

Abstract
Inositol phospholipid signaling pathways have begun to emerge as important players in stem cell biology and bone marrow transplantation [1–4]. The SH2-containing Inositol Phosphatase (SHIP) is among the enzymes that can modify endogenous mammalian phosphoinositides. SHIP encodes an isoform specific to pluripotent stem (PS) cells [5,6] plays a role in hematopoietic stem (HS) cell biology [7,8] and allogeneic bone marrow (BM) transplantation [1,2,9,10]. Here I discuss our current understanding of the cell and molecular pathways that SHIP regulates that influence PS/HS cell biology and BM transplantation. Genetic models of SHIP-deficiency indicate this enzyme is a potential molecular target to enhance both autologous and allogeneic BM transplantation. Thus, strategies to reversibly target SHIP expression and their potential application to stem cell therapies and allogeneic BMT are also discussed.
INTRODUCTION
Inositol phospholipid (IP) signaling pathways have a critical role in the control of cell proliferation, survival, differentiation and effector function [11–13]. The activity of IP modifying enzymes is controlled by protein phosphorylation based signaling pathways with tyrosine phosphorylation being required for either their enzymatic activity (e.g. PI3K) or their efficient recruitment to the plasma membrane (e.g., SHIP). In turn IP modifying enzymes also control the activity of key protein phosphorylation enzymes (e.g., PH domain containing kinases). For instance, the generation of PI(3,4,5)P3 by PI3K allows a cell to avoid programmed cell death or apoptosis through activation of Protein Kinase B (PKB)/Akt [14]. IP signaling is highly integrated with other intracellular signaling pathways and thus has a crucial role in cell fate decisions. A great deal of research has been devoted to understanding how these IP signaling pathways influence effector function and survival in differentiated and mature cell types.
Several different inositol phosphatases such as PTEN, SHIP and SHIP2 have been found to oppose PI3K by their ability to hydrolyze PI(3,4,5)P3 to PI(4,5)P2 or PI(3,4)P2. In the hematopoietic system all three of these enzymes are expressed by most blood cell lineages and may participate in some of the same IP signaling pathways as demonstrated by compound heterozygosity at the PTEN and SHIP loci [15,16]. Understanding how IP modifying enzymes interact in the same pathway and why one inositol phosphatase or kinase takes the lead in certain signaling pathways is a puzzle that the IP signaling field must tackle in the coming years. Nonetheless, genetic analysis of PTEN, SHIP and SHIP2 indicate segregated roles for these IP phosphatases in certain cell types and signaling pathways. Why PTEN, SHIP or SHIP2 are the pivotal players in certain cell types and signaling pathways is likely determined by their individual pattern of expression and/or their relative ability to be recruited to specific receptor complexes at the plasma membrane. A role for IP signaling enzymes (PI3K, PTEN, SHIP, s-SHIP, SHIP2) in undifferentiated stem cells has recently begun to be explored at the gene expression, biochemical and genetic level. There is now a substantial body of evidence that IP kinases [17–21] and phosphatases [3,4,6,7,22,23] play specific roles in regulating self-renewal, proliferation, survival or differentiation of stem cell populations. In this review I will highlight the emerging role that the inositol phospholipid-modifying enzyme SHIP plays in primitive stem cell populations and transplantation.
We and others independently identified a gene currently referred to as the SH2-containing Inositol Phosphatase (SHIP) [24–27]. SHIP was isolated by gene-trapping of LPS-response genes in B-lymphoid cells [24] (referred to as 7a33 in our 1996 PNAS manuscript), for its ability to associate with the PTB domain of Shc [25,27] or the SH3 domain of Grb2 [26]. The sequence of SHIP indicated that it is likely to play a role in several signal transduction pathways due to its SH2 domain, an inositol 5′-phosphatase (IP) domain, a proline rich region (for binding to SH3 domains), NPXY sequences that can be phosphorylated and associate with PTB domains and a YIGM motif that can be recognized by the 85kD regulatory subunit of PI-3-kinase [24–28]. Subsequent to the cloning of the SH2 containing isoforms of SHIP, we identified a stem cell specific isoform of SHIP, s-SHIP, that lacks the SH2 domain [22]. S-SHIP is expressed from a stem cell specific intronic promoter located between exons 5 and 6 [22]. In Fig. 1 we provide a summary of the various isoforms encoded by the SHIP gene.
Fig. (1). Isoforms of the SHIP gene expressed in stem cells and/or differentiated cells.
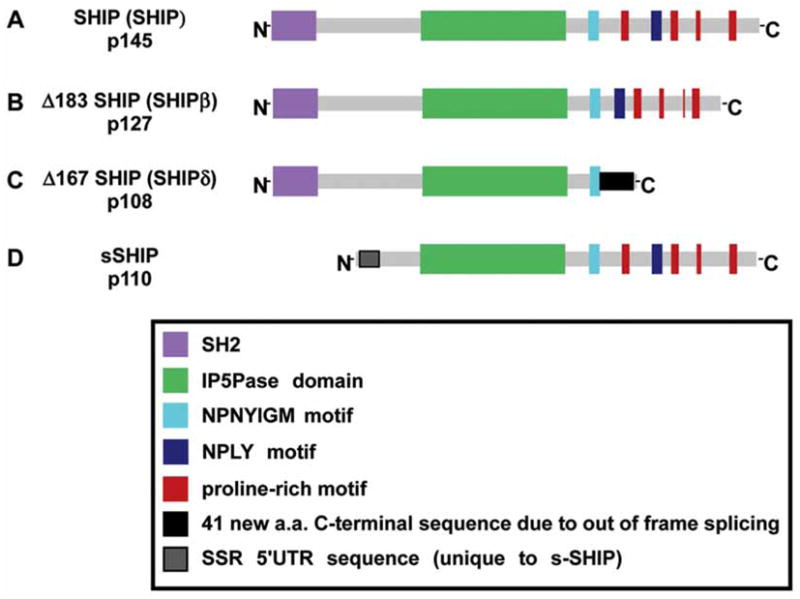
A-C. SH2 containing isoforms that are expressed in differentiated cells, MEF, vascular endothelial cells and HSC. D. s-SHIP isoform that is expressed by ES cells and HSC. (SSR, s-SHIP specific region).
In hematolymphoid cells SHIP can be recruited to a wide variety of receptor complexes including growth factor receptors [25,29–35] and immune receptors such as FcRγIIb, FcγRIII, Ly49A, Ly49B, Ly49C, KLRG1 and 2B4 [1,36–41]. SHIP is recruited to receptor-associated signaling complexes via adapters (e.g. Shc, Grb2, Dok3), scaffold proteins like Gab1/2 or directly via its SH2 domain [22,25,30–35,42–44]. After recruitment to the plasma membrane, SHIP can then hydrolyze PI(3,4,5)P3 and in so doing attenuate several different PI3K effector pathways [11,13]. Hydrolysis of PI(3,4,5)P3 inhibits recruitment of PH domain containing kinases like Akt, Btk, PLC-γ to the plasma membrane. In fact, we were the first group to demonstrate that SHIP limits Akt expression and phosphorylation in vivo in a hematopoietic lineage [1]. Recently Ras, Rab and Arf family proteins that contain polybasic amino acid clusters have been shown to associate with the plasma membrane by binding to negatively charged PI(3,4,5)P3 and PI(4,5)P2 [45], and thus SHIP could potentially cause dissociation of these signaling proteins from the plasma membrane via hydrolysis of phosphate on the D5 position of PI(3,4,5)P3. By antagonizing the plasma membrane recruitment and/or retention of the above signaling proteins or kinases, SHIP can limit the activity of different downstream effectors of PI3K signaling that promote cell survival, migration, differentiation or proliferation. These include such distal kinases as MAP/ERK [46,47], JNK/SAPK [48], p38 MAPK [46,47] and key transcription factors such as NF-κB [46] and NFAT [49]. In Fig. 2 we provide a schema that summarizes potential roles for SHIP in intracellular signaling pathways.
Fig. (2). Role of SHIP in growth factor receptor signal transduction.
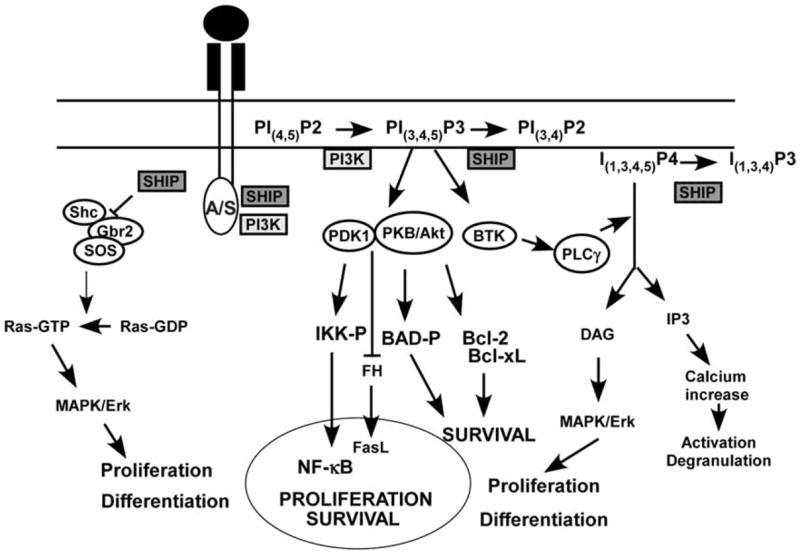
A/S, Anchor/Scaffold proteins that can recruit SHIP to receptor complexes. These include Grb2, Shc, Gab1 and Dok proteins.
SHIP is expressed ubiquitously in differentiated cells of the hematopoietic system [24–26], in endothelial cells [50], hematopoietic stem cells (HSC) [22] and embryonic stem (ES) cells [22]. Because of this rather broad expression pattern it is difficult to predict solely from biochemical studies where and when SHIP could play a role in normal physiology and function. Towards this end genetic analysis of SHIP mutant mice has revealed a pivotal role for SHIP in a wide variety of differentiated hematopoietic cell types. SHIP has been shown to play a role in regulating the receptor repertoire and cytolytic function of Natural Killer (NK) cells [1,2,51], B lymphocyte development and antibody production [52,53], the myeloid cell response to bacterial mitogens [54], development of marginal zone macrophages [55], osteoclast function [56], lymph node recruitment of dendritic cells [9], mast cell degranulation [57] and the homeostasis and function of myeloid immunoregulatory cells [9,10,58]. Whether SHIP participates in these cell processes in a cell autonomous fashion or through extrinsic effects on these cell types remains to be determined. In vitro experiments with highly purified SHIP-deficient NK cells, myeloid suppressor cells (MySC) and mast cells suggest SHIP plays a cell autonomous role in signaling pathways that control the function of these cells. However, because of abnormalities in secretion of cytokines and other molecules that we and others observe in SHIP-deficient mice, the root cause of an abnormality in SHIP-deficient mice could also be due to extrinsic effects on that cell type. In this regard, the analysis of WT/SHIP−/− chimeras or cell type specific deletion of SHIP in vivo is ultimately required before one can conclusively determine which SHIP-deficient cell type causes a specific phenotype in mice with a germline homozygous mutation of SHIP.
A ROLE FOR SHIP IN STEM CELL SIGNALING AND HSC FUNCTION
The first indication that the SHIP gene could play a specific role in stem cell function came with the identification of the s-SHIP isoform [22]. s-SHIP is expressed in undifferentiated murine ES cells and hematopoietic stem cells (HSC) from both fetal and adult sources. The s-SHIP isoform lacks the SH2 domain of SHIP due to internal initiation of transcription in the intron between exons 5 and 6. However, despite the lack of an SH2 domain, s-SHIP is recruited to the plasma membrane in pluripotent stem cells via the adapter Grb2 [22]. This interaction presumably occurs via poly-proline rich regions present in s-SHIP and SH3 domains in Grb2. The s-SHIP-Grb2 interaction does not appear to require phosphorylation of either partner and thus we proposed that s-SHIP is constitutively recruited to the plasma membrane in pluripotent stem cell populations. Indeed this is the case, as we find that the membrane fraction of murine ES cells contains a significant amount of s-SHIP [22]. Given our recent finding that s-SHIP is recruited to gp130 [6], a signal transduction component for key stem cell growth factor receptors for LIF and SCF, we speculate that s-SHIP sets a threshold for tonic growth factor receptor signals that are constantly received by stem cells from the niche. In this manner s-SHIP could limit incidental proliferation of primitive stem cells and thus help to preserve quiescence in the stem cell compartment. Consistent with a steady state role for s-SHIP in stem cell signaling, we were unable to detect tyrosine phosphorylation of s-SHIP in ES cells in response to acute stimulation with either LIF or SCF [22], suggesting its membrane recruitment does not require induction by proximal tyrosine kinases that are activated following ligand engagement of growth factor receptors. This is a key molecular difference between s-SHIP expressed in stem cells and the SH2-containing SHIP isoforms expressed in more differentiated cells. SHIP isoforms are routinely tyrosine phosphorylated in response to essentially all growth factors or antigen receptor stimuli received by differentiated cells - including LIF and SCF. Thus, SHIP recruitment to signaling complexes at the plasma membrane likely requires an acute stimulus and tyrosine phosphorylation, while s-SHIP recruitment occurs in the absence of such receptor mediated events and thus can oppose basal PI3K activity in quiescent stem cells. As both s-SHIP and SHIP are expressed in Sca1+Kit+Lin− HSC [22], it is possible that s-SHIP and SHIP could have distinct signaling roles in quiescent vs. activated HSC, respectively. Below we provide a model of how s-SHIP might participate in primitive stem cell signaling (Fig. 3) which in this example features s-SHIP recruitment to gp130. Whether s-SHIP is also recruited to other growth factor receptor subunits active in stem cells remains to be determined.
Fig. (3). LIFR/gp130 receptor complex signal transduction pathways and how s-SHIP may impact them in pluripotent stem cells.
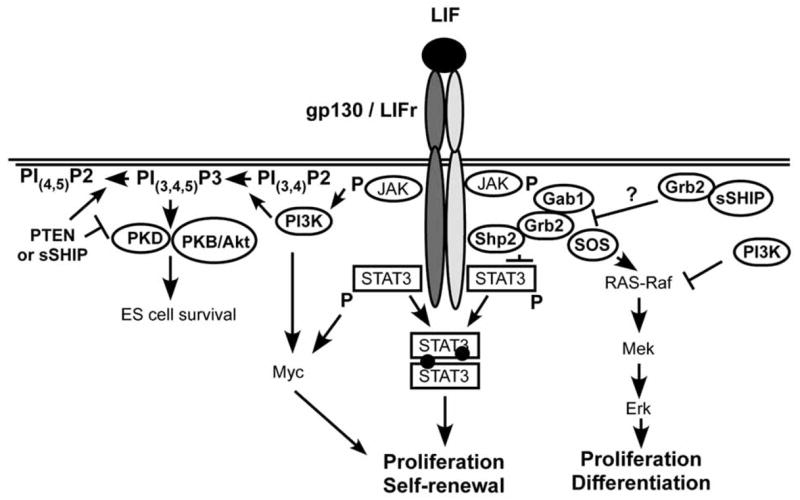
Although s-SHIP does not become phosphorylated after LIF stimulation, s-SHIP is constitutively present at the membrane and thus can impact the signaling pathways downstream of ES cell receptors. Adapted from illustrations by Desponts et al. [6] and Burdon et al. [76].
Whether s-SHIP actually plays a role in limiting pluripotent stem cell proliferation, differentiation or other functions of these primitive cells remains to be determined by genetic approaches. However, it will be important to determine how and when the SHIP locus switches from expression of the s-SHIP isoform in primitive stem cells, expressed from an intronic promoter, to production of the SH2 containing isoforms that predominate in more differentiated cells and which are expressed from the 5′ promoter proximal to the SH2 domain encoding exons. Interestingly, both the s-SHIP and SHIP isoforms are expressed in purified Sca1+Kit+Lin− cells in fetal and adult hematopoiesis. Whether these two isoforms perform different functions in the same HSC subset or are confined to different subsets of HSC remains to be determined. In this regard, Rohrschneider et al recently developed s-SHIP promoter-GFP transgenic mice [59] that should allow single cell detection and purification of HSC expressing s-SHIP. Analysis of SHIP expression in GFP+ HSC from these mice by RT-PCR and the function of this GFP+ HSC subset in transplantation assays would inform the above questions.
The SH2 containing isoforms of SHIP are expressed in both fetal and adult HSC [22]. Genetic analysis showed that the BM HSC compartment in SHIP−/− mice is compromised in its ability to reconstitute multiple blood cell lineages when compared to HSC from WT donors using either whole BM cells in a competitive repopulating unit (CRU) assay [7,8] or highly purified HSC in a direct competition assay (DCA) [7]. The DCA result with purified SHIP−/− HSC transplanted into SHIP-competent hosts suggested a cell autonomous role for SHIP in HSC homing and repopulation. However, our recent analysis of SHIP−/− HSC using a novel HSC assay where gene ablation is induced after HSC are already resident in a SHIP-competent BM microenvironment indicates SHIP-deficient HSC have comparable repopulating function to WT HSC as long as they are rendered SHIP-deficient when resident in a SHIP-competent BM niche. In fact, when SHIP−/− BM HSC are transplanted from this in situ deletion model they repopulate multiple lineages at normal levels for an extended period (Hazen and Kerr, unpublished data). The disparate findings in these distinctly different models can be accounted for by the fact that in germline SHIP-deficient mice both the HSC and the BM microenvironment are SHIP-deficient [7,8] while in the in situ deletion model the BM microenvironment is SHIP-competent. Thus, a SHIP-deficient BM environment fundamentally alters the ability of HSC harvested from this environment to home properly and reconstitute the blood system. A role for SHIP in the BM microenvironment suggests that SHIP is expressed by some cellular components of the BM niche and, moreover, that SHIP is required for the normal support of HSC by the BM niche.
Consistent with the above hypothesis, we find that steady-state production of several soluble factors that influence HSC proliferation, homing or mobilization are altered in SHIP-deficient mice. These include increased TPO, G-CSF, MMP-9, soluble VCAM-1, IL-5 and IL-6 and decreased SDF-1 production (Hazen and Kerr, unpublished data). In accordance with these microenvironmental perturbations HSC in mice with germline or induced systemic SHIP-deficiency have increased numbers of HSC in their BM, a higher proliferative rate and lower apoptotic rate with substantially more HSC mobilized to the blood and spleen [7]; Hazen and Kerr, unpublished data]. Surprisingly, the extramedullary HSC compartment in mice with systemic SHIP-deficiency retains normal HSC activity as SHIP−/− splenocytes possess significant radioprotective activity, long-term multi-lineage repopulating potential and the capacity for self-renewal (Hazen and Kerr, unpublished data) – activities not typically found at significant levels in extramedullary compartments. Thus, SHIP expression in the BM microenvironment is required for a normal BM HSC niche, while in the absence of SHIP expression HSC function is transferred partially or completely to peripheral, extramedullary sites. This finding suggests a potential therapeutic strategy for treatment of BM failure syndromes caused by viruses, bone-seeking radioisotopes, chemotherapy or malignancy. The induction of SHIP-deficiency in such patients could be used to temporarily relocate the HSC compartment to extramedullary sites such as the spleen to provide sufficient blood cell production until such time as the supportive function of the BM returns to normal.
SHIP AND ALLOGENEIC TRANSPLANTATION
A role for SHIP in allogeneic transplantation first emerged with the demonstration that SHIP−/− hosts fail to reject MHC-mismatched BM grafts in an acute fashion and are relatively resistant to graft-versus-host disease (GvHD) [1]. The cellular and molecular basis for a depressed host versus graft (HvG) response and GvHD in the SHIP-deficient host remains an active area of investigation [1,2,9,10,51], but appears to be multi-factoral as summarized in Fig. 4. Compromised acute rejection of BM grafts is caused by a profound disruption of NK receptor (NKR) expression and qualitative changes in signaling at certain NK inhibitory receptors. This results in a skewed balance of activating vs. inhibitory signals in SHIP-deficient NK cells such that inhibitory receptor signals dominate normally robust activating receptor signals – a phenomenon we refer to as “receptor dominance” [1,2,51]. Receptor dominance occurs in the SHIP-deficient NK cell and is caused by one or two inhibitory receptors dominating signaling such that an imbalance toward negative signals squelches activating receptor signals required for lysis of target cells. Receptor dominance appears to specifically hamper cytolysis of complex NK targets that express both self ligands for inhibitory receptors (MHC-I, CD48) and ligands for activating receptors (Rae1, m157) [2,51]. Despite this signaling disruption that impairs killing of complex targets, rejection of simple “missing self” targets (i.e., β2m−/− BM graft) by SHIP-deficient NK cells appears to be intact [1].
Fig. (4). Multiple cellular and molecular mechanisms contribute to reduced HvG and GVHD in SHIP-deficient hosts.
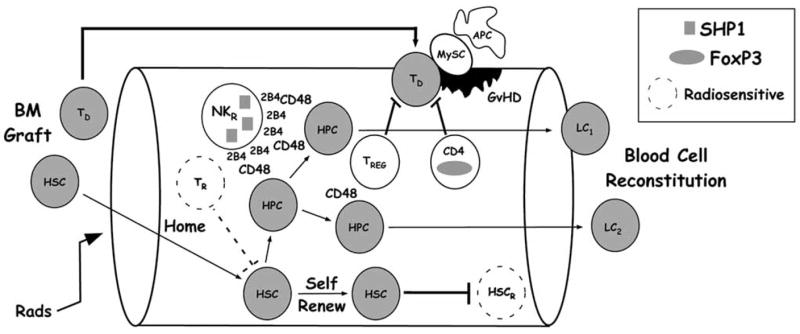
Schema that depicts the major host cells types (white) altered by SHIP-deficiency that facilitate engraftment of donor HSC or reduce GvHD by donor T cells (TD) [donor cells are grey]. The rectangular key indicates molecules whose expression or activities are altered by SHIP-deficiency that contribute to reduced HvG and GvHD in the indicated host cell types.
In our initial report, SHIP−/− mice on a mixed 129/BL6 background were found to have their NKR repertoire dominated by Ly49A and C with all other Ly49 receptors and CD94 significantly under-represented [1]. We provided a direct link between the disruption of the NKR repertoire and engraftment of MHC mismatched BM in SHIP-deficient mice by showing that blockade of the inhibitory Ly49C receptor with F(ab)′2 fragments could partially restore acute rejection of MHC mismatched BM [1]. Subsequently we found that NK cells in SHIP−/− mice backcrossed to a C57BL6 background had over-representation of another inhibitory receptor, the SLAM family receptor 2B4, while CD94 and all Ly49 receptors, including A and C, were significantly under-represented in the SHIP−/− NKR repertoire [2]; Paraiso and Kerr, unpublished data]. FACS and biochemical analysis of SHIP-deficient NK cells showed not only increased 2B4 expression, but also a pronounced bias towards expression of the inhibitory isoform, 2B4L [2,51]. Depending upon the context, 2B4, upon engaging its CD48 ligand, can trigger intracellular signaling cascades that culminate in either target lysis by NK cells [38,60] or inhibition of these signals to prevent cytolysis [2,51,61]. However, in the SHIP-deficient NK cell the 2B4 inhibitory mode predominates due to over-expression and increased recruitment of the inhibitory tyrosine phosphatase SHP1 to 2B4 [2,51]. These qualitative changes in 2B4 signaling lock the SHIP-deficient NK cell into a hypo-responsive state.
Modulation of expression of SHIP occurs in certain hematopoietic lineages [24,54]. Our findings in SHIP-deficient NK cells suggest the possibility that modulation of SHIP expression within the NK lineage could be a molecular determinant of NK cell unresponsiveness. Thus, modulation of SHIP expression could serve as a molecular switch to limit NK activity during education to self [62], licensing [63], or anergy [64]. Consistent with this hypothesis, SHIP expression is reduced in a subset of human lymph node (LN) NK cells that have reduced cytolytic function [65] and 2B4 has inhibitory function in human LN NK cells [66]. Because 2B4 is expressed at all stages of NK cell development and differentiation, our results suggest that control of SHIP expression could serve to control the activating vs. inhibitory mode of 2B4 and thus prevent NK cytolytic activity in the above contexts or when NK activity could be deleterious to the host or a fetus [67]. The implications for allogeneic BMT are that induction of SHIP-deficiency might potentially be used to reduce host NK responses that hamper engraftment of MHC-mismatched BM [1]. Because host NK cells can also contribute to organ graft rejection [68,69], this strategy might also be useful in solid organ transplantation.
The cellular and molecular basis for lethal GvHD resistance by SHIP-deficient hosts is an active area of investigation [1,9,10], but appears to be multi-faceted. It is well accepted that donor T cells cause lethal GvHD [70]; however, antigen presentation by the host is required to initiate the allogeneic T cell attack on host tissues [71]. Professional antigen presenting cells (APC) present in secondary lymphoid tissues survive pre-transplant myeloablation and thus play a particularly prominent role in this process [72,73]. Host APC are present in normal numbers in peripheral lymphoid tissues of SHIP-deficient mice, but there is a profound increase in myeloid suppressor cells that are potent antagonists of allogeneic T cell activation by host APC [9,10]. However, this may not be the only cell type altered by SHIP-deficiency that can antagonize donor T cells that mediate GvHD. We recently found that Treg cells numbers are also significantly increased in peripheral lymphoid tissues of SHIP−/− mice. Moreover, naïve CD4+CD25− T cells inappropriately acquire expression of the FoxP3 transcription factor and consequently can also suppress allogeneic T cell responses (Collazo and Kerr, unpublished data). As host Treg cells can also suppress GvHD [74], this regulatory T cell compartment expansion in SHIP-deficient hosts is also likely to contribute to the GvHD resistance we have observed in these mice.
FUTURE “COURSE HEADINGS” FOR SHIP IN STEM CELL BIOLOGY AND TRANSPLANTATION
That systemic SHIP-deficiency alters the BM microenvironment to disrupt HSC function and homeostasis suggests an unprecedented role for SHIP in non-hematopoietic cells present in BM. As both osteoblast and vascular endothelial cells provide niches for HSC [75], our findings suggest a potential role for SHIP in the control of their function in BM. Although SHIP expression in osteoblasts has not been reported, SHIP is expressed by vascular endothelial cells [50] as well as mouse embryonic fibroblasts which includes stromal elements [6]. In fact in the endothelial study [50], SHIP expression was shown to be induced by VEGF-A. Thus, SHIP expression may be induced in non-hematopoietic cell types found in the BM in response to stress stimuli (e.g., angiogenic factors, inflammatory cytokines) to limit the response of these microenvironment cells to such stressors. In the context of germline or induced systemic SHIP-deficiency such cells are not capable of inducing SHIP expression and consequently they may become locked into an activated, stress-response state. Consistent with this hypothesis we observe that several cytokines and factors are significantly increased in the plasma of SHIP−/− mice relative to WT controls including MMP9, TPO, G-SCF, IL-6 and IL-5. Osteoblasts, endothelial cells and stromal cells in the BM are all potential sources for these factors. Thus, SHIP may oppose PI3K-mediated signaling pathways in these cells to limit production of these factors, and potentially other secreted molecules, during periods of microenvironmental stress.
Despite our findings that SHIP is not a cell autonomous requirement for BM HSC function, we feel there is likely an intrinsic role for SHIP in HSC and that is to restrict them to the BM compartment. We consistently observe extramedullary hematopoiesis and splenomegaly when SHIP-deficient HSC are present in mice, including in the in situ deletion model where the non-hematopoietic microenvironment is SHIP-competent. Thus, when an HSC is SHIP-deficient it is able to mobilize to the blood and rather than rapidly recycling back to the BM compartment, it is capable of establishing itself in an extramedullary site like the spleen. Whether BM retention of HSC is a bona fide physiological role for SHIP remains to be determined. In this regard, analysis of SHIP expression in HSC present in neonatal spleen or during stress-induced extramedullary hematopoiesis may prove informative. Even if this is not a bona fide physiological role for SHIP, our findings suggest that significant HSC activity can be relocated to the spleen by induction of SHIP-deficiency in the MxCreSHIPflox/flox model (Hazen, Desponts and Kerr, unpublished data). Thus, the development of SHIP inhibition strategies (e.g., RNAi, small molecule inhibitors) could prove useful for rescue of blood cell production during life-threatening infections (e.g., parvovirus, EBV, HHV-6), and exposure to bone-seeking radioisotopes or chemotherapies that selectively compromise medullary hematopoiesis. The reversible use of SHIP inhibition could relocate the HSC compartment to the spleen and other extramedullary sites to protect the host from hematopoietic failure until such time as medullary hematopoiesis recovers.
The role of SHIP in allogeneic transplantation is clearly a prominent one, but also one that is not limited to a single cell lineage or one signaling pathway. We have documented a role for SHIP in the control of NK cytolytic function against MHC-mismatched targets through its effect on 2B4 signaling [2,51]. However, the broad NKR repertoire disruption we observe in SHIP−/− mice could be unrelated to SHIP’s role in 2B4 signaling and might even be a consequence of cytokine alterations in the mice. Consistent with this hypothesis, Gays et al found that certain cytokines can disrupt the NKR repertoire in vivo [40]. How SHIP-deficiency causes NKR repertoire alterations and why the quality of these repertoire disruptions varies with genetic background are important questions that require further study. Along these lines how SHIP-deficiency leads to the expansion of immunoregulatory myeloid cells and T-lineage cells in secondary lymphoid tissues and the inappropriate acquisition of FoxP3 expression by naïve CD4 T cells are also critical questions. Because SHIP expression can be modulated by stressors such as VEGF-A and LPS we should consider the possibility that induction of SHIP-deficiency does not coincidentally lead to immunosuppressive function by NK cells, myeloid cells and T cells. I propose that SHIP expression can also be repressed during the stress of an overwhelming immune stimulus and thus serve as a molecular switch to suppress deleterious cytolytic reactions by NK cells and T cells and thus protect the host. This putative immunoregulatory circuit that operates via control of SHIP expression could obviously be harnessed to facilitate allogeneic transplantation of BM, solid organs or potentially MHC-mismatched stem cell populations for tissues other than blood (e.g., neural stem cells) to protect them and the host from deleterious immune attack.
Acknowledgments
The author wishes to thank former and current members of the Kerr lab whose hard work and dedication have defined the role that SHIP plays in allogeneic transplantation, NK cell biology and stem cell biology. I thank Amy Hazen and Michelle Collazo for allowing me to cite their recent unpublished findings. During the course of our studies in this area my research was and continues to be supported by grants from the NIH (R01 HL72523, R21 DK071872, PO1 NS27405 and RO1 DK54767) and academic development funds from the H. Lee Moffitt Comprehensive Cancer Center and the University of South Florida. During most of this research the author was also the Newman Scholar of the Leukemia and Lymphoma Society.
References
- 1.Wang JW, Howson JM, et al. Influence of SHIP on the NK repertoire and allogeneic bone marrow transplantation. Science. 2002;295:2094–7. doi: 10.1126/science.1068438. [DOI] [PubMed] [Google Scholar]
- 2.Wahle JA, Paraiso KH, Costello AL, Goll EL, Sentman CL, Kerr WG. Cutting edge: dominance by an MHC-independent inhibitory receptor compromises NK killing of complex targets. J Immunol. 2006;176:7165–9. doi: 10.4049/jimmunol.176.12.7165. [DOI] [PubMed] [Google Scholar]
- 3.Yilmaz OH, Valdez R, Theisen BK, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–82. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J, Grindley JC, Yin T, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–22. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 5.Tu Z, Ninos JM, Ma Z, et al. Embryonic and hematopoietic stem cells express a novel SH2-containing inositol 5′-phosphatase isoform that partners with the Grb2 adapter protein. Blood. 2001;98:2028–38. doi: 10.1182/blood.v98.7.2028. [DOI] [PubMed] [Google Scholar]
- 6.Desponts C, Ninos JM, Kerr WG. s-SHIP associates with receptor complexes essential for pluripotent stem cell growth and survival. Stem Cells Dev. 2006;15:641–6. doi: 10.1089/scd.2006.15.641. [DOI] [PubMed] [Google Scholar]
- 7.Desponts C, Hazen AL, Paraiso KH, Kerr WG. SHIP deficiency enhances HSC proliferation and survival but compromises homing and repopulation. Blood. 2006;107:4338–45. doi: 10.1182/blood-2005-12-5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helgason CD, Antonchuk J, Bodner C, Humphries RK. Homeostasis and regeneration of the hematopoietic stem cell pool is altered in SHIP-deficient mice. Blood. 2003 doi: 10.1182/blood-2002-12-3939. [DOI] [PubMed] [Google Scholar]
- 9.Ghansah T, Paraiso KH, Highfill S, et al. Expansion of myeloid suppressor cells in SHIP-deficient mice represses allogeneic T cell responses. J Immunol. 2004;173:7324–30. doi: 10.4049/jimmunol.173.12.7324. [DOI] [PubMed] [Google Scholar]
- 10.Paraiso KH, Ghansah T, Costello A, Engelman RW, Kerr WG. Induced SHIP deficiency expands myeloid regulatory cells and abrogates graft-versus-host disease. J Immunol. 2007;178:2893–900. doi: 10.4049/jimmunol.178.5.2893. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, Majerus PW. Phosphatidylinositol signalling reactions. Seminars in Cell & Developmental Biology. 1998;9:153–60. doi: 10.1006/scdb.1997.0220. [DOI] [PubMed] [Google Scholar]
- 12.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 13.Majerus PW, Kisseleva MV, Norris FA. The role of phosphatases in inositol signaling reactions. J Biol Chem. 1999;274:10669–72. doi: 10.1074/jbc.274.16.10669. [DOI] [PubMed] [Google Scholar]
- 14.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate [see comments] Science. 1997;275:665–8. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 15.Moody JL, Pereira CG, Magil A, Fritzler MJ, Jirik FR. Loss of a single allele of SHIP exacerbates the immunopathology of Pten heterozygous mice. Genes Immun. 2003;4:60–6. doi: 10.1038/sj.gene.6363903. [DOI] [PubMed] [Google Scholar]
- 16.Moody JL, Jirik FR. Compound heterozygosity for Pten and SHIP augments T-dependent humoral immune responses and cytokine production by CD(4+) T cells. Immunology. 2004;112:404–12. doi: 10.1111/j.1365-2567.2004.01901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paling NR, Wheadon H, Bone HK, Welham MJ. Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J Biol Chem. 2004;279:48063–70. doi: 10.1074/jbc.M406467200. [DOI] [PubMed] [Google Scholar]
- 18.Lee J, Kanatsu-Shinohara M, Inoue K, et al. Akt mediates self-renewal division of mouse spermatogonial stem cells. Development. 2007;134:1853–9. doi: 10.1242/dev.003004. [DOI] [PubMed] [Google Scholar]
- 19.Armstrong L, Hughes O, Yung S, et al. The role of PI3K/AKT, MAPK/ERK and NFkappabeta signalling in the maintenance of human embryonic stem cell pluripotency and viability highlighted by transcriptional profiling and functional analysis. Hum Mol Genet. 2006;15:1894–913. doi: 10.1093/hmg/ddl112. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe S, Umehara H, Murayama K, Okabe M, Kimura T, Nakano T. Activation of Akt signaling is sufficient to maintain pluripotency in mouse and primate embryonic stem cells. Oncogene. 2006;25:2697–707. doi: 10.1038/sj.onc.1209307. [DOI] [PubMed] [Google Scholar]
- 21.Kim SJ, Cheon SH, Yoo SJ, et al. Contribution of the PI3K/Akt/PKB signal pathway to maintenance of self-renewal in human embryonic stem cells. FEBS Lett. 2005;579:534–40. doi: 10.1016/j.febslet.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 22.Tu Z, Ninos JM, Ma Z, et al. Embryonic and hematopoietic stem cells express a novel SH2-containing inositol 5′-phosphatase isoform that partners with the Grb2 adapter protein. Blood. 2001;98:2028–38. doi: 10.1182/blood.v98.7.2028. [DOI] [PubMed] [Google Scholar]
- 23.Muraille E, Dassesse D, Vanderwinden JM, et al. The SH2 domain-containing 5-phosphatase SHIP2 is expressed in the germinal layers of embryo and adult mouse brain: increased expression in N-CAM-deficient mice. Neuroscience. 2001;105:1019–30. doi: 10.1016/s0306-4522(01)00240-8. [DOI] [PubMed] [Google Scholar]
- 24.Kerr WG, Heller M, Herzenberg LA. Analysis of lipopolysaccharide-response genes in B-lineage cells demonstrates that they can have differentiation stage-restricted expression and contain SH2 domains. Proc Natl Acad Sci USA. 1996;93:3947–52. doi: 10.1073/pnas.93.9.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Damen JE, Liu L, Rosten P, et al. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc Natl Acad Sci USA. 1996;93:1689–93. doi: 10.1073/pnas.93.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lioubin MN, Algate PA, Tsai S, Carlberg K, Aebersold A, Rohrschneider LR. p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes & Development. 1996;10:1084–95. doi: 10.1101/gad.10.9.1084. [DOI] [PubMed] [Google Scholar]
- 27.Kavanaugh WM, Pot DA, Chin SM, et al. Multiple forms of an inositol polyphosphate 5-phosphatase form signaling complexes with Shc and Grb2. Curr Biology. 1996;6:438–45. doi: 10.1016/s0960-9822(02)00511-0. [DOI] [PubMed] [Google Scholar]
- 28.Lucas DM, Rohrschneider LR. A novel spliced form of SH2-containing inositol phosphatase is expressed during myeloid development. Blood. 1999;93:1922–33. [PubMed] [Google Scholar]
- 29.Lamkin TD, Walk SF, Liu L, Damen JE, Krystal G, Ravichandran KS. Shc interaction with Src homology 2 domain containing inositol phosphatase (SHIP) in vivo requires the Shc-phosphotyrosine binding domain and two specific phosphotyrosines on SHIP. J Biol Chem. 1997;272:10396–401. doi: 10.1074/jbc.272.16.10396. [DOI] [PubMed] [Google Scholar]
- 30.Huber M, Helgason CD, Damen JE, Liu L, Humphries RK, Krystal G. The src homology 2-containing inositol phosphatase (SHIP) is the gatekeeper of mast cell degranulation. Proc Natl Acad Sci USA. 1998;95:11330–5. doi: 10.1073/pnas.95.19.11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verdier F, Chretien S, Billat C, Gisselbrecht S, Lacombe C, Mayeux P. Erythropoietin induces the tyrosine phosphorylation of insulin receptor substrate-2. An alternate pathway for erythropoietin-induced phosphatidylinositol 3-kinase activation. J Biol Chem. 1997;272:26173–8. doi: 10.1074/jbc.272.42.26173. [DOI] [PubMed] [Google Scholar]
- 32.Zamorano J, Keegan AD. Regulation of apoptosis by tyrosine-containing domains of IL-4R alpha: Y497 and Y713, but not the STAT6-docking tyrosines, signal protection from apoptosis. J Immunol. 1998;161:859–67. [PubMed] [Google Scholar]
- 33.Zhang S, Broxmeyer HE. p85 subunit of PI3 kinase does not bind to human Flt3 receptor, but associates with SHP2, SHIP, a tyrosine-phosphorylated 100-kDa protein in Flt3 ligand-stimulated hematopoietic cells. Biochem Biophys Res Commun. 1999;254:440–5. doi: 10.1006/bbrc.1998.9959. [DOI] [PubMed] [Google Scholar]
- 34.Zhang S, Mantel C, Broxmeyer HE. Flt3 signaling involves tyrosyl-phosphorylation of SHP-2 and SHIP and their association with Grb2 and Shc in Baf3/Flt3 cells. J Leukocyte Biol. 1999;65:372–80. doi: 10.1002/jlb.65.3.372. [DOI] [PubMed] [Google Scholar]
- 35.Drachman JG, Kaushansky K. Dissecting the thrombopoietin receptor: functional elements of the Mpl cytoplasmic domain. Proc Natl Acad Sci USA. 1997;94:2350–5. doi: 10.1073/pnas.94.6.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature. 1996;383:263–6. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- 37.Galandrini R, Tassi I, Mattia G, et al. SH2-containing inositol phosphatase (SHIP-1) transiently translocates to raft domains and modulates CD16-mediated cytotoxicity in human NK cells. Blood. 2002;100:4581–9. doi: 10.1182/blood-2002-04-1058. [DOI] [PubMed] [Google Scholar]
- 38.Eissmann P, Beauchamp L, Wooters J, Tilton JC, Long EO, Watzl C. Molecular basis for positive and negative signaling by the natural killer cell receptor 2B4 (CD244) Blood. 2005 doi: 10.1182/blood-2004-09-3796. [DOI] [PubMed] [Google Scholar]
- 39.Abramson J, Pecht I. Clustering the mast cell function-associated antigen (MAFA) leads to tyrosine phosphorylation of p62Dok and SHIP and affects RBL-2H3 cell cycle. Immunol Lett. 2002;82:23–8. doi: 10.1016/s0165-2478(02)00013-5. [DOI] [PubMed] [Google Scholar]
- 40.Gays F, Martin K, Kenefeck R, Aust JG, Brooks CG. Multiple cytokines regulate the NK gene complex-encoded receptor repertoire of mature NK cells and T cells. J Immunol. 2005;175:2938–47. doi: 10.4049/jimmunol.175.5.2938. [DOI] [PubMed] [Google Scholar]
- 41.Tessmer MS, Fugere C, Stevenaert F, et al. KLRG1 binds cadherins and preferentially associates with SHIP-1. Int Immunol. 2007;19:391–400. doi: 10.1093/intimm/dxm004. [DOI] [PubMed] [Google Scholar]
- 42.Damen JE, Liu L, Wakao H, et al. The role of erythropoietin receptor tyrosine phosphorylation in erythropoietin-induced proliferation. Leukemia. 1997;11:423–5. [PubMed] [Google Scholar]
- 43.Lecoq-Lafon C, Verdier F, Fichelson S, et al. Erythropoietin induces the tyrosine phosphorylation of GAB1 and its association with SHC, SHP2, SHIP, phosphatidylinositol 3-kinase. Blood. 1999;93:2578–85. [PubMed] [Google Scholar]
- 44.Hunter MG, Avalos BR. Phosphatidylinositol 3′-kinase and SH2-containing inositol phosphatase (SHIP) are recruited by distinct positive and negative growth-regulatory domains in the granulocyte colony-stimulating factor receptor. J Immunol. 1998;160:4979–87. [PubMed] [Google Scholar]
- 45.Heo WD, Inoue T, Park WS, et al. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science. 2006;314:1458–61. doi: 10.1126/science.1134389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalesnikoff J, Baur N, Leitges M, et al. SHIP negatively regulates IgE + antigen-induced IL-6 production in mast cells by inhibiting NF-kappa B activity. J Immunol. 2002;168:4737–46. doi: 10.4049/jimmunol.168.9.4737. [DOI] [PubMed] [Google Scholar]
- 47.Strassheim D, Kim JY, Park JS, Mitra S, Abraham E. Involvement of SHIP in TLR2-Induced Neutrophil Activation and Acute Lung Injury. J Immunol. 2005;174:8064–71. doi: 10.4049/jimmunol.174.12.8064. [DOI] [PubMed] [Google Scholar]
- 48.Robson JD, Davidson D, Veillette A. Inhibition of the Jun N-terminal protein kinase pathway by SHIP-1, a lipid phosphatase that interacts with the adaptor molecule Dok-3. Mol Cell Biol. 2004;24:2332–43. doi: 10.1128/MCB.24.6.2332-2343.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell. 1997;90:293–301. doi: 10.1016/s0092-8674(00)80337-2. [DOI] [PubMed] [Google Scholar]
- 50.Zippo A, De Robertis A, Bardelli M, Galvagni F, Oliviero S. Identification of Flk-1 target genes in vasculogenesis: Pim-1 is required for endothelial and mural cell differentiation in vitro. Blood. 2004;103:4536–44. doi: 10.1182/blood-2003-11-3827. [DOI] [PubMed] [Google Scholar]
- 51.Wahle JA, Paraiso KH, Kendig RD, et al. Inappropriate Recruitment and Activity by the Src Homology Region 2 Domain-Containing Phosphatase 1 (SHP1) Is Responsible for Receptor Dominance in the SHIP-Deficient NK Cell. J Immunol. 2007;179:8009–15. doi: 10.4049/jimmunol.179.12.8009. [DOI] [PubMed] [Google Scholar]
- 52.Brauweile AM, Tamir I, Cambier JC. Bilevel control of B-cell activation by the inositol 5-phosphatase SHIP. Immunol Rev. 2000;176:69–74. doi: 10.1034/j.1600-065x.2000.00612.x. [DOI] [PubMed] [Google Scholar]
- 53.Helgason CD, Kalberer CP, Damen JE, et al. A dual role for Src homology 2 domain-containing inositol-5-phosphatase (SHIP) in immunity: aberrant development and enhanced function of b lymphocytes in ship −/− mice. J Exp Med. 2000;191:781–94. doi: 10.1084/jem.191.5.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sly LM, Rauh MJ, Kalesnikoff J, Song CH, Krystal G. LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity. 2004;21:227–39. doi: 10.1016/j.immuni.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 55.Karlsson MC, Guinamard R, Bolland S, Sankala M, Steinman RM, Ravetch JV. Macrophages control the retention and trafficking of B lymphocytes in the splenic marginal zone. J Exp Med. 2003;198:333–40. doi: 10.1084/jem.20030684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takeshita S, Namba N, Zhao JJ, et al. SHIP-deficient mice are severely osteoporotic due to increased numbers of hyper-resorptive osteoclasts. Nat Med. 2002;8:943–9. doi: 10.1038/nm752. [DOI] [PubMed] [Google Scholar]
- 57.Huber M, Helgason CD, Scheid MP, Duronio V, Humphries RK, Krystal G. Targeted disruption of SHIP leads to Steel factor-induced degranulation of mast cells. EMBO J. 1998;17:7311–9. doi: 10.1093/emboj/17.24.7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rauh MJ, Ho V, Pereira C, et al. SHIP represses the generation of alternatively activated macrophages. Immunity. 2005;23:361–74. doi: 10.1016/j.immuni.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 59.Rohrschneider LR, Custodio JM, Erson TA, Miller CP, Gu H. The intron 5/6 promoter region of the ship1 gene regulates expression in stem/progenitor cells of the mouse embryo. Dev Biol. 2005;283:503–21. doi: 10.1016/j.ydbio.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 60.Tangye SG, Lazetic S, Woollatt E, Sutherland GR, Lanier LL, Phillips JH. Cutting edge: human 2B4, an activating NK cell receptor, recruits the protein tyrosine phosphatase SHP-2 and the adaptor signaling protein SAP. J Immunol. 1999;162:6981–5. [PubMed] [Google Scholar]
- 61.McNerney ME, Guzior D, Kumar V. 2B4 (CD244) - CD48 interactions provide a novel MHC class I-independent system for NK cell self-tolerance in mice. Blood. 2005 doi: 10.1182/blood-2005-01-0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johansson S, Johansson M, Rosmaraki E, et al. Natural killer cell education in mice with single or multiple major histocompatibility complex class I molecules. J Exp Med. 2005;201:1145–55. doi: 10.1084/jem.20050167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim S, Poursine-Laurent J, Truscott SM, et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. 2005;436:709–13. doi: 10.1038/nature03847. [DOI] [PubMed] [Google Scholar]
- 64.Fernandez NC, Treiner E, Vance RE, Jamieson AM, Lemieux S, Raulet DH. A subset of natural killer cells achieves self-tolerance without expressing inhibitory receptors specific for self-MHC molecules. Blood. 2005;105:4416–23. doi: 10.1182/blood-2004-08-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trotta R, Parihar R, Yu J, et al. Differential expression of SHIP1 in CD56bright and CD56dim NK cells provides a molecular basis for distinct functional responses to monokine costimulation. Blood. 2005;105:3011–8. doi: 10.1182/blood-2004-10-4072. [DOI] [PubMed] [Google Scholar]
- 66.Morandi B, Costa R, Falco M, et al. Distinctive lack of CD48 expression in subsets of human dendritic cells tunes NK cell activation. J Immunol. 2005;175:3690–7. doi: 10.4049/jimmunol.175.6.3690. [DOI] [PubMed] [Google Scholar]
- 67.Vacca P, Pietra G, Falco M, et al. Analysis of natural killer cells isolated from human decidua: Evidence that 2B4 (CD244) functions as an inhibitory receptor and blocks NK-cell function. Blood. 2006;108:4078–85. doi: 10.1182/blood-2006-04-017343. [DOI] [PubMed] [Google Scholar]
- 68.Baldwin WM, 3rd, Larsen CP, Fairchild RL. Innate immune responses to transplants: a significant variable with cadaver donors. Immunity. 2001;14:369–76. doi: 10.1016/s1074-7613(01)00117-0. [DOI] [PubMed] [Google Scholar]
- 69.Maier S, Tertilt C, Chambron N, et al. Inhibition of natural killer cells results in acceptance of cardiac allografts in CD28−/− mice. Nat Med. 2001;7:557–62. doi: 10.1038/87880. [DOI] [PubMed] [Google Scholar]
- 70.Storb R. Allogeneic hematopoietic stem cell transplantation--yesterday, today, tomorrow. Exp Hematol. 2003;31:1–10. doi: 10.1016/s0301-472x(02)01020-2. [DOI] [PubMed] [Google Scholar]
- 71.Kosaka H, Surh CD, Sprent J. Stimulation of mature unprimed CD8+ T cells by semiprofessional antigen-presenting cells in vivo. J Exp Med. 1992;176:1291–302. doi: 10.1084/jem.176.5.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shlomchik WD, Couzens MS, Tang CB, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 1999;285:412–5. doi: 10.1126/science.285.5426.412. [DOI] [PubMed] [Google Scholar]
- 73.Teshima T, Ordemann R, Reddy P, et al. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nature Medicine. 2002;8:575–81. doi: 10.1038/nm0602-575. [DOI] [PubMed] [Google Scholar]
- 74.Taylor PA, Panoskaltsis-Mortari A, Swedin JM, et al. L-Selectin(hi) but not the L-selectin(lo) CD4+25+ T-regulatory cells are potent inhibitors of GVHD and BM graft rejection. Blood. 2004;104:3804–12. doi: 10.1182/blood-2004-05-1850. [DOI] [PubMed] [Google Scholar]
- 75.Kiel MJ, Morrison SJ. Maintaining hematopoietic stem cells in the vascular niche. Immunity. 2006;25:862–4. doi: 10.1016/j.immuni.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 76.Burdon T, Smith A, Savatier P. Signalling, cell cycle and pluripotency in embryonic stem cells. Trends Cell Biol. 2002;12:432–8. doi: 10.1016/s0962-8924(02)02352-8. [DOI] [PubMed] [Google Scholar]