

Abstract
Complementary to studies that provided the first yatakemycin total synthesis resulting in its structure revision and absolute stereochemistry assignment, a second generation asymmetric total synthesis is disclosed herein. Since the individual yatakemycin subunits are identical to those of duocarmycin SA (alkylation subunit) or CC-1065 (central and right-hand subunits), the studies also provide an improvement in our earlier total synthesis of CC-1065 and, as detailed herein, have been extended to an asymmetric total synthesis of (+)-duocarmycin SA. Further extensions of the studies provided key yatakemycin partial structures and analogues for comparative assessments. This included the definition of the DNA selectivity (adenine central to a five base-pair AT sequence, eg. 5′-AAAAA), efficiency, relative rate, and reversibility of ent-(−)-yatakemycin and its comparison with the natural enantiomer (identical selectivity and efficiency), structural characterization of the adenine N3 adduct confirming the nature of the DNA reaction, and comparisons of the cytotoxic activity of the natural product (L1210 IC50 = 5 pM) with its unnatural enantiomer (IC50 = 5 pM) and a series of key partial structures including those that probe the role of the C-terminus thiomethyl ester. The only distinguishing features between the enantiomers is that ent-(−)-yatakemycin alkylates DNA at a slower rate (krel = 0.13) and is reversible, whereas (+)-yatakemycin is not. Nonetheless, even ent-(−)-yatakemycin alkylates DNA at a faster rate and with a greater thermodynamic stability than (+)-duocarmycin SA illustrating the unique characteristics of such “sandwiched” agents.
Introduction
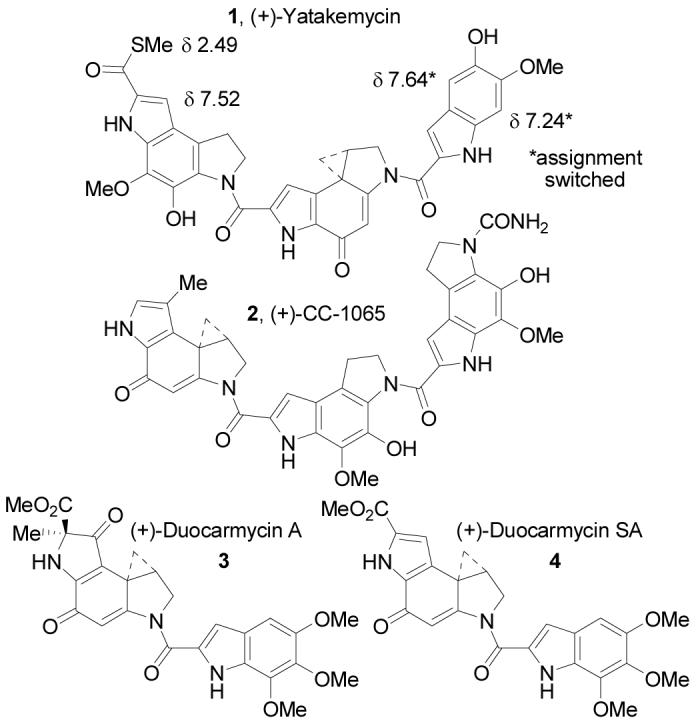
Yatakemycin (1)1 was isolated from the culture broth of Streptomyces sp. TP-AO356 and represents the newest and most potent member of a class of antitumor compounds that includes CC-1065,2 duocarmycin A,3 and duocarmycin SA4 (Figure 1). Each derives its biological properties through a characteristic and distinguishable DNA alkylation reaction.5-9 The structure of (+)-yatakemycin was disclosed in 2003 as 5 based on extensive spectroscopic studies (Figure 2).1 As such, it represented a remarkable hybrid of the preceding natural products containing a central alkylation subunit identical to that found in duocarmycin SA, a right-hand subunit similar to that of the duocarmycins, and a left-hand subunit similar to the central and right-hand subunits of CC-1065. Distinct from the preceding natural products, it represented the first naturally occurring member of this class that contains DNA binding subunits flanking each side of the alkylation subunit. Since our earlier studies established that this “sandwiched” arrangement conveyed remarkable properties to a series of duocarmycin SA analogues (enhanced DNA alkylation rate, uniquely altered alkylation selectivity, more potent cytotoxic activity),10 we became especially interested in yatakemycin. With a sample of natural material provided by Igarashi, we defined natural yatakemycin's DNA alkylation properties which proved characteristic of such a “sandwiched” compound.5
Figure 1.
Natural products.
Figure 2.
Original yatakemycin structure and first structure reformulation.
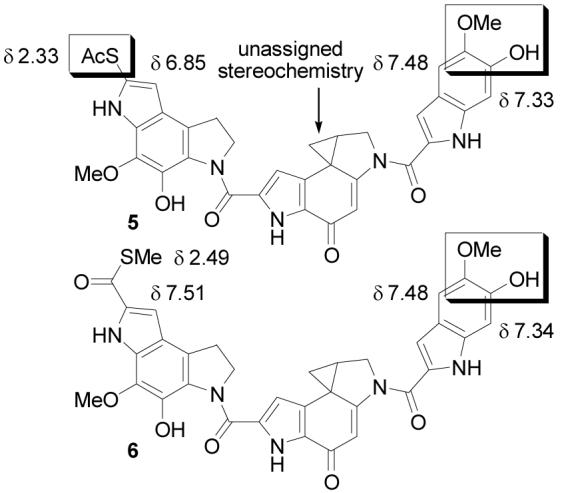
In 2004, we reported the total synthesis of 5 and its lack of correlation with the natural product (Figure 2).11 Based on spectroscopic distinctions between 5 and yatakemycin, the natural product structure was reformulated as 6. Notably, the 1H NMR of 5 did not match that of natural yatakemycin, and the greatest discrepancies occurred in the left-hand subunit. The 1H NMR chemical shift of the yatakemycin left subunit indole C8-H is found at δ 7.52, significantly downfield of the corresponding proton in 5 at δ 6.85, and the thioacetate methyl group was found 0.16 ppm upfield of the corresponding protons in the natural material.1 Model substrates and computer 1H NMR predictions supported a reassignment of the C7-substituent as a thiomethyl ester versus thioacetate. The electron-withdrawing character of a C7-thioester would account for the downfield shift in the indole C8-H and this reformulated structure (6) was more consistent with other members of the natural product family. Most notably, the left-hand subunit was now identical to the central and right-hand subunits of CC-1065, albeit capped as a thiomethyl ester. Diagnostically, aryl thiomethyl esters12a exhibit a methyl 13C NMR chemical shift of roughly δ 11, matching the chemical shift reported for yatakemycin (δ 11.14), whereas aryl thioacetates12b occur at δ 30. Thus, yatakemycin was reformulated as 6 and targeted for synthesis.
Total synthesis of this alternative structure provided a compound nearly identical to, but still subtly distinct from, the natural product. However, the 1H NMR chemical shifts in the left-hand and central subunits of 6 matched those of authentic yatakemycin suggesting this portion of the structure incorporating the reformulated thiomethyl ester was now correct. The remaining discrepancies rested exclusively with subtle perturbations in the 1H NMR chemical shifts in the right-hand subunit of 6. After convincing ourselves that these 1H NMR differences were not due to concentration or solvent effects (e.g., water in pyridine-d5 or partial phenol deprotonation) and that 6 and yatakemycin were chromatographically distinguishable (coelute in most solvents), a reexamination of the 1H NMR and HMBC data disclosed in the structure identification established that the substituent locations, but not their identity, had been defined. Model indole 7 was synthesized14 and found to correlate well with the right-hand subunit of yatakemycin in the same NMR solvent (pyridine-d5), particularly with respect to the most deshielded aromatic proton δ 7.66 (vs. δ 7.62 for yatakemycin) which was absent in both previous candidate structures (Figure 3). Further characterization of 7 by NMR (pyridine-d5, ROESY, HMBC, HMQC, 13C-APT) and correlation with yatakemycin indicated that the reported 1H NMR chemical shifts of C4-H and C7-H of the right-hand subunit most likely had been switched, which could have caused the substituent locations to be misassigned. As a result, 1 was targeted for synthesis and bears this right-hand subunit substituent reassignment as well as the left-hand subunit thiomethyl ester. This further reformulation of the yatakemycin structure was confirmed by total synthesis of (+)- and ent-(−)-1 in studies that additionally established the absolute configuration of the natural product.11
Figure 3.
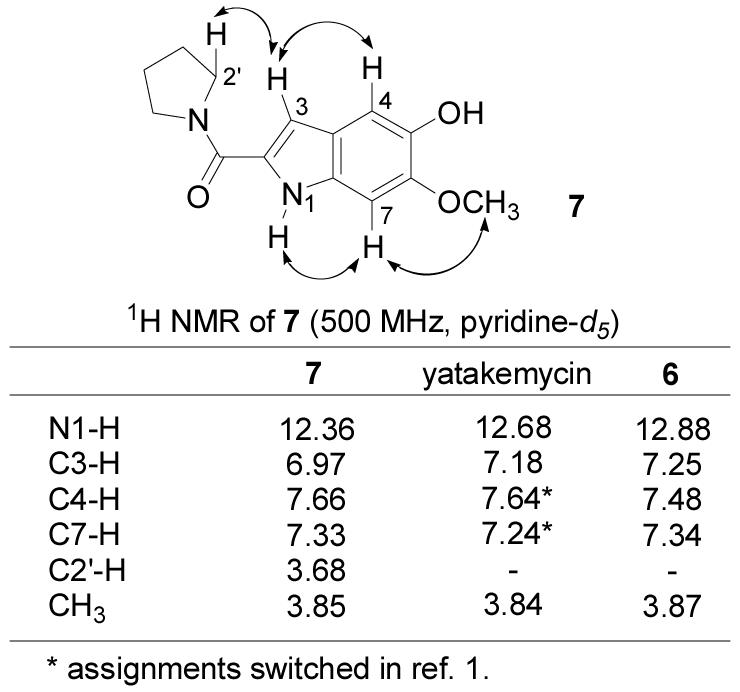
Model comparison.
Herein we report full details of studies providing a second generation, asymmetric total synthesis of (+)- and ent-(−)-yatakemycin,11,13 the extension of our preceding studies to the preparation of key partial structures and analogues, and details of the assessments of their properties. This includes the definition of the DNA alkylation properties of synthetic ent-(−)-yatakemycin and its comparison with those of the natural enantiomer, the isolation and structure determination of the thermally-released DNA adenine adduct confirming the nature of the DNA alkylation reaction, and a comparison of the cytotoxic activity of the natural product (L1210 IC50 = 5 pM) with its unnatural enantiomer (IC50 = 5 pM) and a series of key partial structures and analogues. Since the individual subunits of yatakemycin are identical to those found in duocarmycin SA (alkylation subunit) or CC-1065 (central and right-hand subunits), the studies also provide an improvement in our reported total synthesis of CC-1065 and, as also detailed herein, have provided a second generation, asymmetric total synthesis of (+)-duocarmycin SA.15
Chemistry
Like our original synthesis of yatakemycin, the natural product was accessed from three subunits that compose its structure, using a defined order for their coupling (Scheme 1). Unlike our prior approach, a final transannular Ar-3′ spirocyclization was used to close the activated cyclopropane enlisting a Mitsunobu activation of a precursor secondary alcohol permitting the free phenol 19 to be utilized directly in the coupling sequence without protection and shortening the number of late stage steps. Additionally, an asymmetric synthesis of the central alkylation subunit was developed and incorporates a late stage introduction of the chiral center permitting ready access to either enantiomer. Key elements of the strategic design of the routes used to access the central and left-hand subunits are discussed in the following sections, whereas the right-hand subunit (5-hydroxy-6-methoxyindole-2-carboxylic acid) was readily available in 4 steps from commercially available material.14
Scheme 1.
Synthesis of the Alkylation Subunit
Complementary to our original synthesis of the alkylation subunit first developed for the total synthesis of (+)- and (−)-duocarmycin SA16-21 which relied on a late stage resolution of an advanced intermediate for accessing optically active material,16 an asymmetric synthesis was developed and is disclosed herein. Key to its implementation is a surprisingly effective and regioselective intramolecular epoxide opening inspired by the studies of Sakamoto,22 a uniquely concise synthesis of the requisite iodoindole precursor 16, and a final stage transannular Ar-3′ spirocyclization for introduction of the activated cyclopropane (Scheme 2).17,21,23,24 Importantly, the late stage introduction of the chiral center derived from (R)- or (S)-glycidol permits ready access to either enantiomer.
Scheme 2.
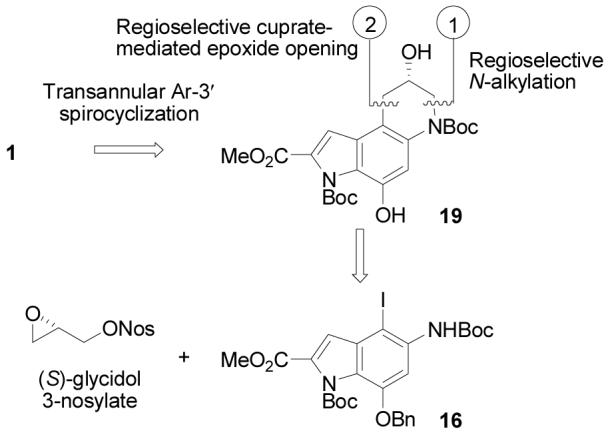
The asymmetric synthesis of the alkylation subunit began with aldehyde 825 which is prepared in a single step from commercially available 3,5-dinitrobenzyl alcohol (Scheme 3). Nucleophilic displacement of one nitro group in 8 using benzaldehyde oxime26 (1.5 equiv, 3 equiv of K2CO3, DMF, 90 °C, 1.25 h) occurred with in situ elimination to the phenol, that was trapped as the benzyl ether (1.6 equiv of BnBr, DMF, 25 °C, 2 h, 86%) to provide 10 in superb overall yield. This one-pot substitution of a benzyloxy group (hydroxy group) for a nitro substituent serves as a superb alternative to a low yielding mono-reduction/diazotization procedure commonly described in the literature27 and merits more widespread recognition and adoption than is presently the case. Condensation of aldehyde 10 with methyl azidoacetate (4 equiv, 5 equiv of NaOMe, MeOH, 4 °C, 48 h, 78%) provided 11. Thermolysis of the resulting styryl azide (xylenes, 140 °C, 7 h, 68%) provided the desired Hemetsberger-Rees product 1214,28 and protection of the indole, nitro reduction, and Boc protection of the resulting amine provided 15 (91% overall).20
Scheme 3.
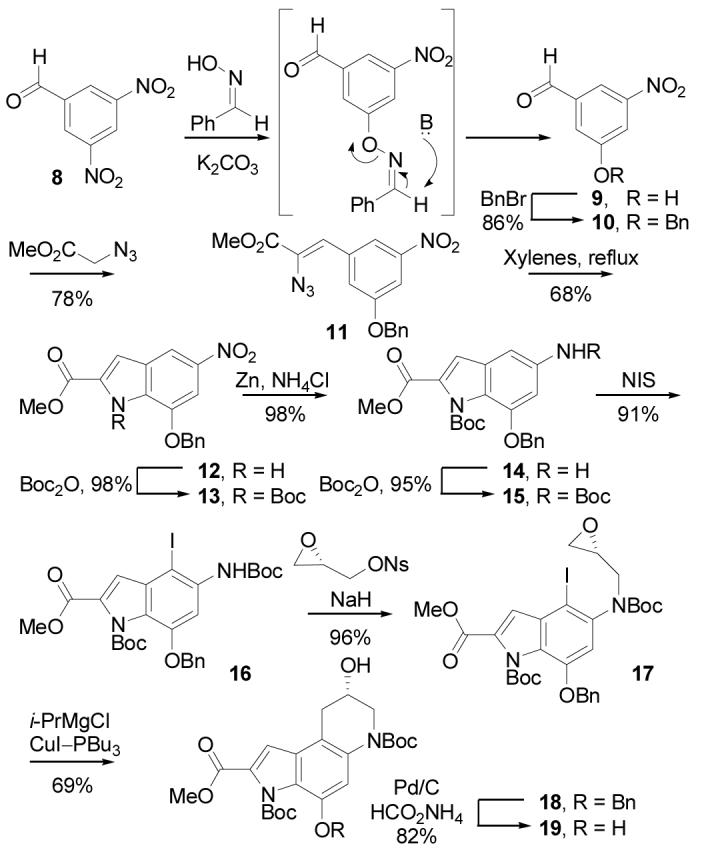
Regioselective iodination of 15 (2 equiv of NIS, toluene–HOAc, 25 °C, 8 h, 91%) set the stage for the key intramolecular epoxide opening and the final stages of the synthesis. Alkylation of 16 by treatment with (S)-glycidyl-3-nosylate29 (1.2 equiv, 1.2 equiv of NaH, DMF, 0 °C, 3 h, 96%) provided 17 (for natural enantiomer) with clean Sn2 displacement of the of the nosylate versus SN2′ epoxide opening and subsequent displacement of the nosylate. Competitive SN2′ displacement would give the enantiomeric epoxide reducing the enantiomeric purity of the product epoxide,29 but 17 was isolated in high, near perfect optical purity confirming the superb alkylation regioselectivity. Formation of the Grignard reagent by metal–halogen exchange (1.1 equiv of i-PrMgCl, −42 °C, 1 h) followed by transmetalation to the cuprate (0.2 equiv of CuI-Bu3P, −78 °C, 2 h, 69%) induced rapid intramolecular ring opening to give exclusively 18 (99% ee, Chiralcel OD HPLC), the result of 6-endo versus 5-exo addition to the epoxide. Notably, the corresponding aryl lithium reagent failed to give any of the desired product due to preferential or competitive reactions of the metalating reagent (n-BuLi, 1.0 equiv, −78 °C) with the methyl ester or Boc group precluding the use of this approach to the generation of an appropriate cuprate reagent. Metal–halogen exchange to the Grignard reagent proceeded without such competitive nucleophilic additions, and the intermediate aryl Grignard was unreactive (at −40 °C) toward the epoxide until treated with a catalytic amount of the copper reagent. In contrast to the present example, analogous intramolecular epoxide additions with both organolithium and organocuprate reagents typically give mixtures of 5-endo and 6-exo products in modest yields.30 Transfer hydrogenolysis of the benzyl ether (excess 25% aqueous HCO2NH4, 10% Pd/C, 9:1 THF–MeOH, 25 °C, 3 h, 82%) gave the alkylation subunit precursor 19.
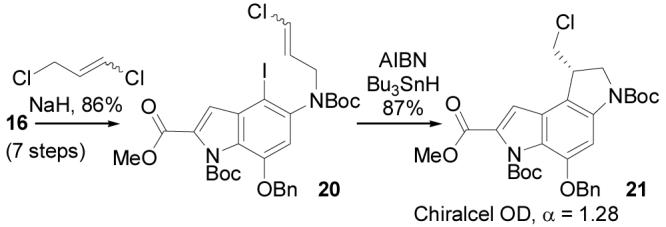
In addition, N-alkylation of 16 with 1,3-dichloropropene followed 5-exo-trig free radical ring closure following a protocol introduced in 199731,32 provides an expedient route to the racemic alkylation subunit precursor 21 (Scheme 4) analogous to the approach enlisted by Tietze and co-workers20 in their total synthesis of (±)-duocarmycin SA. Resolution of 21 by chromatographic separation33 (Chiralcel OD, 2 × 25 cm, 7 mL/min, α = 1.28) provides each enantiomer of the alkylation subunit (≥99.9% ee), suitable for elaboration to optically active yatakemycin or duocarmycin SA. Notably, the route to 16 detailed herein (7 steps) reduces this approach to a simple 9-step synthesis of an alkylation subunit precursor, resolvable at step nine.
Scheme 4.
Total Synthesis of (+)-Duocarmycin SA
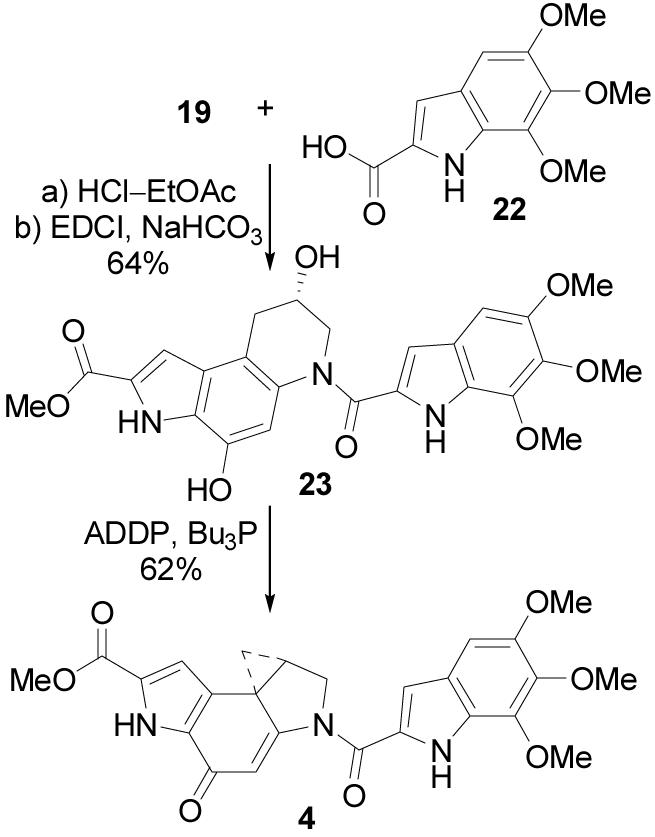
The asymmetric alkylation subunit synthesis described herein was applied to a short and efficient preparation of duocarmycin SA. Boc deprotection of 19 (4 N HCl–EtOAc, 25 °C, 3 h) followed by coupling with 5,6,7-trimethoxyindole-2-carboxylic acid7,34 (22, 1.5 equiv, 4 equiv of EDCI, DMF, 0 °C, 3 h, 64%) provided seco-duocarmycin SA 23 (Scheme 5). The final Ar-3′ spirocyclization was achieved using a Mitsunobu activation and displacement of the secondary alcohol (6 equiv of ADDP, 6 equiv of Bu3P, THF, 0.5 h, 25 °C, 62%) to give (+)-4. This asymmetric synthesis of (+)-duocarmycin SA was accomplished in 12 steps from 3,5-dinitrobenzaldehyde, and constitutes one of the most efficient reported to date.16-21
Scheme 5.
Synthesis of the Left-Hand Subunit
In our initial studies,11 the left-hand subunit of yatakemycin was established to possess a structure identical to the subunits found in CC-1065, albeit capped as a thiomethyl ester. Thus, the originally disclosed C7-thioacetate was reassigned as the C7-thiomethyl ester following the noncorrelation of 5 with yatakemycin and an assessment of the spectroscopic discrepancies. In this initial work, the total synthesis of 1 and its successful correlation with the natural product relied on a preparation of this subunit enlisting a late-stage intermediate used in the synthesis of the CC-1065 central and right-hand subunits. Herein, we report an improved synthesis of this intermediate and its incorporation into yatakemycin. The approach represents an extension of our earlier yatakemycin studies that provided the misassigned left-hand subunit in which the amide that was used
to introduce the thioacetate (via the thioamide) is now enlisted to introduce a methyl ester via its conversion to the corresponding bromoindole and subsequent Pd(0)-catalyzed carbonylation (Equation 1).
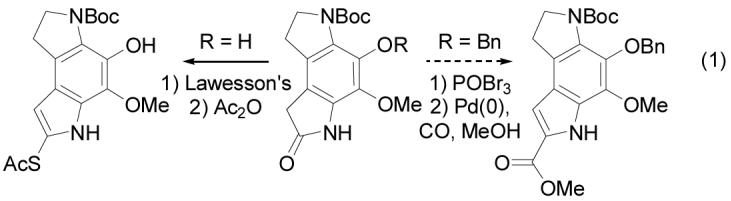
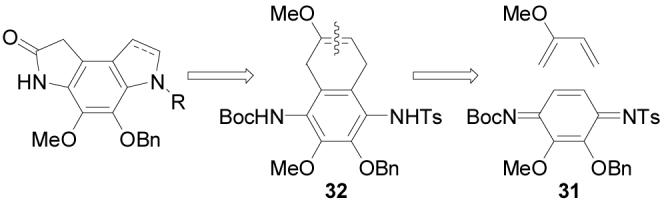
Key to conception of this concise synthesis of the left-hand subunit of first 5 and now 1 was a regioselective Diels–Alder reaction of a selectively-activated p-quinodiimide in which it is the toluenesulfonyl (versus Boc) group of 31 that expectantly controls the regioselectivity35 of the [4 + 2] cycloaddition reaction (Scheme 6). Subsequent oxidative cleavage of the enol ether in the cycloadduct 32 was anticipated to provide the appropriately substituted dihydropyrroloindole skeleton of the left-hand subunit.
Scheme 6.
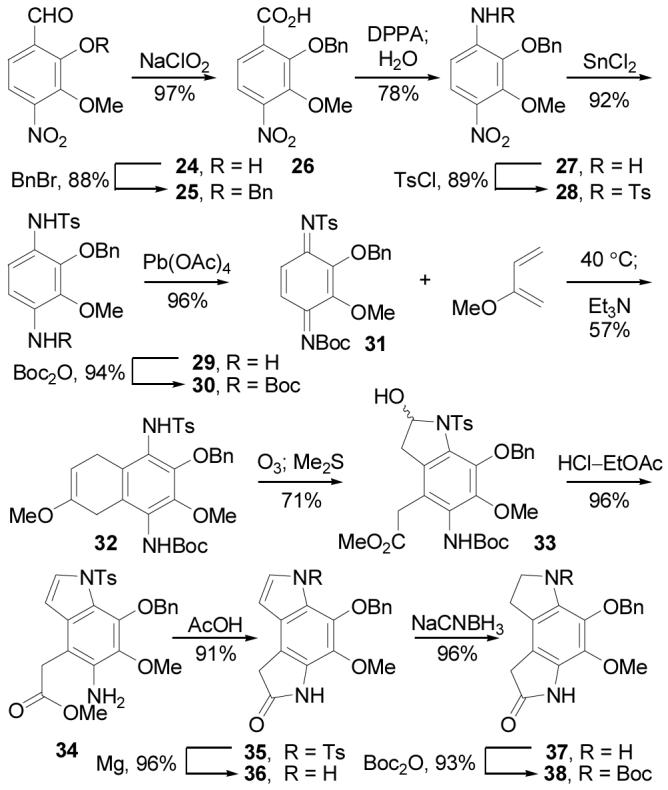
This synthesis of the left-hand subunit began with 24 which is readily available from o-vanillin.35 Phenol protection as the benzyl ether (1.5 equiv of BnBr, 2.0 equiv of K2CO3, DMF, 25 °C, 14 h, 88%) followed by aldehyde oxidation of 25 (1.2 equiv of NaClO2, t-BuOH, 25 °C, 2 h, 97%) provided 26 (Scheme 7). Subsequent Curtius rearrangement (1.05 equiv of DPPA, 3 equiv of Et3N, THF, 25 °C, 3 h) with water addition to the intermediate isocyanate provided amine 27 (78% overall), which was N-tosylated (1.5 equiv of p-TsCl, pyridine, 25 °C, 2 h, 89%) to give 28. Nitro reduction (5 equiv of SnCl2-2H2O, 1:1 dioxane–EtOH, 85 °C, 0.5 h, 92%), Boc protection of the resulting amine 29 (1.2 equiv of Boc2O, THF, 65 °C, 14 h, 94%) and subsequent oxidation of 30 (1.05 equiv of Pb(OAc)4, CHCl3, 25 °C, 2 h, 96%) provided the selectively activated p-quinodiimide 31. Throughout this preparation of 31, the easily purified crystalline intermediates permitted the multigram synthesis of advanced intermediates. The key Diels–Alder reaction of 31 with 2-methoxy-1,3-butadiene (20 equiv, 40 °C, 48 h) provided a single cycloadduct regioisomer that was treated with Et3N to effect in situ aromatization to 32 (57% overall).36 As expected, the cycloaddition regioselectivity was dominated by the stronger electron-withdrawing character of the N-tosylimine and set the stage for cleavage of the enol ether with release of two differentially oxidized side chains suitable for closure to an appropriately functionalized dihydropyrroloindole.37 Thus, ozonolysis of 32 (O3, 1:1 CH2Cl2–MeOH, −78 °C) followed by reductive workup (10 equiv of Me2S, 71%) provided the labile 2-hydroxyindoline 33 which in turn was treated with 4 N HCl–EtOAc (25 °C, 0.6 h, 96%) to induce aromatization and N-Boc deprotection. Further cyclization of 34 to lactam 35 was achieved upon treatment with HOAc (25 °C, 24 h, 91%). The indole N-tosyl group was reductively cleaved using Mg in MeOH (20 equiv of Mg, sonication, 25 °C, 1.5 h, 96%). Subsequent indole reduction (2.0 equiv of NaCNBH3, AcOH, 25 °C, 2 h, 96%) afforded 37 that was protected (2.0 equiv of Boc2O, THF, 65 °C, 1.5 h, 93%) to provide 38.
Scheme 7.
Direct conversion of lactam 38 to the bromoindole 39 was effected by treatment with POBr3 (6 equiv, 4 equiv of imidazole, 3 equiv of anisole, dioxane, 25 °C, 24 h, 72%) in the presence of imidazole (Scheme 8). The anisole additive to the reaction mixture avoided additional C8 bromination and conducting the reaction in the presence of imidazole38 precluded competitive O-debenzylation and N-Boc deprotection. Palladium-catalyzed carbonylation (0.1 equiv of PdCl2(PPh3)2, 3 equiv of Et3N, CO (1 atm), 20% MeOH–toluene, 90 °C, 36 h, 97%) installed the C7 methyl ester providing 40 in superb yield. Benzyl ether hydrogenolysis (1 atm H2, 10% Pd/C, THF, 2 h, 92%), followed by ester hydrolysis of 41 (4 equiv of LiOH, THF–MeOH–H2O, 25 °C, 14 h, 95%) provided carboxylic acid 42, that was coupled with CH3SH (excess, 4 equiv of EDCI, DMF, 0 °C, 2 h, 82%)11 to provide the key thiomethyl ester 43. Deprotection of 43 (4 N HCl–EtOAc, 25 °C, 0.5 h, 100%) provided the indoline hydrochloride salt which was used immediately and directly in subsequent coupling reactions.
Scheme 8.
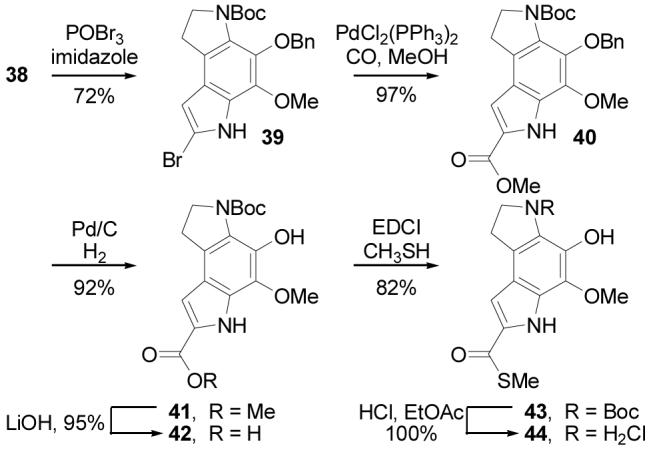
This route to 43, by virtue of proceeding through 41, also represents an improved, alternative synthesis of the central and right-hand subunits of CC-1065 (PDE-I and II).39 As such, it also constitutes an improvement in our total synthesis of (+)-andent-(−)-CC-106540 that was disclosed at the start of our interests in this class of natural products.
Completion of the Total Synthesis of (+)- and ent-(−)-Yatakemycin
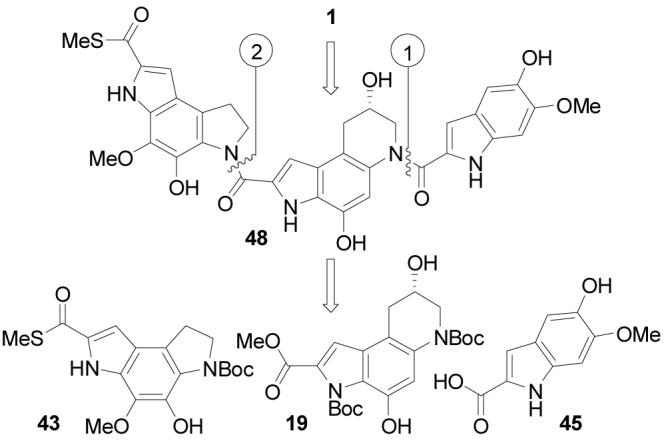
The asymmetric alkylation subunit synthesis and improved left-hand subunit synthesis were combined to provide an efficient synthesis of (+)- andent-(−)-yatakemycin (Scheme 9). Boc deprotection of 19 (4 N HCl-EtOAc, 25 °C, 3 h) followed by coupling of the resulting amine hydrochloride with 5-hydroxy-6-methoxyindole-2-carboxylic acid14 (45, 1.5 equiv, 4 equiv of EDCI, DMF, 0 °C, 3 h, 58%) provided 46. Notably, the successful coupling in the presence of the free secondary alcohol and two free phenols reduces the number of late stage protecting group manipulations. Saponification of the methyl ester (excess LiOH, 3:2:1 THF–MeOH–H2O, 25 °C, 94%) gave carboxylic acid 47, which was coupled with amine hydrochloride 44 (2 equiv, 4 equiv of EDCI, DMF, 25 °C, 5 h, 45%). The final transannular Ar-3′ spirocyclization was achieved using Mitsunobu activation and intramolecular displacement of the secondary alcohol (3 equiv of DEAD, 9 equiv of Ph3P, 10 min, THF, 25 °C, 70%) and proceeded extraordinarily rapidly to give (+)- and ent-(−)-1.
Scheme 9.
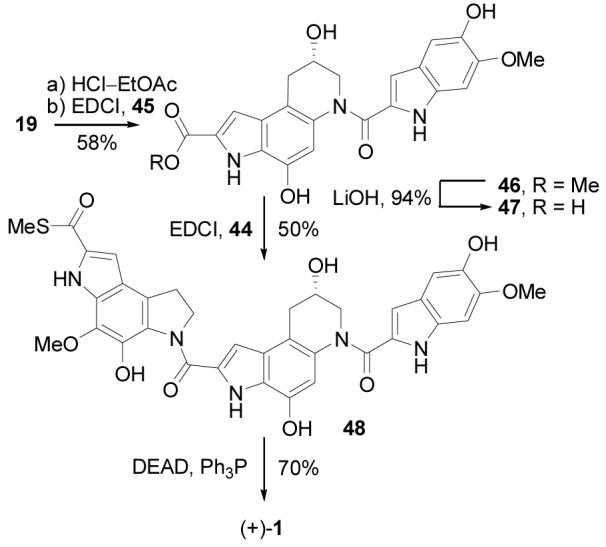
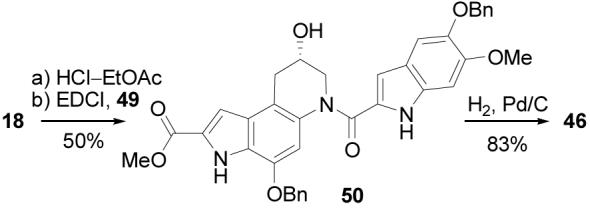
Several variations on the coupling protocol used to link the central and right-hand subunits were examined. The direct coupling of 19 and 45 was much less successful if conducted in the presence of base to liberate the free amine from its hydrochloride salt (4 equiv of EDCI, 4 equiv of NaHCO3, DMF, 3 h, 25 °C, 9%) or when conducted at room temperature (4 equiv of EDCI, DMF, 3 h, 25 °C, 29%) due principally to competitive or subsequent secondary alcohol O- versus N-acylation. In addition, the coupling of the O-benzyl ethers 18 and 49 (Scheme 10) in the presence of added base proceeded cleanly and rapidly (4 equiv of EDCI, 4 equiv NaHCO3, DMF, 3 h, 25 °C, 50%) to provide 50, and subsequent O-debenzylations (10% Pd/C, 9:1 THF–MeOH, 25 °C, 3 h, 83%) also provided 46.
Scheme 10.
Key Partial Structures and Analogues
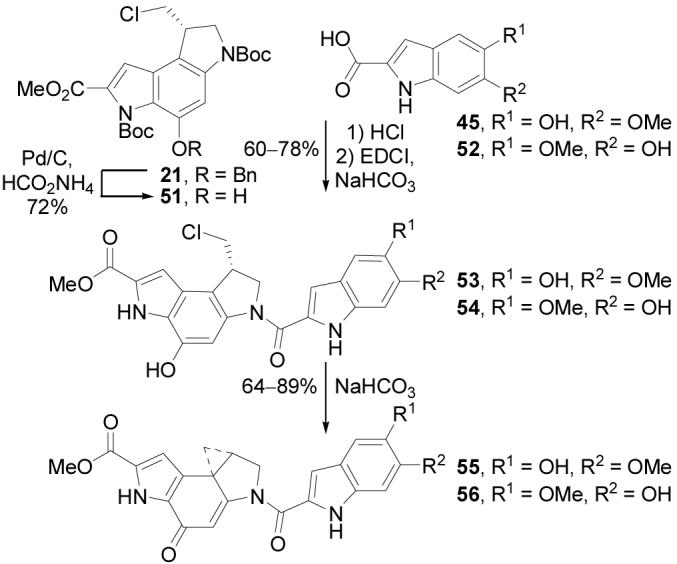
The alkylation subunit precursor 21 was deprotected by transfer hydrogenolysis (excess 25% aqueous HCO2NH4, Pd/C, 25 °C, 3 h, 72%) to provide 51. Concurrent with our original studies,11 the right-hand partial structures 55 and 56 were prepared by acid-catalyzed deprotection of 51 (natural or unnatural enantiomer, 4 N HCl–EtOAc, 70 °C, 1 h) followed by coupling with 5-hydroxy-6-methoxyindole-2-carboxylic acid (45)14 or 6-hydroxy-5-methoxyindole-2-carboxylic acid (52,37 1.5 equiv, 4 equiv of EDCI, DMF, 25 °C, 12 h, 60–78%) to provide 53 and 54 (Scheme 11). Base-induced spirocyclization (NaHCO3, 2:1 DMF–H2O, 1 h, 25 °C, 64–89%) gave the key partial structures 55 and 56, the former of which bears the authentic yatakemycin right-hand subunit and the latter which incorporates the original misassigned indole.
Scheme 11.
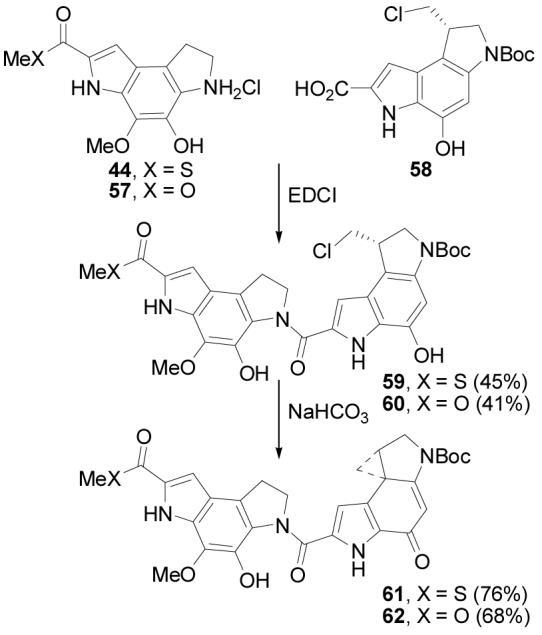
The left-hand partial structures 61 and 62 were prepared by acid-catalyzed deprotection of 43 and 41 to give 44 and 57, followed by coupling with the alkylation subunit precursor 5810b (1.0 equiv, 4 equiv of EDCI, DMF, 25 °C, 24 h, 41–45%). Spirocyclization was accomplished under mild, basic conditions (NaHCO3, 2:1 DMF–H2O, 25 °C, 1 h, 68–76%) to provide 61 or 62 (Scheme 12).
Scheme 12.
Analogues that Address the Impact of the Thiomethyl Ester
A unique feature of yatakemycin that is not found in the preceding natural products is the C-terminus thiomethyl ester. As a consequence, four key analogues or partial structures were prepared that might shed insight into its role or impact on the properties of 1. The first (65) represents the C-terminus methyl versus thiomethyl ester of yatakemycin (Scheme 13), the second and third (70 and 72) represent the thiomethyl versus methyl ester of duocarmycin SA (Scheme 14) and N-Boc-DSA (Scheme 15), and the fourth (62, presented in Scheme 12) represents the methyl versus thiomethyl ester of the key partial structure 61 that also lacks the right-hand subunit.
Scheme 13.
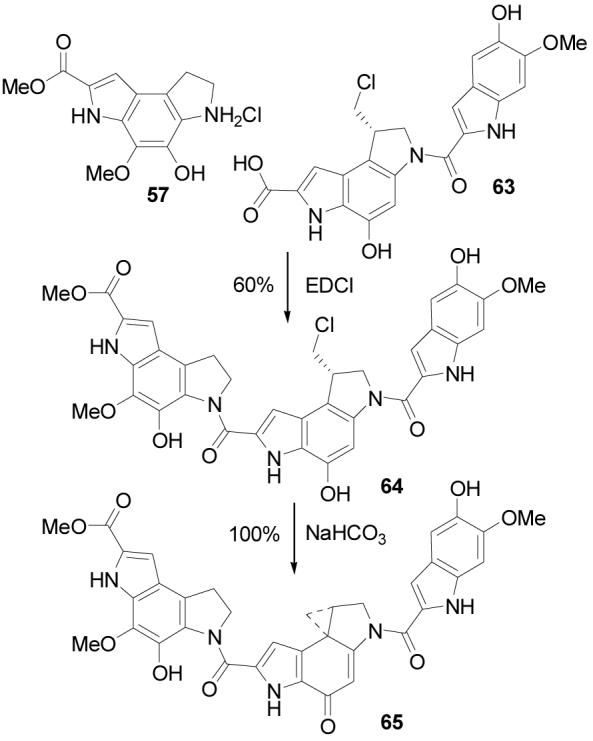
Scheme 14.
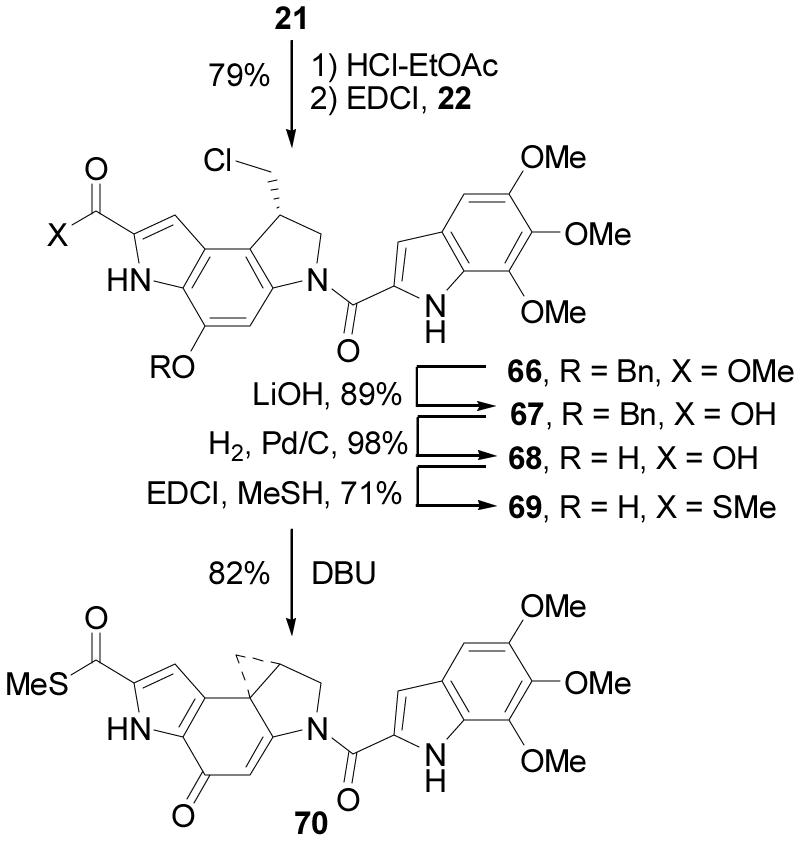
Scheme 15.
The yatakemycin methyl ester analogue 65 was prepared by coupling of a late-stage precursor of yatakemycin (63)11 with 57 (1.5 equiv, 4 equiv of EDCI, DMF, 25 °C, 24 h, 60%) to give 64, which was spirocyclized under basic conditions (NaHCO3, 2:1 DMF–H2O, 25 °C, 1 h, 100%) to provide 65 (Scheme 13). The thiomethyl ester of duocarmycin SA (70) was prepared from 21 by acid-catalyzed deprotection (4 N HCl–EtOAc, 70 °C, 1 h) followed by coupling with 5,6,7-trimethoxyindole-2-carboxylic acid acid7,34 (22, 1.5 equiv, 4 equiv of EDCI, 2 equiv of NaHCO3, DMF, 25 °C, 24 h, 79%) to provide 66. Saponification of the methyl ester (4 equiv of LiOH, THF–MeOH–H2O, 25 °C, 3 h, 89%) followed by hydrogenation of the benzyl ether (1 atm H2, 10% Pd/C, THF, 2 h, 98%) provided 68. Coupling with methyl thiol (excess, 4 equiv of EDCI, DMF, 0 °C, 2 h, 71%) installed the thiomethyl ester, and subsequent spirocyclization (10 equiv of DBU, CH3CN, 1 h, 25 °C, 82%) gave 70 (Scheme 14). The thiomethyl ester of the alkylation subunit (72) was prepared by an analogous sequence of coupling 5810b with methyl thiol (excess, 4 equiv of EDCI, DMF, 0 °C, 2 h, 67%) followed by spirocyclization (10 equiv of DBU, 1 h, 25 °C, 70%) to give 72 (Scheme 15).
DNA Alkylation Studies and Cytotoxic Activity
Isolation, Quantitation, and Characterization of the (+)-Yatakemycin Adenine Adduct
With limited amounts of naturally occurring yatakemycin available (1–2 mg), our initial studies carried out in the collaboration with Igarashi focused on defining its DNA alkylation selectivity, efficiency, reversibility, and relative rate.5 With larger quantities of the natural product acquired through its total synthesis now being available to us, we had the opportunity to address additional features of the reaction not yet examined. The first of these was the isolation and characterization of the thermally-released adenine adduct in efforts that confirm the site of nucleophilic attack (adenine N3) and the structure of the DNA alkylation product (addition to the least substituted cyclopropane carbon). The structural similarity of yatakemycin to duocarmycin SA, for which this has been established,8 suggested that the two compounds would behave in an analogous manner and this expectation was confirmed with the studies detailed below.
Analogous to the studies conducted with CC-1065,6 duocarmycin A,7b and duocarmycin SA,8 a solution of calf thymus DNA (100 bp equiv) in pH 7.4 phosphate buffer was treated with (+)-yatakemycin under conditions (25 °C, 6 h) that achieve complete DNA alkylation. The alkylated DNA was isolated by precipitation (EtOH) and no residual unreacted yatakemycin was observed in the supernatant. Thermal depurination of the alkylated calf thymus DNA (pH 7.4 phosphate buffer, 100 °C, 30 min, 2×) followed by n-butanol extraction and subsequent purification by precipitation (DMF–CH2Cl2) afforded the adenine adduct 73 in 72% yield (Figure 4). Because of the lability of the thiomethyl ester, we were not able to establish whether the remaining balance of material represents alternative non-thermally labile DNA alkylation events that are not detected including potential intra- or interstrand cross-linking reactions, or simply an instability of the thiomethyl ester of the adduct 73 to the depurination conditions leading to its diminished recovery. Complementary studies with duocarmycin SA (adenine adduct recovery 90–95%)8 suggests that alternative, non-thermally labile DNA alkylation events are unlikely and additional studies detailed herein failed to detect interstrand DNA cross-links via the thiomethyl ester. At present, it appears that the non quantitative recovery of 73, which is also extraordinarily insoluble making its isolation and purification challenging, may simply reflect its stability to the thermal depurination reaction conditions or our ability to quantitatively recover it from the reaction. Nonetheless, the 72% recovery of 73 accounts for the majority of the compound, and indicates that it constitutes the predominant alkylation event.
Figure 4.
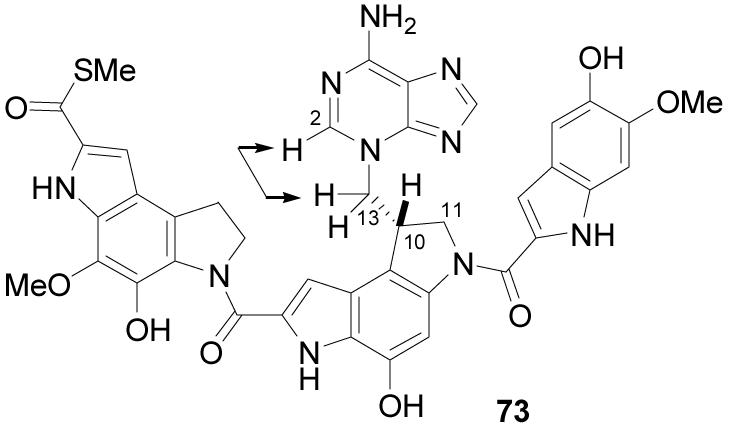
Adenine adduct.
The full NMR spectroscopic characterization (1H, 13C-APT, HMQC, ROESY) of the adenine adduct led to the unambiguous assignment of the structure 73 in which adenine N3 addition to the unsubstituted cyclopropane carbon of yatakemycin was established. This was first evident from the spectroscopic similarity to the duocarmycin SA–adenine adduct, most importantly the 1H NMR C10-H chemical shift at δ 4.52 (vs δ 4.48, duocarmycin SA)8 and the 13C NMR C10 chemical shift at δ 41.1 (vs δ 41.1, duocarmycin SA).8 The assignment was corroborated by analysis of the alkyl carbon 13C chemical shifts, in which the C10 methine (δ 41.1) is located upfield of the adjacent C11 and C13 methylene peaks (δ 54.8 and 54.3). This indicates that both methylenes are bound to heteroatoms and is consistent only with the five-membered (vs ring expansion 6-membered) ring structure. The carbon shifts corresponding to the adenine portion were assigned based on the high degree of correlation with the carbon shifts of 3-methyladenine.8 A ROESY 1H–1H cross peak between the C2-H of adenine (δ 8.28), identified by the associated C2 carbon shift at δ 154.3 (vs δ 153.6, 3-methyladenine), and the C13-H methylene of the adduct established the adenine N3 the alkylation site.
Exploratory studies were also performed with the methyl ester analogue 65 of yatakemycin, which lacks the reactive thiomethyl ester and was expected to be more stable to the thermal depurination conditions. Alkylation of calf thymus DNA and isolation as described above yielded the adenine adduct in 69% yield. The spectroscopic characteristics of its adenine adduct were analogous to those of the yatakemycin adenine adduct. It, like 73, was remarkably insoluble and challenging to work with. Unlike 73, it would not be expected to be unstable to the thermal depurination conditions and yet it was isolated with comparable recovery suggesting the quantitation of the recovered adduct 73 (72%) reflects the challenges of its isolation rather than reactivity intrinsic to the thiomethyl ester.
DNA Alkylation Selectivity of ent-(−)-Yatakemycin
With ample supplies of the synthetic samples of both enantiomers of yatakemycin available, the DNA alkylation properties of the unnatural enantiomer were established for comparison with those of the natural product.5 Thus, the DNA alkylation of ent-(−)-yatakemycin was examined in five 150 base-pair segments of DNA42 for which the alkylation sites of the natural enantiomer5 as well as an extensive range of related compounds (e.g., 2–4) have been established. After exposure to the compounds and thermally-induced depurination of the adenine N3 adducts in singly 5′ 32P-end-labeled duplex DNA, denaturing PAGE alongside Sanger dideoxynucleotide sequencing standards permitted alkylation site identification used in the subsequent assessment of the alkylation selectivity. This DNA alkylation selectivity was assessed and established under a range of reaction conditions (25 °C, 1–8 days, typically 18 h) and agent concentrations (1 × 10−4 – 1 × 10−7 M). A statistical treatment of the alkylation sites also considering those sites not alkylated was performed in establishing a consensus sequence of DNA alkylation for ent-(−)-1 and highlighted features that would otherwise be missed upon examination of only the alkylated sites.
One of the exciting features of yatakemycin was that it represented the first naturally occurring sandwiched agent containing an alkylation subunit flanked with DNA binding subunits on each side. As previously disclosed, this provides compounds that not only alkylate DNA with extraordinary rates and efficiencies that exceed those of the more classical compounds including duocarmycin SA and CC-1065, but it occurs remarkably and predictably with both enantiomers alkylating the same sites exhibiting essentially the same selectivity.10 Consistent with this, ent-(−)-yatakemycin alkylated DNA in a manner essentially identical to the selectivity of (+)-yatakemycin, and distinct from 2–4 (Figure 5). The consensus alkylation sequence for ent-(−)-1 is detailed in Table 1, the sequence preference for alkylation is outlined in Table 2, and illustration of the DNA alkylation sites within the DNA sequences is provided in the Supporting Information. All alkylation sites observed were at adenine exclusively, and no minor guanine N3 or N7 alkylation was detected. This lack of guanine alkylation is characteristic of the more stable compounds (also not observed with duocarmycin SA or (+)-yatakemycin), but is increasingly more prevalent with the more reactive natural products (duocarmycin A > CC-1065).8,43 All the observed adenine N3 alkylation sites were flanked on both sides by an A or T base. As shown in Table 2, the preference for this three base-pair sequence was 5′-AAA > 5′-AAT ≅ 5′-TAA > 5′-TAT.44 This A/T sequence preference extended outward an additional base, revealing a strong preference for the second 5′ and 3′ base to be A or T. In general, it was observed that typically one, but not both of these positions could be G or C in a given alkylation sequence highlighting the preference for ent-(−)-yatakemycin to alkylate sites central to an extended AT-rich tract. Thus, alkylation was observed at adenine-N3 central to a five base-pair A/T tract (e.g. 5′-AAAAA) as summarized in Table 1. The consensus alkylation sequence and sequence preference data for (+)- and ent-(−)-1 were found to be essentially indistinguishable.
Figure 5.
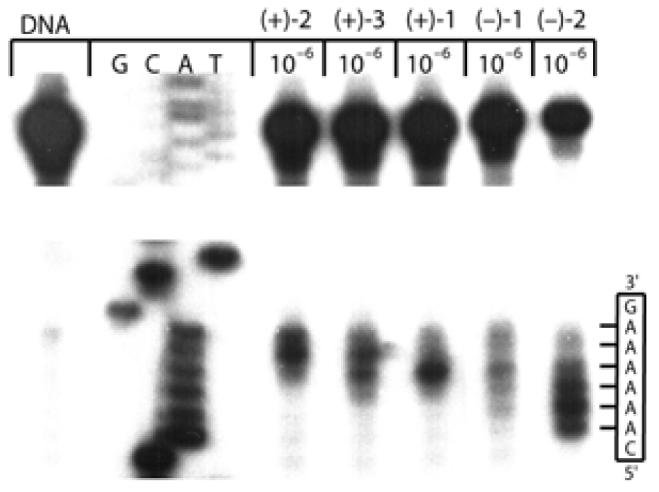
Thermally-induced strand cleavage of w836 DNA (146 bp, nucleotide 5189–91) after DNA–agent incubation at 25 °C (24 h), removal of unbound agent by EtOH precipitation, thermal depurination (100 °C, 30 min), denaturing 8% PAGE, and autoradiography. Lane 1, control DNA; lanes 2–4, Sanger G, C, A, T sequencing standards; lane 5, (+)-CC-1065 (1 × 10−6 M); lane 6, (+)-duocarmycin SA (1 × 10−6 M); lane 7, (+)-yatakemycin (1 × 10−6 M); lane 8, ent-(−)-yatakemycin (1 × 10−6 M); lane 9, ent-(−)-CC-1065 (1 × 10−6 M).
Table 1.
(−)-Yatakemycin Consensus DNA Alkylation Sequence (5′→3′)
| Basea | −3 | −2 | −1 | 0 | 1 | 2 | 3 |
|---|---|---|---|---|---|---|---|
| A(30)b | 56 | 59 | 88 | 100 | 91 | 59 | 44 |
| T(26) | 12 | 19 | 12 | 0 | 9 | 19 | 19 |
| G(21) | 19 | 12 | 0 | 0 | 0 | 19 | 22 |
| C(23) | 12 | 9 | 0 | 0 | 0 | 3 | 15 |
| A/T(56) | 69 | 78 | 100 | 100 | 100 | 78 | 63 |
| composite | A/T≥G/C | A/T>G/C | A/T | A/T | A/T | A/T>G/C | A/T≥G/C |
Percentage of the indicated base located at the designated position relative to the adenine-N3 alkylation site.
Percentage composition within the DNA examined.
Table 2.
(−)-Yatakemycin Sequence Preferences
| Sequence | no. ASa | no. TSb | %c |
|---|---|---|---|
| 5′-(NAAAN)-3′ | 25 | 39 | 64 |
| 5′-(NTAAN)-3′ | 4 | 18 | 22 |
| 5′-(NAATN)-3′ | 3 | 15 | 20 |
| 5′-(NTATN)-3′ | 0 | 15 | 0 |
Number of alkylated sites in the DNA examined (AS = alkylated sites).
Total number of sites in the DNA examined (TS = total sites).
No. AS/No. TS × 100, % of sites alkylated in the DNA examined.
Illustration of these alkylation properties in w836 DNA is provided in Figure 5. This 6 base-pair polyA sequence in w836 provides a unique setting in which all compounds can be compared and for which the relative alkylation selectivity can be nicely contrasted. (−)-Yatakemycin preferentially alkylates adenine central to this polyA tract as was found for (+)-yatakemycin, but contrasts the 3′ terminal adenine alkylation observed for the natural enantiomers, (+)-CC-1065 and (+)-duocarmycin SA, as well as the 5′ terminal adenine alkylation observed for their unnatural enantiomers (represented by ent-(−)-CC-1065).6d Although a slight erosion in selective alkylation of the central adenine by ent-(−)-1 relative to (+)-1 was observed, a clear and smooth trend from 3′ to 5′ selective alkylation can be observed simply by changing the DNA alkylation subunit position and cyclopropane stereochemistry in a controlled fashion. As was observed with (+)-1, the differences in DNA alkylation selectivity between ent-(−)-1, 2 and 3 are more pronounced than depicted in Figure 5. As a result of alkylation central to AT sequences, many alkylation sites observed for 2 and 3 do not overlap with those for ent-(−)-1, whereas this unnatural enantiomer exhibits an essentially identical alkylation site selectivity to its natural enantiomer. In summary, ent-(−)-1 was found to alkylate adenine central to a five base-pair A/T sequence (preference: 5′-AAA > 5′-AAT ≅ 5′-TAA > 5′-TAT)44 in agreement with the selectivity established for the natural enantiomer (+)-1, but differing from the 3′ terminal adenine alkylation of DNA by (+)-CC-1065 (five base-pair sequence, e.g. 5'-AAAAA) and (+)-duocarmycin SA (3–4 base pair sequence, e.g. 5'-AAAA), and the offset 5′ terminal adenine alkylation observed for their unnatural enantiomers including ent-(−)-CC-1065 (five base-pair sequence, e.g. 5'-AAAAA). Models of the natural and unnatural enantiomer alkylation of a w794 high affinity site are shown in Figure 6 and illustrate nicely these features. Both enantiomers alkylate and bind across the same 5′-TAATT site with adenine alkylation central to this five base-pair AT site, but with reversed binding orientations and as diastereomeric complexes. In these models, it is easy to recognize that the thiomethyl ester lies on the outer face of the complexes sandwiched in the minor groove. Its positioning in either syn or anti orientation appears distal from any nucleophilic site in DNA that might provide inter- or intrastrand DNA cross-linking, but it is ideally located to capture external nucleophiles (e.g., Lys or Cys in proteins) when adopting its anti conformation.
Figure 6.
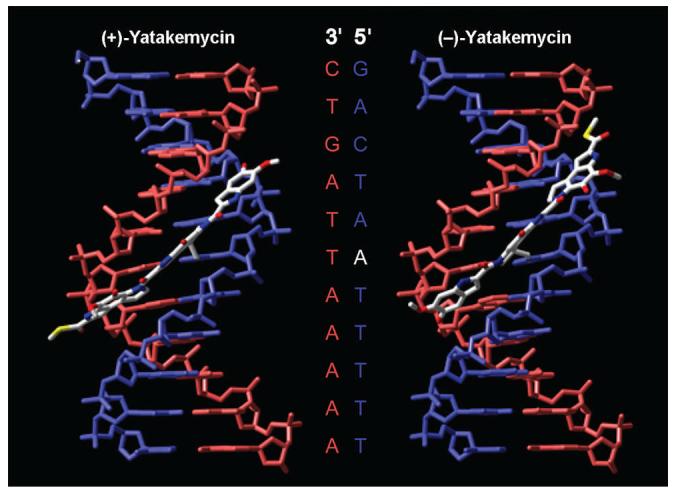
Models of the yatakemycin natural (left, anti thioester shown) and unnatural (right, syn thioester shown) enantiomer DNA alkylation of a w794 high affinity site. Both enantiomers alkylate and bind across the site 5′-TAATT, but with reversed binding orientations.
Relative Efficiency and Rate of DNA Alkylation
The efficiency of DNA alkylation was indistinguishable for (+)- and ent-(−)-1 under the conditions examined (25 °C, 1–8 days). The lowest concentration at which alkylation was observed was 10−6–10−7 M in our studies, the reactions were extraordinarily fast even at 4 °C, and even subtle differences in the DNA alkylation efficiencies were not detected when the reactions were taken to completion. The relative rates of alkylation (krel at 1 × 10−6 M) were established by observation of alkylation in w836 DNA (25 °C). Impressively, (+)-1 consumed >50% of the DNA in less than 30 sec at room temperature. Under identical conditions, ent-(−)-1 was found to alkylate DNA with minimal loss of rate (∼8 fold), an observation in accord with previous work with sandwiched agents where (+)-CDPI-DSA-CDPI exhibited a ∼1.6 fold faster rate of alkylation than its enantiomer (Figure 7).10
Figure 7.
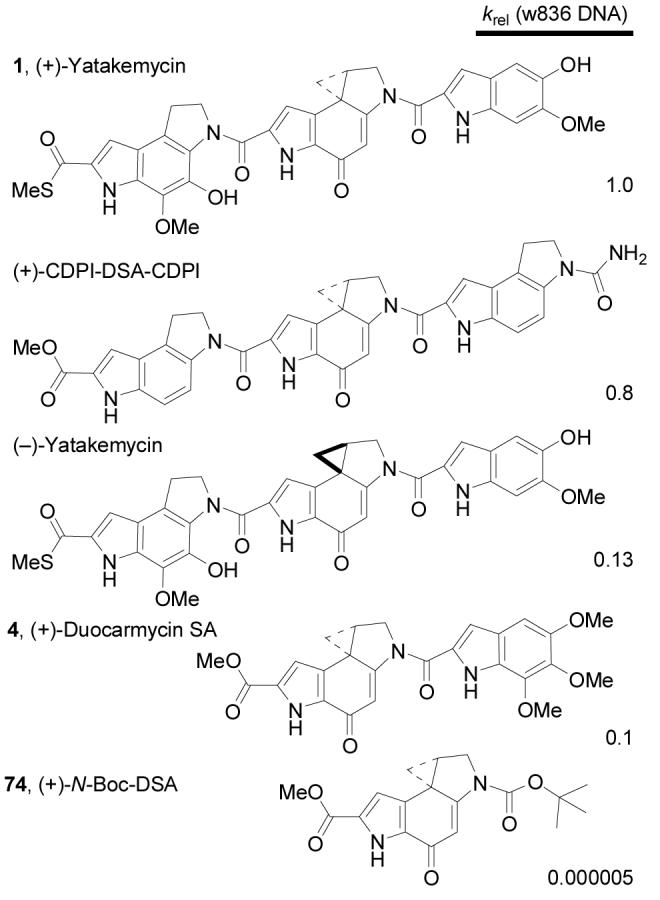
Relative rates of DNA alkylation.
Reversibility of the DNA Alkylation Reaction
Previous studies established that (+)-CC-1065 and (+)-yatakemycin constituted irreversible DNA alkylating agents,5,45 whereas smaller (e.g., duocarmycin SA) compounds were found to be slowly reversible under select conditions.8,46 Moreover, no study on the reversibility of an unnatural enantiomer in this class has been reported, and this is likely the result of the decreased rates and efficiency of DNA alkylation and the decreased cytotoxic potency typically exhibited by such analogues. Since ent-(−)-1 was found to be identical to its natural enantiomer in nearly all of these respects, we elected to examine its DNA alkylation reversibility.
The reversibility of DNA alkylation by ent-(−)-1 was examined under a variety of conditions (pH 6.0–8.4, 25–47 °C, 1–8 days) alongside duocarmycin SA8 as a positive control and (+)-yatakemycin5 as a negative control. The compounds were incubated with unlabeled w836 DNA at 25 °C for 24 h (∼90% DNA alkylation under identical conditions with labeled DNA) to ensure complete alkylation before any remaining unbound compound was extracted using an exhaustive protocol, which was followed by EtOH precipitation of the DNA to ensure complete compound removal. Covalently modified unlabeled DNA was mixed with singly 5′-end-labeled w836 DNA (1:1 ratio) and incubated together under a range of conditions. The reversibility of DNA alkylation was established by observation of compound transfer from the unlabeled to labeled DNA as detected using denaturing PAGE following thermal depurination with the results visualized by autoradiography.
No reversibility of the ent-(−)-yatakemycin DNA alkylation was observed when the transfer incubation was conducted at the lower pH and temperature conditions (pH 6.0, 25–47 °C, and pH 7.4, 25 °C) even when conducted for up to eight days. Under intermediate incubation conditions including those close to physiological conditions (pH 7.4, 37 °C and pH 8.4, 25 °C), minimal (∼10%) alkylation reversibility was observed but only after extended reaction times (8 days). However, under more forcing conditions enlisting higher incubation temperatures and pH (pH 7.4, 47 °C and pH 8.4, 37 or 47 °C), more extensive reversibility of alkylation was observed with the transfer being complete at pH 8.4 and 47 °C at the extended times (4–8 days). The quantitation of transfer of ent-(−)-1 from unlabeled to labeled DNA is shown in Figure 8 for these more extreme incubation conditions.
Figure 8.
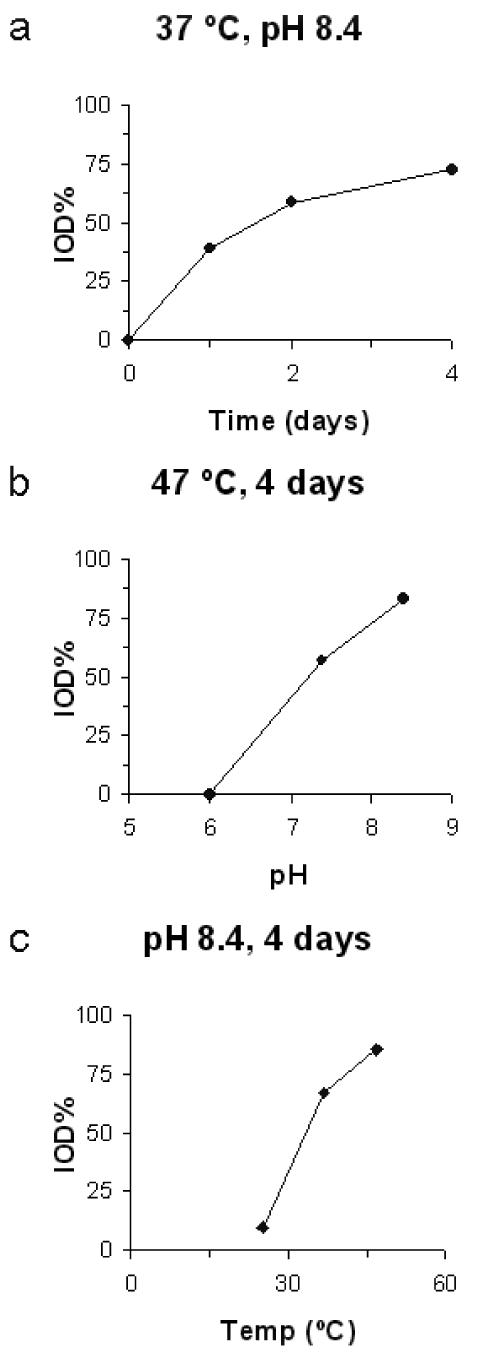
Time (a), pH (b), and temperature (c) dependence of the reversible ent-(−)-yatakemycin DNA alkylation reaction. No reversibility was detected at pH 6.0 (25–47 °C) or pH 7.4 (25 °C) and minimal reversibility was observed at pH 7.4 (37 °C) and pH 8.4 (25 °C).
This alkylation reversibility was not observed with the natural enantiomer and was slower than that of (+)-duocarmycin SA, which were examined alongside ent-(−)-1. Consequently, the diastereomeric relationships of the DNA adducts for ent-(−)-1 versus (+)-1 are sufficient to manifest itself in a detectibly reversible versus irreversible DNA alkylation reaction. Although this diastereomeric nature of the resulting adducts has often been correlated with relative rates or efficiencies of DNA alkylation (natural ≥ unnatural enantiomers) and inferences have been made on the relative stabilities of the resulting adducts,8,9 it has not been previously exemplified. As such, the comparison of (+)- and ent-(−)-yatakemycin illustrate an important and third structural feature that affects the reversibility of the DNA alkylation reaction. From previous studies, we established that the compound relative reactivity (duocarmycin A > SA) correlates with a slower adenine-N3 adduct retroalkylation,8 and the extent of noncovalent binding stabilization (size of compound) also results in less effective adduct reversibility (1 vs 3)5 Herein, we additionally demonstrate that the diastereomeric interaction of enantiomeric compounds with duplex DNA affects the reversibility (stability) of such adducts, illustrating that the unnatural enantiomers not only alkylate DNA at slower rates (kinetic effect), but they also provide thermodynamically less stable adducts (thermodynamic effect). Moreover, even the unnatural enantiomer of yatakemycin alkylates DNA at a faster rate and with a greater thermodynamic stability than the natural enantiomer of duocarmycin SA illustrating the unique characteristics of such “sandwiched” agents.
Interstrand DNA Cross-link
(+)-Yatakemycin represents the first member of this class of antitumor agents to contain a thioester. Although an analogous methyl ester has been encountered in many structures of this class (e.g. duocarmycin SA), the corresponding thiomethyl ester in 1 represents a unique second, modestly reactive electrophile susceptible to nucleophilic attack.47 With this in mind, a series of conditions were examined (25–37 °C, 1–9 days) to probe formation of DNA interstrand cross-links. The DNA cross-linking studies were performed on both singly 5′-end-labeled w836 DNA48 and a 146 base-pair segment (α-satellite) of DNA.49 Following incubation with (+)-1 under standard conditions (1 × 10−5 M) where typically one alkylation site/DNA stand is occupied or saturating conditions (1 × 10−4 M) where complete consumption of DNA occurs and multiple alkylation sites/DNA strand are observed, removal of unbound agent by EtOH precipitation, no thermal depurination, denaturation, followed by denaturing PAGE (w836 DNA) or non-denaturing PAGE (α-satellite DNA) failed to reveal evidence of detectable quantities of a DNA–DNA cross-link (Supporting Information Figure S1).
Cytotoxic Activity
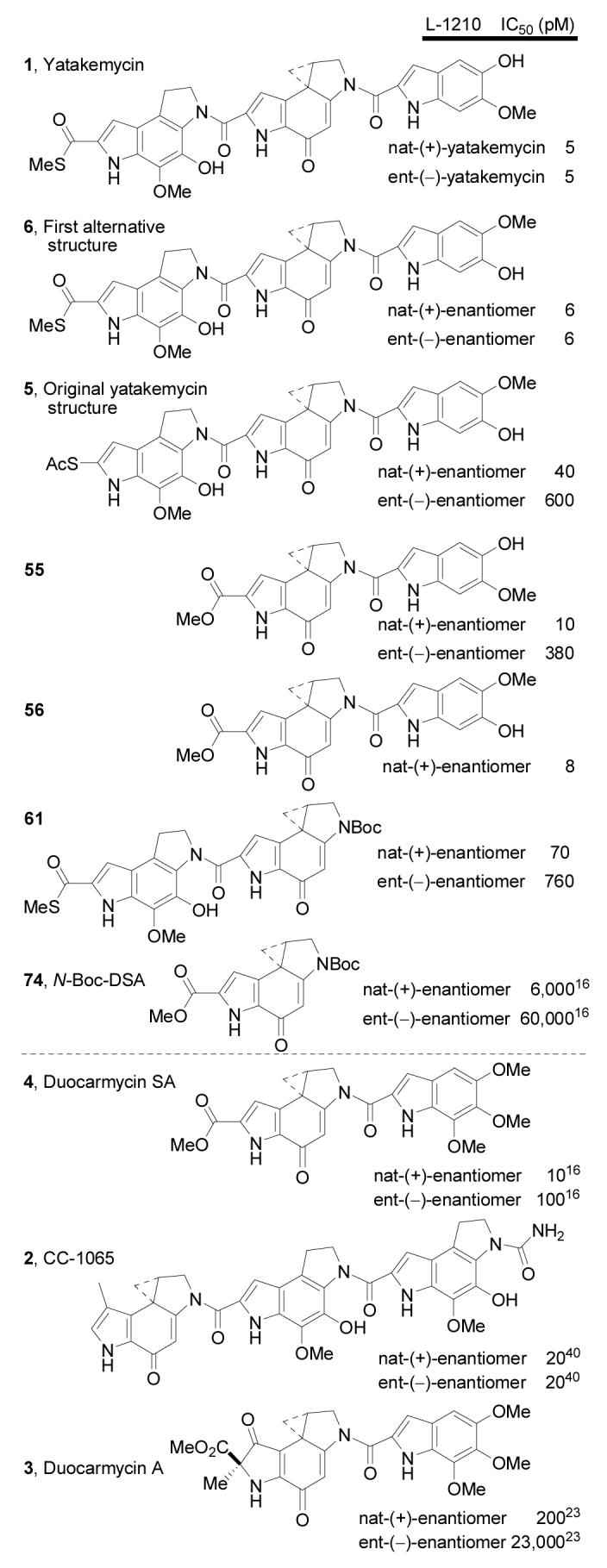
A key element of the studies detailed herein was the opportunity the synthetic studies provide for assessing the properties of not only the natural product which has been available in only limited quantities, but also the unnatural enantiomer, key partial structures and analogues, as well as the misassigned structures 5 and 6. Most central to these is synthetic ent-(−)-1 which was anticipated and found to be essentially indistinguishable from the natural enantiomer itself (Figure 9). Summarized in Figure 9 is the cytotoxic activity of the key series of compounds against a cell line (L1210 mouse leukemia cell line) that has been used historically to compare compounds in this class. Included in this study are several key partial structures (55, 56, 61) that were prepared (Scheme 11, 12) independent of the yatakemycin total synthesis in efforts to define the role of each subunit.
Figure 9.
Cytotoxic activity.
Both natural (+)- and synthetic ent-(−)-yatakemycin proved to be exceptionally potent cytotoxic compounds (IC50 = 5 pM) indistinguishable from one another in this functional cellular assay.50,51 Both are roughly 2-fold more active than (+)-duocarmycin SA (IC50 = 8–10 pM),16 20-fold more potent than ent-(−)-duocarmycin SA (IC50 = 100 pM),16 and 4–5 times more potent than (+)- or ent-(−)-CC-1065 (IC50 = 20 pM).40 Like CC-1065, the two enantiomers of yatakemycin exhibit indistinguishable cytotoxic potency and this serves as a contrast to the behavior of duocarmycin SA where the unnatural enantiomer is substantially less active than the natural enantiomer. On the surface, these observations may appear unusual, but they fit a pattern that has emerged in the studies to date. The unnatural enantiomers of compounds in this class that contain a single DNA binding subunit exhibit significantly less potent cytotoxic activity than the natural enantiomers, whereas those that contain two or more DNA binding subunits approach or match the activity of the corresponding natural enantiomers. These observations mirror the relative differences in their efficiencies of DNA alkylation.
The original structure 5 disclosed for yatakemycin was found to be much less active than the natural product. Although perhaps surprising on the surface, we found that this compound is not very stable and undergoes ready hydrolysis of the thioacetate. Most likely its reduced activity is related to this reduced stability highlighting the fact that the reformulation of the yatakemycin structure as 1 also entailed the removal of a structural liability in the molecule (thioacetate) and its replacement with a structural asset (thiomethyl ester). In contrast and consistent with this, both enantiomers of the first alternate structure 6 prepared11 that differs from yatakemycin only in the right-hand subunit substituent locations (substitution switched) exhibited cytotoxic activity indistinguishable from the natural product and its unnatural enantiomer.
Significantly, removal of the left-hand subunit with 55 (or its isomer 56) provided a key partial structure that was slightly less potent than yatakemycin (2-fold for natural enantiomer) and whose unnatural enantiomer was now 75-fold less potent than ent-(−)-yatakemycin. Nonetheless, the activity of the natural and unnatural enantiomers mirrors that of the two enantiomers of duocarmycin SA, albeit with the unnatural enantiomer being even less potent (4-fold). Most significantly, removal of the right-hand subunit with 61 resulted in a >10-fold loss in activity for the natural enantiomer and a >100-fold loss in activity for the unnatural enantiomer. The alkylation subunit N2-acyl substituent composed of an extended heterocyclic chromophore has been shown to contribute to and be largely responsible for catalysis of the DNA alkylation reaction.9,10 Consequently, it is not surprising that its removal with 61 provided a much less effective compound. Typically, the loss in activity with such “reversed” analogues of duocarmycin SA has been greater than 20-fold (ca 100-fold)10 suggesting that either the unique left-hand subunit of61 (PDE) or the terminal thiomethyl ester is now productively enhancing the activity of 61. Finally, the comparison of yatakemycin and the preceding partial structures 55 and 61 with N-Boc-DSA (74) illustrate that removal of both DNA binding domains reduce the activity 1000-fold (natural enantiomer) and 10,000-fold (unnatural enantiomer) relative to yatakemycin, 600 and 200 fold relative to 55, and 50 to 100-fold relative to61.
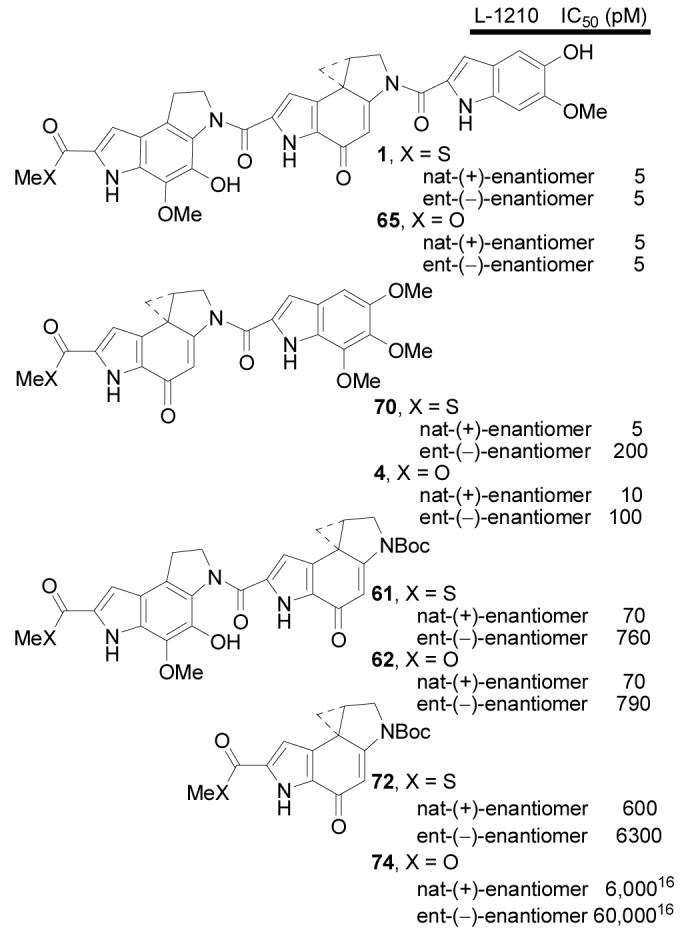
Additionally, four important comparisons were conducted in efforts to establish a functional role for the C-terminus thiomethyl ester (Figure 10). The first, the comparison of both enantiomers of yatakemycin with their corresponding methyl esters 65, revealed that the cytotoxic activity was indistinguishable (IC50 = 5 pM). Thus, both enantiomers of the yatakemycin methyl ester (65) exhibited identical cytotoxic activity to one another independent of their absolute configuration and at a potency that was indistinguishable from the natural product and its unnatural enantiomer.
Figure 10.
Cytotoxic activity.
The second entailed the examination of the duocarmycin SA thiomethyl ester 70 and its comparison with duocarmycin SA itself which possesses a C-terminus methyl ester. Here, interestingly, the natural enantiomer of the modified C-terminus thiomethyl ester was found to be reproducibly 2-fold more potent (IC50 = 5 pM) than the natural product (IC50 = 10 pM) and essentially equipotent with yatakemycin. In this case, the C-terminus thiomethyl ester enhanced the cytotoxic potency of the compound in this functional assay 2-fold. Provocatively, given the results below and this behavior of 70 and its structural relationship with 1, we would suggest that it may be simply a matter of time before 70 is identified as a natural product in its own right. In contrast, the unnatural enantiomer of 70 (IC50 = 200 pM) was 2-fold less potent than the unnatural enantiomer of duocarmycin SA (IC50 = 100 pM) and 40-fold less active than the natural enantiomer of 70.
In contrast to naïve expectations, the comparison of 61 with 62 similarly revealed little or no apparent role for the thiomethyl ester. Thus, the natural enantiomer of the methyl ester 62 exhibited a cytotoxic potency equivalent to that displayed by the thiomethyl ester 61, both being >10-fold less active than yatakemycin. Similarly, the unnatural enantiomers exhibited indistinguishable cytotoxic activities (IC50 = 760–790 pM), both being >100-fold less active than either enantiomer of yatakemycin and 10-fold less active than their respective natural enantiomers. As such, the surprising potency of 61 relative to typical reversed analogues of duocarmycin SA10 (ca. 100-fold less potent) is not attributable to the thiomethyl ester, but appears to be unique to the incorporation of the yatakemycin left-hand subunit.
The final comparison was that of 72 with 74 (N-Boc-DSA) and it proved to be the most interesting. Here both enantiomers of the thiomethyl ester of the simple alkylation subunit (72) were found to be 10-fold more potent than the respective enantiomer of N-Boc-DSA (74) which bears the C-terminus methyl ester. Moreover, this level of activity (IC50 = 600 pM or 0.6 nM) for the natural enantiomer is remarkable for such a simple derivative and it is only 100-fold less potent than duocarmycin SA or yatakemycin. To put this in perspective, N-Boc-DSA (74, IC50 = 6 nM) itself is the most potent of such simplified N-Boc derivatives (IC50 = 300 nM for N-Boc-CPI and 80 nM for N-Boc-CBI),50 a correlation that mirrors the relative stability of the alkylation subunits,50 and its thiomethyl ester is now even 10-fold more potent. This effect is larger than one could expect of a simple substituent effect and suggests a special role for the thioester. Of those that can be envisioned, protein conjugation is most attractive. Whether this entails the intervention of other or additional biological targets or whether this might represent protein conjugation before or following DNA alkylation cannot be distinguished from the studies to date, but the latter would provide DNA-protein cross-links, and investigations to probe such possibilities are in progress.
Conclusions
Complementary to our earlier studies that provided the structural reassignment of yatakemycin and its first total synthesis,11 herein we detailed a second generation, asymmetric total synthesis of (+)-and ent-(−)-yatakemycin enlisting a final transannular Ar-3′ spirocyclization of the free alcohol 48 to close the activated cyclopropane within the full trimer structure. In turn, the left-hand subunit was assembled using a key, regioselective Diels–Alder reaction of a selectively-activated p-quinodiimide followed by oxidative cleavage of a resulting enol ether with release of two differentially oxidized side chains suitable for closure to an appropriately functionalized dihydropyrroloindole. Extending our preceding studies, the lactam 38 used to prepare the misassigned thioacetate of the original structure 5now served as a key precursor to the C-terminus thiomethyl ester via its conversion to 2-bromoindole 39, followed by an effective Pd(0)-catalyzed carbonylation. The central alkylation subunit was prepared enlisting a surprisingly effective regioselective intramolecular 6-endo epoxide addition in which the late stage introduction of the chiral center derived from (R)- or (S)-glycidol permits ready access to either enantiomer.
With the larger quantities of the natural product now being available through its total synthesis and completing our characterization of the (+)-yatakemycin DNA alkylation reaction,5 the isolation, characterization, and quantitation of the thermally-released adenine adduct was accomplished confirming the exclusive adenine N3 addition (no guanine N3 or N7 addition) to only the least substituted cyclopropane carbon of yatakemycin and establishing that this represents the predominant (≥72%), perhaps exclusive, alkylation event.
Just as significantly and with the synthetic unnatural enantiomer now available, the DNA alkylation properties of ent-(−)-yatakemycin were established. Although unusual on the surface, the unnatural enantiomer was found to alkylate DNA with essentially the same selectivity (adenine central to a five-base AT-rich sequence; e.g. 5′-AAAAA) and efficiency as the natural enantiomer consistent with prevailing models.8-10 Differentiating the two enantiomers and consistent with the models, the unnatural enantiomer alkylates DNA at a slower rate (8-fold) and in a reaction that is reversible under forcing conditions whereas that of the natural enantiomer is not, illustrating that the unnatural enantiomer diastereomeric adducts are both kinetically and thermodynamically less favorable than those of the natural enantiomer. Nonetheless, even the unnatural enantiomer of yatakemycin alkylates DNA at a faster rate and with a thermodynamic stability that exceeds that of the natural enantiomer of duocarmycin SA illustrating the special characteristics such “sandwiched” structures possess.
Finally, the synthetic studies provided the opportunity to assess the cytotoxic properties of not only the natural product, but its unnatural enantiomer, key partial structures and analogues as well as those of the misassigned structures 5 and 6. Both synthetic (+)- and ent-(−)-yatakemycin were found to be exceptionally potent and indistinguishable in the functional cellular assay (L1210 IC50 = 5 pM)51 being 2-fold more potent than (+)-duocarmycin SA, 20-fold more potent than ent-(−)-duocarmycin SA and 4–5 fold more potent than the two enantiomers of CC-1065. Each of the flanking subunits contribute to the cytotoxic potency of the natural product with the effect of the right-hand subunit being more pronounced than the left-hand subunit especially with regard to the activity of the unnatural enantiomers. Significantly and surprising on the surface, the thiomethyl ester in yatakemycin and a series of key partial structures was shown not to have apparent impact on their cytotoxic properties. In contrast, the enhanced properties of the thiomethyl ester of duocarmycin SA (2-fold enhancement vs duocarmycin SA methyl ester for the natural enantiomer) suggests that it may well constitute a natural product itself and the remarkable potentiation of the cytotoxic activity of the simple N-Boc derivative 72 of the alkylation subunit (IC50 = 600 pM, 10-fold enhancement vs N-Boc-DSA) implies there may be a special role that the thiomethyl ester can play in enhancing their properties. Additional studies of these and related structural features of yatakemycin continue to be explored and the results of such studies will be disclosed in due time.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA41986 and CA42056), the Skaggs Institute for Chemical Biology, and predoctoral fellowship support from the American Chemical Society (2005–2006 M.S.T., sponsored by Roche Pharmaceuticals) and American Society for Engineering Education (2003–2005 J.D.T., NDSEG). We wish to thank Professor Igarashi of the Toyama Perfectural University for authentic samples of yatakemycin. J.D.T, D.B.K., and M.S.T. are Skaggs Fellows.
Footnotes
Supporting Information Available: Full experimental details, comparison 1H NMR spectra of natural and synthetic 1, and figures illustrating the DNA alkylation sites. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Igarashi Y, Futamata K, Fujita T, Sekine A, Senda H, Naoki H, Furumai T. J. Antibiot. 2003;56:107–113. doi: 10.7164/antibiotics.56.107. [DOI] [PubMed] [Google Scholar]
- 2.Martin DG, Biles C, Gerpheide SA, Hanka LJ, Krueger WC, McGovren JP, Mizsak SA, Neil GL, Stewart JC, Visser J. J. Antibiot. 1981;34:1119–1125. doi: 10.7164/antibiotics.34.1119. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi I, Takahashi K, Ichimura M, Morimoto M, Asano K, Kawamoto I, Tomita F, Nakano H. J. Antibiot. 1988;41:1915–1917. doi: 10.7164/antibiotics.41.1915. [DOI] [PubMed] [Google Scholar]
- 4.Ichimura M, Ogawa T, Takahashi K, Kobayashi E, Kawamoto I, Yasuzawa T, Takahashi I, Nakano H. J. Antibiot. 1990;43:1037–1038. doi: 10.7164/antibiotics.43.1037. [DOI] [PubMed] [Google Scholar]
- 5.Yatakemycin: Parrish JP, Kastrinsky DB, Wolkenberg SE, Igarashi Y, Boger DL. J. Am. Chem. Soc. 2003;125:10971–10976. doi: 10.1021/ja035984h.Trzupek JD, Gottesfeld JM, Boger DL. Nature Chem. Biol. 2006;2:79–82. doi: 10.1038/nchembio761.
- 6.(a) Hurley LH, Lee C-S, McGovren JP, Warpehoski MA, Mitchell MA, Kelly RC, Aristoff PA. Biochemistry. 1988;27:3886–3892. doi: 10.1021/bi00410a054. CC-1065: [DOI] [PubMed] [Google Scholar]; (b) Hurley LH, Warpehoski MA, Lee C-S, McGovren JP, Scahill TA, Kelly RC, Mitchell MA, Wicnienski NA, Gebhard I, Johnson PD, Bradford VS. J. Am. Chem. Soc. 1990;112:4633–4649. [Google Scholar]; (c) Boger DL, Johnson DS, Yun W, Tarby CM. Bioorg. Med. Chem. 1994;2:115–135. doi: 10.1016/s0968-0896(00)82007-6. [DOI] [PubMed] [Google Scholar]; (d) Boger DL, Coleman RS, Invergo BJ, Sakya SM, Ishizaki T, Munk SA, Zarrinmayeh H, Kitos PA, Thompson SC. J. Am. Chem. Soc. 1990;112:4623–4632. [Google Scholar]
- 7.Duocarmycin A: Boger DL, Ishizaki T, Zarrinmayeh H, Munk SA, Kitos PA, Suntornwat O. J. Am. Chem. Soc. 1990;112:8961–8971.Boger DL, Ishizaki T, Zarrimayeh H. J. Am. Chem. Soc. 1991;113:6645–6649.Boger DL, Yun W, Terashima S, Fukuda Y, Nakatani K, Kitos PA, Jin Q. Bioorg. Med. Chem. Lett. 1992;2:759–765.
- 8.Duocarmycin SA: Boger DL, Johnson DS, Yun W. J. Am. Chem. Soc. 1994;116:1635–1656.
- 9.Reviews: Boger DL, Johnson DS. Angew. Chem. Int. Ed. Engl. 1996;35:1438–1474.Boger DL. Acc. Chem. Res. 1995;28:20–29.Boger DL, Johnson DS. Proc. Natl. Acad. Sci. U.S.A. 1995;92:3642–3649. doi: 10.1073/pnas.92.9.3642.Boger DL, Garbaccio RM. Acc. Chem. Res. 1999;32:1043–1052.Boger DL, Garbaccio RM. Bioorg. Med. Chem. 1997;5:263–276. doi: 10.1016/s0968-0896(96)00238-6.
- 10.(a) Boger DL, Bollinger B, Hertzog DL, Johnson DS, Cai H, Mesini P, Garbaccio RM, Jin Q, Kitos PA. J. Am. Chem. Soc. 1997;119:4987–4998. [Google Scholar]; (b) Boger DL, Hertzog DL, Bollinger B, Johnson DS, Cai H, Goldberg J, Turnbull P. J. Am. Chem. Soc. 1997;119:4977–4986. [Google Scholar]
- 11.Tichenor MS, Kastrinsky DB, Boger DL. J. Am. Chem. Soc. 2004;126:8396–8398. doi: 10.1021/ja0472735. [DOI] [PubMed] [Google Scholar]
- 12.(a) Barbero M, Cadamuro S, Degani I, Fochi R, Gatti A, Regondi V. Synthesis. 1988:300–302. [Google Scholar]; (b) Jordan F, Kudzin Z, Witczak Z, Hoops P. J. Org. Chem. 1986;51:571–573. [Google Scholar]
- 13.Okano K, Tokuyama H, Fukuyama T. J. Am. Chem. Soc. 2006;128:7136–7137. doi: 10.1021/ja0619455. [DOI] [PubMed] [Google Scholar]
- 14.Synthesis of 5-hydroxy-6-methoxyindole-2-carboxylic acid (45): 3-benzyloxy-4-methoxybenzaldehyde was condensed with N3CH2CO2Me (4 equiv, 4 equiv of NaOMe, MeOH, −15 °C, 3 h, then 0 °C, 24 h, 87%) and then subjected to cyclization in xylenes (130 °C, 12 h, 72%). Methyl ester hydrolysis (4 equiv of LiOH, 25 °C, 14 h, 93%) and benzyl ether deprotection (1 atm H2, 10% Pd/C, 25 °C, 1 h, 98%) provided 45. The conversion of 45 to 7 accomplished by coupling with pyrrolidine (3 equiv, 3 equiv of EDCI, 25 °C, 4 h, 69%). See:MacKenzie AR, Moody CJ, Rees CW. J. Chem. Soc., Chem. Commun. 1983;22:1372–1373.Bolton RE, Moody CJ, Pass M, Rees CW, Tojo G. J. Chem. Soc. Perkin Trans. 1988;I:2491–2499.
- 15.Review of synthetic studies: Boger DL, Boyce CW, Garbaccio RM, Goldberg JA. Chem. Rev. 1997;97:787–828. doi: 10.1021/cr960095g.
- 16.(a) Boger DL, Machiya K. J. Am. Chem. Soc. 1992;114:10056–10058. [Google Scholar]; (b) Boger DL, Machiya K, Hertzog DL, Kitos PA, Holmes D. J. Am. Chem. Soc. 1993;115:9025–9036. [Google Scholar]
- 17.(a) Muratake H, Matsumura N, Natsume M. Chem. Pharm. Bull. 1995;43:1064–1066. doi: 10.1248/cpb.46.559. [DOI] [PubMed] [Google Scholar]; (b) Muratake H, Abe I, Natsume M. Chem. Pharm. Bull. 1996;44:67–79. [Google Scholar]; (c) Muratake H, Tonegawa M, Natsume M. Chem. Pharm. Bull. 1998;46:400–412. doi: 10.1248/cpb.46.559. [DOI] [PubMed] [Google Scholar]
- 18.Fukuda Y, Terashima S. Tetrahedron Lett. 1997;38:7207–7208. [Google Scholar]
- 19.Yamada K, Kurokawa T, Tokuyama H, Fukuyama T. J. Am. Chem. Soc. 2003;125:6630–6631. doi: 10.1021/ja035303i. [DOI] [PubMed] [Google Scholar]
- 20.Tietze LF, Haunert F, Feuerstein T, Herzig T. Eur. J. Org. Chem. 2003;3:562–566. [Google Scholar]
- 21.Hiroya K, Matsumoto S, Sakamoto T. Org. Lett. 2004;6:2953–2956. doi: 10.1021/ol0489548. [DOI] [PubMed] [Google Scholar]
- 22.(a) Uchiyama M, Kameda M, Mishima O, Yokoyama N, Koike M, Kondo Y, Sakamoto T. J. Am. Chem. Soc. 1998;120:4934–4946. [Google Scholar]; (b) Kondo Y, Matudaire T, Sato J, Murata N, Sakamoto T. Angew. Chem. Intl. Ed. Engl. 1996;35:736–738. [Google Scholar]; (c) Uchiyama M, Koike M, Kameda M, Kondo Y, Sakamoto T. J. Am. Chem. Soc. 1996;118:8733–8734. [Google Scholar]
- 23.Boger DL, McKie JA, Nishi T, Ogiku T. J. Am. Chem. Soc. 1996;118:2301–2302. [Google Scholar]
- 24.Boger DL, McKie JA, Boyce CE. Synlett. 1997:515–517. [Google Scholar]
- 25.Barrett AG, Braddock DC, McKinnell RM, Waller FJ. Synlett. 1999:1489–1490. The Swern oxidation is a more effective alternative to synthesize 8 (1.5 equiv of (ClCO)2, 3 equiv of DMSO, CH2Cl2, −78 °C, 1 h; then 5 equiv of Et3N, −30 °C, 1 h, 95%).
- 26.(a) Shevelev SA, Vatsadze IA, Dutov MD. Mendeleev Commun. 2002;5:196–198. [Google Scholar]; (b) Knudsen RD, Snyder HR. J. Org. Chem. 1974;39:3343–3346. [Google Scholar]
- 27.Abraham DJ, Gazze DM, Kennedy PE, Mokotoff M. J. Med. Chem. 1984;27:1594–1559. doi: 10.1021/jm00378a005. [DOI] [PubMed] [Google Scholar]
- 28.The undesired regioisomer, methyl 5-benzyloxy-7-nitroindole-2-carboxylate, was also produced in 18% (3.8:1 selectivity favoring 12).
- 29.Hanson RM. Chem. Rev. 1991;91:437–475. [Google Scholar]
- 30.Erdik E. Tetrahedron. 1984;40:641–657. [Google Scholar]
- 31.Boger DL, Boyce CW, Garbaccio RM, Searcey M. Tetrahedron Lett. 1998;39:2227–2230. [Google Scholar]
- 32.Patel VF, Andis SL, Enkema JK, Johnson DA, Kennedy JH, Mohamadi F, Schultz RM, Soose DJ, Spees MM. J. Org. Chem. 1997;62:8868–8874. [Google Scholar]
- 33.Boger DL, Yun W. J. Am. Chem. Soc. 1994;116:7996–8006. [Google Scholar]
- 34.Boger DL, Ishizaki T, Zarrinmayeh H, Kitos PA, Suntornwat O. J. Org. Chem. 1990;55:4499–4502. [Google Scholar]
- 35.(a) Adams R, Reifschneider W. Bull. Soc. Chim. Fr. 1958;1:23–65. Review: [Google Scholar]; (b) Boger DL, Zarrinmayeh H. J. Org. Chem. 1990;55:1379–1390. Regioselectivity: [Google Scholar]
- 36.(a) Kraus GA, Yue S, Sy J. J. Org. Chem. 1985;50:283–284. [Google Scholar]; (b) Zawada PV, Banfield SC, Kerr MA. Synlett. 2003:971–974. [Google Scholar]
- 37.Kato S, Morie T. J. Heterocyclic Chem. 1996;33:1171–1178. [Google Scholar]
- 38.Chen JJ, Wei Y, Drach JC, Townsend LB. J. Med. Chem. 2000;43:2449–2456. doi: 10.1021/jm990320x. [DOI] [PubMed] [Google Scholar]
- 39.(a) Boger DL, Coleman RS. J. Am. Chem. Soc. 1987;109:2717–2727. [Google Scholar]; (b) Boger DL, Coleman RS. J. Org. Chem. 1986;51:3250–3252. [Google Scholar]; (c) Carter P, Fitzjohn S, Halazy S, Magnus P. J. Am. Chem. Soc. 1987;109:2711–2717. [Google Scholar]; (d) Rawal VH, Cava MP. J. Am. Chem. Soc. 1986;108:2110–2112. [Google Scholar]; (e) Bolton RE, Moody CJ, Rees CW, Tojo G. J. Chem. Soc., Perkin Trans. 1987;1:931–935. [Google Scholar]; (f) Komoto N, Enomoto Y, Tanaka Y, Nitanai K, Umezawa H. Agric. Biol. Chem. 1979;43:559–561. [Google Scholar]; (g) Boger DL, Coleman RS. J. Org. Chem. 1984;49:2240–2245. see also: [Google Scholar]
- 40.Boger DL, Coleman RS. J. Am. Chem. Soc. 1988;110:1321–323. [Google Scholar]
- 41.6-Hydroxy-5-methoxyindole-2-carboxylic acid (52) was prepared by benzyl deprotection (H2, 10% Pd/C, MeOH, 1 h, 99%) of 6-benzyloxy-5-methoxyindole-2-carboxylic acid (Spectrum Chemical).
- 42.(a) Boger DL, Munk SA, Zarrinmayeh H, Ishizaki T, Haught J, Bina M. Tetrahedron. 1991;47:2661–2682. [PubMed] [Google Scholar]; (b) Boger DL, Munk SA. J. Am. Chem. Soc. 1992;114:5487–5496. [Google Scholar]
- 43.Guanine N3 alkylation even for duocarmycin A and CC-1065 is detected only upon exhaustion of the available adenine N3 alkylation sites and generally requires excess compound to detect. See: ref 8.
- 44.Unlike 5′-AAA, the mixed sequences can contain competitive alkylation sites on the complementary unlabeled strand that diminishes the apparent alkylation efficiency on the labeled strand. The majority of the observed three base A/T selectivity is simply a statistical preference exaggerated by competitive unlabeled strand alkylation rather than unique inherent characteristics present in individual sequences.
- 45.Li LH, Swenson DH, Schpok SLF, Kuentzel SL, Dayton BD, Krueger WC. Cancer Res. 1982;42:999–1004. [PubMed] [Google Scholar]; Swenson DH, Li LH, Hurley LH, Rokem JS, Petzold GL, Dayton BD, Wallace TL, Lin AH, Krueger WC. Cancer Res. 1982;42:2821–2828. [PubMed] [Google Scholar]
- 46.(a) Boger DL, Yun W. J. Am. Chem. Soc. 1993;115:9872–9873. [Google Scholar]; (b) Warpehoski MA, Harper DE, Mitchell MA, Monroe TJ. Biochemistry. 1992;31:2502–2508. doi: 10.1021/bi00124a009. [DOI] [PubMed] [Google Scholar]; (c) Lee C-S, Gibson NW. Biochemistry. 1993;32:9108–9114. doi: 10.1021/bi00086a015. [DOI] [PubMed] [Google Scholar]
- 47.(a) Wolkenberg SE, Boger DL. Chem. Rev. 2002;102:2477–2496. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]; (b) Tse WC, Boger DL. Chem. Biol. 2004;11:1607–1617. doi: 10.1016/j.chembiol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 48.Boger DL, Johnson DS, Palanki MSS. Bioorg. Med. Chem. 1993;1:27–38. doi: 10.1016/s0968-0896(00)82100-8. [DOI] [PubMed] [Google Scholar]
- 49.Luger K, Maeder AW, Richmond RK, Sargent DF, Richmond TJ. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 50.Parrish JP, Hughes TV, Hwang I, Boger DL. J. Am. Chem. Soc. 2004;126:80–81. doi: 10.1021/ja038162t. [DOI] [PubMed] [Google Scholar]
- 51.The IC50 (L1210) for (+)-yatakemycin ranges from 3–5 pM and has been tested ≥ 20 times over several years, side-by-side with (+)-duocarmycin (IC50 = 8–10 pM). The 2–3 fold difference in potency is always observed and the absolute potencies always fall in this narrow range indicated. We have developed highly refined, reproducible conditions for conducting such cytotoxic assays and are confident in other similarly close comparisons made herein. We would be happy to share our insights with others who might experience typically more variable results with such assays.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.