

Abstract
The protiodesilylation of unactivated C(sp3)–SiMe2Ph bonds proceeds efficiently by treatment with tetrabutylammonium fluoride in wet DMF or THF via isolable dimethylsilanol intermediates
An important strategy for the stereoselective synthesis of highly substituted tetrahydrofurans involves the [3+2]-annulation of chiral crotyl- and allylsilanes with aldehydes.1 Our laboratory has contributed to this area by demonstrating the predictable stereochemical outcome of [3+2]-annulation reactions of chiral allylsilanes via a formal three-component coupling of two aldehydes and the chiral γ-silyl substituted allylborane 1 (Figure 1).2,3 Initial coupling of chiral allylborane 1 with an aldehyde (R1CHO) followed by exposure of the protected α-hydroxy allylsilane 2 to a Lewis acid and a second aldehyde (R2CHO), selectively affords either c i s - or trans-substituted tetrahydrofurans 3 or 4 in good yields. Importantly, the stereochemistry of the [3+2]-annulation reaction is determined by the nature of the Lewis acid employed (Figure 1).2a The 2,5-cis tetrahydrofuran 3 is obtained typically with ≥20 : 1 selectivity when BF3•OEt2 is employed, while the diastereomeric 2,5-trans disubstituted tetrahydrofuran 4 is obtained, also typically with ≥20 : 1 d.s., by using SnCl4 (via a chelate controlled transition state, requiring that R2CHO be capable of chelate formation). In principle, manipulation of the –SiMe2Ph substituent in 3 or 4 via Tamao-Fleming oxidation4 or protiodesilylation5 should allow for general access to tetrahydrofurans 5 and 6, respectively.
Figure 1.
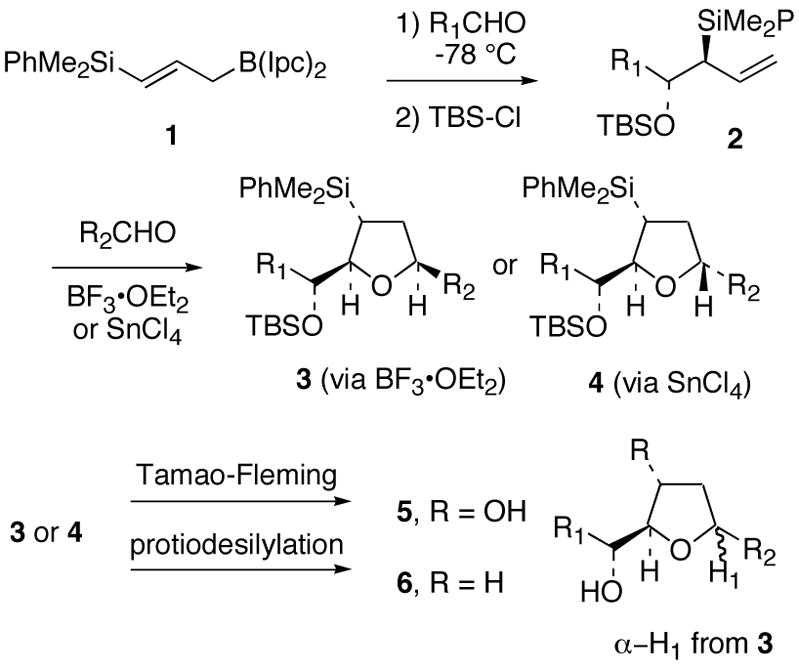
Three-Components Coupling Strategy for the Stereocontrolled Synthesis of Tetrahydrofurans
The established literature procedure for protiodesilylation of unactivated C(sp3)–SiMe2R bonds (i.e. RCH2SiMe3 or RCH2SiMe2Ph → RCH3) involves extended basic hydrolysis (DMSO/H2O, 5–10% KOtBu, 18-crown-6, 95 °C, 2–7 days).2,5 Although tetrahydrofurans 6 can be obtained from 3 or 4 using this procedure,2 the extended reaction times and extremely harsh conditions severely limit the potential applications of this method, with protiodesilylation generally failing for substrates with any reasonably complex R1 or R2 (vida infra).2c
During the course of several ongoing studies in natural product synthesis, it became paramount that a mild method for accomplishing this protiodesilylation (e.g., 3 or 4 → 6) reaction be developed. In particular, in connection with studies on the synthesis of amphidinolide F,6 we demonstrated that protiodesilylation of highly functionalized tetrahydrofurans of general structure 4 could be effected by treatment with TBAF in DMF (Scheme 1).2c We report herein a much wider range of examples of this process, which serve to define the scope of this mild and efficient protiodesilylation reaction.
Scheme 1.
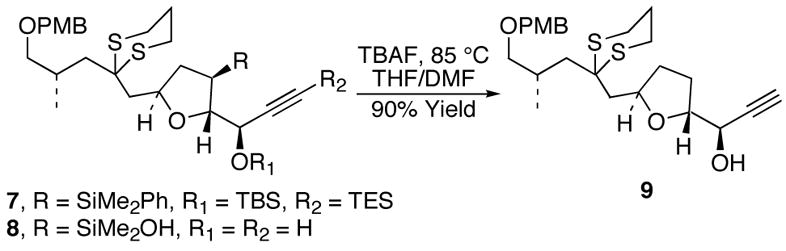
Key TBAF-mediated Protiodesilylation of an Amphidinolide F Precursor (7)3c
We began with a careful exploration of the original Hudrlik-type conditions5a–c by using tetrahydrofurans 1 1 and 14α/β (Scheme 2).7 Initially, we anticipated that a neighboring hydroxyl group was required to activate the–SiMe2Ph group toward protiodesilylation by analogy to the trimethylsilane substrates investigated by Hudrlik,5a–c the diphenylsilyl analogs explored by Landais,5d and the isolated siloxanes investigated by Hoveyda and Stork.5e,8 Accordingly, tetrabutylammonium fluoride (TBAF) was added to the standard Hudrlik reaction conditions (DMSO/H2O, 5–10% KOtBu, 18-crown-6, 95 °C) to effect in situ desilylation of the TBS ethers present in both substrates. Unfortunately, these reactions were highly irreproducible, requiring reaction times from 1 day to 1 week for complete conversion. The long reaction times necessitated that these experiments be performed in sealed pressure tubes (to prevent evaporation of solvent), which proved highly inconvenient for reaction monitoring. In addition, significant decomposition of even the relatively simple tetrahydrofuran 11 was observed.
Scheme 2.
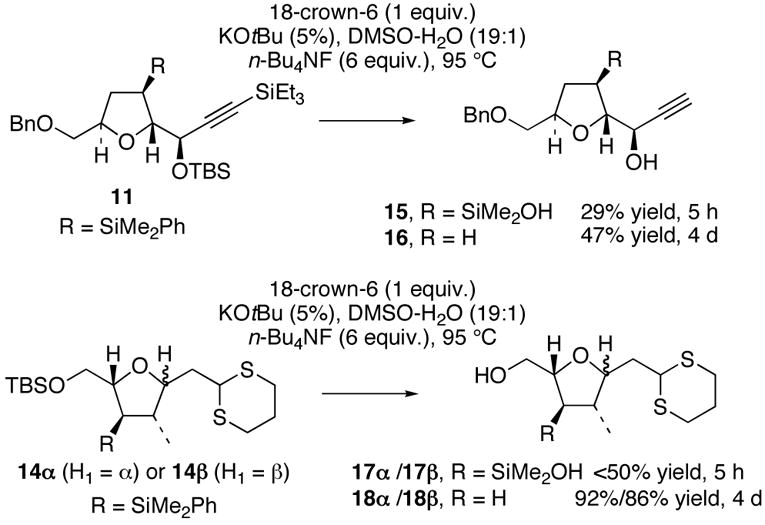
Protiodesilylations of Tetrahydrofurans 11 and 14α/β by using TBAF-modified Literature Conditions
Interestingly, brief treatment of both 11 and 1 4 α or 14β under the TBAF-modified Hudrlik conditions led to the generation of the sensitive but isolable silanols 15, 17α, and 17β after aqueous work-up (Scheme 2).9 Both 15 and 17α/β were competent in the further conversion to 16 and 18α/β upon exposure to the reaction conditions. These silanol intermediates are likely not accessible from the corresponding trimethylsilyl derivatives explored by Hudrlik.5 This suggested to us that the protiodesilylation of –SiMe2Ph groups might occur via a different mechanistic pathway compared to the –SiMe3 derivatives, and that a cyclic silicate or siloxane may not be a required intermediate.
Significant differences in substrate scope for the present process compared to the –SiMe3 substrates studied by Hudrlik quickly became evident. Tetrahydrofurans 19–2110 undergo smooth carbon-silicon bond cleavage to afford protiodesilylated adducts 22–24 in good yields (entries 1–3, conditions A, Table 1). Interestingly, the protiodesilylation of 20 and 21 proceeds smoothly even though they lack a proximal hydroxyl group—clearly indicating that a neighboring hydroxyl group is not required for the protiodesilylation of –SiMe2Ph groups 11
Table 1.
Probing the Role of Neighboring Hydroxyl Assistance in the Protiodesilylation of 19–21
| entry | substrate R = SiMe2Ph | Product R = H | Cond. A | cond. B |
|---|---|---|---|---|
| 1 |
19 |
22 | 85% | 83% |
| 2 |
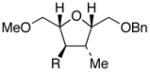
20 |
23 | 92% | 82% |
| 3 |
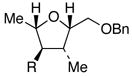
21 |
24 | 91% | 83% |
| 4 | 11 | 16 | 47% | 86% |
| 5 | 14α | 18α | 92% | 99% |
A: 18-crown-6 (1 equiv.), KOtBu (5%), DMSO/H2O (19:1), 95 °C, TBAF (6 equiv.), 1–7 d
B: TBAF (3 equiv.), DMF/THF, 75 °C, 4–16 hr
A systematic study of the reagents employed for the conversion of 21 to 24 indicated that TBAF played a role beyond simple in situ desilylation of the silicon protecting groups. In fact, commercial (wet) TBAF alone12 (added as a solution in tetrahydrofuran) to 19–21 in either wet DMF or THF led to rapid and clean conversion to the corresponding protiodesilylated products 22–24, again via the intermediacy of the corresponding silanols (entries 1–3, conditions B, Table 1).13 Importantly, a substantial improvement in the isolated yield of 1 6 from the protiodesilylation of 11 was realized under these new conditions (entry 4, Table 1).
The reproducible isolation of silanol intermediates in all the systems studied, and the competence of these silanols toward further protiodesilylation, suggests that the Ph–SiMe2R bond undergoes rapid protiodesilylation as an initial step.12a–c The deuterium labeling study illustrated in Scheme 3 indicates that the Si–substrate bond in a subsequently formed silicate intermediate (e.g., 25)14 is sufficiently nucleophilic to undergo efficient and stereoselective protonolysis with complete retention of stereochemistry in the one case studied (i.e. 2 5 → 26, Scheme 3). The significant enhancement of reaction rate (5 d → 4 h) of the TBAF-mediated protiodesilylation reaction (new conditions) compared to the original hydroxide-mediated reaction conditions5 indicates that a fluorosilicate intermediate analogous to 25 with –F replacing one or more –OD groups in 25 may be a key intermediate in the TBAF-mediated process.
Scheme 3.
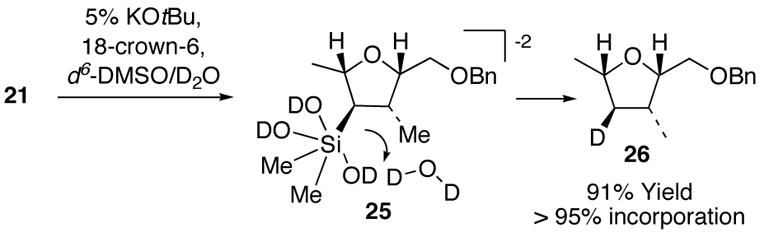
Protonation of Intermediate Silanols Proceeds with Retention of Stereochemistry
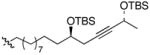
This TBAF-mediated procedure for protiodesilylation of unactivated C(sp3)–SiMe2Ph bonds has proven to be crucial in our efforts to apply the [3+2]-annulation reaction strategy in a variety of ongoing total synthetic endeavors. Specifically, amphidinolide E precursors 27 and 28, which could only be coaxed into slow decomposition using the original Hudrlik-type protocol,5 now undergo efficient protiodesilylation in >90% yield (entries 1–2, Table 2). Furthermore, global desilylation of 7 and 2 9 afford versatile C(15)–C(26)2c and C(1)–C(9) fragments of amphidinolide F (entries 3–4, Table 2). Bistetrahydrofurans 30–32, assembled using sequential [3+2] annulations,15 can be efficiently protiodesilylated using this modified protocol (entries 5–7, Table 2), and represent important steps in our ongoing efforts toward asimicin and a variety of other Annonaceous acetogenins. The protiodesilylation of the benzhydryldimethylsilane 3 3 (entry 8, Table 2) is noteworthy in that the reaction proceeded with comparable efficiency using either method A or B. Interestingly, the conversion of 33 to 40 was found to proceed through a stable cyclic siloxane intermediate, the only such example uncovered during the course of these studies.16
Table 2.
Key Protiodesilylation Reactions Directed Toward Natural Product Syntheses
| Entry | Substrate | product (all Si→ H) | cond.A | cond.B |
|---|---|---|---|---|
| 1 |
27 |
34 | <10% | 91% |
| 2 |
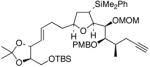
28 |
35 | <10% | 97% |
| 3 |
7 |
9 | 0–48% | 90% |
| 4 |
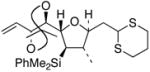
29 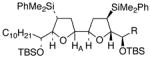
|
36 | 41% | 87% |
| 5 | 30,β–HA,R=C10H21 | 37 | < 20% | 60% |
| 6 |
31,β–HA,R=
|
38 | 0–40% | 55% |
| 7 |
32,α–HA,R=
|
39 | 0–50% | 72% |
| 8 |
 33 33
|
40 | 76% | 77% |
A: 18-crown-6 (1 equiv.), KOtBu (5%), DMSO/H2O (19:1), 95 °C, nBu4NF (6 equiv.), 1–4 d
B: TBAF (6 equiv.), DMF/THF, 75 °C, 4–16 hr
In conclusion, a systematic investigation of the protiodesilylation reactions of Me2PhSi–substituted tetrahydrofurans has revealed that (i) free hydroxyl groups adjacent to the silicon substituent are not required for activation of the C(sp3)–SiMe2Ph bond (20→23, 21→ 24, 28→35 and 29→36) (ii) silanols (i.e. RSiMe2OH) are isolable intermediates and are competent for conversion to protiodesilylated products when resubjected to the reaction conditions, and (iii) use of TBAF (wet) rather than KOtBu and 18-crown-6 leads to a substantial increase in reaction rate, functional group tolerance, and overall efficiency in the protiodesilylation of –SiMe2Ph groups Applications of this method in the total synthesis of natural products will be reported in due course.
Supplementary Material
Supporting Information Available: Experimental procedures and full spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pbs.acs.org.
Acknowledgments
This work was supported by the National Institutes of Health (GM 38907). J. B. S., E. M., and W. T. L. gratefully acknowledge the National Cancer Institute for postdoctoral fellowships (CA 103339, CA 103507 and CA 108191, respectively). C. L. H. thanks the University of Michigan REU program for a summer fellowship (NSF Grant No. 0353553).
References
- 1.(a) Panek J, Yang M. J Am Chem Soc. 1991;113:9868. [Google Scholar]; (b) Panek J, Beresis R. J Org Chem. 1993;58:809. [Google Scholar]; (c) Masse CE, Panek JS. Chem Rev. 1995;95:1293. [Google Scholar]; (d) Peng ZH, Woerpel KA. Org Lett. 2002;4:2945. doi: 10.1021/ol026343e. [DOI] [PubMed] [Google Scholar]; (e) Peng ZH, Woerpel KA. J Am Chem Soc. 2003;125:6018. doi: 10.1021/ja034865z. [DOI] [PubMed] [Google Scholar]
- 2.(a) Micalizio GC, Roush WR. Org Lett. 2000;2:461. doi: 10.1021/ol9913082. [DOI] [PubMed] [Google Scholar]; (b) Micalizio GC, Roush WR. Org Lett. 2001;3:1949. doi: 10.1021/ol0160250. [DOI] [PubMed] [Google Scholar]; (c) Shotwell JB, Roush WR. Org Lett. 2004;6:3865. doi: 10.1021/ol048381z. [DOI] [PubMed] [Google Scholar]
- 3.Roush W, Pinchuk A, Micalizio G. Tetrahedron Lett. 2000;41:9413. [Google Scholar]
- 4.Jones GR, Landais Y. Tetrahedron. 1996;52:7599. [Google Scholar]
- 5.(a) Hudrlik P, Hudrlik A, Kulkarni A. J Am Chem Soc. 1982;104:6809. [Google Scholar]; (b) Hudrlik P, Holmes P, Hudrlik A. Tetrahedron Lett. 1988;29:6395. [Google Scholar]; (c) Hudrlik P, Gebreselassie P, Tafesse L, Hudrlik A. Tetrahedron Lett. 2003;44:3409. [Google Scholar]; (d) Landais Y, Mahieux C. Tetrahedron Lett. 2005;46:675. [Google Scholar]; (e) Stork G, Sofia MJ. J Am Chem Soc. 1986;108:6826. [Google Scholar]
- 6.Kobayashi J, Ishibashi M, Wälchli M, Nakamura H, Hirata Y, Sasaki T, Ohizumi Y. J Am Chem Soc. 1988;110:490. [Google Scholar]
-
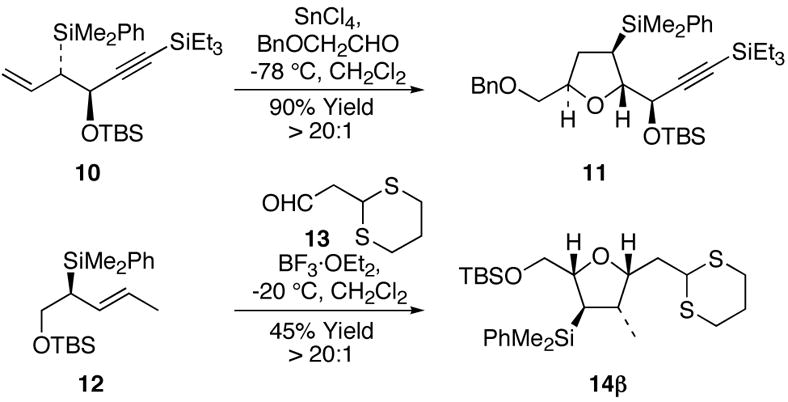
7.Tetrahydrofurans 11 and 14β were prepared as summarized below.
- 8.Hale M, Hoveyda A. J Org Chem. 1992;57:1643. [Google Scholar]
- 9.See: Murakami M, Suginome M, Fujimoto K, Nakamura H, Andersson P, Ito Y. J Am Chem Soc. 1995;115:6487. In our hands silanols 17α/β show a marked propensity toward oligomerization upon attempted isolation (see Supporting Information). These oligomeric mixtures are competent intermediates toward further protiodesilylation.
- 10.Available from the [3+2]-annulation of 1 2 with α-benzyloxyacetaldehyde under non-chelate conditions and subsequent standard transformations (see Supporting Information).
- 11.Silanols corresponding to 20 and 21 (R = SiMe2OH) have been isolated and fully characterized. These silanols are easily handled, suggesting that the oligomerization of 17α/β proceeds via condensation of the C(7) hydroxyl and silanol (see Supporting Information).
- 12.For protiodesilylations of stabilized or C(sp2) systems via silanol intermediates see: (a) Anderson J, Flaherty A. J Chem Soc, Perkin Trans 1. 2000:3025.Anderson JC, Munday RH. J Org Chem. 2004;69:8971. doi: 10.1021/jo048746t.Anderson JC, Anguille S, Bailey R. Chem Commun. 2002:2018. doi: 10.1039/b205765d.Where silanols have not been implicated: Hulme AN, Henry SS, Meyers AI. J Org Chem. 1995;60:1265.Ni Y, Montgomery J. J Am Chem Soc. 2004;126:11162. doi: 10.1021/ja046147y.
- 13.TBAF is apparently unique in promoting this reaction, as screening of several other fluoride sources for protiodesilylation of 21 (CsF, KF, TAS-F) in various solvent/temperature combinations (MeCN, DMF, DMSO, 23 °C→90 °C, pressure tube) led only to recovered starting materials. Additionally, tetrabutylammonium hydroxide does not promote this transformation (see ref. 2c).
- 14.Chuit C, Corriu RJP, Reye C, Young JC. Chem Rev. 1993;93:1371. [Google Scholar]
- 15.Tinsley JM, Roush WR. J Am Chem Soc. doi: 10.1021/ja051986l. submitted. [DOI] [PubMed] [Google Scholar]
-
16.Siloxane 41 was formed cleanly upon brief exposure of 33 to excess TBAF at room temperature:
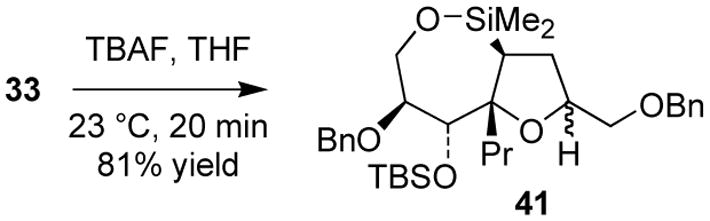 Siloxane 41 was efficiently protiodesilylated to give 40 upon exposure to the reagent combination of method B (89% yield).
Siloxane 41 was efficiently protiodesilylated to give 40 upon exposure to the reagent combination of method B (89% yield).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Experimental procedures and full spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pbs.acs.org.