

Abstract
Nuclear receptors are ligand-dependent transcription factors that are of interest as potential tools to artificially regulate gene expression. Ligand binding induces a conformational change involving helix-12 which forms part of the dimerization interface used to bind transcriptional co-activators. When triiodothyronine (T3) binds the thyroid hormone receptor (TR) it indirectly contacts helix-12 through intermediary residues His(435) and Phe(451) termed a His-Phe switch. The mutant TRβ(H435A) is non-responsive to physiological concentrations of T3 but can be activated by the synthetic hormone analog QH2 which potently activates His435→Ala mutant at concentrations that do not activate the wild-type receptors TRα and TRβ. QH2 does not show antagonist behavior with the wild-type TRs. QH2’s functionally orthogonal behavior with TRβ(H435A) is preserved on the three consensus thyroid hormone response elements.
Nuclear hormone receptors (NHRs) are ligand-dependent transcription factors that regulate gene transcription in response to small molecule hormones. NHRs directly bind DNA sequences in hormone responsive genes and activate transcription in response to hormone. NHRs that have been engineered to uniquely respond to synthetic ligands are of interest as artificial gene regulatory systems. Several approaches have been used to engineer NHRs including screening molecular libraries for ligands that match non-functional receptor mutants or by selecting libraries of receptor mutations that impart response to otherwise inactive ligand analogs. 1–5 The highly flexible nature of NHR ligand binding domains make it difficult to construct highly specific ligand-receptor pairs as mutant receptors often retain significant activity towards their natural hormones and ligand analogs often have activities with endogenous receptors. Here we describe a highly selective, “functionally-orthogonal,” ligand-thyroid hormone receptor (TR)-ligand pair generated by targeting receptor residues directly involved in the receptors ligand-dependent transactivation allosterism. The site of receptor modification, identified by examining naturally occurring mutations to TR associated with genetic disease, should provide a general strategy for generating highly selective ligand-receptor pairs from NHRs.
NHRs are composed of two fundamental functional domains; a DNA binding domain that uniquely targets an NHR to a specific subset of hormone-responsive genes and a ligand-binding domain which selectively binds hormone and activates gene transcription. Upon binding hormone, the NHR ligand-binding domain undergoes a conformational change that leads to the release of co-repressor proteins and the recruitment of transcriptional co-activators.6–8 Helix-12 of the ligand-binding domain plays a central role in controlling the ligand-induced receptor allosterism as it has been observed to undergo a substantial conformational change on ligand binding and to form a part of the receptor/co-activator interface. The proper positioning of helix-12 is required to create a proper dimerization interface for “LXXLL” NR-box sequences of transcriptional co-activators (Scheme 1A). The co-crystal structures of NHR ligand-binding domains with antagonists suggest that many antagonists block transcriptional activation by preventing helix-12 from adopting its active agonist conformation.9,10
Scheme 1. Ligand-induced conformational change of nuclear receptors.
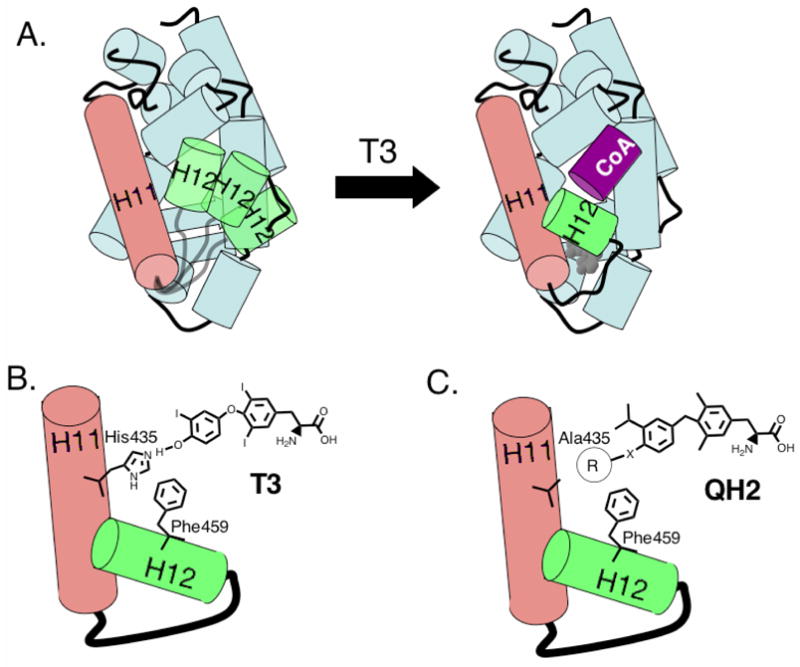
A. Hormone binding repositions helix-12 (H12, green) to create a dimerization interface for transcriptional co-activators containing a conserved “LXXLL” NR-box (purple). B. In the His-Phe switch of TRβ, T3 hydrogen bonds to His435, which interacts directly with Phe459 on helix-12. C. The transducer role of His435 may be replaced with appropriate hydrophobic groups.
Although the conformation of helix-12 is highly dependent on the structure of the bound ligand, surprisingly, most of the known NHR ligand-binding domain structures indicate that the ligand does not directly contact helix-12 but rather the ligand interacts with helix-12 through intermediary residues that bridge between the ligand and helix-2. For example, in the T3-TRβ co-crystal structure, the 4′-hydroxyl of T3 hydrogen bonds to His435 that in turn forms favorable interactions (either pi-pi or edge-face interactions) with Phe459 a critical residue on helix-12 (Scheme 1B). This general motif was first recognized in LXR involving His435 and Trp457, and was termed a His-Trp switch or in the case of TR, a His-Phe switch.11 A similar His-Phe switch is found in TRα and in VDR involving His397 and Phe422 (Figure 1).12,13 Mutations to these residues that are directly involved in the mechanism of ligand-induced allosterism can have a dramatic effect on hormone responsiveness to these receptors. For example, mutation to the His member of His-Trp/His-Phe switches as in LXRβ(H435A), LXRα(H421A) and VDR(H305Q) or mutation to the Trp/Phe residues as in LXRα(W443F) and TRβ(F459C), TRβ(F459A) cause inactivity or show severely reduced activity compared to wild-type.11,14–16 The rickets-associated mutant VDR(H305Q) is of particular interest because the Gln residue retains the potential to hydrogen bond to the ligand. Although the natural ligand, 1,25-dihydroxy vitamin D3, binds VDR(H305Q) with near wild-type affinity in vitro, it is substantially less potent (80-fold less potent) in cellular transcription assays indicating that the transduction mechanism is specifically impaired.16
Figure 1.
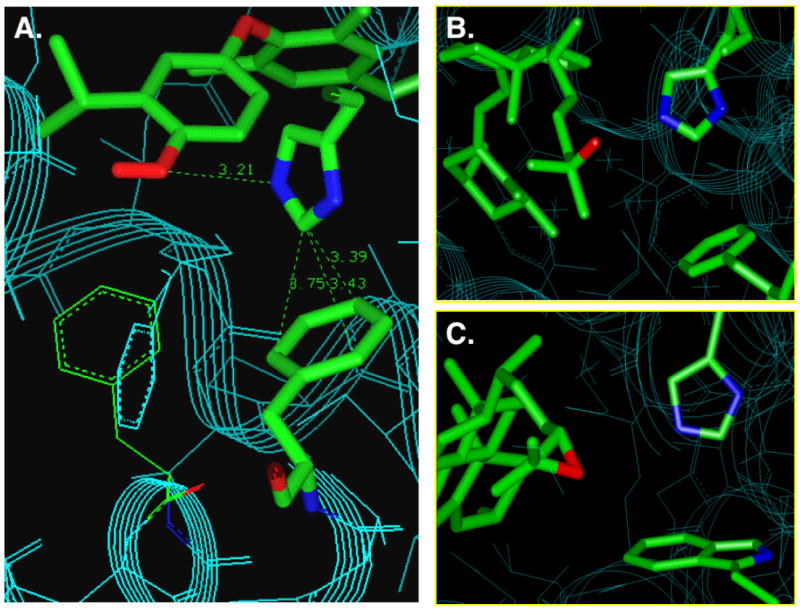
The His-Trp and His-Phe switch serves as a transducer to relay ligand binding information to the AF-2 domain of nuclear receptors. a. TRα,13 b. VDR,12 and c. LXR.17
Several groups have developed analog-selective forms of hormone receptors as ligand-inducible regulators of gene expression by screening ligand libraries to match mutant receptors or by applying selection/directed evolution methods to identify receptors that respond to otherwise inactive ligand analogs.2,4,5,18,19 One of the challenges associated with generating orthogonal ligand-receptor pairs from NHRs is finding mutations that efficiently prevent activation by the endogenous hormone without otherwise compromising the receptor’s structure or intrinsic ability to activate gene transcription. A few groups have used genetic selections in yeast to identify mutant receptors that selectively respond to synthetic ligands, however, the ligand’s selectivity for the mutant versus the wild-type receptor can change substantially when these ligand-receptor pairs are transferred to mammalian cell lines.1,4,5 In some cases, receptors selected for their ability to respond to synthetic ligands, may also retain significant activity for their natural ligands.4
For consideration as artificial transcriptional regulators, engineered receptors should not be responsive to physiological concentrations of the natural hormone but respond to concentrations of a synthetic ligand analog that does not activate wild-type (or other endogenous) receptors. We have termed ligand-receptor pairs that match this practical definition for functioning independent of the endogenous hormone-receptor pair as “functionally orthogonal.”2,20 Though of potential academic interest, the change in the receptor’s selectivity for the synthetic versus natural ligand (i.e. the receptor’s “ligand selectivity”) will have little practical importance if the concentrations of synthetic ligand needed to activate the engineered receptors also activate endogenous receptors. Provided that a modified receptor is no longer responsive to endogenous concentrations of the natural ligand, the synthetic ligand’s receptor selectivity, defined as EC50(mutant)/EC50(wt) for a given ligand represents a practical measurement of selectivity of a system. By this criterion ligand-receptor pairs developed by directed evolution strategies do not show better receptor selectivity than the best systems developed by rational design.2 This suggests that judicious choice of the site of receptor modification is necessary for any ligand-receptor engineering strategy.
As part of our program to generate small molecules that rescue function to mutant NHRs associated with human genetic diseases,21–24 we have developed thyroid hormone receptor analogs that complement mutant forms of TRβ associated with the genetic disease RTH (resistance to thyroid hormone). RTH is characterized by a hyposensitivity to thyroid hormone, triiodothyronine (T3), in peripheral tissues. The vast majority of RTH-associated mutations show dominant-negative inheritance indicating that the presence of one mutant copy of the receptor is able to compete with, and directly interfere with the function of wild-type copies of the receptor through the formation of inactive receptor dimers.8,25 The dominant negative activity of RTH-associated TR mutants imply that these receptors have a relatively stable structure that can bind DNA and form receptor dimers but are functionally impaired in their ability to bind ligand and recruit co-activators.
Surprisingly, many TRβ mutants associated with RTH are only mildly less responsive to T3 than wild-type TRβ(TRβ(wt)). For example, some of the most common RTH-associated mutants, TRβ(R320C), TRβ(R320H) and TRβ(M310T), under investigation in our laboratory show less than a 10-fold reduction in responsiveness to T3 compared to wild-type, TRβ(wt).21,22,26 Evidently, even relatively modest changes in hormone responsiveness are sufficient to upset the normal balance of peripheral tissue responsiveness and hormone synthesis needed to cause RTH.
In general, nuclear receptors appear to accommodate a wide range of mutations without gross impairment of receptor function. Even when ligand potency is significantly reduced by receptor mutations, ligands often show full efficacy though at higher ligand concentrations.27 Similarly, NHRs can often accommodate a range of ligand structures that would initially appear to cause steric clashes with receptor side-chains.28,29 In contrast, some RTH-associated mutations are known to have dramatic effects on receptor function. Mutations to His435, His435→Tyr, His435→Leu and His435→Gln, have been shown to have large effects on hormone responsiveness and have been identified in RTH patients as well as in TSH secreting pituitary adenoma (TSHoma) tumors.30,31 TRβ(H435Y) has strongly refractory response to T3 and is 230-times less responsive towards T3 than TRβ(wt). TRβ(H435L) is completely devoid of cellular activity within experimentally accessible concentrations of T3.30 Examination of the T3-TRβ(wt) co-crystal structure,32 does not suggest any obvious structural perturbations associated with these mutations. Rather, mutations to His435, a part of TR’s His-Phe switch, may disrupt the normal molecular transduction of the ligand-binding signal to helix-12. Similar magnitude effects were observed with mutations to Phe459 which is the interacting partner of His435 located on helix-12.14,15 The low responsiveness of the mutations at His435 prompted us to exploit this critical transducer residue to create highly selective ligand-receptor pairs from TRβ.
We have generated the His435→Ala mutation in TRβ (pSG5hTRβ) using oligonucleotide directed site-specific mutagenesis and evaluated its activity in transiently transfected HEK293 cells that were cotransfected with pSG5hTRβ(H435A), the hormone responsive promoter DR4-Luc+ and pRLbasic, a renilla luciferase control. The alanine substitution was selected as a cavity forming mutation that would maintain a high helical propensity at position 435, which is centrally located in helix-11. Reporter gene assays conducted with this mutation demonstrate that TRβ(H435A) is 769-fold less responsive to T3 (EC50=392±84 nM), than TRβ(wt) (EC50 = 0.51±0.05 nM). The synthetic halogen-free thyromimic GC-1 is more than 2380-fold less potent with TRβ(H435A) (EC50 >5000 nM) than with TRβ(wt) (EC50 = 2.1 nM) (Figure 2). Although the TRβ(H435A) is still responsive to extremely high concentrations of T3, the mutant is no longer responsive to physiological concentrations of T3, which are generally well below 2 nM (total T3 = 2 nM; free T3 3–8 pM),6 suggesting that TRβ (H435A) is non-responsive to physiological concentrations of T3.
Figure 2.
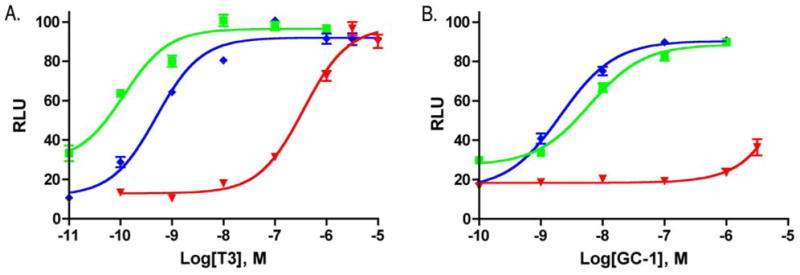
Cellular dose response behavior of
 TRα(wt),
TRα(wt),
 TRβ(wt) and τ TRβ(H435A) towards A. T3 and B. GC-1.
TRβ(wt) and τ TRβ(H435A) towards A. T3 and B. GC-1.
We envisioned that the His-Phe switch found in TRβ(wt) which uses both hydrogen bonding and edge-pi aromatic interactions could be replaced by an appropriate hydrophobic cluster (Scheme 1C). Based on a site-model generated from an appropriately modified TRβ/T3 co-crystal structure, we designed two series of potential, mutant-selective analogs of the halogen-free thyromimetic GC1. The general synthetic strategy is based on the synthesis of Scanlan et al.33,34 Three 4′-O-alkyl substituted ethers of GC-1, QH1, QH2 and QH3 were synthesized from the corresponding 2-isopropylphenol ethers (Scheme 2).
Scheme 2. Synthesis of alkoxy analogs.
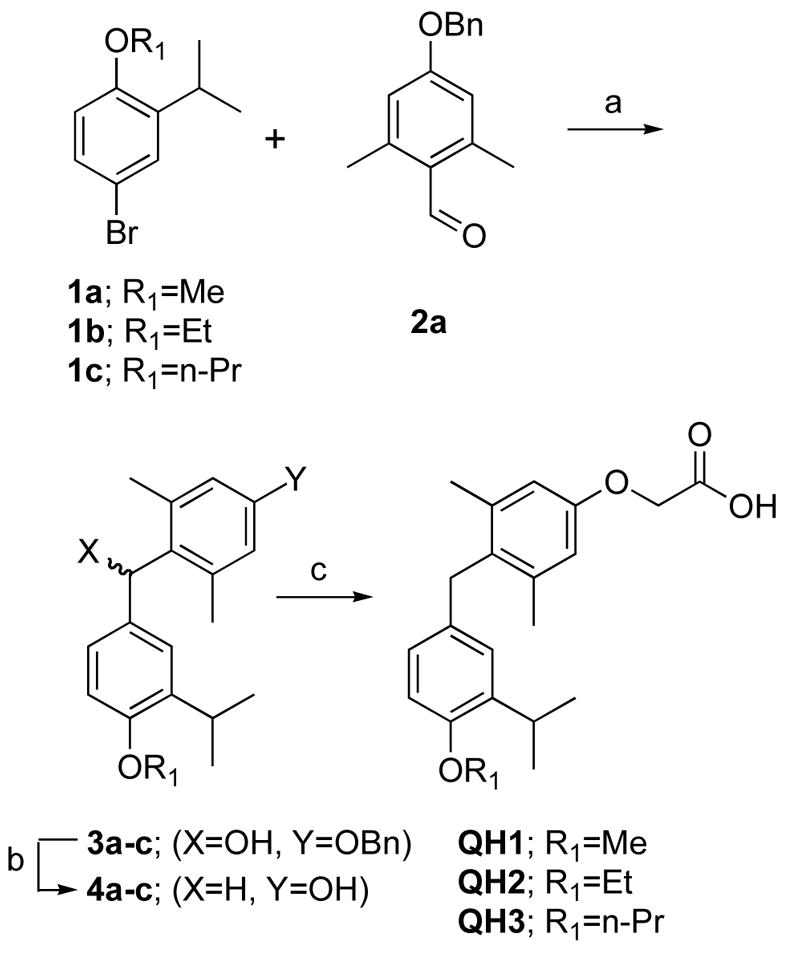
(a) n-BuLi, THF, −78°C (b) H2, 10%Pd/C, 9% AcOH/EtOH (c) Cs2CO3, BrCH2CO2R3, DMF, then NaOH, MeOH.
Similarly, 4′-alkyl substituted analogs of GC-1 were synthesized via Kumada coupling of the intermediate aryl triflate 7 (Scheme 3). While the synthesis of QH4, QH5 and QH7 by this route was straight-forward, Kumada coupling with the more stericly demanding isopropyl grignard afforded 8c which contained the proteo-analog 8e as an inseparable impurity. The “benzene” analog QH9 was synthesized independently by another route and was found to be inactive in cell based assays with TRβ(H3435A) (Scheme 3). Therefore, QH6 was synthesized and evaluated as an 85% mixture of QH6 and QH9.
Scheme 3. Synthesis of alkyl substituted analogs.
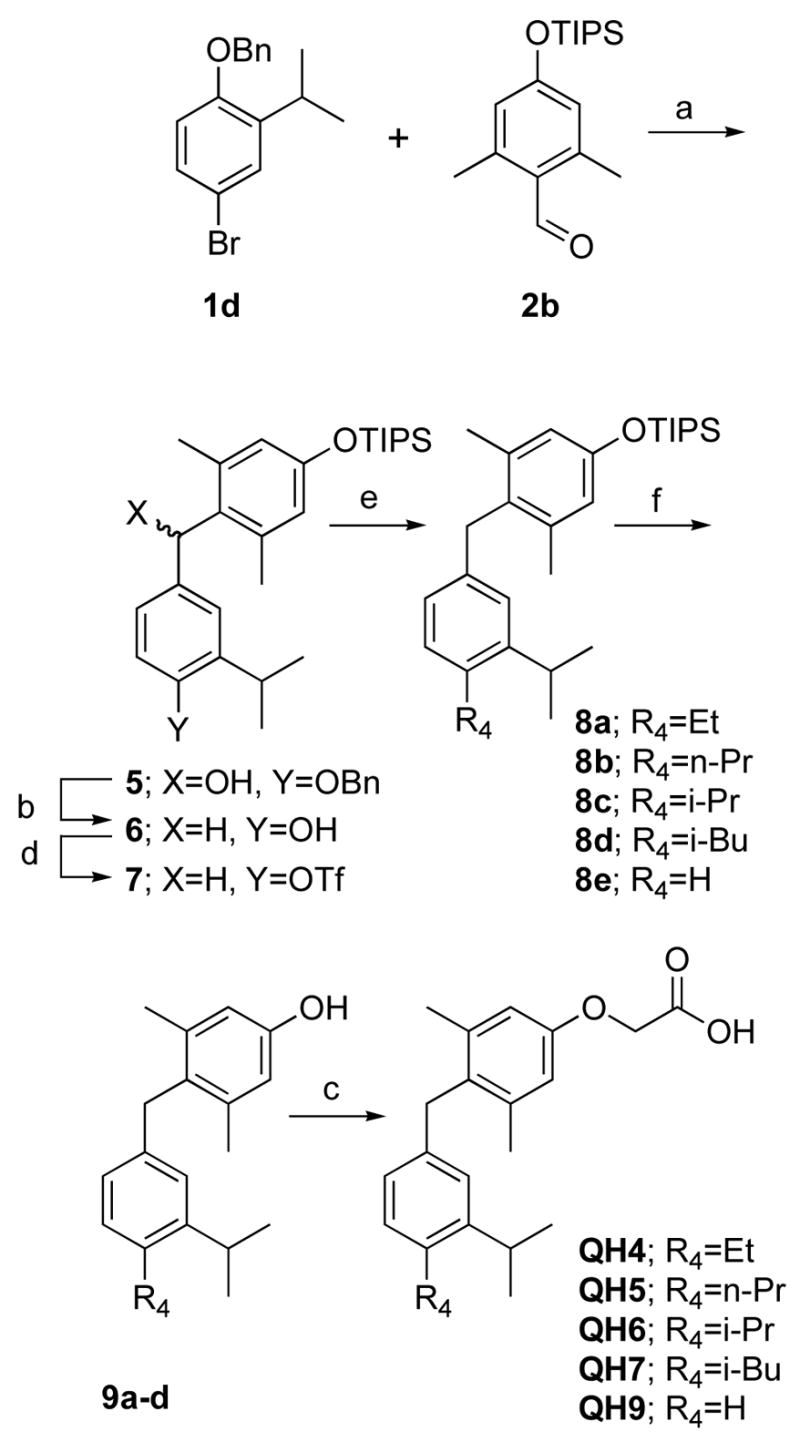
a) n-BuLi, THF, −78°C (b) H2, 10%Pd/C, 9% AcOH/EtOH (c) Cs2CO3, BrCH2CO2R3, DMF, then NaOH, MeOH (d) K2CO3, DMF, p-NO2PhOTf (e) PdCl2(dppf), R4MgCl (f) TBAF, THF.
The analogs QH1–QH7 were evaluated in cell based reporter gene assays. All of the analogs show some activity below one micromolar with the mutant TRβ(H435A), however, only QH2, the O-ethyl ether of GC1, has low nanomolar activity (EC50=6.4±0.57) with the Ala mutation (Figure 3). QH2 is 61-fold more potent than T3 and 778-fold more potent than the parent analog GC1 with the mutant TRβ(H435A). The structure appears to be quite sensitive to the exact size of the alkoxy subtituent at the 4′ position as the methoxy substituted analog QH1 (EC50= 55 nM) is 9-times less potent with half the efficacy (45%). The propoxy substituted analog QH3 (EC50=130 nM) is also significantly less active than QH2. Interestingly, the isosteric 4′-propyl analog QH5 is not very potent suggesting that the ether oxygen may still be important for high affinity binding to the mutant. The propyl and isopropyl substituted analogs, QH5 and QH6, are very poor agonists for TRβ(H435A), however, the isobutyl analog, QH7 (EC50 = 123 nM), is slightly more active.
Figure 3.
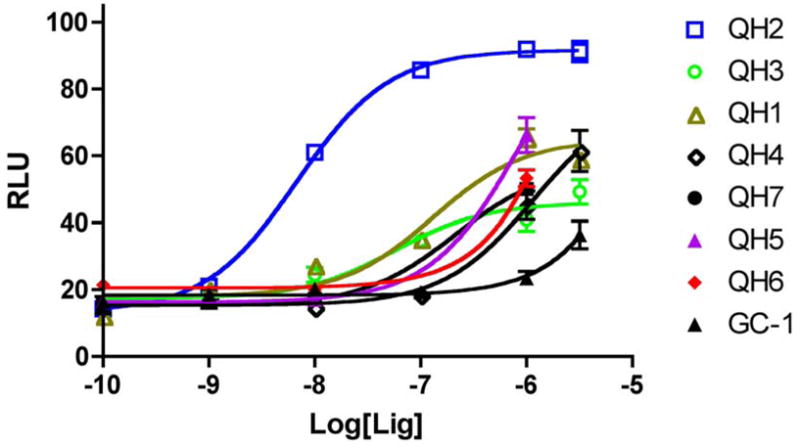
Cellular response of synthetic ligands towards TRβ(H435A).
Our ability to rescue the His435→Ala mutant of TRβ prompted us to examine if these same ligands may be able to rescue related RTH mutants, TRβ(H435L), TRβ (H435Y). Cell based reporter gene assays indicate that QH1 thru QH7 were devoid of activity below 5 uM (data not shown). The lack of activity observed with any of our ligands with these related His-to-hydrophobic mutations further suggest that successful rescue at this position requires a precise match and size, shape and functionality.
The mutant TRβ(H435A) is not responsive to physiological concentrations of T3. As a potential tool for independently regulating gene expression, it is important to further demonstrate that the concentrations of QH2 used to fully activate the mutant TRβ(H435A) do not activate either of the two known TR subtypes; TRβ(wt) or TRα(wt). Cell based reporter gene assays show that QH2 is a weak agonist for both TRα(wt) (EC50 = 737 nM) and TRβ(wt) (EC50 = 341 nM). QH2 is therefore 53-fold selective for the mutant TRβ(H435A) over the TRβ(wt) and 115-times more potent than hTRα(wt). Concentrations of QH2 that almost fully activate with TRβ(H435A) do not significantly activate the endogenous receptor TRβ(wt) or TRα (wt) (Figure 4a, Table 1).
Figure 4.
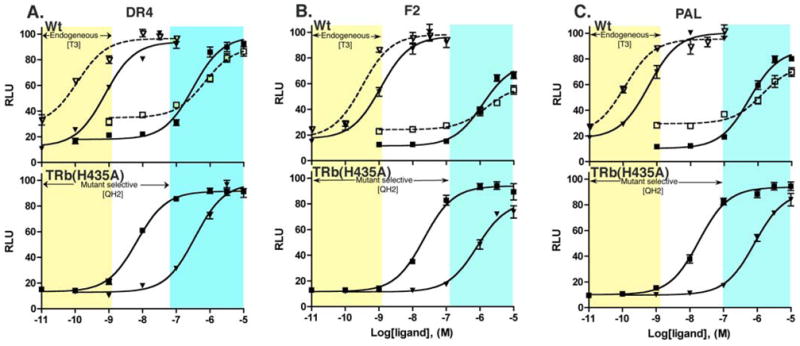
Comparative dose response behavior of QH2 with TRα (wt), TRβ(wt) and TRβ(H435A) on three consensus TRE reporters. ∇: TRα+T3, ○: TRα+QH2, τ: TRβ+T3, ν: TRβ+QH2.
Table 1.
Potencies and Efficacies of T3 and Synthetic Ligands for TRβ(H435A)
| Ligand | Structure | EC50 nM (% efficacy) | Ligand | Structure | EC50 nM (% efficacy) |
|---|---|---|---|---|---|
| T3 |
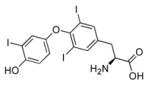
|
392±84 (100) | QH4 |
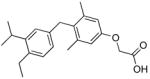
|
>1000 (~75) |
| GC-1 |
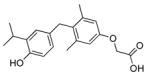
|
>5000 (~40) | QH5 |
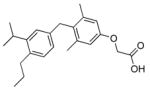
|
>1000 (~68) |
| QH1 |
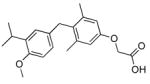
|
55±9.3* (45) | QH6 |
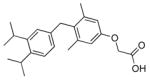
|
>1000 (~52) |
| QH2 |
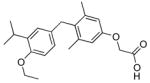
|
6.4±0.6 (93) | QH7 |
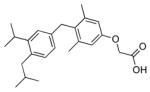
|
123±16* (69) |
| QH3 |
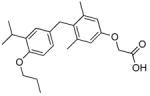
|
130±13* (65) | QH9 |
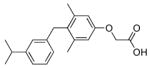
|
>10000 (~36) |
Cellular reporter gene assays in HEK293 cells transiently transfected with pSG5TRβ(H435A), DR4-Luc+. Potencies are measured as ± SEM except as indicated.
Potencies of weak agonists are reported as ± min/max from two separate experiments performed in triplicate.
Thyroid hormone receptor can act on hormone responsive genes bearing several consensus types of thyroid hormone response elements in their promoters.8 The DR4 promoter is the most common thyroid hormone response element (TRE), however, it is also important to evaluate the potency, efficacy and selectivity of our compounds on the other common TREs, PAL and F2. Reporter gene assays conducted using F2-luc and PAL-luc promoters show that QH2 retains mutant selectivity, potency and efficacy on genes controlled by these common TREs. The actions of QH2 on the TRβ(H435A) mutant were also found to be functionally orthogonal on two other common thyroid hormone response elements, F2 and PAL (Figure 4B and 4C). Furthermore, QH2 does not compete with T3 in TRβ(wt) demonstrating that QH2 is not an antagonist and only serves as a potent and selective agonist for the engineered mutant (Figure 5, Table 2).
Figure 5.
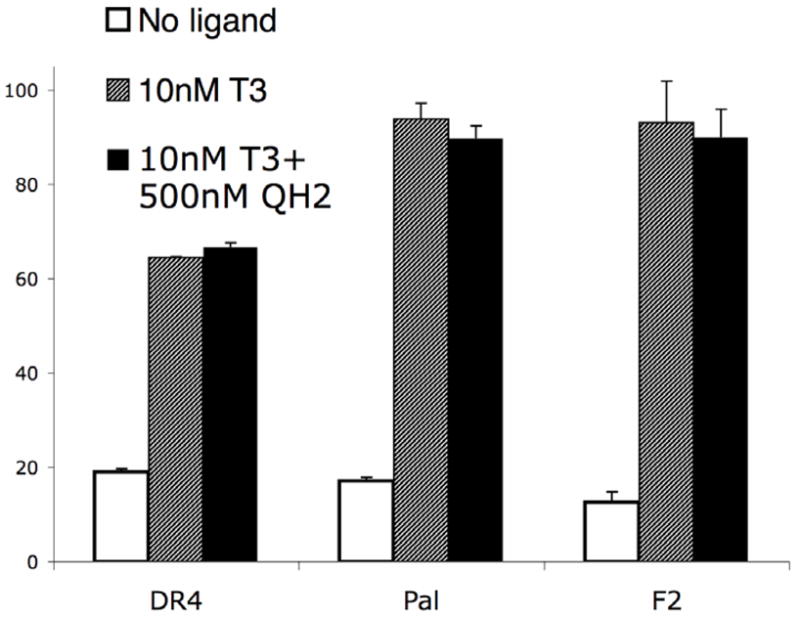
Cellular response of T3 activated TR towards QH2.
Table 2.
Summary of activities on three consensus promotors.
| TRE | Receptor | T3, nM (% efficacy) | QH2, nM (% efficacy) | GC-1, nM (% efficacy) |
|---|---|---|---|---|
| DR4 | TRα | 0.12±0.03 (100) | 737±59.0 (90) | 6.2±1.6 (88) |
| TRβ | 0.51±0.05 (100) | 341±25 (98) | 2.1±0.9 (90) | |
| TRβ(H435A) | 392±84 (97) | 6.43±0.57 (93) | >5000 (40) | |
|
| ||||
| F2 | TRα | 0.25±0.08 (100) | 1596±84 (76) | n.d. |
| TRβ | 0.86±0.11 (100) | 1240±122 (58) | n.d. | |
| TRβ(H435A) | 774±53 (82) | 27.9±7.1 (93) | n.d. | |
|
| ||||
| Pal | TRα | 0.12±0.04 (100) | 1439±142 (75) | n.d. |
| TRβ | 0.44±0.05 (100) | 594±37 (87) | n.d. | |
| TRβ (H435A) | 747±119 (90) | 22.9±5.9 (93) | n.d. | |
% efficacy: maximum inducible activity compared to TR(wt) with T3. n.d.: not determined
Compared to other known mutations in the TRβ ligand binding pocket, mutations to His435 which disrupt the receptors “His-Phe switch” represent a hypersensitive region of the ligand binding pocket that can be effectively manipulated to generate highly selective ligand-receptor pairs. In this example, receptor function is greatly impaired (769-fold) by His435→Ala mutation, which is almost completely rescued by the synthetic ligand QH2 which is more than 777-fold more potent than the parent analog GC-1. Significantly, though a similar motif is used to relay ligand binding to helix-12 in several other nuclear receptors, the use of hydrogen bonding and edge-pi aromatic interactions to act as transducers of ligand binding to AF-2 domain is not essential and can be replaced with a hydrophobic cluster. Subtle structural modifications of the ligand at this position lead to large changes in ligand potencies and efficacies.
His435-Phe459 switch of TRβ is only part of a complex network of molecular interactions involved in ligand-dependent switching of TRβ to its active conformation. Therefore, even though T3 is 769-fold less potent in TRβ(H435A) than TRβ(wt), it is still a full agonist in the mutant. It is important to recognize that ligand binding and activity are energetically coupled as has been shown that the presence of co-activators can affect ligand off-rates 35 and observed cellular potencies (EC50’s).36 Here we target residues involved in transactivation but are in principle effecting binding activity as well. The low affinity of T3 for TRβ(H435A) made determination of QH2’s binding affinity by standard radio-ligand displacement assays intractable. His435 plays a role in both ligand binding and transactivation function and QH2 is a very weak agonist towards TRβ(wt) and does not show antagonist activity with TRβ(wt).
The selectivites observed with QH2 are not as good as the best analogs identified through multiple rounds of selection which have reported selectivites of 10–300 fold or the best systems developed by design (up to 400-fold). For example, Schwimmer et al., identified a RXR mutant that was 300-times more responsive to the synthetic ligand LG335 than wild-type RXR, however the selected receptor retains substantial activity with the natural ligand 9-cis-retinoic acid.37 Zhao et al. reported that in HEC-1 cells, dihydroxybenzil (DHB) had a receptor selectivity of 178-fold with a modified estrogen receptor (ER) mutant identified through multiple rounds of selection. This mutant identified through selection also exploited residues at the interface between helix-11 and helix-12. Interestingly, this same ligand exhibited only 8-fold receptor selectivity between this same mutant and wild-type in the original yeast strain from which it was identified. The 53-fold receptor selectivity of QH2 for TRβ(H435A) is above the median selectivity reported for other single point mutation rescues reported in the literature.
Mutant-selective ligands (or substrates) for engineered proteins are an important chemical biological tool for the study of cellular function. The design of functionally orthogonal ligand-receptor pairs from flexible proteins such as NHRs presents a unique challenge in molecular design. Steric-based strategies or “bump-and-hole” engineered ligand-receptor pairs often fail to provide receptors that no longer respond to endogenous ligands or ligands that do not also activate endogenous receptors.20
A variety of strategies have been used to develop mutant selective ligands including structure-based ligand design as well as selection/directed evolution strategies. Regardless of the design strategy, it is critical to judiciously select the site of receptor modification so as to effectively abolish response to the natural ligand without disrupting the intrinsic function of the receptor.
Here we describe a new ligand-receptor design strategy based on targeting critical residues involved in the ligand-dependent transactivation mechanism. Of note, His435 mutants of TRβ have been reported in human pituitary cancer as well as resistance to thyroid hormone, suggesting that similar ligand design strategies might someday be used to rescue receptor function associated with these important disease states.
Supplementary Material
Supporting Information Available: Experimental procedures and spectroscopic characterization of compounds available.
Acknowledgments
We thank the National Institutes of Health for financial support of this work (NIH RO1DK54257). Q.H. was supported in part by a University of Delaware Competitive Graduate Fellowship.
References
- 1.Whelan J, Miller N. J Steroid Biochem Mol Biol. 1996;58:3–12. doi: 10.1016/0960-0760(96)00010-6. [DOI] [PubMed] [Google Scholar]
- 2.Shi YH, Koh JT. J Am Chem Soc. 2002;124:6921–6928. doi: 10.1021/ja016897x. [DOI] [PubMed] [Google Scholar]
- 3.Tedesco R, Thomas JA, Katzenellenbogen BS, Katzenellenbogen JA. Chem Biol. 2001;8:277–287. doi: 10.1016/s1074-5521(01)00006-0. [DOI] [PubMed] [Google Scholar]
- 4.Schwimmer LJ, Rohatgi P, Azizi B, Seley KL, Doyle D. Proc Natl Acad Sci U S A. 2004;101:14707–14712. doi: 10.1073/pnas.0400884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chockalingam K, Chen ZL, Katzenellenbogen JA, Zhao HM. Proc Natl Acad Sci USA. 2005;102:5691–5696. doi: 10.1073/pnas.0409206102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hulbert AJ. Biol Rev. 2000;75:519–631. doi: 10.1017/s146479310000556x. [DOI] [PubMed] [Google Scholar]
- 7.Katzenellenbogen JA, Katzenellenbogen BS. Chem Biol. 1996;3:87–94. doi: 10.1016/s1074-5521(96)90143-x. [DOI] [PubMed] [Google Scholar]
- 8.Yen PM. Physiol Rev. 2001;81:1097–1142. doi: 10.1152/physrev.2001.81.3.1097. [DOI] [PubMed] [Google Scholar]
- 9.Wurtz JM, Bourguet W, Renaud JP, Vivat V, Chambon P, Moras D, Gronemeyer H. Nat Struct Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- 10.Weatherman RV, Fletterick RJ, Scanlan TS. Annu Rev Biochem. 1999;68:559–581. doi: 10.1146/annurev.biochem.68.1.559. [DOI] [PubMed] [Google Scholar]
- 11.Williams S, Bledsoe RK, Collins JL, Boggs S, Lambert MH, Miller AB, Moore J, McKee DD, Moore L, Nichols J, Parks D, Watson M, Wisely B, Willson TM. J Biol Chem. 2003;278:27138–27143. doi: 10.1074/jbc.M302260200. [DOI] [PubMed] [Google Scholar]
- 12.Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. Mol Cell. 2000;5:173–179. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]
- 13.Wagner R, Apriletti JW, McGarth ME, West BL, Baxter JD, Fletterick RJ. Nature. 1995;378:690–697. doi: 10.1038/378690a0. [DOI] [PubMed] [Google Scholar]
- 14.Cheng SY, Ransom SC, Mcphie P, Bhat MK, Mixson AJ, Weintraub BD. Biochemistry. 1994;33:4319–4326. doi: 10.1021/bi00180a028. [DOI] [PubMed] [Google Scholar]
- 15.Tone Y, Collingwood TN, Adams M, Chatterjee VK. J Biol Chem. 1994;269:31157–31161. [PubMed] [Google Scholar]
- 16.Gardezi SA, Nguyen C, Malloy PJ, Posner GH, Feldman D, Peleg S. J Biol Chem. 2001;276:29148–29156. doi: 10.1074/jbc.M100898200. [DOI] [PubMed] [Google Scholar]
- 17.Wilkinson JR, Towle HC. J Biol Chem. 1997;272:23824–23832. doi: 10.1074/jbc.272.38.23824. [DOI] [PubMed] [Google Scholar]
- 18.Koh JT, Putnam M, Tomic-Canic M, McDaniel CM. J Am Chem Soc. 1999;121:1984–1985. [Google Scholar]
- 19.Doyle DF, Braasch DA, Jackson LK, Weiss HE, Boehm MF, Mangelsdorf DJ, Corey DR. J Am Chem Soc. 2001;123:11367–11371. doi: 10.1021/ja0164632. [DOI] [PubMed] [Google Scholar]
- 20.Koh JT. Chemistry & Biology. 2002;9:17–23. doi: 10.1016/s1074-5521(02)00087-x. [DOI] [PubMed] [Google Scholar]
- 21.Ye HF, O’Reilly KE, Koh JT. J Am Chem Soc. 2001;123:1521–1522. doi: 10.1021/ja003442j. [DOI] [PubMed] [Google Scholar]
- 22.Shi Y, Ye H, Link KH, Putnam MC, Hubner I, Dowdel S, Koh JT. Biochem J. 2005;44:4612–4626. doi: 10.1021/bi0482349. [DOI] [PubMed] [Google Scholar]
- 23.Swann SL, Bergh J, Farach-Carson MC, Ocasio CA, Koh JT. J Am Chem Soc. 2002;124:13795–13805. doi: 10.1021/ja0268377. [DOI] [PubMed] [Google Scholar]
- 24.Swann SL, Bergh JJ, Farach-Carson MC, Koh JT. Org Lett. 2002;4:3863–3866. doi: 10.1021/ol0266931. [DOI] [PubMed] [Google Scholar]
- 25.Yen PM. Trends Endocrin Met. 2003;14:327–333. doi: 10.1016/s1043-2760(03)00114-0. [DOI] [PubMed] [Google Scholar]
- 26.Furlanetto TW, Kopp P, Peccin S, Gu WX, Jameson JL. Mol Genet Metab. 2000;71:520–526. doi: 10.1006/mgme.2000.3088. [DOI] [PubMed] [Google Scholar]
- 27.Shi XB, Ma AH, Xia L, Kung HJ, White RWD. Cancer Res. 2002;62:1496–1502. [PubMed] [Google Scholar]
- 28.Katzenellenbogen JA, Muthyala R, Katzenellenbogen BS. Pure Appl Chem. 2003;75:2397–2403. [Google Scholar]
- 29.Koh JT, Biggins JB. Curr Top Med Chem. 2005;5:413–420. doi: 10.2174/1568026053828420. [DOI] [PubMed] [Google Scholar]
- 30.Nomura Y, Nagaya T, Tsukaguchi H, Takamatsu J, Seo H. Endocrinology. 1996;137:4082–4086. doi: 10.1210/endo.137.10.8828460. [DOI] [PubMed] [Google Scholar]
- 31.Ando S, Sarlis NJ, Oldfield EH, Yen PM. J Clin Endocrinol Metab. 2001;86:5572–5576. doi: 10.1210/jcem.86.11.7984. [DOI] [PubMed] [Google Scholar]
- 32.Wade CB, Robinson S, Shapiro RA, Dorsa DM. Endocrinology. 2001;142:2336–2342. doi: 10.1210/endo.142.6.8071. [DOI] [PubMed] [Google Scholar]
- 33.Chiellini G, Nguyen NH, Yoshihara HAI, Scanlan TS. Bioorg Med Chem Lett. 2000;10:2607–2611. doi: 10.1016/s0960-894x(00)00531-x. [DOI] [PubMed] [Google Scholar]
- 34.Chiellini G, Apriletti JW, al Yoshihara H, Baxter JD, Ribeiro RC, Scanlan TS. Chem Biol. 1998;5:299–306. doi: 10.1016/s1074-5521(98)90168-5. [DOI] [PubMed] [Google Scholar]
- 35.Gee AC, Carlson KE, Martini PGV, Katzenellenbogen BS, Katzenellenbogen JA. Mol Endocrinol. 1999;13:1912–1923. doi: 10.1210/mend.13.11.0373. [DOI] [PubMed] [Google Scholar]
- 36.Szapary D, Huang Y, Simons SS., Jr Mol Endocrinol. 1999;13:2108–2121. doi: 10.1210/mend.13.12.0384. [DOI] [PubMed] [Google Scholar]
- 37.Schneekloth JS, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM. J Am Chem Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Experimental procedures and spectroscopic characterization of compounds available.