (Fig. 3E).
FIGURE 3. Klotho enhances FOXO transcriptional activity and increases SOD2 protein levels.

A, measurement of FOXO transcriptional activity by using the luciferase reporter 3xFHRE-Luc (left panel), which contains three canonical FOXO-binding sites, or SOD2-Luc (right panel), which contains the human SOD2 gene promoter. HeLa cells were transfected with the reporter plasmids and incubated in the presence (+) or absence (−) of 400 pm Klotho protein before the luciferase assay. The error bars indicate S.D. *, p < 0.05 by ANOVA (n = 6). B, the effect of Klotho protein on FOXO-binding to the SOD2 promoter. HeLa cells were transfected with FOXO1-myc expression vector and then stimulated with 400 pm Klotho protein or left untreated in the presence or absence of 50 nm wortomannin before chromatin immunoprecipitation assay. Wortmannin treatment activates FOXO1 through inhibiting PI3K and serves as a positive control. A representative result from four independent experiments is shown. C, the effect of Klotho protein on SOD2 protein levels. HeLa cells were incubated with the indicated concentrations of Klotho protein for 16 h and subjected to immunoblot analysis using anti-SOD2 antibody (SOD2) and anti-actin antibody (Actin). D, quantification of relative amounts of SOD2 protein in cultured cells (COS, HeLa, CHO) by reverse-phase protein microarray. Upper left, microarray layout. The numbers indicate hours after Klotho stimulation (400 pm). The cell lysates were arrayed in duplicate in a serial dilution format as indicated. A representative blot with anti-SOD2 antibody from two independent experiments is shown. The graph shows a representative plot between spot signal intensity (y axis) and relative sample concentration (x axis, log scale) with a fitted curve and 99% confidence bands. The regression line at the half-maximal intensity (EC50) is indicated. The amount of SOD2 protein in the sample was determined as the dilution factor at EC50. The amount of actin was quantified in the same way for loading controls. The coefficient of variation was <0.9% and R2 > 0.996 for all regression lines. E, a time course of Klotho-induced increase in SOD2 protein levels quantified by protein microarray analysis. COS, HeLa, and CHO cells were incubated with 400 pm Klotho protein for 6 or 16 h. The fold increase in the SOD2 protein amount from time 0 is indicated. All data were normalized to the actin level. The average of two independent experiments is shown. Each dilution was spotted in duplicate. F, Klotho overexpression increases SOD2 mRNA in muscle. Total RNA isolated from muscles of four 8-week-old male KL−/− mice (KL−/−), wild-type mice (WT), or EFmKL48 mice (EFmKL) were pooled, and 20-μg aliquots were loaded on each lane. G, Klotho overexpression increases SOD2 protein in mouse tissues. Muscle of 8-week-old male KL−/− mice (KL−/−), wild-type mice (WT), and EFmKL48 mice (EFmKL) were analyzed as described for C. H, quantification of SOD2 protein by the protein microarray analysis. Protein extracted from the hind limb muscle of 8-week-old male KL−/− mice (KL−/−), wild-type mice (WT), and EFmKL48 mice (EFmKL) was analyzed as in D. The error bars indicate S.D. *, p < 0.05 by ANOVA (n = 4). Each dilution was spotted in duplicate.
Consistent with these observations in cultured cells, SOD2 mRNA levels in muscle samples from KL−/−, wild-type, and Klotho-overexpressing transgenic mice (EFmKL48) determined by Northern blot analysis positively correlated with circulating Klotho levels (Fig. 3F). Accordingly, SOD2 protein levels in muscles increased as the blood Klotho level increased (Fig. 3G). Quantitative protein microarray analysis revealed that the SOD2 protein level in muscles of KL−/− mice and EFmKL48 mice was 77 ± 16% and 234 ± 6% (mean ± S.D., n = 4), respectively, when compared with that of wild-type mice (Fig. 3H). All of these findings indicate that Klotho protein increases SOD2 expression both in vitro and in vivo.
DISCUSSION
Our results reveal a signaling pathway through which the anti-aging hormone Klotho increases resistance to oxidative stress. Binding of Klotho protein to cell-surface Klotho receptors signals inhibition of FOXO phosphorylation and promotes its nuclear translocation. The nuclear FOXO then directly binds to the SOD2 promoter and up-regulates its expression, thereby facilitating removal of reactive oxygen species and conferring resistance to oxidative stress. Although it remains to be determined whether or not the ability of Klotho to confer oxidative stress resistance is entirely dependent on the induction of SOD2 expression, this activity may be crucial for the anti-aging properties of Klotho, because overexpression of another enzyme that removes reactive oxygen species (catalase) in mitochondria extends the life span in mice (26). We propose that the Klotho protein suppresses aging via at least two distinct anti-aging mechanisms that are evolutionarily conserved: 1) the inhibition of insulin-like signaling and 2) an increase in resistance to oxidative stress. Our results also suggest that Klotho serves as an important link between these two anti-aging mechanisms.
We speculate that the ability of Klotho to activate FOXO is primarily dependent on its ability to inhibit the insulin/IGF-1/PI3K/Akt signaling cascade. However, Klotho protein may have other unknown activities that could also contribute to the FOXO activation. Recent studies have demonstrated that FOXO can be activated by inhibition of serum- and glucocorticoid-inducible kinase (SGK) (27) and by activation of c-Jun NH2-terminal kinase (JNK) (28) or β-catenin (29), raising the possibility that Klotho activates FOXO not only through inhibiting Akt but also through inhibiting SGK and/or activating JNK or β-catenin. Further studies on the Klotho signaling pathway are expected to promote a better understanding of the mechanism by which Klotho activates FOXOs, induces oxidative stress resistance, and eventually suppresses aging.
Acknowledgment
We thank E. C. Friedberg at the University of Texas Southwestern Medical Center for critical reading of the manuscript.
Footnotes
This work was supported in part by grants from the Endowed Scholar Program at the University of Texas Southwestern Medical Center (to M. K.), Pew Scholars Program in Biomedical Science (to M. K.), Eisai Research Fund (to M. K.), High Impact/High Risk Research Program at the University of Texas Southwestern Medical Center (to M. K.), and National Institutes of Health Grants R01AG19712 (to M. K.) and R01AG25326 (to M. K. and K. P. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article was selected as a Paper of the Week.
The abbreviations used are: IGF-1, insulin-like growth factor-1; IRS, insulin receptor substrates; PI3K, phosphatidylinositol 3-kinase; FOXO, FoxO forkhead transcription factor; SOD2, manganese superoxide dismutase; DMEM, Dulbecco's modified Eagle's medium; CHO, Chinese hamster ovary; 8-OHdG, 8-hydroxydeoxyguanosine; ANOVA, analysis of variance; FITC, fluorescein isothiocyanate.
REFERENCES
- 1.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima Y. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 2.Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi H, Kawaguchi H, Shimomura I, Yakayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kenyon C. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Arking DE, Krebsova A, Macek M, Sr., Macek M, Jr., Arking A, Mian IS, Fried L, Hamosh A, Dey S, McIntosh I, Dietz HC. Proc. Natl. Acad. Sci. U. S. A. 2002;99:856–861. doi: 10.1073/pnas.022484299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arking DE, Becker DM, Yanek LR, Fallin D, Judge DP, Moy TF, Becker LC, Dietz HC. Am. J. Hum. Genet. 2003;72:1154–1161. doi: 10.1086/375035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogata N, Matsumura Y, Shiraki M, Kawano K, Koshizuka Y, Hosoi T, Nakamura K, Kuro-o M, Kawaguchi H. Bone. 2002;31:37–42. doi: 10.1016/s8756-3282(02)00786-x. [DOI] [PubMed] [Google Scholar]
- 7.Kawano K, Ogata N, Chiano M, Molloy H, Kleyn P, Spector TD, Uchida M, Hosoi T, Suzuki T, Orimo H, Inoue S, Nabeshima Y, Nakamura K, Kuro-o M, Kawaguchi H. J. Bone Miner. Res. 2002;17:1744–1751. doi: 10.1359/jbmr.2002.17.10.1744. [DOI] [PubMed] [Google Scholar]
- 8.Yamada Y, Ando F, Niino N, Shimokata H. J. Mol. Med. 2004;83:50–57. doi: 10.1007/s00109-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 9.Arking DE, Atzmon G, Arking A, Barzilai N, Dietz HC. Circ. Res. 2005;96:412–418. doi: 10.1161/01.RES.0000157171.04054.30. [DOI] [PubMed] [Google Scholar]
- 10.Finkel T, Holbrook NJ. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 11.Daitoku H, Yamagata K, Matsuzaki H, Hatta M, Fukamizu A. Diabetes. 2003;52:642–649. doi: 10.2337/diabetes.52.3.642. [DOI] [PubMed] [Google Scholar]
- 12.Geho D, Lahar N, Gurnani P, Huebschman M, Herrmann P, Espina V, Shi A, Wulfkuhle J, Garner H, Petricoin E, 3rd, Liotta LA, Rosenblatt KP. Bioconjug. Chem. 2005;16:559–566. doi: 10.1021/bc0497113. [DOI] [PubMed] [Google Scholar]
- 13.Pap EH, Drummen GP, Winter VJ, Kooij TW, Rijken P, Wirtz KW, Op den Kamp JA, Hage WJ, Post JA. FEBS Lett. 1999;453:278–282. doi: 10.1016/s0014-5793(99)00696-1. [DOI] [PubMed] [Google Scholar]
- 14.Higami Y, Shimokawa I. Cell Tissue Res. 2000;301:125–132. doi: 10.1007/s004419900156. [DOI] [PubMed] [Google Scholar]
- 15.Shigenaga MK, Gimeno CJ, Ames BN. Proc. Natl. Acad. Sci. U. S. A. 1989;86:9697–9701. doi: 10.1073/pnas.86.24.9697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 17.Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- 18.Lin K, Hsin H, Libina N, Kenyon C. Nat. Genet. 2001;28:139–145. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- 19.Henderson ST, Johnson TE. Curr. Biol. 2001;11:1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- 20.Giannakou ME, Goss M, Junger MA, Hafen E, Leevers SJ, Partridge L. Science. 2004;305:361. doi: 10.1126/science.1098219. [DOI] [PubMed] [Google Scholar]
- 21.Hwangbo DS, Gershman B, Tu MP, Palmer M, Tatar M. Nature. 2004;429:562–566. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- 22.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 23.Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM. Nature. 1999;398:630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- 24.Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. J. Biol. Chem. 1999;274:17179–17183. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 25.Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 26.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 27.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Mol. Cell. Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter, ND, Bos JL, Burgering BM. EMBO J. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Science. 2005;308:1181–1184. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]