

Abstract
The human nuclear receptor pregnane X receptor (PXR) responds to a wide variety of potentially harmful chemicals and coordinates the expression of genes central to xenobiotic and endobiotic metabolism. Structural studies reveal that the PXR ligand binding domain (LBD) uses a novel sequence insert to form a homodimer unique to the nuclear receptor superfamily. Terminal β-strands from each monomeric LBD interact in an ideal antiparallel fashion to bury potentially exposed surface β-strands, generating a ten-stranded intermolecular β-sheet. Conserved tryptophan and tyrosine residues lock across the dimer interface and provide the first tryptophan-zipper (Trp-Zip) interaction observed in a native protein. We show using analytical ultracentrifugation that the PXR LBD forms a homodimer in solution. We further find that removal of the interlocking aromatic residues eliminates dimer formation but does not affect PXR's ability to interact with DNA, RXRα, or ligands. Disruption of the homodimer significantly reduces receptor activity in transient transfection experiments, however, and effectively eliminates the receptor's recruitment of the transcriptional coactivator SRC-1 both in vitro and in vivo. Taken together, these results suggest that the unique Trp-Zip-mediated PXR homodimer plays a role in the function of this nuclear xenobiotic receptor.
The human pregnane X receptor PXR plays a important role in controlling the expression of genes central to drug and endobiotic metabolism, including those encoding cytochrome P450s (CYPs), UDP-glucuronosyl-transferases, glutathione-S-transferases, and drug efflux pumps (1-5). PXR is considered to be a master regulator of the expression of CYP 3A4 isoform, which metabolizes more than 50% of human drugs (6). PXR is expressed largely in the liver and intestines and responds to a wide variety of structurally distinct endobiotic and xenobiotic compounds, including pregnenolone, progesterone, lithocholic acid, paclitaxel, rifampicin, and the St. John's wort constituent hyperforin (7-11). The activation of this xenobiotic sensor has also been linked to clinically-relevant drug interactions. For example, in patients taking the unregulated herbal antidepressant St. John's wort, which contains the potent PXR agonist hyperforin (10, 11), the upregulation of drug metabolism genes has been observed to generate significant decreases in the serum levels of therapeutics including oral contraceptives, anti-viral compounds, and immunosuppressant (12-15).
PXR is a member of the nuclear receptor (NR) superfamily of ligand-activated transcription factors, which includes receptors for estrogen, progesterone, retinoid and thyroid hormones as well as retinoids, cholesterol metabolites and vitamins. Many nuclear receptors bind to dual DNA response elements of various arrangements as either homodimers or as heterodimers with the retinoid X receptor-alpha (RXRα) (16, 17). In the absence of activating ligand, NRs have been shown to associate with transcriptional corepressors, which down-regulate gene expression by a variety of mechanisms including histone deacetylation (17, 18). In response to an activating ligand, however, NRs interact with transcriptional coactivators that up-regulate target gene expression in part by histone acetylation and by facilitating the recruitment of the basal transcriptional machinery (17, 18). PXR functions as a heterodimer with RXRα and has been shown to bind to a variety of dual DNA response elements arranged as direct and everted repeats. Upon ligand activation, PXR recruits several of the p160-class of transcriptional coactivators, including the steroid receptor coactivator-1 (SRC-1) (7-9, 19). For its potent control of CYP3A4 expression, PXR has been shown to employ two DNA response elements, one proximal (bases −172 to −149) and one distal (bases −7836 to −7607) relative to the start site of transcription. Both are required for maximal induction of gene expression in response to ligands (20). PXR has also been shown to regulate the expression of MDR1 and CYP isoform 2B6 by using a combination of proximal and distal DNA response elements (3, 21).
The PXRs of known sequence contain a ∼50-amino acid insert unique to members of the nuclear receptor superfamily. This region is located between helices 1 and 3 within the canonical NR LBD fold, and adds a novel helix 2 and two β-strands adjacent to PXR's ligand binding cavity. Numerous crystal structures of the human PXR LBD have also revealed that the novel β-turn-β motif of this insert extends the two- to three-stranded antiparallel β-sheet common to NRs to a five-stranded β-sheet in PXR (22-25). It is the terminal β-strands in each of these β-sheets that associate in an antiparallel fashion to generate the PXR homodimer, which produces a ten-strand intermolecular antiparallel β-sheet (Figure 1A). No other nuclear receptor has been observed to homodimerize in this fashion. In this work, structural, biophysical and functional features of this PXR homodimer are examined. Using sedimentation equilibrium experiments, the PXR LBD is shown to form a homodimer in solution with a Kd of 4.5 μM. Key residues at the dimer interface are also mutated and shown to disrupt formation of the PXR dimer, which significantly reduces transcriptional activity and coactivator recruitment without impacting other necessary receptor actions like RXRα, DNA and ligand binding. Taken together, the data presented suggest that the Trp-Zip-mediated PXR homodimer interface plays a potential role in receptor function.
Figure 1.


A. The PXR LBD homodimer as observed in the crystal structure of the complex of the protein with SR12813 (24). B. Detailed view of the PXR LBD homodimer interface, rotated 180° about the vertical axis relative to Figure 1A. Novel β1′-strands from each monomer are shown in green and cyan. Inter-locking tryptophan and tyrosine residues from each monomer are rendered in green and cyan, respectively. Hydrogen bonding interactions are indicated (dashed or dotted lines), as are Van der Waals contacts (solid arrows).
EXPERIMENTAL PROCEDURES
PXR Expression and Purification
Wild-type human PXR LBD (residues 130−434) was coexpressed with a fragment of SRC-1 (residues 623−710) in E. coli BL21 (DE3) and purified using nickel-affinity chromatography as previously described (22). The Trp223Ala/Tyr225Ala PXR LBD double-mutant was generated using the QuikChange mutagenesis kit (Stratagene), and expressed under the same conditions as wild-type PXR, but formed inclusion bodies in E. coli. The inclusion body pellet was washed twice with buffer containing 0.5% Triton X-100, 20 mM Tris-Cl pH 7.5, 250 mM NaCl, 50 mM imidazole, and 5% glycerol. Following the Triton X-100 wash, the pellet was resuspended in 6 M guanidinium hydrochloride pH 7.5 with the addition of 10 mM β-mercaptoethanol (BME), and stirred at 4 °C for 30 min. The denatured protein was ultra-centrifuged at 28.8K rpm for 30 min, diluted 1:3 with buffer (20 mM Tris-Cl pH 7.5, 250 mM NaCl, 50 mM imidazole, 10 mM BME and 5% glycerol) and then refolded by dialysis against this buffer with four changes. The refolded mutant protein was then purified under the same conditions as wild-type PXR LBD. In preparation for analytical ultracentrifugation, protein samples were concentrated to ∼2.0 mg / ml and dialyzed (1:1000 (v/v) protein to dialysate) overnight with two buffer changes. The dialysis buffer (20mM Tris-Cl pH 7.5, 250mM NaCl, 2.5 mM EDTA, 5mM BME and 5% glycerol) was used to dilute protein to relevant concentrations. The cholesterol drug SR12813 (Sigma) was added at a 4-fold molar excess.
Analytical Ultracentrifugation
Sedimentation equilibrium experiments were performed using a Beckman XL-A analytical ultracentrifuge equipped with scanning absorption optics. Equilibrium measurements were obtained at three different rotor speeds (9,000, 13,000 and 16,000 rpm) and three concentrations (8.6, 17.3 and 21.7 μM) for wild-type PXR LBD and Trp223Ala/Tyr225Ala PXR LBD in triplicate. Baseline absorbance offsets were established by increasing the rotor speed to 45,000 rpm for 6 hrs. Sedimentation equilibrium data was analyzed using the Beckman XL-A/XL-I data Analysis Software Version 4.0 which uses a nonlinear curve fitting procedure to determine the weight-average monomer molecular weight M and the association constant Ka according to the following equation: where cr is the concentration at radial position r, cmon,r0 is the concentration of the monomer at the reference radius r0 , ω is the angular velocity in radians per second, R is the universal gas constant (8.314 × 107 erg·mol−1·K−1), T is the temperature in Kelvin, M is the monomer molecular weight, is the partial specific volume, ρ is the density of the solvent, and Ka is the association constant, and E is the baseline offset. The association constant, Ka was converted to the dissociation constant Kd by the following equation: where b is the path length (1.2 cm) and ε is the molar extinction coefficient (28,390 M−1 cm−1 for PXR LBD) determined using the program Protean ™.
Circular Dichroism Spectropolarimetry
To confirm that the Trp223Ala/Tyr225Ala double-mutant form of the PXR LBD was properly folded, circular dichroism spectropolarimetry (CD) was performed using an Applied Photophysics PiStar-180 CD spectropolarimeter. The ellipticity from 210−300 nm was measured for wild-type PXR LBD and for the Trp223Ala/Tyr225Ala PXR LBD double-mutant. Both proteins were at 0.2 mg ml−1 in 100 mM phosphate buffer, pH 7.8, 100 mM NaCl and 5% glycerol. To examine thermal melting temperatures, the temperature was ramped from 20 to 98 °C while monitoring the ellipticity at 222 nm. Plots of fraction denatured versus temperature were produced by defining the upper and lower temperature baselines as 0 and 100%, respectively. Melting temperatures (Tm's) were defined as the point at which 50% of the sample denatured. Trials were performed in triplicate, and Tm's for individual runs were averaged and standard errors calculated.
Transient Transfection Assays
Mutations in full-length PXR were generated with the Stratagene QuikChange site directed mutagenesis kit according to the manufacturer's instructions. Transfections were performed as described previously (20, 22). Briefly, CV-1 cells were plated in 96-well plates in phenol red-free Dulbecco's modified Eagle's medium containing high glucose and supplemented with 10% charcoal/dextran treated fetal bovine serum (HyClone, Logan, UT). Transfection mixes contained 5 ng of receptor expression vector, 20 ng of reporter plasmid, 12 ng of β-actin SPAP as internal control, and 43 ng of carrier plasmid. Plasmids for wild-type and mutant forms of human PXR and for the XREM-CYP3A4-LUC reporter, containing the enhancer and promoter of the CYP3A4 gene driving Luciferase expression, were as previously described (20). Transfections were performed with LipofectAMINE (Life Technologies, Inc., Grand Island, NY) essentially according to the manufacturer's instructions. Drug dilutions of rifampicin (Sigma, St. Louis, MO) and SR12813 (synthesized in-house) were prepared in phenol red-free Dulbecco's modified Eagle's medium/F-12 medium with 15 mM HEPES supplemented with 10% charcoal-stripped, delipidated calf serum (Sigma, St. Louis, MO) which had previously been heat-inactivated at 62°C for 35 minutes. Serial drug dilutions were performed in triplicate to generate 11-point concentration response curves. Cells were incubated for 24 hours in the presence of drugs, after which the medium was sampled and assayed for alkaline phosphatase activity. Luciferase reporter activity was measured using the LucLite assay system (Packard Instrument Co., Meriden, CT) and normalized to alkaline phosphatase activity. EC50 values were determined by standard methods.
Immunocytochemistry
CV-1 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) plus 10% charcoal-stripped calf serum (Hyclone, Logan, UT). The day before transfection, cells were plated at 2 × 105 cells per well of a 6 well plate (Becton Dickinson, Franklin Lakes, NJ) on ethanol-washed glass cover slips (Fisher Scientific, Pittsburgh, PA). Transfection was carried out using Effectene (Qiagen, Valencia, CA) according to the manufacturer's specifications, with plasmids expressing wild-type PXR, Trp223Ala/Tyr225Ala PXR, or carrier DNA. Transfection complexes were suspended in phenol red-free DMEM/F12 plus 10% charcoal-stripped, delipidated calf serum (Sigma, St. Louis, MO), and left in contact with the cells overnight. The medium was then replaced, drugs [10 μM rifampicin (Sigma, St. Louis, MO) or 2 μM SR12813 (synthesized in house)] or 0.1% DMSO (Sigma, St. Louis, MO) were added, and the incubation was continued for a further 6 hours. For immunofluorescent staining of exogenous proteins, cultures were placed on ice for 5 minutes, rinsed 3 times with cold PBS, then the cover slips were immersed in ice-cold acetone for 5 minutes and air-dried. Nonspecific antibody binding was blocked by incubating for 10 minutes in PBS containing 10% normal donkey serum (Jackson Immunoresearch Labs, West Grove, PA). Goat anti-PXR (Santa Cruz Biotechnology, Santa Cruz, CA) was applied in blocking buffer for 1 hour at a dilution of 1:100. The secondary antibody, donkey anti-goat IgG labeled with ALEXA 488 (Molecular Probes, Eugene, OR), was also applied for one hour in blocking buffer, but at a dilution of 1:1,000. Cultures stained without primary antibody were also obtained. Cover slips were mounted in 90% glycerol (Sigma, St. Louis, MO), 10% PBS, 4% n-propyl gallate (Sigma, St. Louis, MO) and 0.2 μM Hoechst 33258 (Molecular Probes, Eugene, OR). Fluorescent images were obtained on a Zeiss Axiovert 100 TV inverted microscope with a 100x objective under oil immersion, using Zeiss Axiovision software (Carl Zeiss, Inc., Thornwood, NY).
Gel Mobility Shift Assays
Gel mobility shift assays were performed as described before (26). Full-length human wild-type PXR, double-mutant (Trp223Ala/Tyr225Ala) PXR, and RXRα proteins were synthesized using the TNT quick-coupled in vitro transcription/translation system (Promega). Probes NR3 and ER6 from CYP2B6 and CYP3A4, respectively, were labeled with [γ-32P]dATP and purified by Microspin G-25 columns (Amersham Biosciences). Typically, 10 μL of binding reactions contained 10 mM HEPES (pH 7.6), 0.5 mM dithiothreitol, 15% glycerol, 0.05% Nonidet P-40, 50 mM NaCl, 2 μg of poly(dI-dC), with 0.1, 0.5, or 1 μL of in vitro translated nuclear receptor protein, and 4 X 104 cpm of labeled probe. After incubation at room temperature for 10 min, reaction mixtures were resolved on 5% acrylamide gels in 1 X Tris-acetic acid, EDTA buffer at 180 V for 1.5 h. Afterward, gels were dried, and autoradiography was performed overnight at −70 °C.
Competition Ligand Binding Assay
Polylysine YiO imaging beads (Amersham, GE Healthcare) were coated with histidine-tagged WT PXR LBD or Trp223Ala/Tyr225Ala PXR LBD, by mixing for 60 minutes at room temperature, in Tris buffer pH 8.0. Non-specific binding sites were blocked with a ten-fold excess of BSA for an additional 60 minutes at room temperature. The bead/receptor mix was washed and reconstituted in fresh assay buffer (50 mM Tris pH 8.0 containing 10% glycerol, 200mM KCl, 50uM CHAPS, 0.1 mg/mL BSA and 2mM DTT). Biotin (0.1 mM) was added to the suspension and allowed to mix for a further 60 minutes. The blocked receptor bead-mix was centrifuged at 1000 × g for 10 minutes at 4°C. The supernatant was discarded and the receptor-bead pellet was re-suspended in the appropriate volume of assay buffer. [N-methyl-3H]-GW0438X (synthesized at GSK and custom labeled at Amersham Biosciences, UK) was added to this suspension to achieve a final concentration of 10 nM. The receptor/imaging bead/radioligand mix was added directly to test compounds in 384-well plates in a one step addition. Test compounds were prepared from powder stocks by dissolving in DMSO and then serially diluted for displacement curves. Displacement of 10 nM [N-methyl-3H]-GW0438 was measured in a Viewlux 1430 ultraHTS microplate imager (Perkin Elmer Wallac Inc). Non-specific binding was determined in the presence of 10 μM GW0438. A similar competitive ligand binding assay method is described elsewhere (27). Data analysis was achieved using a 3 parameter fit, assuming a slope of 1. The data were calculated as pIC50's, but because the ligand concentration was well below the Kd for the receptor, this value was not different from the pKi. The pKi is the – log of Ki, the inhibitor concentration at which 50% inhibition is observed. Ki is calculated from the IC50 using the Cheng-Prusoff equation:
where L = concentration of free radioligand used in the assay and Kd = dissociation constant of the radioligand for the receptor .
Pull-Down Assays
Pull-down studies were performed using a Profound Pull-Down kit (Promega) at 4 °C according to the manufacturer's instructions. For the PXR-RXRα interaction, the RXRα-LBD (residues 225−462) was cloned into the pMALCH10T vector, expressed in E.coli BL21pLysS cells and purified by nickel affinity chromatography as described elsewhere (28). Purified RXRα-LBD was biotinylated using an EZ-Link Sulfo-NHS-LC-Biotinylation kit (Promega) according to the manufacturer's instructions. Biotinylated RXRα-LBD at 0.2 mg / mL was immobilized on streptavidin-agarose beads. The beads were washed with TBS buffer (25 mM Tris-Cl pH 7.0, 150mM NaCl), then blocked with biotin solution. The beads were equilibrated with binding buffer (50mM Tris-Cl pH 7.8, 250mM NaCl, 2.5 mM EDTA, and 5% glycerol) and wild-type (WT) PXR LBD or Trp223Ala/Tyr225Ala PXR LBD at 0.2 mg / mL was added to the beads and incubated for 12 hrs. Following prey capture, the beads were washed with binding buffer, eluted with elution buffer at pH 2.8, and examined by SDS-PAGE. For the PXR-SRC-1 peptide interaction, biotinylated SRC-1 or random peptides at 0.2 mg / mL were immobilized on streptavidin-agarose beads. The beads were washed with TBS buffer (25 mM Tris-Cl pH 7.0, 150mM NaCl), then blocked with biotin solution. The beads were equilibrated with binding buffer (50mM Tris-Cl pH 7.8, 250mM NaCl, 10% glycerol, 0.5% Triton X-100) and WT PXR LBD or Trp223Ala/Tyr225Ala PXR LBD at 0.2mg / mL was added to the beads and incubated for 12 hrs. Following prey capture, the beads were washed with binding buffer, eluted with buffer at pH 2.8, and examined by SDS-PAGE.
Mammalian Two-Hybrid Studies
HepG2 cells were cultured in minimal essential medium (MEM) (Invitrogen) containing 10% fetal bovine serum albumin supplemented with 0.1 mM non-essential amino acids and 1 mM sodium pyruvate. 1000 ng of VP16_PXR (full-length WT PXR or Trp223Ala/Tyr225Ala PXR), 1000 ng of pM_SRC-1 NRID (nuclear receptor interaction domain I; SRC-1 residues 621−765), 900 ng 5xGal4Luc3 reporter plasmid (29) and 100 ng pCMVB-gal (for normalization) were used for transfections performed in triplicate. Transfections were achieved with Lipofectin according to the manufacturer's instructions (Invitrogen). Equal levels of PXR and RXRα protein expression in transfected HepG2 cells was confirmed by Western analysis (data not shown). Cells were treated with 1 μM SR12813 sixteen hrs after transfection. Twenty-four hours after ligand treatment, cells were lysed and assayed for luciferase and β-galactosidase activity (30).
RESULTS
The PXR LBD Forms a Unique Homodimer
The human PXR ligand binding domain (PXR LBD) forms either a crystallographic or non-crystallographic homodimer in all structures determined to date (22-25). The homodimer interface is formed in large part by the β1′ strands from each monomer, which interact in an ideal anti-parallel fashion to generate a ten-stranded intermolecular β-sheet (Figures 1A,B) (24). The β1 and β1′ strands of the PXR LBD are part of the ∼50 amino acid insert novel to the PXRs relative to other members of the nuclear superfamily. In addition to six main-chain to main-chain intermolecular hydrogen bonds, interdigitating tryptophan (Trp223) and tyrosine (Tyr225) residues from each monomer lock across the dimer interface (Figure 1B). It has been shown that tryptophan and tyrosine residues tend to cluster at protein-protein interaction “hot spots” (31, 32). Pro175 from the loop that follows α1 helps to bury these aromatic side chains, and forms a hydrogen bond between its main-chain carbonyl oxygen and the indol nitrogen on Trp223. The residues involved in this dimer interface are largely conserved in the PXRs of known sequence, including those from human, rhesus monkey, pig, dog, rabbit, mouse and rat. The only exception is dog PXR, which contains a glutamine in place of Trp223; however, glutamine in this position could still hydrogen bond with Pro175 and interdigitate with Tyr225. The formation of the PXR homodimer buries 1,610 Å2 of solvent accessible surface area, which is sufficient to suggest physiological relevance (33).
The PXR LBD Forms a Homodimer in Solution
Sedimentation equilibrium experiments were performed with wild-type PXR LBD to determine whether the homodimer observed in crystal structures is also formed in solution. Experimental data were collected at three speeds (9,000, 13,000 and 16,000 rpm) and three protein concentrations (8.6, 17.3 and 21.7 μM) using a Beckman XL-A analytical ultracentrifuge. When data were fit to a single species model, the molecular weight determined was 67.2 kDa (for n = 9 data sets), which is nearly 2 times the molecular weight of the monomer PXR LBD (36.2 kDa; Table I). The subsequent application of a monomer-dimer equilibrium model produced more random residuals and provided the optimal fit for the experimental data, and generated a dissociation constant of 4.5 ± 0.8 μM (Figure 2A). Experiments repeated in the presence of the PXR agonist SR12813 yielded a similar Kd value of 3.9 ± 1.2 μM using the same monomer-dimer equilibrium model.
Table I.
Sedimentation Equilibrium Results for PXR LBDs
| Mw,calc for PXR LBD = 36,200 Da | ||
|---|---|---|
| Mw,app (Da) | Kd (× 10−6 M) | |
| WTPXR* | 67,200 ± 3500 | 4.5 ± 0.8 |
| WTPXR + SR12813 | 71,100 ± 2100 | 3.9 ± 1.2 |
| WTPXR + 10-fold peptide | 70,050 ± 1397 | 5.3 ± 0.9 |
| WTPXR + 20-fold peptide | 50,317 ± 3022 | 42.8 ± 13.9 |
| Trp223Ala/Tyr225Ala PXR | 36,300 ± 981 | N.A. |
WTPXR: wild-type PXR.
N.A.: not applicable.
Figure 2.


Sedimentation equilibrium data for wild-type and Trp223Ala/Tyr225Ala forms of the human PXR LBD. Each graph indicates a single concentration (left: 8.6 μM, center: 17.3 μM, right: 21.7 μM) collected at 9,000 rpm (circles), 13,000 rpm (squares) and 16,000 rpm (triangles). A. Sedimentation equilibrium data for wild-type PXR. The data in the figure was fit to a monomer-dimer equilibrium model (solid lines) with the residuals for each fit shown in the upper panels. B. Sedimentation equilibrium data obtained for Trp223Ala/Tyr225Ala mutant PXR. The data in the figure were fit to a singles species model (solid lines) with the residuals for each fit shown above.
We next examined the impact that replacing the interlocking aromatic residues at the dimer interface with alanines would have on receptor LBD dimerization. Thus, a Trp223Ala/Tyr225Ala PXR LBD double-mutant was tested by analytical ultracentrifugation using the same speeds and protein concentrations. These data fit well to a single species model and indicated a measured molecular weight of 36.3 kDa, nearly identical to the calculated molecular weight for the PXR LBD of 36.2 kDa (Figure 2B). Possibly due to its inability to form a stabilizing dimer, the Trp223Ala/Tyr225Ala double-mutant form of the PXR LBD formed inclusion bodies during protein expression in E. coli cells and had to be refolded using guanidinium hydrochloride to conduct these ultracentrifugation studies. To confirm that this refolded double-mutant form of the PXR LBD was properly folded, we performed circular dichroism (CD) spectropolarimetry experiments. The CD spectrum from 210−300 nm for the Trp223Ala/Tyr225Ala PXR LBD double-mutant was identical to the spectrum of the wild-type PXR LBD (data not shown), indicating that both proteins have the same secondary structural features. We also measured the melting temperatures (Tm) of the wild-type and double-mutant forms of the PXR LBD using CD spectropolarimetry monitored at 222 nm. The Tm's of wild-type PXR LBD and the Trp223Ala/Tyr225Ala PXR LBD double-mutant were 43.0 ± 0.8 and 39.8 ± 0.5, respectively (data not shown). These data indicate that the overall fold and stability of the double-mutant and wild-type forms of the PXR LBD are similar. Indeed, as shown below, the same refolded PXR double-mutant LBD was able to bind to RXRα LBD in vitro, which further supports the conclusion that it retains a wild-type structure overall. Taken together, these results establish that mutation of the interdigitating aromatic residues at the PXR dimer interface eliminates dimer formation in solution.
We also conducted sedimentation equilibrium experiments in the presence of a peptide of the sequence NH3-GSVWNYKP-CO2, which mimics the dimer interface in PXR. Data analysis indicated that PXR dimerization was partially inhibited by the presence of this peptide. The molecular weight of the PXR LBD measured in the presence of 10-fold molar excess peptide was 70.0 kDa, whereas the molecular weight determined in the presence of 20-fold molar excess peptide (172, 346, or 434 μM) was 50.3 kDa (Table I). Thus, the PXR LBD homodimer interaction can be disrupted in solution using relatively high concentrations of an eight amino acid peptide corresponding in sequence to the dimer interface.
Dimer Interface Residues and Transcription
To examine the impact of dimer interface mutations on PXR function, Trp223Ala and Tyr225Ala alterations were introduced into full-length PXR and the activation of a luciferase reporter gene under control of the CYP3A4 promoter was examined in CV-1 cells. As expected, robust up-regulation in these transient transfection experiments was observed for wild-type PXR in the presence of the agonists SR12813 and rifampicin (Figures 3A,B). However, all mutant forms of the receptor were found to be significantly reduced in their ability to respond to ligands, and exhibited no basal (ligand-independent) transcriptional activation. While the single-site mutant Trp223Ala was found to be more responsive to SR12813 and rifampicin than the Tyr225Ala single mutant, the Trp223Ala/Tyr225Ala double-mutant exhibited little response to SR12813 and essentially no response to rifampicin (Figures 3A,B). These results indicate that mutations that eliminate PXR homodimer formation significantly reduce the ability of the receptor to upregulate gene expression in ligand-dependent and -independent fashions.
Figure 3.


Transient transfections in CV-1 cells using a luciferase reporter construct and wild-type or mutant forms of full-length human PXR. Responses in the presence of increasing concentrations of (A) SR12813, and (B) rifampicin, are shown.
To confirm the proper sub-cellular trafficking of the Trp223Ala/Tyr225Ala double-mutant form of PXR, immunocytochemistry techniques were employed in CV-1 cells, the same cell type used for transfection assays. Wild-type full-length PXR translocated to the nucleus of CV-1 cells both in the absence of agonist and in the presence of either rifampicin or SR12813 (Figure 4A). Similarly, the Trp223Ala/Tyr225Ala double-mutant PXR was also found to translocate to the nucleus in the absence of ligand and in the presence of either rifampicin or SR12813 (Figure 4A). These results confirm that the dramatic changes in transcriptional activity observed for the mutant forms of PXR in transient transfection assays are not caused by improper subcellular localization relative to wild-type PXR.
Figure 4.

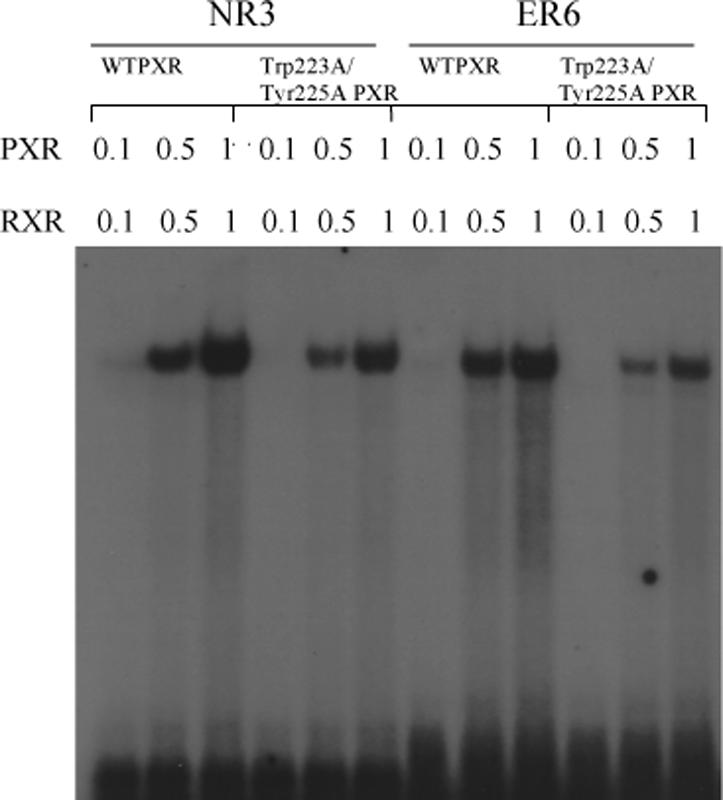

A. Full-length wild-type PXR (WTPXR) and Trp223Ala/Tyr225Ala double-mutant PXR are competent for nuclear translocation in CV-1 cells. Fluorescence due to specific staining of exogenous WTPXR or Trp223Ala/Tyr225Ala PXR in transfected CV-1 cells was concentrated in nuclei, as confirmed by colocalization with the nuclear stain Hoechst 33258 (not shown). Neither untransfected cells nor transfected cells treated with secondary antibody only were stained (not shown). The general distribution of PXR and Trp223Ala/Tyr225Ala PXR within the nuclei was not observed to differ in the presence of either rifampicin or SR12813. B. Full-length PXR/RXRα heterodimers containing wild-type or double mutant (Trp233Ala/Tyr225Ala) PXR bind to CYP2B6 and CYP3A4 responsive elements. In vitro translated wild-type or Trp223Ala/Tyr225Ala PXR (0.1, 0.5 or 1 μL) were combined with equal amounts of RXRα protein and incubated with NR3 (caTGGACTttccTGACCCca) or ER6 (ataTGGACTcaaaggAGGTCAgtg) elements from CYP2B6 and CYP3A4, respectively. Oligonucleotides were labeled with [γ-32P]dATP, and mobility shift assays were performed as described in “Experimental Procedures”. C. Trp223Ala/Tyr225Ala double-mutant PXR LBD is competent to bind to the LBD of RXRα. Wild-type or double-mutant PXR LBD's were incubated with biotin-labeled RXRα LBD in the absence and presence of the PXR agonist SR12813 at 50 μM. RXR was then immobilized on streptavidin beads, and the beads extensively washed. Bound proteins were eluted and examined by SDS-PAGE, and the bands for the 36.2 kDa PXR LBD are shown.
Monomeric PXR Binds Ligands, DNA and RXRα
We next examined whether the Trp223Ala/Tyr225Ala double-mutant form of PXR, which is incapable of homodimerizing and activating transcription, was able to perform basic molecular functions critical to nuclear receptor action. First, the ability of the double-mutant PXR LBD to interact with ligands was examined in a radioligand competition assay. Both wild-type and double-mutant PXRs bound equally well to the agonists SR12813, rifampicin, estradiol and 5β-pregnane-3,20-dione (Table II). Second, gel mobility shift assays were employed to investigate DNA binding. Heterodimeric complexes of full-length RXRα with either full-length wild-type or double-mutant PXRs bound strongly to NR3 and ER6 DNA elements (7, 8, 19, 21) (Figure 4B). Third, the ability of the double-mutant PXR LBD to interact with the LBD of its physiological heterodimer partner RXRα was examined using in vitro pull-down assays (Figure 4C). Both wild-type and double-mutant PXR LBDs were found to form complexes with biotinylated RXRα LBDs. Taken together, these results demonstrate that the Trp223Ala/Tyr225Ala double-mutant PXR retains many of its key molecular functions: its ability to associate with ligands, with its functional heterodimer partner RXRα, and with DNA.
Table II.
Ligand Binding to PXR LBDs
| Wild-Type PXR | W223A/Y225A PXR | |
|---|---|---|
| pKi | pKi | |
| SR12813 | 5.7 | 5.6 |
| Rifampicin | 5.3 | 5.6 |
| Estradiol | 5.8 | 5.7 |
| 5β-pregnane-3,20-dione | 5.0 | 5.0 |
Monomeric PXR Cannot Recruit Coactivator
Because mutant forms of PXR were transcriptionally compromised both alone and in the presence of agonists, the recruitment of the transcriptional coactivator SRC-1 by the double-mutant PXR was examined using two approaches. First, in mammalian two-hybrid studies, the ability of wild-type and Trp223Ala/Tyr225Ala double-mutant full-length PXRs to interact with the nuclear receptor interaction domain (NRID) of SRC-1 in HepG2 cells was tested. It was found that wild-type PXR efficiently complexed with SRC-1, an interaction that was enhanced by the presence of agonist SR12813; however, the Trp223Ala/Tyr225Ala double-mutant PXR failed to interact with SRC-1 either alone or with SR12813 (Figure 5A). Second, the same interaction was examined in vitro using pull-down assays with a biotin-labeled SRC-1 peptide. Similar to the in vivo experiments, only wild-type PXR LBD was observed to complex with the SRC-1 peptide, and this interaction was significantly improved by the presence of SR12813. In contrast, double-mutant PXR LBD was incapable of interacting with the same peptide, even in the presence of SR12813 (Figure 5B). Thus, the inability of Trp223Ala/Tyr225Ala double-mutant PXR to activate transcription appears to be the result of a defect in binding to p160-type coactivators like SRC-1. These results suggest that the unique PXR homodimer formed is involved in coactivator recruitment by the receptor.
Figure 5.


A. Mammalian two-hybrid examination of the interaction between full-length PXR and the nuclear receptor interaction domain (NRID) of the transcriptional coactivator SRC-1. Wild-type and Trp223Ala/Tyr225Ala double-mutant PXR were examined alone and in the presence of the PXR agonist SR12813. B. In vitro examination of the interaction between PXR LBDs and an SRC-1 peptide. Only wild-type PXR LBD interacted with a biotinylated SRC-1 peptide, while a Trp223Ala/Tyr225Ala double-mutant PXR LBD was not observed to bind to the same peptide.
DISCUSSION
In all crystal structures of the ligand binding domain of human PXR examined to date, the protein forms a homodimer involving amino acids unique to PXR. The dimerization interface is essentially created by the association of the β1’ strands of each monomer in an ideal antiparallel fashion, which generates a ten-stranded antiparallel inter-molecular β-sheet (Figure 1). The β1 and β1’ strands of PXR are on a ∼50-residue insert that is unique in sequence and structure in the nuclear receptor superfamily. In this report, we show that the LBD of human PXR forms a homodimer in solution by sedimentation equilibrium studies, and that a double-mutant form of PXR, in which key aromatic residues at the dimer interface are eliminated, is an obligate monomer (Table I; Figure 2). The mutations at the interface (Trp223Ala and Tyr225Ala) also severely impact the response of full-length human PXR to the agonists SR12813 and rifampicin in transient transfections (Figure 3). These mutations do not prevent full-length PXR from entering the nucleus, or from binding to DNA, ligands or RXRα (Figure 4; Table II). Significantly, however, they do prevent PXR from associating with the transcriptional coactivator SRC-1 in both mammalian two-hybrid studies in vivo and in pull-down experiments in vitro (Figure 5). Indeed, the loss of basal activity by the Trp223Ala and Tyr225Ala variant forms of the receptor likely reflects the inability of the unliganded mutant PXR-RXRα heterodimers to recruit sufficient coactivator to promote a low level of gene expression. Similar “long-range” effects have recently been observed in the monomeric nuclear receptor human liver receptor homologue-1, in which disruption of the position of a non-DNA binding helix in the DNA binding domain of this receptor significantly impacts coactivator recruitment by the distantly-located ligand binding domain (34). In summary, the accumulated data presented here suggest that the PXR homodimer may play a role in the proper physiological function of this nuclear xenobiotic receptor.
The interlocking tryptophan and tyrosine residues that form the PXR dimer interface represent the first tryptophan zipper (TrpZip) observed in a native protein. TrpZips have been examined extensively in the design of stable peptide sequences that form predictable secondary structures (35, 36), and it was found that Trp-Trp pairs placed in designed β-hairpins formed the most stable structures of all combinations of amino acids examined (36). We superimposed the homodimer interface of human PXR with the nuclear magnetic resonance (NMR) structure of TrpZip4, a designed β-hairpin structure containing tryptophan zippers. The two tryptophans and two tyrosines in PXR line up well with the four tryptophans in TrpZip4; in addition, the main-chain regions of this native protein dimer and this designed β-hairpin also superimpose well (Figure 6A) (36). A search of the Protein Data Bank (http://www.rcsb.org/pdb/) yielded only one other possible naturally occurring TrpZip in a β-sheet dimer interface, the E2 DNA binding domain of papillomavirus-1. Trp360 from each monomer of the E2 DNA binding domain is in van der Waals contact and contributes a major stabilizing effect (37). However, the tryptophans in this structure are orthogonal and face to face but do not interdigitate like the aromatic residues observed in PXR (37). Richardson and Richardson have shown that it is rare for monomeric proteins to expose terminal β-strands of a β-sheet (38). Exposed β-strands have the potential to form dangerous interactions with other β-strands, leading to intra- or intercellular aggregates like amyloid fibers. Proteins employ a variety of techniques to cap the terminal strands of a β-sheet, including covering loops, β-bulges, and the central placement of charged residues (38). Nuclear receptors, which typically contain a two- to three-stranded antiparallel β-sheet, use several of these methods to cap their terminal β-strands. The PPARs, for example, use a short terminal β-strand, a proline that introduces a kink just prior to the β-strand, and a capping α-helix (39). Similarly, CAR, which is structurally and functionally related to PXR, caps its terminal β-strand with a short α-helix (Figure 6B) (40-42). RXRα, LXR and VDR all employ loops or helices that cap their terminal β-strands, and VDR further places two charged residues in the center of the terminal strand to disrupt potential non-specific contacts with other β-structures (43-45). The observations that PXR leaves the terminal strand in its five-stranded antiparallel β-sheet uncapped, and places residues able to form a TrpZip-like structure on this exposed strand, support the conclusion that the PXRs evolved to form a homodimer . We note, however, that the region of the PXR LBD between residues 178 and 192 has not been visualized structurally to date (Figure 1A). It is possible that this stretch of amino acids caps the terminal β1’ strand but is displaced when the protein forms a homodimer.
Figure 6.


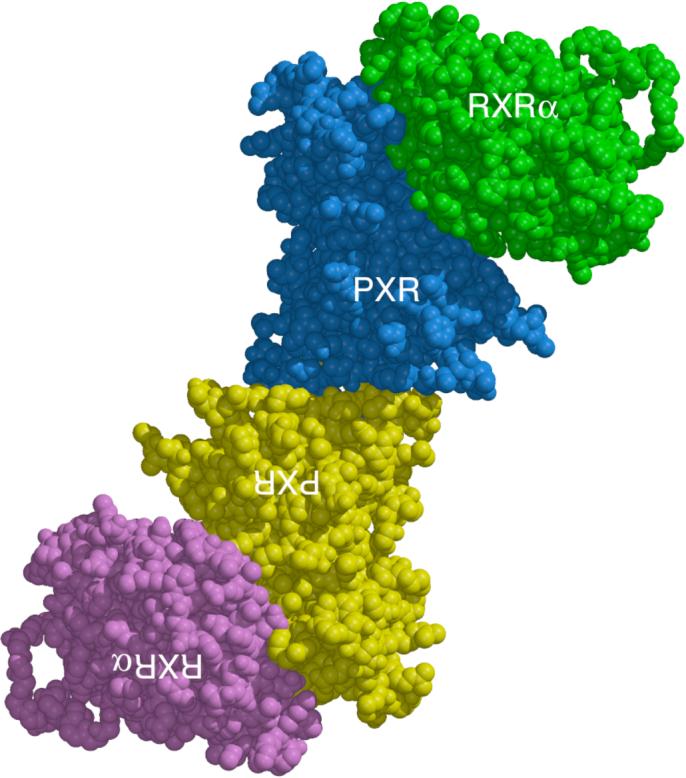
A. Superposition of the PXR LBD homodimer interface on TrpZip4 (30). PXR residues 222−226 are shown in cyan. Residues 41−56 of Trpzip4 are rendered in magenta. B. A side-by-side representation of the LBDs of PXR and CAR (40-42). The extended β-sheet region of PXR is rendered in green. The β-sheet region of CAR is depicted in yellow. Unlike PXR, CAR contains a capping α-helix (shown in magenta) that protects its edge β-strand from non-specific interactions. C. A model for the PXR/RXRα heterotetramer complex of ligand binding domains. The PXR-RXRα heterotetramer was generated using the structure of PPARγ-RXRα complex as a template (46). The PXR LBD homodimer is shown in blue and yellow with the RXRα-LBDs in green and magenta. The PXR LBDs in this figure are viewed in the same orientation shown in Figure 1A.
PXR forms a heterodimer with RXRα to control the transcription of target genes. We examined whether the PXR homodimer would interfere with the formation of the PXR-RXRα heterodimer. Using the crystal structure of the PPARγ-RXRα ligand binding domain heterodimer, we replaced the PPARγ LBD with the PXR LBD to generate a model of the PXR-RXRα LBD heterodimeric complex that maintained most of the key hydrophobic and electrostatic contacts at the interface (46). We also noted that the surfaces used for PXR-RXRα heterodimerization and PXR homodimerization do not overlap; thus, a structurally-compelling model for a PXR-RXRα heterotetramer can be generated (Figure 6C). It is of interest that no PXR-RXRα heterotetramer was observed in gel mobility shift assays (Figure 4B), although this is perhaps due to the moderate strength of the dissociation constant (μM) for the PXR homodimer interface. There are several potential ways that a PXR-RXRα heterotetramer could be involved in receptor function. First, a heterotetramer could form between the two PXR-RXRα heterodimers bound to both the proximal and distal DNA elements in the regulatory regions of genes. Recall that two PXR-RXRα binding elements exist in the CYP3A4 promoter, at bases −172 to −149 (proximal) and −7836 to −7607 (distal), and that both elements are required for maximal transcriptional activation (20). In this model, both PXR-RXRα heterodimers would be bound to DNA, and the DNA would be expected to form a long-range loop to generate the heterotetramer. Second, only one PXR-RXRα heterodimer could bind to a DNA element and the second heterodimer may simply associate with the first (but not with DNA) to form the heterotetramer. In this case, the role of a PXR-RXRα heterotetramer may be to enhance the initial recruitment (and local concentration) of transcriptional coactivators. Third, the heterotetramer may be a crucial trafficking form of the complex required to position the proteins appropriately within the nucleus and/or adjacent to euchromatin. The structural basis of the impact that the homodimer interface has on coactivator binding may be through the indirect stabilization of αAF and the AF-2 surface. Ordering of the β1-β1’ region by dimerization would be expected to stabilize the pseudohelix α2 (which starts at residue 198) that bridges the space between the β-sheet and α-helices 10 and AF in PXR (Figure 1A). Note that β-strands 1 and 1’, as well as α-helix 2, are all on the novel sequence insert unique to PXR and, as we have shown, the contacts they make appear to be involved in receptor function. These observations raise the possibility that disruption of the PXR homodimer interface by small molecule modulators could provide a mechanism to control specifically the regulation of drug metabolism gene expression by PXR during the therapeutic treatment of disease.
ACKNOWLEDGEMENTS
The authors wish to thank members of the Redinbo and McDonnell laboratories for experimental assistance and helpful discussions.
ABBREVIATIONS
- PXR
pregnane X receptor
- LBD
ligand binding domain
- Trp-Zip
tryptophan zipper
- CYP
cytochrome P450
- NR
nuclear receptor
- RXRα
retinoid X receptor-alpha
- SRC-1
steroid receptor coactivator-1
- BME
β-mercaptoethanol
- CD
circular dichroism
- Tm
melting temperature
- WT
wild-type
- NRID
nuclear receptor interaction domain
- NMR
nuclear magnetic resonance
- PPAR
peroxisome proliferator activated receptor
- CAR
consitutive androstane receptor
- LXR
liver X receptor
- VDR
vitamin D receptor
- AF
activation function.
Footnotes
This work was supported by NIH grant DK62229 (M.R.R.), NIH ATLAS grant DK62434 (D.P.M.), and a National Science Foundation Graduate Research Fellowship (V.E.C.).
REFERENCES
- 1.Gardner-Stephen D, Heydel JM, Goyal A, Lu Y, Xie W, Lindblom T, Mackenzie P, Radominska-Pandya A. Human PXR variants and their differential effects on the regulation of human UDP-glucuronosyltransferase gene expression. Drug Metab Dispos. 2004;32:340–7. doi: 10.1124/dmd.32.3.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie W, Evans RM. Orphan nuclear receptors: the exotics of xenobiotics. J Biol Chem. 2001;276:37739–42. doi: 10.1074/jbc.R100033200. [DOI] [PubMed] [Google Scholar]
- 3.Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem. 2001;276:14581–7. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- 4.Gerbal-Chaloin S, Daujat M, Pascussi JM, Pichard-Garcia L, Vilarem MJ, Maurel P. Transcriptional regulation of CYP2C9 gene. Role of glucocorticoid receptor and constitutive androstane receptor. J Biol Chem. 2002;277:209–17. doi: 10.1074/jbc.M107228200. [DOI] [PubMed] [Google Scholar]
- 5.Kliewer SA. The nuclear pregnane X receptor regulates xenobiotic detoxification. Journal of Nutrition. 2003;133:2444S–2447S. doi: 10.1093/jn/133.7.2444S. [DOI] [PubMed] [Google Scholar]
- 6.Maurel P. In: Cytochromes P450: metabolic and toxicological aspects. Ioannides C, editor. CRC Press, Inc.; Boca Raton, FL: 1996. pp. 241–270. [Google Scholar]
- 7.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–23. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, Sydow-Backman M, Ohlsson R, Postlind H, Blomquist P, Berkenstam A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:12208–12213. doi: 10.1073/pnas.95.21.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, Perlmann T, Lehmann JM. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 10.Moore LB, Goodwin B, Jones SA, Wisely GB, Serabjit-Singh CJ, Willson TM, Collins JL, Kliewer SA. St. John's wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:7500–2. doi: 10.1073/pnas.130155097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wentworth JM, Agostini M, Love J, Schwabe JW, Chatterjee VK. St John's wort, a herbal antidepressant, activates the steroid X receptor. J Endocrinol. 2000;166:R11–6. doi: 10.1677/joe.0.166r011. [DOI] [PubMed] [Google Scholar]
- 12.Ernst E. Second thoughts about safety of St John's wort. Lancet. 1999;354:2014–6. doi: 10.1016/S0140-6736(99)00418-3. [DOI] [PubMed] [Google Scholar]
- 13.Fugh-Berman A. Herb-drug interactions. Lancet. 2000;355:134–8. doi: 10.1016/S0140-6736(99)06457-0. [DOI] [PubMed] [Google Scholar]
- 14.Piscitelli SC, Burstein AH, Chaitt D, Alfaro RM, Falloon J. Indinavir concentrations and St John's wort. Lancet. 2000;355:547–8. doi: 10.1016/S0140-6736(99)05712-8. [DOI] [PubMed] [Google Scholar]
- 15.Ruschitzka F, Meier PJ, Turina M, Luscher TF, Noll G. Acute heart transplant rejection due to Saint John's wort. Lancet. 2000;355:548–9. doi: 10.1016/S0140-6736(99)05467-7. [DOI] [PubMed] [Google Scholar]
- 16.Giguere V. Orphan nuclear receptors: from gene to function. Endocr Rev. 1999;20:689–725. doi: 10.1210/edrv.20.5.0378. [DOI] [PubMed] [Google Scholar]
- 17.Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81:1269–304. doi: 10.1152/physrev.2001.81.3.1269. [DOI] [PubMed] [Google Scholar]
- 18.Rosenfeld M, Glass C. Coregulator codes of transcriptional regulation by nuclear receptors. J Biol Chem. 2001;276:36865–8. doi: 10.1074/jbc.R100041200. [DOI] [PubMed] [Google Scholar]
- 19.Blumberg B, Sabbagh W, Jr., Juguilon H, Bolado J, Jr., van Meter CM, Ong ES, Evans RM. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes & Development. 1998;12:3195–205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodwin B, Hodgson E, Liddle C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Molecular Pharmacology. 1999;56:1329–39. doi: 10.1124/mol.56.6.1329. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Faucette S, Sueyoshi T, Moore R, Ferguson S, Negishi M, LeCluyse EL. A novel distal enhancer module regulated by pregnane X receptor/constitutive androstane receptor is essential for the maximal induction of CYP2B6 gene expression. J Biol Chem. 2003;278:14146–52. doi: 10.1074/jbc.M212482200. [DOI] [PubMed] [Google Scholar]
- 22.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–33. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 23.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR. 2.1 angstrom crystal structure of human PXR in complex with the St. John's wort compound hyperforin. Biochemistry. 2003;42:1430–1438. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 24.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. Coactivator binding promotes the specific interaction between ligand and the pregnane X receptor. J Mol Biol. 2003;331:815–28. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 25.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, Redinbo MR. Structural disorder in the complex of human pregnane x receptor and the macrolide antibiotic rifampicin. Mol Endocrinol. 2005;19:1125–34. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 26.Honkakoski P, Moore R, Washburn KA, Negishi M. Activation by diverse xenochemicals of the 51-base pair phenobarbital-responsive enhancer module in the CYP2B10 gene. Mol Pharmacol. 1998;53:597–601. doi: 10.1124/mol.53.4.597. [DOI] [PubMed] [Google Scholar]
- 27.Nichols JS, Parks DJ, Consler TG, Blanchard SG. Development of a scintillation proximity assay for peroxisome proliferator-activated receptor gamma ligand binding domain. Anal Biochem. 1998;257:112–9. doi: 10.1006/abio.1997.2557. [DOI] [PubMed] [Google Scholar]
- 28.Ortlund EA, Lee Y, Solomon IH, Hager JM, Safi R, Choi Y, Guan Z, Tripathy A, Raetz CR, McDonnell DP, Moore DD, Redinbo MR. Modulation of human nuclear receptor LRH-1 activity by phospholipids and SHP. Nat Struct Mol Biol. 2005;12:357–63. doi: 10.1038/nsmb910. [DOI] [PubMed] [Google Scholar]
- 29.Chang C, Norris JD, Gron H, Paige LA, Hamilton PT, Kenan DJ, Fowlkes D, McDonnell DP. Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libraries: discovery of peptide antagonists of estrogen receptors alpha and beta. Mol Cell Biol. 1999;19:8226–39. doi: 10.1128/mcb.19.12.8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Norris J, Fan D, Aleman C, Marks JR, Futreal PA, Wiseman RW, Iglehart JD, Deininger PL, McDonnell DP. Identification of a new subclass of Alu DNA repeats which can function as estrogen receptor-dependent transcriptional enhancers. J Biol Chem. 1995;270:22777–82. doi: 10.1074/jbc.270.39.22777. [DOI] [PubMed] [Google Scholar]
- 31.DeLano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol. 2002;12:14–20. doi: 10.1016/s0959-440x(02)00283-x. [DOI] [PubMed] [Google Scholar]
- 32.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 33.Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–98. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 34.Solomon IH, Hager JM, Safi R, McDonnell DP, Redinbo MR, Ortlund EA. Crystal Structure of the Human LRH-1 DBD-DNA Complex Reveals Ftz-F1 Domain Positioning is Required for Receptor Activity. J Mol Biol. 2005 doi: 10.1016/j.jmb.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 35.Russell SJ, Cochran AG. Designing stable beta-hairpins: Energetic contributions from cross-strand residues. Journal of the American Chemical Society. 2000;122:12600–12601. [Google Scholar]
- 36.Cochran AG, Skelton NJ, Starovasnik MA. Tryptophan zippers: stable, monomeric beta -hairpins. Proc Natl Acad Sci U S A. 2001;98:5578–83. doi: 10.1073/pnas.091100898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hegde RS, Grossman SR, Laimins LA, Sigler PB. Crystal structure at 1.7 A of the bovine papillomavirus-1 E2 DNA-binding domain bound to its DNA target. Nature. 1992;359:505–12. doi: 10.1038/359505a0. [DOI] [PubMed] [Google Scholar]
- 38.Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci U S A. 2002;99:2754–9. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–43. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 40.Shan L, Vincent J, Brunzelle JS, Dussault I, Lin M, Ianculescu I, Sherman MA, Forman BM, Fernandez EJ. Structure of the murine constitutive androstane receptor complexed to androstenol: a molecular basis for inverse agonism. Mol Cell. 2004;16:907–17. doi: 10.1016/j.molcel.2004.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suino K, Peng L, Reynolds R, Li Y, Cha JY, Repa JJ, Kliewer SA, Xu HE. The nuclear xenobiotic receptor CAR: structural determinants of constitutive activation and heterodimerization. Mol Cell. 2004;16:893–905. doi: 10.1016/j.molcel.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 42.Xu RX, Lambert MH, Wisely BB, Warren EN, Weinert EE, Waitt GM, Williams JD, Collins JL, Moore LB, Willson TM, Moore JT. A structural basis for constitutive activity in the human CAR/RXRalpha heterodimer. Mol Cell. 2004;16:919–28. doi: 10.1016/j.molcel.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 43.Gampe RT, Jr., Montana VG, Lambert MH, Wisely GB, Milburn MV, Xu HE. Structural basis for autorepression of retinoid X receptor by tetramer formation and the AF-2 helix. Genes & Development. 2000;14:2229–41. doi: 10.1101/gad.802300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams S, Bledsoe RK, Collins JL, Boggs S, Lambert MH, Miller AB, Moore J, McKee DD, Moore L, Nichols J, Parks D, Watson M, Wisely B, Willson TM. X-ray crystal structure of the liver X receptor beta ligand binding domain: regulation by a histidine-tryptophan switch. J Biol Chem. 2003;278:27138–43. doi: 10.1074/jbc.M302260200. [DOI] [PubMed] [Google Scholar]
- 45.Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Molecular Cell. 2000;5:173–9. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]
- 46.Gampe RT, Jr., Montana VG, Lambert MH, Miller AB, Bledsoe RK, Milburn MV, Kliewer SA, Willson TM, Xu HE. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Molecular Cell. 2000;5:545–55. doi: 10.1016/s1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]