

Abstract
Several bacterial systems show behavior interpreted as evidence for stress-induced mutagenesis (adaptive mutation), a postulated process by which nongrowing cells temporarily increase their general mutation rate. Theoretical considerations suggest that periodic stress-induced general mutagenesis would not be advantageous in the long term, due to the high cost of deleterious mutations. Alternative explanations have been tested for very few of the systems used as evidence for stress-induced mutation. In one prominent system, mutants resistant to rifampicin (RifR; rpoB; RNA polymerase) accumulate in cell populations that “age” on solid medium with little net growth. Mutant accumulation was initially attributed to stress-induced general mutagenesis in nongrowing cells. Evidence is presented that these RifR mutants accumulate because they grow faster than parent cells during the aging period. Direct tests revealed no increase in the frequency of other mutant types during the aging period.
Classic experiments have led to the conclusion that mutations arise as errors in replication or repair and show a frequency and site specificity that is random with respect to phenotypic consequences or need (1, 2). Once formed, mutants may change in frequency, depending on their growth rate relative to that of the parent strain. Mutations can also be induced by external conditions: chemical mutagens or DNA-damaging agents (3).
The possibility of a third source of mutations has been entertained for over 150 years but never demonstrated conclusively. That is, do cells possess mechanisms to generate mutations in their own genome in response to environmental stresses that are not directly mutagenic? Stress-induced mutation lost favor after classic demonstrations that bacterial mutants form before exposure to the selective conditions used to detect them (1, 2). Interest was rekindled when it was pointed out that these experiments used lethal selections that could detect only mutants formed before selection, leaving open the possibility of some stress-induced mutations (4–6).
Several bacterial systems show behavior consistent with a mechanism that up-regulates the general mutation rate during stress, thereby contributing new beneficial mutations that circumvent growth limitation. In opposition to this interpretation, the vast excess of deleterious mutations has been cited as evidence that such a mechanism is unlikely to be of long-term benefit (7).
In the Cairns system (8), an Escherichia coli mutant defective in lactose catabolism regains the ability to use lactose during exposure to selective conditions, a behavior initially attributed to stress-induced mutagenesis (adaptive mutation). The behavior of this system can now be fully explained by selection alone (9), although the original interpretation retains enthusiastic support (10, 11). Resolution of the question of stress-induced mutagenesis requires that each system be tested for the possibility that its behavior reflects cryptic growth and selection (rather than mutagenesis).
Prominent support for stress-induced mutagenesis is the increase in the frequency of rifampicin-resistant mutants (RifR, rpoB, RNA polymerase) seen in aging, nongrowing colonies of E. coli (12–14). A colony forms during 1 day of growth on rich solid medium. During the following 6 days, the colony population increases very little (1.2-fold), but its frequency of RifR mutant cells increases substantially (10–100-fold). The increase in number of RifR cells was attributed to stress-induced mutagenesis in nongrowing cells (12). This observation is re-examined here in both E. coli and Salmonella enterica. The original results are confirmed: RifR mutant frequency increases with time and depends on RpoS (a sigma factor of RNA polymerase active in stationary phase). Evidence is presented that the accumulation of RifR mutants is due to selection; RifR mutants arising before growth limitation grow faster than parent cells during the aging period. No general mutagenesis occurs, as direct tests revealed no accumulation of auxotrophic mutations during aging.
Results
Increases in the Frequency of RifR Mutants in Aging Colonies of S. enterica and E. coli.
The accumulation of RifR mutants in aging colonies was first described for E. coli (13, 14). The extent to which RifR mutants accumulated varied widely from one E. coli isolate to the next (12) and depended on a functional rpoS gene (encoding a stationary phase sigma factor of RNA polymerase). Results below confirm the original results for E. coli and demonstrate the same accumulation in S. enterica.
Colonies of two well characterized strains of S. enterica serovar Typhimurium, LT2 and the more virulent strain 14028s, were initiated by spotting 100–1,000 cells on nitrocellulose filters resting on rich agar plates (LA; Materials and Methods). During the first day, the colony population grew to 109 viable cells, defined as colony forming units (cfu). The population increased an additional 10-fold (to 1010 cells) by Day 2, but showed no further increase or decrease during the next 5 days, as reported earlier for E. coli (14).
Because so few cells were used to initiate the colony, no RifR mutants were present in the initial inoculum. Thus any RifR mutants found later within the colony must have arisen during the time on solid medium. Table 1 describes the time-dependent increase in the frequency of RifR mutants within a colony of S. enterica. Between Day 1 and Day 7, this frequency increased 21-fold for strain 14028s, but only 4-fold for LT2 (the standard genetic wild-type strain). Because accumulation of RifR mutants in E. coli was originally shown to depend on RpoS (12), it seemed that differences in RpoS levels might explain the different accumulation rates in the two S. enterica strains. The rpoS gene of strain LT2 is known to be partially defective due to its atypical start codon, UUG (15), whereas the rpoS gene of strain 14028s has a standard AUG start codon.
Table 1.
Frequency (×109) of RifR mutants per colony
| RifR/Day 1 colony* | Dev† | N‡ | RifR/Day 7 colony* | Dev.† | N‡ | Day 7/Day 1§ | P¶ | |
|---|---|---|---|---|---|---|---|---|
| S. enterica | ||||||||
| 14028s rpoS+ | 6.8 | 9.4 | 36 | 150 | 58 | 25 | 21 | 0.0001 |
| LT2 rpoS | 4.9 | 7.7 | 18 | 21 | 3.5 | 21 | 4 | 0.0015 |
| 14028s rpoS | 3.1 | 7.1 | 49 | 17 | 2.2 | 27 | 5 | <0.0001 |
| LT2 rpoS+ | 7.0 | 3.8 | 12 | 240 | 2.4 | 12 | 34 | 0.0006 |
| E. coli | ||||||||
| MG1655 | 3.7 | 2.5 | 10 | 34 | 2.0 | 10 | 9 | 0.0058 |
| CFT073 | 3.3 | 19 | 5 | 11 | 4.5 | 8 | 3 | 0.3421 |
| Nu14 | 3.2 | 7.2 | 5 | 8.7 | 11 | 8 | 3 | 0.093 |
| C1181 | 2.7 | 3.2 | 5 | 45 | 4.3 | 8 | 16 | 0.0067 |
| C1197 | 2.3 | 12 | 5 | 530 | 4.7 | 8 | 230 | 0.0044 |
*Median frequency (× 109) of RifR cells/colony on day 1 and day 7.
†Dev. is the absolute average deviation from the median (see Materials and Methods).
‡N is the number of independent colonies assayed for RifR mutants.
§Day 7/Day 1 is the ratio of median frequency of RifR mutants in day 7 colonies to frequency in day 1 colonies.
¶Two-tailed P values. (The Day 7/Day 1 ratio is significantly different from 1 as measured by the Mann–Whitney nonparametric test).
To test dependence on RpoS, we introduced an rpoS mutation into strain 14028s, and found that it reduced the Day 7 level of RifR mutants to that seen in LT2 (Table 1). Conversely, introduction of a wild-type rpoS locus from 14028s into LT2 increased the Day 7 RifR mutant frequency to at least the level observed in 14028s. Thus the time-dependent increase in RifR mutant frequency is strongly dependent on RpoS in S. enterica, as shown previously for E. coli.
The original demonstration that RifR mutants accumulate in E. coli was confirmed for E. coli strains MG1655 and CFT073, whose genomes have been sequenced, and for 51 clinical isolates previously characterized for their antibiotic resistance phenotypes (16, 17). See Table 1 for two examples. A ratio of the RifR mutant frequency on Day 7 to the frequency on Day 1 ranged from 0.08 to 400, with a median value of 8 (and an absolute average deviation from median of 5). This is similar to the ratio of 7 found previously for 787 natural isolates of E. coli (12).
Logarithmic Accumulation of RifR Mutants Suggested Growth Under Selection.
The time course of RifR mutant accumulation was measured in both wild-type (14028s) and rpoS mutant cells over 28 days, well beyond the period of the original experiment (7 days) (See Fig. 1.) For both wild-type and the rpoS parent strains, the RifR mutant frequency increased logarithmically and fit well to exponential curves (R2 values of 0.93 and 0.91, respectively) with exponents of 0.41 and 0.21 respectively. The increase in RifR mutant frequency depended heavily on RpoS.
Fig. 1.
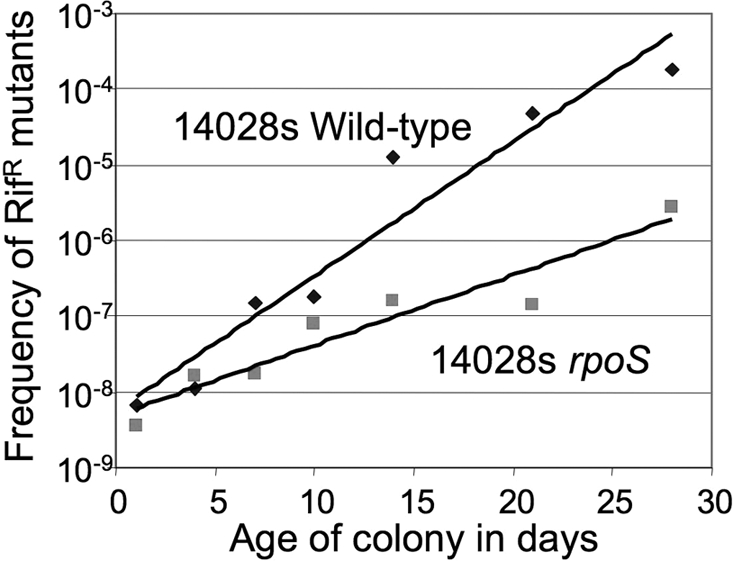
Increase in the frequency of RifR mutants as a function of colony age. Colonies aging on LA plates were assayed for their frequency of RifR mutant cells. The two strains tested differ only by substitution of an AmpR determinant for rpoS (rpoS::AmpR).
The logarithmic frequency increase suggested that RifR mutants accumulate during aging because they grow faster than the parental strain (or they grow logarithmically while the parent fails to grow) and that this growth advantage depends on RpoS. Assuming that the parent population shows no growth, these data are consistent with RifR cells growing with a doubling time of 1.7 days in the presence of RpoS and 3.2 days with the rpoS mutation. Thus very little growth is needed to explain the observed increase the frequency of RifR cells.
If RifR mutant accumulation were due to mutagenesis, as originally proposed, the mutant frequency would be expected to increase linearly with time (assuming a nongrowing population and a constant mutation rate). To explain the observed logarithmic increase by mutagenesis of nongrowing cells requires the mutation rate to increase logarithmically, which seems unlikely. To explain the 104-fold increase in RifR mutant frequency over 28 days by mutagenesis requires an intensity higher than any demonstrated experimentally for a nongrowing population. Standard laboratory mutagenesis of nongrowing cells can increase mutant frequency 100–1,000 fold; larger increases are prevented by loss of viability due to lethal mutations. Larger increases in mutant frequency can be obtained only by mutagenizing a growing population so as to continuously counterselect deleterious mutations. Heavy mutagenesis would predict accumulation of dead cells which was not seen (see Materials and Methods), and a large increase in secondary nonlethal mutations in RifR mutants, also not observed (see next paragraph).
RifR Mutants Show No Evidence of Intensive Mutagenesis.
Colonies of wild-type 14028s were aged on rich medium for 1, 7, or 21 days. Day 1 colonies were suspended and spread on LA-Rif selective media. Day 7 and Day 21 colonies were replica-plated directly onto LA-Rif. RifR colonies were picked and purified, then patched onto minimal medium and LA. The frequency of auxotrophs among RifR mutants (Table 2) were in the range of 1/1,000 as expected for loss-of-function mutations in the roughly 200-gene target used to detect auxotrophs. (Null mutations in a single gene are typically present at 10−5 in an unselected overnight culture.) The small increase in the auxotroph frequency over time is similar to that reported initially for resistance to a variety of antimicrobial agents (12). In contrast to this very small increase in auxotroph frequency, the frequency of RifR mutants increased 21-fold by Day 7, and 6,800-fold by Day 21 (Fig. 1). The rarity of RifR mutants with an associated auxotrophy makes it unlikely that the RifR mutants owed their appearance to intense mutagenesis.
Table 2.
Autotroph frequency in aging wild type (14028s)
| Age of population | RifR mutants tested | No. of auxotrophs | Auxotroph frequency | Fold increase |
|---|---|---|---|---|
| Day 1 | 2169 | 14 | 3.8 × 10−3 | 1 |
| Day 7 | 794 | 3 | 6.5 × 10−3 | 1.7 |
| Day 21 | 459 | 4 | 8.7 × 10−3 | 2.3 |
Evidence for Clonal Growth of RifR Cells in Aging Colonies.
If RifR mutants accumulate because the few mutants cells that arise during growth (Day 1–2) grow faster than the parent population during the aging period of growth-limiting selection (Days 2–7), then the mutant cells that accumulate under selection should be found as localized clones (papillae) at a few sites within the aging colony. In contrast, if growth limitation (aging) induces mutagenesis in a nongrowing cell population as suggested previously (12), then individual RifR cells should arise independently and be broadly distributed throughout the aged colony.
To test these predictions, we divided aged colonies into 16 sectors (4 × 4) and assayed each sector individually for its frequency of RifR mutants. Sectors were tested from 30 colonies of strain 14028s on Day 7 and from 5 colonies on Day 14. In each case, ≤95% of the RifR mutants were contained within 1 or a few of the 16 sectors (see two examples in Fig. 2 and additional data in Fig. 3). The median number of RifR mutants per sector on Day 7 was 4. Sectors with many RifR mutants (50–10,000) are called “jackpot” sectors. This uneven distribution of accumulated RifR mutant cells is expected if preexisting RifR mutants (arising by Day 1) form clones that expand during the aging period (Day 1 to Day 7).
Fig. 2.
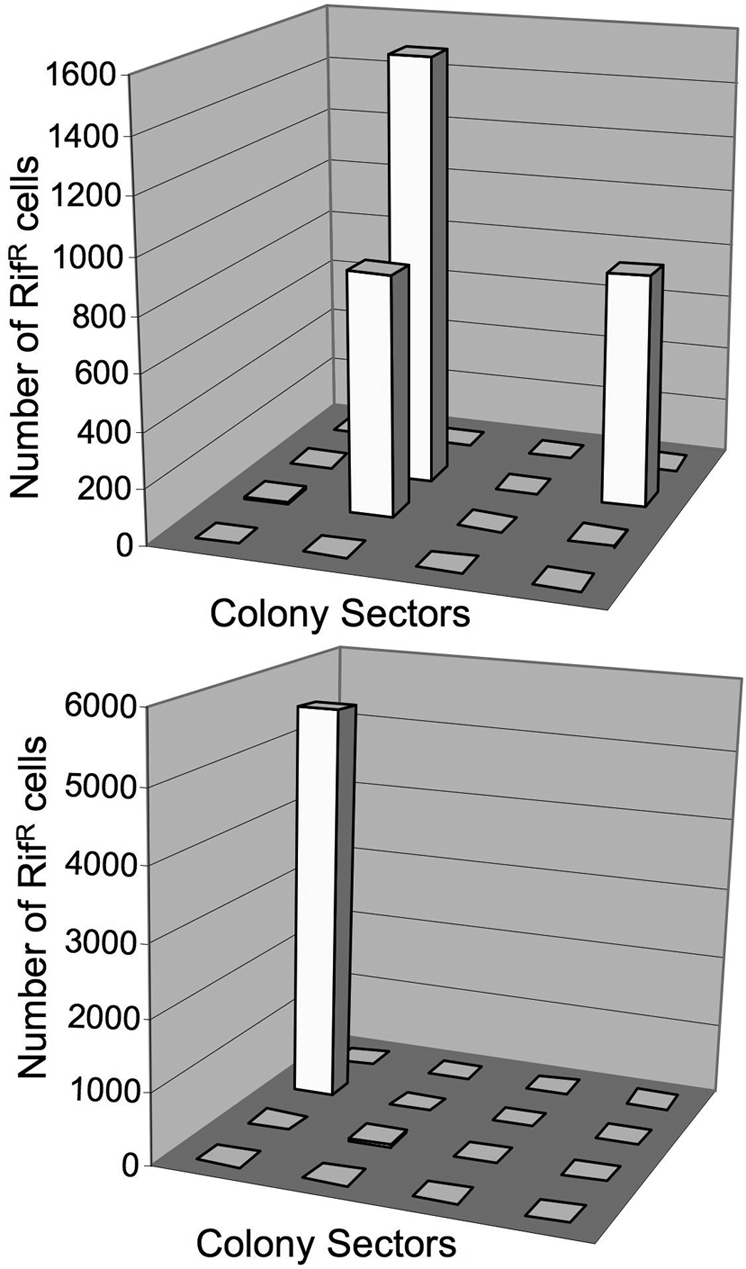
Distribution of RifR cells in two Day 7 colonies of strain 14028s. Each colony was cut into 16 equal sectors, which were assayed for RifR cells. More data are in Fig. 3.
Fig. 3.
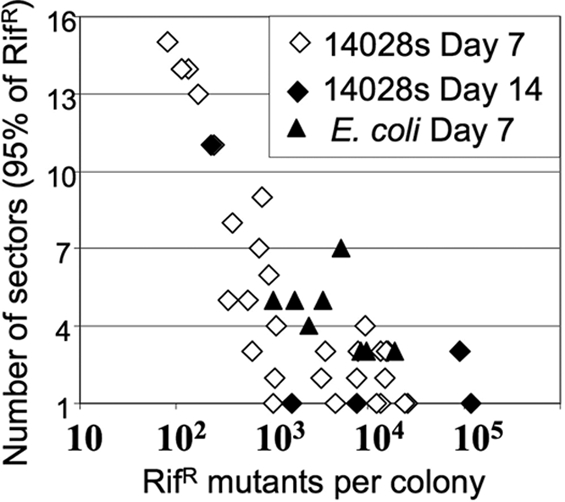
Colonies with more RifR mutants have fewer, but larger, clones. Each point in the graph represents one aged colony. The total RifR mutants per colony (sum of all sectors) is plotted against the number of sectors that together include 95% of the mutants. Strain 14028s is an S. enterica isolate. The E. coli natural isolates were described previously (16).
Relationship Between Spatial Distribution and Mutant Number.
The uneven spatial distribution of RifR mutants is a general feature of this system. Two colonies are described in Fig. 2 and additional colonies in Fig. 3. In each colony, the bulk of the mutant cells (>95%) were found in a few of the 16 sectors tested.
If the accumulation of RifR mutants is due primarily to the growth of clones, then variation in the number of mutants from one colony to the next is expected to depend most heavily on differences in mutant clone size (which increases logarithmically) and less on variability in the number of clones present. Colonies with more mutants are predicted to have larger clones, allowing fewer sectors to contain the bulk of the mutants (>95%). That is, the more mutants a colony contains, the more uneven is the spatial distribution of mutant cells. Conversely, if mutagenesis of nongrowing cells were responsible for the accumulation of RifR mutants, one would expect the spatial distribution of mutants to become more even as the total number of mutant cells increases. Data in Fig. 3 support the idea that an increasing number of mutants is associated with more uneven distribution, as predicted by growth under selection.
In E. coli, as in S. enterica, the accumulated RifR mutants were found in a few separated local sectors within the colony. Colonies of the clinical isolates C1181 and C1197 were divided into 16 sectors, and the frequency of RifR mutants was found to vary widely from one sector to the next. These results are included in Fig. 3, which portrays the small number of highly populated sectors and the reduced number of sectors required to account for 95% of the RifR mutants (as seen in S. enterica).
Evidence That RifR Cells from a Single Sector Are Clonally Related.
The DNA base sequence of the rpoB gene (where RifR mutations occur) was determined for at least five RifR mutants from each of several colony sectors. In almost all cases (see below), the five sequenced mutants from an individual sector carried the same rpoB sequence change. Mutants from different sectors seldom shared the same sequence change. This argues strongly that the many RifR cells in a single sector (above) belong to one clone that expanded during the selection period. These sequence changes are described in more detail below.
The Nature of RifR (rpoB) Mutations That Provide a Growth Advantage in Aging Colonies.
Although the physiological basis of the growth advantage of RifR mutants is not known, it seemed possible that only a particular subset of RifR mutations might enhance growth during aging. Mutations of rpoB that confer a RifR phenotype affect a large number of sites in the rpoB gene (18) and can have a wide variety of effects on other aspects of cell physiology (19, 20). Thus one might expect that certain RifR (rpoB) mutations are more likely than others to be found in the jackpot sectors of Day 7 colonies.
Two factors are expected to contribute to the likelihood of a particular RifR mutation appearing in a jackpot clone. One is the magnitude of its conferred growth advantage over wild type during the aging period. Another factor is the frequency with which a particular mutation arises. This frequency influences not only the number of times a particular mutation appears, but also the time in the history of the colony at which the RifR mutation arises. Any RifR mutation that arises early during growth is expected to have more time in which to grow and achieve a large clone size, both before and during the aging period. Frequent mutations are likely to appear early in the rapid growth period (106-fold) on Day 1. Thus growth advantage and frequency of occurrence are expected to contribute independently to the likelihood of particular mutations being found in large clones after the aging period.
To test these expectations, we sequenced the rpoB gene from 271 different RifR mutants from the following sources:
From Day 1 populations (that experienced very little selection), one RifR mutant was sequenced from each of 46 independent filter colonies. This set included 12 different mutant rpoB alleles (see Table 3).
From each of 55 nonjackpot sectors of Day 7 colonies (median 4 RifR cells/sector), a single randomly chosen RifR mutant was tested. This yielded 20 different rpoB alleles.
From each of 32 jackpot sectors (with 50–10,000 RifR cells), five RifR mutants were tested. This collection included 8 different sequence changes. For 30 of the 32 sectors, all five mutants tested had the same rpoB sequence change, demonstrating clonality of the population. The two other sectors each included two mutant types; both of these sectors had a very small jackpot (<200 mutants).
Mutants from different jackpot sectors of the same colony (6/7 colonies with multiple jackpots) showed different rpoB sequence changes.
Table 3.
Nature and selective value of mutations arising during aging
|
rpoB mutation |
No. of occurrences |
Competitive index (CI) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Substitution | Day 1 | Day 7, nonjackpot | Day 7, jackpot | CI | Dev* | N† | P value‡ | |
| 1 | D516G | 14 | 8 | 14 | 2.7 | 2.3 | 11 | 0.0002 | |
| 2 | S531F | 10 | 3 | 0 | 1.0 | 0.4 | 12 | 0.1362 | |
| 3 | H526Y | 5 | 3 | 8 | 1.1 | 7.6 | 14 | 0.6312 | |
| 4 | S512P | 3 | 5 | 3 | 1.1 | 0.4 | 16 | 0.8103 | |
| 5 | R529H | 3 | 4 | 0 | 5.2 | 3.6 | 8 | <0.0001 | |
| 6 | P564L | 3 | 4 | 4 | 27.8 | 93.2 | 11 | <0.0001 | |
| 7 | S522F | 2 | 3 | 0 | 0.2 | 0.7 | 12 | 0.0014 | |
| 8 | I572F | 2 | 1 | 0 | 1.7 | 0.7 | 5 | 0.1188 | |
| 9 | K504N | 1 | 1 | 0 | 3.0 | 1.0 | 11 | <0.0001 | |
| 10 | L511P | 1 | 1 | 0 | 3.1 | 1.5 | 11 | <0.0001 | |
| 11 | S512F | 1 | 4 | 2 | 0.9 | 0.3 | 17 | 0.2891 | |
| 12 | R529C | 1 | 6 | 1 | 17.2 | 7.2 | 16 | <0.0001 | |
| 13 | S512Y | 0 | 2 | 0 | 0.8 | 0.6 | 18 | 0.3628 | |
| 14 | Q513L | 0 | 2 | 0 | 5.7 | 3.7 | 13 | 0.0061 | |
| 15 | Q513H | 0 | 1 | 0 | 12.9 | 5.9 | 15 | 0.0001 | |
| 16 | D516Y | 0 | 2 | 0 | 2.2 | 0.9 | 23 | <0.0001 | |
| 17 | S522Y | 0 | 0 | 1 | 4.6 | 8.0 | 13 | 0.0105 | |
| 18 | H526L | 0 | 2 | 0 | 1.4 | 1.0 | 12 | 0.2041 | |
| 19 | H526P | 0 | 1 | 0 | 1.3 | 2.3 | 25 | 0.2225 | |
| 20 | R529S | 0 | 1 | 0 | 40.6 | 45 | 12 | <0.0001 | |
| 21 | R529L | 0 | 1 | 1 | 8.1 | 9.4 | 18 | <0.0001 | |
*Dev is the absolute average deviation from the median (see Materials and Methods). Note: the CI was positive for all competitions.
†N is the number of independent colony competitions for each mutant.
‡Two-tailed P values (CI of mutant significantly different from CI of wild-type) measured by Mann–Whitney nonparametric test.
From all sources combined, 21 different mutant rpoB sequences were found among the 271 mutant genes sequenced. Thus many mutant types recurred. Results are described below and in Table 3.
The 8 different rpoB mutations recovered from 32 distinct jackpot sectors could be either types that conferred the largest growth advantage during aging or types that conferred a smaller advantage but arose early during growth and had the most time to expand their clones both before and during aging. The 12 rpoB mutant types that recurred repeatedly among RifR mutants identified before selection (Day 1) are inferred to arise commonly (top of Table 3). To test whether there is a correlation between conferred growth advantage and likelihood of being found in a jackpot sector, we tested the 21 different rpoB mutations in reconstruction experiments for their ability to provide improved growth within an aging colony.
Demonstrating the Selective Advantage of RifR Mutants in Reconstruction Experiments.
Strains carrying each of the 21 different rpoB mutations (described above) were marked with a chromosomal insertion of the transposition-defective Tn10-TetR. Cells of wild-type 14028s were grown as a colony for 1 day, after which a mixture of TetR-marked RifR mutants and CamR-marked (wild-type) cells were spotted onto the colony (see Materials and Methods). These constructed colonies were incubated for a further 7 days, then assayed for the relative frequency of TetR (marking the RifR mutant being tested) and CamR (marking the control wild-type cells). After 7 days, the ratio of TetR to CamR cells was determined and compared to the ratio seen on Day 1. The factor by which the ratio changes over the course of the 7-day aging period is defined as the competitive index (CI), which expresses the magnitude of the growth advantage of RifR cells over parent cells. Results (Table 3) suggest that most of the RifR mutant types (12/21) have a significant growth advantage over the wild type (Fig. 4). A small intrinsic effect of the TetR versus CamR markers on competitive advantage was measured (1.8-fold over 7 days) and was taken into account to normalize the RifR/wild-type competition data (see Materials and Methods).
Fig. 4.

The competitive advantage of 21 selected RifR mutants over the parent strain. Above each column is the number of independent occurrences in Day 7 jackpots. Below the columns are the rpoB mutation numbers (Table 3). Open columns indicate CI values statistically significant from wild type; hatched columns indicate values not statistically significant from wild type.
Four of the 21 mutant types showed a very large growth advantage, with a CI > 10. The magnitude of the growth advantage of a mutant type in the competition assay does not correlate well with the frequency of these rpoB mutations in Day 7 jackpot sectors. This supports the hypothesis that the particular RifR alleles found in Day 7 jackpots are also biased in favor of those that arose early and had the longest time to expand their clones during both the growth and aging periods rather than solely in favor of those with the largest growth advantage. Both of the two mutations (numbers 17 and 21) found in Day 7 jackpots, but not among mutants recovered on Day 1, have a large selective advantage (4- and 8-fold respectively). Conversely the 4 jackpot mutant types with the lowest selective advantage (mutations 1, 3, 4, and 11) were frequent enough to appear (some repeatedly) among the Day 1 mutants.
Demonstrating that the Characterized rpoB (RifR) Mutation Is Sufficient to Provide a Growth Advantage During Aging.
The data in Fig. 4 show that most RifR mutants recovered from aged colonies have a growth advantage in colony competition against the wild type. The growth advantage during aging is actually conferred by the same mutation (rpoB) that confers the RifR phenotype. This was shown by moving three RifR rpoB mutations (mutants 6, 15, and 20) into a wild-type strain (14028s) that had never experienced selection. Each of these three constructed RifR strains was then used in a competition experiment with wild-type cells. In each case, the constructed RifR mutant strain showed a clear growth advantage over the isogenic wild-type strain in the aging colony (Table 4). We conclude that each of these three rpoB mutations is sufficient to confer a growth advantage over wild-type cells in an aging colony.
Table 4.
Mutations (rpoB) fully explain growth advantage
| Mutation | Selected rpoB mutant |
Constructed rpoB mutant |
||||
|---|---|---|---|---|---|---|
| Median CI | (Dev)† | N‡ | Median CI | (Dev.)† | N‡ | |
| Q513H | 12.9 | (5.9) | 15 | 5.5 | (10) | 16 |
| R529S | 40.6 | (45) | 12 | 73.9 | (100) | 11 |
| P564L | 27.8 | (93.2) | 11 | 42.3 | (18.4) | 16 |
*P values (CI of mutant significantly different from CI of wild type by Mann–Whitney nonparametric test) were <0.0001 for all competitions.
†Dev. is the absolute average deviation from the median (see Materials and Methods). Note: the CI was positive for all competitions.
‡N is the number of independent colony competitions for each mutant.
Discussion
An increase in the frequency of RifR cells within aging colonies was initially attributed to stress-induced general mutagenesis (14) and has been cited as support for the idea that cells mutagenize their own genome when growth is prevented (10, 11). Evidence is presented here that the accumulation of RifR mutants is due to growth, not mutagenesis; RifR cells grow during the aging period, whereas the parent population does not. In this system, as in the Cairns lac system (9), accumulation of mutants in stressed cells can be explained by growth under selection and requires no increase in mutation rate.
The following lines of evidence support selection (and argue against mutagenesis):
The frequency of RifR mutants increased logarithmically during the aging period. A linear increase is expected if mutants arise by mutagenesis of a nongrowing population.
Mutant cells were found in localized sectors within the aging colony, consistent with growth of the mutant population from individual precursor cells. An even spatial distribution is predicted if mutants accumulate due to mutagenesis of a nongrowing population.
Mutant cells within a single sector of the colony carried the same sequence alteration, demonstrating their clonal relatedness.
In colonies with more mutant cells, mutants (95% of the total) were found in a smaller number of localized subclones, consistent with variation due to differences in clone growth rate rather than differences in the number of clones.
The RifR mutants that accumulate during aging showed no increase in the frequency of secondary mutations, suggesting that they were not made by induced general mutagenesis.
The growth advantage of RifR mutants during aging can be demonstrated in reconstruction experiments and is conferred on naïve cells that receive the particular rpoB (RifR) mutation.
Part of the evidence initially used to support stress-induced general mutagenesis was an increase in the number of cells resistant to other antibiotics during aging. However, the increase in RifR was much larger than that of the other resistances. The particular E. coli isolate (C4750) showed a 77-fold increase in RifR mutant frequency between Days 1 and 7, but no increase in the frequency of resistance to streptomycin or nalidixic acid. In the same experiment, mutants resistant to 5-fluorouracil, mecillinam, or fosfomycin increased only 3.4-, 1.9-, and 4.8-fold, respectively (12). If general mutagenesis were responsible, one might expect a similar magnitude of increase for all resistant mutants. We suggest that the small increases in other drug-resistant mutants seen previously (like the small increases in auxotroph frequency seen here) are likely to reflect the additional rounds of replication enjoyed by a subset of the population during the aging process.
Although it is not clear why many RifR mutations have a growth advantage during aging, it seems likely that some mutant forms of RNA polymerase may allow expression of genes that promote growth under the aging conditions. Many RifR mutations are known to affect transcription elongation or termination (19–25), including two of the rpoB mutations shown here to enhance growth during aging. Mutation 20 (R529S) increases the probability of transcription through Rho-dependent terminators (23). Mutation 6 (P564L) causes constitutive expression of the pyrimidine biosynthetic gene pyrE by preventing attenuation (26).
The importance of RpoS for the accumulation of RifR mutants was noted initially (12) but was attributed to a role of RpoS in regulating a proposed mechanism for “stress-induced” mutagenesis. We suggest instead that RpoS and the mutant RNA polymerase (RpoB) produced by RifR mutants act together to enhance expression of genes that facilitate growth in the aging colony. This fits with evidence that RpoS is up-regulated during growth limitation and serves to enhance expression of many genes during strong growth limitation (27).
The majority of the different RifR mutants (12/21) found in jackpot clones after selection showed a statistically significant growth advantage when retested. Some of the other jackpot clones may reflect frequent mutations that arise early and expand during the growth period, requiring little or no advantage over the wild type. Other clones may have RifR mutations that confer a growth advantage only when the rpoB gene is selectively amplified. The rpoB gene is located between rrn repeats and is subject to frequent amplifications, which would be subject to loss during unselected growth before retesting.
The phenomenon described here may represent a situation in which a cellular mechanism limits growth under particular conditions. The RifR mutants can be viewed as cheaters that ignore or override the growth controls (28) and continue growing within the quiescent parent colony. The behavior of these RifR mutants may parallel the loss of growth control in cancer cells that proliferate in a multicellular body, where most cells are quiescent.
Materials and Methods
Bacterial Strains and Growth Conditions.
Strains of S. enterica serovar Typhimurium were derived from wild-type strains ATCC 14028s and LT2. E. coli strains are described in the text. Transduction crosses used phage P22 HT (high transducing). Bacteria were grown at 37°C in Luria-Bertani broth (LB) and on plates of LB medium solidified with 1.5% agar (Oxoid), 0.2% glucose, and 3 mM CaCl2 (LA plates). Rifampicin was in media at 100 μg/ml, tetracycline at 15 μg/ml, and chloramphenicol at 50 μg/ml.
Determination of RifR Frequency in Aging Colonies.
Colonies were initiated by spotting 2 μl (100–1,000 cfu) of a diluted fresh overnight culture onto a nitrocellulose filter (82-mm diameter, 0.2-μm pore size, Protran BA83; Whatman, Schleicher & Schuell) on an LA plate. A maximum of four colonies were grown per filter. Plates with filters were incubated in sealed plastic bags at 37°C for 1 or 7 days typically, but up to 28 days in some cases. After incubation, colonies were cut from the filter and suspended in 1 ml 0.9% NaCl. Appropriate dilutions were spread on LA and LA–rifampicin plates and incubated for 24 h.
In the earlier work by Bjedov et al., it was claimed that mutagenesis occurred in a nongrowing population (12). To determine whether the rather constant cell number during aging reflected a balance between more substantial rates of cell growth and death, we assessed the number of dead cells in the colony. This number did not increase between Day 1 and Day 7 colonies based on microscopy using the LIVE/DEAD BacLight bacterial viability kit (Molecular Probes).
Colony Sectoring.
An aged colony (typically 1.5 cm in diameter after 7 or 14 days) was cut into 16 pieces by using a sharp sterile scissors and fine tweezers, and cells on each piece were suspended in 500 μl of 0.9% NaCl solution. Less than 10% of the cells in a colony were lost during the cutting and processing of colonies. Dilutions were plated on LA and LA–rifampicin plates and incubated 24 h at 37°C to determine total cells and number of RifR mutant cells.
Colony Competition Assay.
RifR mutants isolated from colonies were assayed for their growth competitiveness in reconstructed colonies. A filter colony was initiated as described above. After 24 h incubation, 4 μl of a competition mix was spotted onto the Day 1 colony. This mixture contained equal numbers of RifR competitor cells (marked with a tetracycline resistance marker, zhe-8953::Tn10dTet) and wild-type cells (marked with a chloramphenicol resistance marker, zcd-3677::Tn10dCam). To test the effect of these markers on fitness, we conducted 59 competition experiments obetween Tet-marked and Cam-marked wild-type strains. The zhe-8953::Tn10dTet marker confers a small advantage over 7 days of competition: median TetR/CamR = 1.8 (Mann–Whitney: P = 0.0017), which was used to normalize the reported competitive index for TetR-marked mutant versus CamR-marked wild-type strains.
Mixtures with either ≈102 or ≈103 cfu of each competitor were tested in parallel (Day 0). After 7 days of additional incubation (Day 7) the colony was suspended in 1 ml of 0.9% NaCl and appropriate dilutions plated on LA, LA–tetracycline, and LA–chloramphenicol plates. The CI was calculated for each RifR mutant from the change in the RifR/wild-type ratio from Day 0 to Day 7.
Statistical Analysis.
Median and average absolute deviations from the median (AADM) were calculated using a descriptive statistics package available online (http://www.physics.csbsju.edu/stats/descriptive2.html). Note that an AADM larger than the median is caused by the non-normal distribution of the CI values (the presence of outliers, colonies in which the positive CI was very large) and does not imply negative CI values in any competitions. The significance of the differences in CI between mutants and wild-type (P values, two-tailed 95% confidence value) were calculated using the Mann–Whitney nonparametric test available on the VassarStats Web Site for Statistical Computation (http://faculty.vassar.edu/lowry/VassarStats.html).
PCR Amplification and DNA Sequencing.
To prepare DNA, we suspended a single bacterial colony in 100 μl sterile water and boiled it for 5 min. PCR was performed with Ready-2-Go PCR beads (Amersham Bioscience) in a 25-μl volume containing 0.4 μM forward and reverse primers and 1 μl DNA sample. PCR conditions were as follows: denaturation at 95°C for 5 min, then 30 cycles of 95°C (for 15 s), 55°C (20 s), and 72°C (2 min). Primers for amplifying rpoB were 1421F: 5′-CGGTGAAAGAGCGTCTGTCT and 2170R: 5′-CAGTACCGCCACGTTTAGCT. Each sequencing reaction (Uppsala Genome Center, Rudbeck Laboratory, Uppsala, Sweden) contained ≈10 ng purified PCR product and 1.6 pmol sequencing primer 1453F: 5′-GATACCCTGATGCCTCAG.
Acknowledgments.
This work was supported by grants from the Swedish Research Council and the EU 6th Framework Program (Contract LSHM-CT-2005-518152) (to D.H.), and by NIH Grant GM27068 (to J.R.R.).
Footnotes
The authors declare no conflict of interest.
References
- 1.Lederberg J, Lederberg EM. Replica plating and indirect selection of bacterial mutants. J Bacteriol. 1952;63:399–406. doi: 10.1128/jb.63.3.399-406.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luria SE, Delbrück M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mortelmans K, Zeiger E. The Ames Salmonella/microsome mutagenicity assay. Mutat Res. 2000;455:29–60. doi: 10.1016/s0027-5107(00)00064-6. [DOI] [PubMed] [Google Scholar]
- 4.Cairns J, Overbaugh J, Miller S. The origin of mutants. Nature. 1988;335:142–145. doi: 10.1038/335142a0. [DOI] [PubMed] [Google Scholar]
- 5.Hall BG. Selection-induced mutations. Curr Opin Genet Dev. 1992;2:943–946. doi: 10.1016/s0959-437x(05)80120-0. [DOI] [PubMed] [Google Scholar]
- 6.Shapiro JA. Observations on the formation of clones containing araB-lacZcistron fusions. Mol Gen Genet. 1984;194:79–90. doi: 10.1007/BF00383501. [DOI] [PubMed] [Google Scholar]
- 7.Roth JR, et al. Regulating general mutation rates: Examination of the hypermutable state model for Cairnsian adaptive mutation. Genetics. 2003;163:1483–1496. doi: 10.1093/genetics/163.4.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cairns J, Foster PL. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics. 1991;128:695–701. doi: 10.1093/genetics/128.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roth JR, Kugelberg E, Reams AB, Kofoid E, Andersson DI. Origin of mutations under selection: The adaptive mutation controversy. Annu Rev Microbiol. 2006;60:477–501. doi: 10.1146/annurev.micro.60.080805.142045. [DOI] [PubMed] [Google Scholar]
- 10.Galhardo RS, Hastings PJ, Rosenberg SM. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol. 2007;42:399–435. doi: 10.1080/10409230701648502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster PL. Stress-induced mutagenesis in bacteria. Crit Rev Biochem Mol Biol. 2007;42:373–397. doi: 10.1080/10409230701648494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bjedov I, et al. Stress-induced mutagenesis in bacteria. Science. 2003;300:1404–1409. doi: 10.1126/science.1082240. [DOI] [PubMed] [Google Scholar]
- 13.Taddei F, Halliday JA, Matic I, Radman M. Genetic analysis of mutagenesis in aging Escherichia coli colonies. Mol Gen Genet. 1997;256:277–281. doi: 10.1007/s004380050570. [DOI] [PubMed] [Google Scholar]
- 14.Taddei F, Matic I, Radman M. cAMP-dependent SOS induction and mutagenesis in resting bacterial populations. Proc Natl Acad Sci USA. 1995;92:11736–11740. doi: 10.1073/pnas.92.25.11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee IS, Lin J, Hall HK, Bearson B, Foster JW. The stationary-phase sigma factor sigma S (RpoS) is required for a sustained acid tolerance response in virulent Salmonella typhimurium. Mol Microbiol. 1995;17:155–167. doi: 10.1111/j.1365-2958.1995.mmi_17010155.x. [DOI] [PubMed] [Google Scholar]
- 16.Komp Lindgren P, Karlsson A, Hughes D. Mutation rate and evolution of fluoroquinolone resistance in Escherichia coli isolates from patients with urinary tract infections. Antimicrob Agents Chemother. 2003;47:3222–3232. doi: 10.1128/AAC.47.10.3222-3232.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marcusson LL, et al. Mutant prevention concentrations of the fluoroquinolone ciprofloxacin for urinary tract infection isolates of Escherichia coli. J Antimicrob Chemother. 2005;55:938–943. doi: 10.1093/jac/dki136. [DOI] [PubMed] [Google Scholar]
- 18.Garibyan L, et al. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair (Amst) 2003;2:593–608. doi: 10.1016/s1568-7864(03)00024-7. [DOI] [PubMed] [Google Scholar]
- 19.Jin DJ, Gross CA. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J Mol Biol. 1988;202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- 20.Yanofsky C, Horn V. Rifampin resistance mutations that alter the efficiency of transcription termination at the tryptophan operon attenuator. J Bacteriol. 1981;145:1334–1341. doi: 10.1128/jb.145.3.1334-1341.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher RF, Yanofsky C. Mutations of the beta subunit of RNA polymerase alter both transcription pausing and transcription termination in the trp operon leader region in vitro. J Biol Chem. 1983;258:8146–8150. [PubMed] [Google Scholar]
- 22.Jin DJ, Gross CA. RpoB8, a rifampicin-resistant termination-proficient RNA polymerase, has an increased Km for purine nucleotides during transcription elongation. J Biol Chem. 1991;266:14478–14485. [PubMed] [Google Scholar]
- 23.Singer M, Jin DJ, Walter WA, Gross CA. Genetic evidence for the interaction between cluster I and cluster III rifampicin resistant mutations. J Mol Biol. 1993;231:1–5. doi: 10.1006/jmbi.1993.1251. [DOI] [PubMed] [Google Scholar]
- 24.Landick R, Stewart J, Lee DN. Amino acid changes in conserved regions of the beta-subunit of Escherichia coli RNA polymerase alter transcription pausing and termination. Genes Dev. 1990;4:1623–1636. doi: 10.1101/gad.4.9.1623. [DOI] [PubMed] [Google Scholar]
- 25.Sagitov V, Nikiforov V, Goldfarb A. Dominant lethal mutations near the 5′ substrate binding site affect RNA polymerase propagation. J Biol Chem. 1993;268:2195–2202. [PubMed] [Google Scholar]
- 26.Hammer K, Jensen KF, Poulsen P, Oppenheim AB, Gottesman M. Isolation of Escherichia coli rpoB mutants resistant to killing by lambda cII protein and altered in pyrE gene attenuation. J Bacteriol. 1987;169:5289–5297. doi: 10.1128/jb.169.11.5289-5297.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hengge-Aronis R. Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol Mol Biol Rev. 2002;66:373–395. doi: 10.1128/MMBR.66.3.373-395.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vulic M, Kolter R. Evolutionary cheating in Escherichia coli stationary phase cultures. Genetics. 2001;158:519–526. doi: 10.1093/genetics/158.2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]