

Abstract
We investigated the properties of several peptides with sequences related to LWYIK, a segment found in the gp41 protein of HIV and believed to play a role in sequestering this protein to a cholesterol-rich domain in the membrane. This segment fulfills the requirements to be classified as a CRAC motif that has been suggested to predict those proteins that will partition into cholesterol-rich regions of the membrane. All of the peptides were studied with the terminal amino and carboxyl groups blocked, i.e. as N-acetyl-peptide-amides. Effects of cholesterol on the intensity of W emission generally parallel DSC evidence of sequestration of cholesterol. Modeling studies indicate that all of these peptides tend to partition with their mass center at the membrane interface at the level of the hydroxyl of cholesterol. Interaction with cholesterol is dual: van der Waals interactions between mainly hydrophobic surfaces and, electrostatic stabilization of the cholesterol OH group. Thus, both experiments and modeling studies indicate that preference of CRAC motifs for cholesterol-rich domains might be related to a membrane interfacial preference of the motif, to a capacity to wrap and block the cholesterol polar OH by H-bond interactions and to a capacity for peptide aromatic side chains to stack with cholesterol. These results were supported by studies of single mutations in the gp41 protein of HIV-1, in which L679 is replaced with I. Despite the similarity of the properties of these amino acid residues, this single substitution resulted in a marked decrease in the capability of JC53-BL HeLa-based HIV-1 indicator cells to form syncytia.
It has been demonstrated that peptides and proteins can cause the redistribution of cholesterol in membranes (1). Triggering domain formation may play an important role in signal transduction (2). One class of domains thought to be present in biological membranes has been termed “rafts”. Although there is controversy about the nature and prevalence of such domains, it is well established that caveolae are distinct domains in the membrane with many chemical and physical properties similar to those proposed for “rafts”.
Several features have been identified that facilitate the partitioning of proteins into raft-like domains. These features do not result in a specific stoichiometric binding between the protein and a specific lipid component, but rather a preferential interaction of a protein with certain lipid components. Among the features that promote more favorable partition into cholesterol-rich domains of membranes are certain types of protein lipidation (3). Some integral membrane proteins, devoid of lipidation, also partition into cholesterol-rich domains. Tryptophan at the flanking regions of these helices has been suggested to be important for this partitioning (4). In addition, several proteins that interact with cholesterol have an amino acid sequence in the juxtamembrane region conforming to the pattern -L/V-(X)(1-5)-Y-(X)(1-5)-R/K-, in which (X)(1-5) represents between one to five residues of any amino acid (5). This motif, defined by a peptide segment -L/V-(X)(1-5)-Y-(X)(1-5)-R/K-, has been termed the CRAC motif, corresponding to the Cholesterol Recognition/interaction Amino acid Consensus.
One protein that has a CRAC motif adjacent to its transmembrane helix is the HIV-1 fusion protein, gp41. This segment contains the sequence LWYIK. The role of the short sequence LWYIK for binding to cholesterol has been demonstrated by measuring the binding of maltose binding protein fusion proteins to cholesterol-hemisuccinate agarose (6) and by MAS/NMR and DSC studies (7). A longer juxtamembrane region of gp41 is W-rich and contains the LWYIK sequence at the carboxyl terminal end. It has been shown to promote membrane fusion both by mutational studies of the intact viral protein (8) as well as with the use of a 20 amino acid synthetic peptide (9). The amino terminal segment of this peptide is thought to facilitate oligomerization of gp41 (10). Cholesterol was found to be required for HIV infection (11-13) as well as for fusion promoted by the synthetic peptide (9). Depletion of cholesterol from HIV results in loss of infectivity (14). Although the segment LWYIK of the gp41 protein of HIV has a CRAC motif and there is good evidence that this region of the protein interacts with cholesterol, the general requirements for the CRAC motif are too flexible to be accurate. For example, there are homologous segments of the W-rich, juxtamembrane region of gp41 proteins of SIV and HIV-2 that interact with cholesterol but do not fulfill the requirements of a CRAC motif (15). The present study was designed to systematically vary certain positions of the LWYIK segment and to determine which of the molecular features of the peptide are important for its ability to sequester cholesterol.
Materials and Methods
Materials
The peptides are all N-acetyl-peptide-amides, i.e. blocked at the terminal amino and carboxyl groups. The peptides were synthesized by SynPep Corporation (Dublin, CA) and purified by HPLC to >95% purity. Phospholipids and cholesterol were purchased from Avanti Polar Lipids (Alabaster, AL).
Preparation of samples for DSC
Phospholipid and cholesterol were codissolved in chloroform/methanol (2/1, v/v). For samples containing peptide, an aliquot of a solution of the peptide in methanol was added to the lipid solution in chloroform/methanol. The solvent was then evaporated under a stream of nitrogen with constant rotation of a test tube so as to deposit a uniform film of lipid over the bottom third of the tube. Last traces of solvent were removed by placing the tube under high vacuum for at least two hours. The lipid film was then hydrated with 20 mM PIPES, 1 mM EDTA, 150 mM NaCl with 0.002% NaN3, pH 7.40 and suspended by intermittent vortexing and heating to 50°C over a period of 2 minutes under argon.
Differential Scanning Calorimetry (DSC)
Measurements were made using a Nano Differential Scanning Calorimeter (Calorimetry Sciences Corporation, American Fork, UT). The scan rate was 2°C/min and there was a delay of 5 minutes between sequential scans in a series to allow for thermal equilibration. The features of the design of this instrument have been described (16). DSC curves were analyzed by using the fitting program, DA-2, provided by Microcal Inc. (Northampton, MA) and plotted with Origin, version 5.0.
Tryptophan fluorescence studies
The peptide was dissolved in methanol or in 10 mM Hepes buffer, 0.14 M NaCl, 0.1 mM EDTA, pH 7.4 to a final concentration of 20 μM. The purity of the peptide was determined by absorbance at 280 nm using an extinction coefficient calculated from the amino acid content (17). Only for these fluorescence studies, SUVs were used in order to reduce contributions from light scattering. SUVs were made from lipid films dried from chloroform:methanol (2:1, v/v). Films were hydrated with Hepes buffer by vortexing and then sonicated. Peptide solutions (20 μM) were then added to the partially clarified liposome preparation before measuring the fluorescence. The final lipid to peptide molar ratio was 10. The excitation wavelength was 280 nm, with a 4 nm bandwidth in excitation and 8 nm in emission. Polarizers, set to 90 ° in excitation and 0 ° in emission, were used to reduce stray light. Fluorescence emission spectra were measured using ultrasensitive quartz mirror microcuvettes (N. L. Vekshin, Institute of Cell Biophysics, Pushchino, Russia). Spectra were determined at 25 °C using an SLM Aminco Bowman AB-2 spectrofluorimeter. The spectra were corrected for instrumental factors and a buffer blank was subtracted. Inner filter effect corrections were applied. When the fluorescence of the peptide was measured in the presence of lipid, the same concentration of SUVs without peptide was used as a blank.
Membrane partitioning studies
Films containing 10 mol% of peptide and SOPC:cholesterol (1:1), were prepared by solvent evaporation and subsequently hydrated, as described for the preparation of samples for DSC. The final lipid concentration was 2.5 mg/mL in 20 mM PIPES, 0.14 M NaCl, 1 mM EDTA, pH 7.4. The suspension was vortexed well and were freeze-thawed two times between liquid nitrogen and ∼50 °C to remove lipid still on the walls of the tube. The suspension was then incubated for 30 minutes at 38 °C. After further vortexing, the MLVs were transferred to 1 mL silanized polycarbonate tubes and centrifuged at 200,000xg (100,000 rpm) for 150 minutes at 25 °C in a Sorvall RC M120 centrifuge. A clear supernatant was collected in silanized glass tubes and the absorbance read by scanning between 350 nm and 250 nm in a Varian Cary 50Bio spectrophotometer, using the supernatant from a suspension of lipid without peptide as a blank. The absorbance at 280nm, corrected for the blank and for light scattering was used to calculate amount of peptide remaining in supernatant, compared with the total concentration of peptide determined by absorbance before the addition of lipid.
Modeling
Structure Properties
A search was made for the most favorable conformation of each of the peptides by selecting the lowest energy combinations of favorable dihedral angles (18-20). The blocked pentapeptides have 5 peptide bonds, each of which is allowed to have any of 6 different sets of dihedral angles, resulting in 65 or 7776 different structures. This stereoalphabet method was used previously to study the loop conformation for helix-helix interactions (21). The energy of peptide conformations are calculated by an all atom description of structures and the addition of van der Waals, electrostatic, internal and external hydrophobicity energy terms. The van der Waals contribution was calculated using the 6-12 Lennard-Jones description of the energy of interactions between unbonded atoms (21). The Coulomb’s equation was used for electrostatic interactions between unbonded charged atoms with a dielectric ‘constant’ sigmoidally varying from 1 to 80 with distance between atoms and, using FCPAC partial atomic charges (22). The intramolecular hydrophobicity contribution to stability was calculated using atomic Etr (energy of transfer) and the fractions of atomic surface covered by the atom in interactions. Seven atomic types are used for Etr (20). When structures are calculated in water, the contribution of solvent is accounted for by an external hydrophobicity energy term where the solvent-accessible surface of atoms is calculated by the method of Shrake and Rupley with 162 points (23;24). From the 7776 calculated structures, the 100 most stable are saved (50 in water, 50 in water-free medium).
Membrane insertion
Insertion of all these structures into the membrane was tested with the IMPALA systematic program (25). The minimal restraint energy profile of insertion of each peptide in membrane was determined by inserting separately the 100 previously-selected structures in membrane. Each molecule was moved every 1 Å step across the bilayer (Z axis) and was rotated in the zx plane (5,000 positions over 360°) at each step. The insertion profile of one peptide in membrane was set as the combination of the minima at all steps; hence the profile could account for a different structure and a different angle of insertion at each level of the membrane.
Cholesterol-peptide interactions
Determining the interactions of the most stable conformation of each peptide in membrane with cholesterol extended the analysis. The best IMPALA position was used for each peptide conformation (100 conformations per peptide) and the best position for the cholesterol in the membrane. The two molecules were tested in the calculation varying the position of cholesterol in the vertical (z axis) and horizontal (around the CRAC peptide) directions as well as by self-rotation around the long axis of cholesterol. All together 5,720,400 relative positions between each peptide conformation and sterol were tested to finally retain the conformation of maximal intermolecular interactions that was refined by angular dynamics (32). The best complex among all complexes formed was evaluated on having optimal (i.e. minimal) values of intra and inter-molecule energies.
Cell-cell fusion
Cell culture
JC53-BL HeLa-based HIV-1 indicator cells (26) that show a high susceptibility to HIV-1 infection were kindly provided by Tranzyme Inc., Birmingham, Ala. These cells contain reporter genes, luciferase and β-galactosidase, that are expressed in the presence of Tat, under the influence of HIV-1 LTR. African green monkey kidney cells (Cos-1) cells were purchased from American Type Culture Collection. Cells were subcultured every 3 to 4 days by trypsinization and maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics.
Mutagenesis and plasmid vectors
An NL4.3 (laboratory-adapted X4 strain) KpnI-BamHI envelope fragment was cloned into vector pSP72. Mutant L679I of the HIV-1 membrane-proximal external region (MPER) was performed using QuikChange (Stratagene, La Jolla, CA). The mutated envelope fragment was subsequently subcloned into an SV-40-based vector (pSRHS) that expresses Tat and Env. (8).
Cell - cell fusion assays
Cos-1 cells were transfected with pSRHS constructs expressing wild-type and mutant HIV-1 envelope using Fugene 6 transfection reagent (Roche Diagnostics Corporation, Indianapolis, IN). Twenty hours post-transfection, they were mixed in a 5:1 ratio with JC53-BL cells and replated in 24-well plates. After 20 hours, the cells were microscopically examined for fusion by counting the blue syncytia and the number of nuclei/syncytium following addition of 5-bromo-4-chloro-3-indolyl-beta-D-galactopyranoside (X-gal; Promega, Madison, WI) as described by Kimpton and Emerman (27). Fusion was also evaluated by measuring luciferase activity (Luciferase Assay System, Promega) according to the manufacturer’s protocol. All fusion assays were done in triplicate.
Results
DSC
DSC can be used to assess the miscibility of cholesterol and SOPC. With increasing mol fractions of cholesterol, the melting transition of SOPC will become broader and have a reduced transition enthalpy. If a peptide promotes the rearrangement of cholesterol into cholesterol-rich domains, the remaining SOPC-rich domains will exhibit a more cooperative (sharper) phase transition of higher enthalpy. This is most easily discerned in the cooling scans, because of the low transition enthalpy of this phospholipid. The phase behavior of SOPC with several mol fractions of cholesterol in the presence of N-acetyl-LWYIK-amide is known (7). In this work we study several other related blocked pentapeptides. In this new group of peptides we kept positions 2 and 4 of LWYIK as W and I, respectively, but have made substitutions in the other three positions. Some of the homologs fulfilled the requirements of the CRAC motif and others did not.
A pair of peptides that differ only by a single OH group on Y is N-acetyl-LWFIK-amide and N-acetyl-LWYIK-amide. However, although N-acetyl-LWFIK-amide is not an example of a CRAC motif, it does have similar effects to N-acetyl-LWYIK-amide in redistributing cholesterol (Fig. 1). In contrast, substitution of Y with another aromatic amino acid, W, resulted in much less sequestering of cholesterol (Fig. 2). A third similar peptide that does not fulfill the CRAC formula is N-acetyl-IWYIK-amide, since an L or V residue is required in the first position. Unlike the non-CRAC peptide N-acetyl-LWFIK-amide, the peptide N-acetyl-IWYIK-amide is not effective in depleting a domain of cholesterol (Fig. 3).
Figure 1.

Differential scanning calorimetry of SOPC with 0 (A-D), 30 (E-H), 40 (I-L) and 50 (M-P) mol% cholesterol as well as 0 (A, E, I, M), 5 (B, F, J, N), 10 (C, G, K, O) and 15 (D, H, L, P) mol % N-acetyl-LWFIK-amide. Scan rate 2K/min. Lipid concentration 2.5 mg/mL in 20 mM PIPES, 1 mM EDTA, 150 mM NaCl with 0.002% NaN3, pH 7.40. Sequential heating and cooling scans between 0 and 50 °C. Numbers are the order in which the scans were carried out, with scans 1, 3 and 5 being heating scans, each of which was followed by one of the cooling scans 2, 4 or 6. Scans were displaced along the y-axis for clarity of presentation.
Figure 2.

Differential scanning calorimetry of SOPC with 30 (A-C), 40 (D-F) and 50 (G-I) mol% cholesterol as well as 5 (A, D,G), 10 (B, E, H) and 15 (C,F, I) mol % N-acetyl-LWWIK-amide. Other details, as for Figure 1.
Figure 3.

Differential scanning calorimetry of SOPC with 30 (A-C), 40 (D-F) and 50 (G-I) mol% cholesterol as well as 5 (A, D,G), 10 (B, E, H) and 15 (C,F, I) mol % N-acetyl-IWFIK-amide. Other details, as for Figure 1.
A peptide that requires cholesterol to bind to membranes should not greatly affect the phase transition of pure SOPC. In general the peptides used in this study do not lower the enthalpy of the phase transition of SOPC (Table 1, left-hand column, under “Pure SOPC”). One peptide, N-acetyl-IWYIK -amide, however, does cause the enthalpy of SOPC to decrease.
Table 1.
Enthalpy of the Chain Melting Transition of SOPC
| Pure SOPC | SOPC: cholesterol 7:3 | SOPC: cholesterol 6:4 | |||
|---|---|---|---|---|---|
| Peptide | ΔH (cal/mol) | Peptide | ΔH (cal/mol) | Peptide | ΔH (cal/mol) |
| None | 3,800 | None | 520 | None | 100 |
| 5% LWYIK | 3,800 | 5% LWYIK | 1,000 | 5% LWYIK | 100 |
| 10% LWYIK | 3,800 | 10% LWYIK | 1,100 | 10% LWYIK | 300 |
| 15% LWYIK | 3,800 | 15% LWYIK | 1,200 | 15% LWYIK | 500 |
| 5% LWFIK | 3,750 | 5% LWFIK | 380 | 5% LWFIK | 65 |
| 10% LWFIK | 3,800 | 10% LWFIK | 440 | 10% LWFIK | 160 |
| 15% LWFIK | 3,700 | 15% LWFIK | 1,300 | 15% LWFIK | Broad |
| 5% LWYIR | 4,100 | 5% LWYIR | 550 | 5% LWYIR | 160 |
| 10% LWYIR | 3,600 | 10% LWYIR | 440 | 10% LWYIR | 250 |
| 15% LWYIR | 3,900 | 15% LWYIR | 440 | 15% LWYIR | 205 |
| 5% LWYIH | 3,700 | 5% LWYIH | 520 | 5% LWYIH | 200 |
| 10% LWYIH | 3,900 | 10% LWYIH | 450 | 10% LWYIH | Broad |
| 15% LWYIH | 3,400 | 15% LWYIH | 325 | 15% LWYIH | Broad |
| 5% LWWIK | 3,700 | 5% LWWIK | 148 | 5% LWWIK | ∼0 |
| 10% LWWIK | 3,900 | 10% LWWIK | 245 | 10% LWWIK | 135 |
| 15% LWWIK | 3,700 | 15% LWWIK | 340 | 15% LWWIK | 195 |
| 5% LWLIK | 4,000 | 5% LWLIK | 210 | 5% LWLIK | ∼0 |
| 10% LWLIK | 3,900 | 10% LWLIK | 330 | 10% LWLIK | 110 |
| 15% LWLIK | 3,500 | 15% LWLIK | 280 | 15% LWLIK | 165 |
| 5% IWYIK | 3,600 | 5% IWYIK | ∼0 | 5% IWYIK | ∼0 |
| 10% IWYIK | 3,200 | 10% IWYIK | ∼0 | 10% IWYIK | 140 |
| 15% IWYIK | 2,600 | 15% IWYIK | ∼0 | 15% IWYIK | 160 |
It is well known that cholesterol broadens and lowers the main transition enthalpy of PC (Table 1). Peptides that preferentially bind to cholesterol-rich domains in mixtures with PC will deplete cholesterol from a region of the membrane, resulting in a peptide-induced increase in the enthalpy of the phospholipid chain melting transition (1). The peptide N-acetyl-LWYIK-amide is the only peptide of this group that is very effective in doing this at 30% cholesterol (Fig. 4). When the amount of cholesterol in the membrane exceeds 30%, several peptides cause an increase in the enthalpy of the SOPC phase transition enthalpy (Table 1 and Fig. 4). Hence, analysis of the DSC data supports the suggestion of a cholesterol-sequestering effect that is selective for LWYIK at 30% cholesterol and occurs with other peptides at higher cholesterol concentrations.
Figure 4.
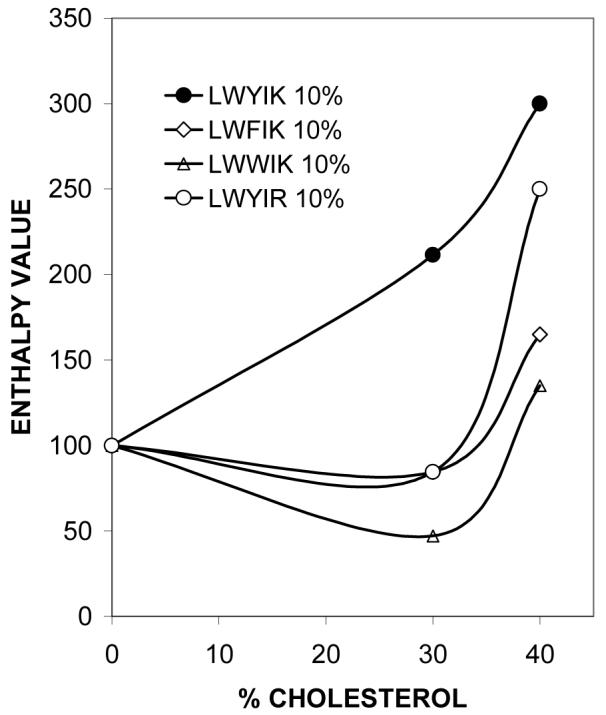
Graphic representation of the change in the SOPC phase transition enthalpy caused by the presence of selected peptides. The relative enthalpy value, defined as the enthalpy of the SOPC transition in the presence/absence of 10 mol% peptide times 100 is plotted as a function of the cholesterol content in the membrane. N-acetyl-LWYIK-amide (●), N-acetyl-LWFIK-amide (◊), N-acetyl-LWWIK-amide (∆), N-acetyl-LWYIR-amide (○).
Another criterion for the formation of cholesterol-rich domains is that the sterol passes its solubility limit in a region of the membrane and can form crystallites. Anhydrous crystalline cholesterol is known to undergo a crystalline polymorphic phase transition at 38 °C on heating and at 23 °C on cooling at a scan rate of 2°/min (28), with an enthalpy of 910 cal/mol (29). At a SOPC/cholesterol molar ratio of 7/3 no cholesterol crystals, or at most traces of crystalline cholesterol, are found with any of the peptides. However, at a SOPC/cholesterol molar ratio of 6/4, anhydrous cholesterol crystals can be detected with all of the peptides, except for N-acetyl-LWYIH-amide (not shown). At a 1:1 mixture of SOPC and cholesterol there is still no deposition of cholesterol in the absence of peptide, but all of the peptides used in this study cause some cholesterol crystals to form (Table 2). There are two mechanisms by which peptides can promote the formation of cholesterol-rich domains: one is by preferentially binding to these domains and stabilizing them and the other is by being excluded from these domains and preferentially binding to cholesterol-depleted regions of the membrane, forcing cholesterol into another region of the membrane (1). From the formation of cholesterol crystallites it is not possible to distinguish between these two mechanisms. In the case of peptides that sequester into cholesterol-rich domains, they will only cause the formation of cholesterol crystallites from the fraction of cholesterol that the peptide is not directly bound to. Cholesterol that is bound to peptide has a lower probability of forming crystals. Generally, the amount of cholesterol crystals formed does not increase proportionally to peptide concentration (Table 2). This is particularly striking in the case of N-acetyl-LWYIH-amide that has fewer cholesterol crystals with each successive increment in peptide concentration; this may be a consequence of more cholesterol being directly bound to peptide as the peptide concentration increases.
Table 2.
Enthalpy of the Polymorphic Transition of Anhydrous Cholesterol Present in Mixtures of Peptides with SOPC:cholesterol (1:1)
| Peptide | ΔH (cal/mol) |
|---|---|
| 5% LWYIK | 25 |
| 10% LWYIK | 80 |
| 15% LWYIK | 60 |
| 5% LWFIK | 120 |
| 10% LWFIK | 125 |
| 15% LWFIK | 170 |
| 5% LWYIR | 20 |
| 10% LWYIR | 70 |
| 15% LWYIR | 65 |
| 5% LWYIH | 180 |
| 10% LWYIH | 90 |
| 15% LWYIH | 60 |
| 5% LWWIK | 125 |
| 10% LWWIK | 80 |
| 15% LWWIK | 140 |
| 5% LWLIK | 60 |
| 10% LWLIK | 65 |
| 15% LWLIK | 120 |
| 5% IWYIK | 100 |
| 10% IWYIK | 140 |
| 15% IWYIK | 175 |
Tryptophan fluorescence studies
Fluorescence emission spectra of each of the peptides was determined in methanolic solution, in aqueous buffer and in the presence of SUVs of SOPC or SOPC:cholesterol (1:1) at a lipid to peptide molar ratio of 10. There is little difference in the maximum emission wavelength among the seven peptides in any of the four conditions (Table 3).
Table 3. W Emission Properties.
| Peptide | Condition | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Methanol | Buffer | SOPC | SOPC/cholesterol (1:1) | Ratio1 | |||||
| λ (nm) | Relative Intensity | λ (nm) | Relative Intensity | λ (nm) | Relative Intensity | λ (nm) | Relative Intensity | ||
| LWYIK | 336 | 1.7 | 341 | 0.74 | 340 | 0.71 | 340 | 0.43 | 1.7 |
| LWFIK | 336 | 1.3 | 341 | 0.75 | 340 | 0.61 | 340 | 0.43 | 1.4 |
| LWYIR | 335 | 2.0 | 340 | 1.0 | 340 | 0.73 | 340 | 0.69 | 1.1 |
| LWYIH | 335 | 1.6 | 341 | 0.91 | 342 | 0.82 | 342 | 0.60 | 1.4 |
| LWWIK | 336 | 1.95 | 340 | 0.95 | 339 | 0.90 | 341 | 0.75 | 1.2 |
| LWLIK | 335 | 2.4 | 340 | 1.3 | 340 | 1.0 | 340 | 0.87 | 1.1 |
| IWYIK | 336 | 2.0 | 340 | 0.87 | 339 | 0.67 | 339 | 0.66 | 1.0 |
The ratio is the ratio of the maximal emission intensity of the peptide in SOPC divided by the emission intensity of the peptide in SOPC/cholesterol (1:1).
Membrane partitioning
The results of partitioning of N-acetyl-LWYIK-amide into SOPC:cholesterol (1:1) (Fig. 5) are in reasonable agreement with our previously published results (7), particularly taking into account the differences in protocol in the two cases. However, there was some variation in the partitioning of other peptides into liposomes of. SOPC:cholesterol (1:1) (Fig. 5). In particular, N-acetyl-LWLIK-amide showed lower membrane partitioning.
Figure 5.

Partitioning of peptides into MLVs of SOPC:cholesterol (1:1) containing 10 mol% peptide. Error bars are the standard error of the mean of experiments repeated two times.
Modeling
Because of their small size all these peptides have limited possibilities of stabilization via intramolecular interactions and hence might have a large structural diversity. This diversity will be restricted by the presence of bulky residues and by the proximity in the sequence of polar and hydrophobic residues. The RMS of every structure was calculated on the basis of an all atom fit with the peptide conformation of lower energy (the best structure). The values are ranked and analysis of the plot of increasing values demonstrates either a stepped or a constant increase of RMS values (Fig.6). A constant variation of RMS (see LWFIK in Fig.6) indicates small discrete variations between structures, whereas a stepped increase (see LWWIK in Fig.6) indicates more distinct subpopulations. The number of structures within a 3.5 RMS value difference is a subjective index that we used to evaluate this difference. For instance, changing L for I, two residues with the same number of carbons but an elongated instead of a bulky branched side chain, restricts structural diversity from the 55 conformations for LWYIK to only 27 conformations for IWYIK (Fig.6). The same is true when Y is changed for W (LWYIK as compared to LWWIK) whereas changing Y for F (LWYIK as compared to LWFIK) diversifies the panel of conformations, suggesting more rigidity of the LWYIK structure. Changing K for R (LWYIK as compared to LWYIR) also rigidifies the structure most likely because R is much more polar than K and tends to point away from the rest of the peptide. This analysis suggests a rough classification of increasing structure flexibility from LWYIR<LWWIK< IWYIK< LWYIH<LWYIK to LWFIK.
Figure 6.
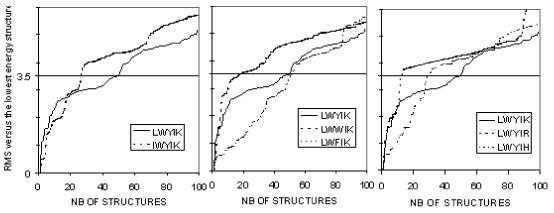
Analysis of peptide conformation diversity. For each peptide, 7776 conformation were calculated twice, once in a hydrophobic medium, once in water, in each condition, the 50 best conformations were saved. Diversity of these structures was analyzed by calculating the all atom RMS deviation with reference to the lowest energy conformation. Structures were ranked by increasing RMS. For the sake of analysis, a critical value, arbitrarily set at RMS 3.5, was used to analyze structural rigidity: the lower the number of conformations at that RMS 3.5 value, the more rigid the structure.
Calculating the energy for insertion for the 100 most stable conformations of each of these peptides (50 structures calculated in water plus 50 calculated in hydrophobic environment) supports the conclusion that all are more stable in a membrane than in water because all peptides have minimal restraint energy in a membrane (Fig. 7). Moreover all favor partitioning in the lipid acyl chain/polar interface. Some, such as LWWIK, might cross the membrane more easily than others such as LWYIR because the energy barrier is lower.
Figure 7.
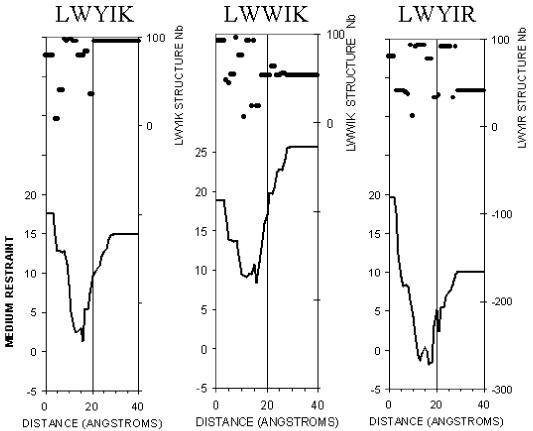
Restraint profile of insertion of LWYIK, LWWIK and LWYIR in the membrane. The 100 conformations saved from stereoalphabet were independently tested for a systematic screening of insertion in a DPPC membrane. Then, a unique insertion profile was made by choosing at each membrane level the conformation that gave the minimal insertion restraint (lower curve). The membrane center is at x axis 0, at 20 Å is the membrane-water surface, 0 to 15 Å is the acyl chain layer, 15 to 20 Å is the membrane-water interface (lipid polar headgroups) and over 20 Å is the water-medium. At each level, the peptide is located by a point at its mass center. The rank number of the conformations giving the best insertion is plotted on the top of the figure to demonstrate that optimal insertion involves different conformations of peptides. The restraint values are calculated as in (28) as the sum of two restraints, one simulating the bilayer hydrophobicity (Epho), and the other, the lipid perturbation (Elip). The lowest restraint values correspond to the best stability. Peptides with lower restraint value in the membrane than in water should be able to cross the membrane. Peptides with a lower restraint value in the lipid polar head interface than in the membrane core will more easily remain at the interface, rather than cross the membrane.
The best structure and position of each peptide in membrane was used to test the interaction with cholesterol as described in methods. The final angular dynamics optimization of the procedure gave a series of complexes of low intramolecular and/or medium-restraint energies. Comparative analyses of the non-covalent bonding occurring between the family of peptides and cholesterol show that the two parts of cholesterol (the fused ring structure and the hydroxyl) can be involved in the interaction. The hydroxyl can be implicated in two interactions, one as an H-donor and a second as an H acceptor. The ring can be involved in van der Waals contacts. We find that there is a unique feature of the interaction of LWYIK and cholesterol that is not seen with any of the other peptides of this series (Fig. 8): the cholesterol hydroxyl can be the H-donor of the lysine terminal C=O and the H acceptor of the OH of tyrosine side chain. This fact is unique since in other complexes, the cholesterol hydroxyl is either an H-donor or an H-acceptor but not both and sometimes it is not involved in interaction as illustrated for LWWIK (Fig. 9). The interaction between cholesterol and LWYIK optimally satisfies the cholesterol polar head requirement and it leaves the cholesterol rings rather free. With other peptides, lowering the energy of interaction results in the two molecules having significant van der Waals interactions, as occurs for the LWWIK-cholesterol complex (Fig. 9), where the W indole ring lies parallel to the planar face of cholesterol.
Figure 8.
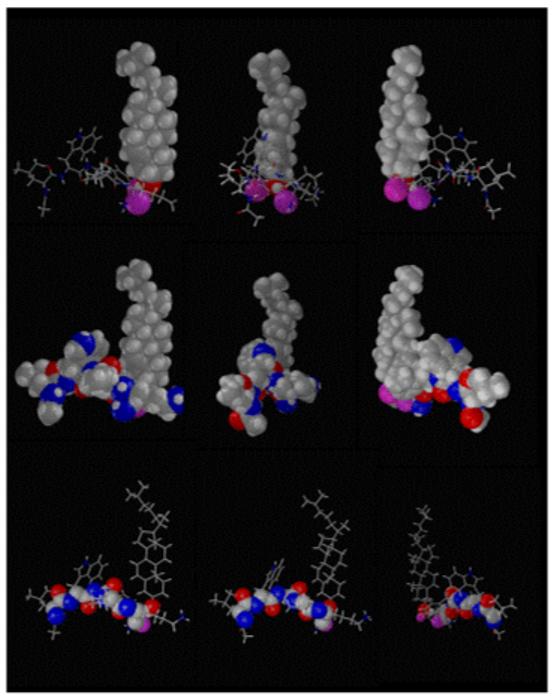
Illustration of the lowest energy complex between cholesterol and LWYIK in the membrane. Positions of the two molecules were systematically tested and minimized by angular dynamics (32). The complex of lower energy for intra and intermolecule interactions is here shown in three different positions (A, B and C) with three different outlook -1 cholesterol is drawn in CPK, the peptide bonds are sticks; -2 all atoms (cholesterol and peptide) are in CPK -3 backbone atoms (N-C-CO) are drawn in CPK. All other bonds are sticks. In all cases the oxygen of tyrosine OH and of lysine backbone CO are colored in pink.
Figure 9.
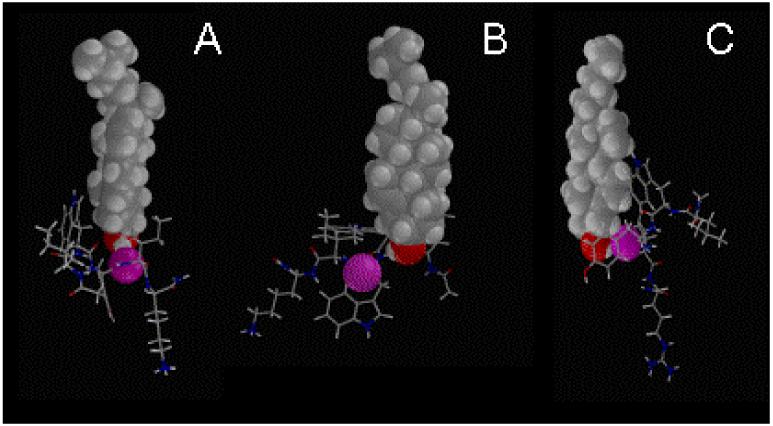
Illustration of the lowest energy complexes between cholesterol and IWYIK (A), LWWIK (B) and LWYIR (C) and, cholesterol in the membrane. The molecules of lower energy for intra and intermolecule interactions are shown here with CPK image of the cholesterol atoms, and the closest peptide oxygen in pink-colored CPK (in each case, it was the third residue backbone C=O).
Mutagenesis of the membrane-proximal external region (MPER) of HIV-1 gp41
Our goal was to examine the effects of mutations in the LWYIK pentapeptide in the context of the entire HIV-1 Env. This peptide is part of the HIV-1 MPER in the gp41 ectodomain whose crucial role in viral fusion has been established (8). We employed the QuikChange mutagenesis method to substitute Leucine (L) at position 679 with Isoleucine (I). The mutation was introduced in the KpnI - BamHI NL4.3 envelope fragment that had been subcloned into vector pSP72. The mutated envelope fragment was then subcloned into the envelope expression vector pSRHS. All mutations were confirmed by sequencing.
Effects on cell-cell fusion
Cos-1 cells were transfected with WT and mutant PSRHS vectors and 48h later were mixed with JC53BL cells. Introduction of Tat through fusion results in the expression of firefly luciferase and ß-galactosidase under transcriptional control of a minimal HIV LTR. The luciferase-based fusion assay showed that the L679I mutant fused cells 27% less efficiently than the wild-type construct (WT) (Fig. 10). β-galactosidase assays were performed by counting the number of blue syncytia per well and the average number of nuclei per syncytium. The latter provides a more reproducible measure of Env function since large syncytium formation requires multiple rounds of fusion (8). The average number of syncytia per well was reduced by 34% for the L679I mutant compared to WT, while the average number of nuclei per syncytium was reduced further to 48% that of WT (Fig. 10).
Figure 10.

Cell-cell fusion assays of wild-type and MPER mutant L679I. Data shown is the average of three runs, for each of which samples were run in duplicate. The number of syncytia per well was determined by counting the syncytia in each well (24-well plate). The number of nuclei per syncytium was determined by counting 20 syncytia per well. Luciferase readings (RLU, relative light units) were taken as per the manufacturer’s instructions.
Discussion
The requirements for the CRAC motif are so flexible that there are many possible combinations to test for their ability to sequester cholesterol. According to the CRAC algorithm, a CRAC motif can vary in length anywhere from 5 to 13 amino acids. For the longer peptides there are 10 positions that can be any one of the 20 naturally occurring amino acids. This feature alone can generate 1.024 × 1013 possible sequences and if one includes the variability in length, the number gets much larger. It is unlikely that such a vast number of possible sequences can all be equally efficient to sequester cholesterol. In fact it has been pointed out that the CRAC motif occurs very frequently in a few genomes (30). In this work we have focused on a specific pentapeptide CRAC motif, LWYIK, that has been shown to sequester cholesterol and is present in a protein, the gp41 protein of HIV, which associates with cholesterol-rich domains. We have blocked the terminal amino and carboxyl groups as they would be blocked in the intact protein. In this group of LWYIK analogs we have not varied the W or I residues in the second and fourth position since these positions can be varied according to the CRAC algorithm, -L/V-(X)(1-5)-Y-(X)(1-5)-R/K-, i.e. they are the “X”. We have, however, made peptides in which the first, third and fifth residues of LWYIK are substituted. These three positions for a pentapeptide are required by the CRAC motif to be L/V, Y and K/R, respectively.
The peptide effects on the enthalpy of the phospholipid chain melting transition in the presence of cholesterol (Table 1) provide a good criterion for identifying peptides that deplete a region of the membrane from cholesterol, resulting in an increase in the cooperativity and enthalpy of this transition. The suggestion that cholesterol is redistributed in the membrane and is sequestered in regions where it surpasses its solubility limit is supported by the observation that anhydrous cholesterol crystallites are formed (Table 2). Among the peptides used in the present study, N-acetyl-LWYIK-amide is the most potent in increasing the enthalpy of the SOPC transition, and appears to be the only one to do so at 30% cholesterol (Fig. 4). With higher cholesterol concentration LWYIK has the strongest cholesterol-sequestering effect, followed by N-acetyl-LWFIK-amide and then N-acetyl-LWYIR-amide and N-acetyl-LWYIH-amide that have comparable potencies (Table 1). N-acetyl-LWWIK-amide and N-acetyl-LWLIK-amide have still weaker potency and N-acetyl-IWYIK-amide is the poorest in forming regions in the membrane devoid of cholesterol.
Differences in membrane partitioning among the peptides (Fig. 5) should not account for their different effects on the phase transitions of SOPC:cholesterol mixtures. Indeed, all peptides prefer to be at the membrane interface. Interestingly N-acetyl-IWYIK-amide has the strongest effect in lowering the enthalpy of the transition for the pure SOPC, indicating that the weaker effect of this peptide on cholesterol-containing membranes is not a consequence of its inability to partition into membranes.
There is also little difference in the maximum emission wavelength among the seven peptides in the presence of lipids (Table 3). This indicates that the differences in the extent of sequestering of cholesterol observed by DSC is not a consequence of a difference in hydrophobicity of the peptides that would result in a change in the depth of insertion of the peptide into the membrane. However, although the emission wavelength does not change, in some cases the intensity of emission does change. The peptides with Y can undergo resonance energy transfer to W. N-acetyl-LWWIK-amide has the lowest emission intensity per W of all of the peptides (given that it has two W). There can be resonance energy transfer between the two W but this will not result in a change in emission intensity, if both W emit at the same wavelength and have the same quantum yields. The peptide N-acetyl-LWLIK-amide has the highest emission intensity, even though this peptide cannot exhibit resonance energy transfer. This is true even with solutions of the peptide in methanol or in buffer. The fact that the quantum yields of these peptides vary indicates that even though these peptides are small and likely very flexible, there is sufficient difference in their preferred folding that results in differences in the environment of the W residue.
In general the fluorescence emission intensity from the W residue of these peptides is lower in the presence of membranes containing cholesterol than it is in pure SOPC vesicles (Table 3). Interestingly the ratio of fluorescence emission intensity of each peptide in the presence of SOPC vs. SOPC/cholesterol (1:1) is generally related to the ability of the peptide to sequester cholesterol into domains. Thus, N-acetyl-LWYIK-amide, which is the most potent in sequestering cholesterol, exhibits the largest difference in W emission intensity between membranes with or without cholesterol. Conversely, the W emission of the peptide N-acetyl-IWYIK-amide, that shows no tendency to preferentially partition into cholesterol-rich domains, is unaffected by cholesterol (Table 3). We suggest that N-acetyl-IWYIK-amide sequesters into cholesterol-depleted domains and therefore its fluorescence is not affected by the presence of cholesterol. The DSC results support this suggestion because N-acetyl-IWYIK-amide lowers the transition enthalpy of pure SOPC as well as that of SOPC in mixtures with cholesterol. The fluorescence study provides another independent parameter, in addition to the DSC results, to show which peptides are affected by cholesterol in the membrane.
The peptide that causes the greatest rearrangement of cholesterol in membranes is N-acetyl-LWYIK-amide. This peptide has a CRAC motif. Major factors contributing to the differences among the peptides used in this work include their structural flexibility, their position at the interface and their non-covalent bonding with cholesterol. A comparison of the cholesterol sequestering action of these analog peptides provides good support for the concept of the CRAC motif. The peptide N-acetyl-LWFIK-amide is not formally a CRAC peptide and it is not as selective as N-acetyl-LWYIK-amide in sequestering cholesterol at low concentration. N-acetyl-LWFIK-amide differs from N-acetyl-LWYIK-amide by only a single OH group. This OH group is involved as an H-donor in the LWYIK-cholesterol complex. The H-bond interaction is between the C=O of the terminal lysine of LWYIK which acts as an H-acceptor in the complex. N-acetyl-LWWIK-amide has much lower cholesterol-sequestering activity and it is not a CRAC motif. In addition, the weakest peptide, N-acetyl-IWYIK-amide, has the same tyrosine groups as the best cholesterol-sequestering peptide, N-acetyl-LWYIK-amide. The IWYIK does not correspond to a CRAC motif. In this latter case, the reduced conformational flexibility of the peptide may inhibit the formation of stable interactions with cholesterol. Thus, aromatic groups are likely important for stacking with cholesterol, but the conformation of the peptide and its presentation to the membrane are also important in determining its ability to preferentially bind to cholesterol-rich regions of the membrane. The central Y residue in LWYIK may have a particular role in stabilizing interactions with cholesterol by forming H-bonds (Fig. 10). However, as expected because of the short length and flexibility of these peptides, this feature is necessary for cholesterol selectivity but is not sufficient. Changing K for R decreases the capacity to interact with cholesterol because the polar head of R tends to force the peptide out towards the water interface and in addition it decreases the motif’s structural flexibility.
On the basis of NMR evidence, it was suggested that one of the aromatic groups of N-acetyl-LWYIK-amide stacks with the A ring of cholesterol (7). The results of modeling support that, in the best energy conditions, the LWYIK motif is located within the interface wrapped around the polar head of cholesterol. However, in less favorable conditions, more frequent with other peptides, aromatic groups stack with the A ring of cholesterol. More in concordance with modeling are the NMR and mutational evidence showing that the Y residues of the benzodiazepine receptor are important for interaction with cholesterol (31).
We further tested the segment IWYIK in the context of an intact gp41 molecule. The L679I mutant of gp41 had significantly less fusogenic activity compared with the wild type protein (Fig. 9). This mutation did not completely eliminate the ability of gp41 to induce syncytia formation but it did lower the activity despite the fact that the substituted amino acid, isoleucine, has similar properties to the native residue, leucine. Further studies are currently underway to determine the cause for the lower activity of the L679I mutant. From the results of the peptide studies, it seems likely that the decreased infectivity of this mutant is a result of its diminished interaction with raft domains.
This is the first study to systematically investigate the properties of a family of closely related peptides in terms of their ability to promote rearrangement of cholesterol in membranes. It is found that their effectiveness is very dependent on small changes in their amino acid composition. It can result in the formation of cholesterol-rich domains either by being excluded from cholesterol-rich domains, as is the case for N-acetyl-IWYIK-amide or by preferentially binding to these domains as occurs with N-acetyl-LWYIK-amide. There is some relationship to the rules for a CRAC motif in that peptides that fulfill the requirements of this motif are often found to sequester cholesterol by binding to cholesterol-rich domains. However, this criterion is not exact. Furthermore, there may be differences between the properties of the isolated small peptide and the behavior of the same sequence when it is part of a larger protein, although in this present example, the L679I mutant of gp41 also exhibits decreased biological activity. It seems likely that no general rule will be able to predict with exactitude if a protein segment will recruit cholesterol in a membrane, although the presence of a CRAC motif adjacent to a transmembrane helix is suggestive of such an event taking place.
Abbreviations used
- CRAC
Cholesterol Recognition/interaction Amino acid Consensus
- HIV
human immunodeficiency virus
- PC
phosphatidylcholine
- SOPC
1-stearoyl-2-oleoyl phosphatidylcholine
- SUV
sonicated unilamellar vesicles
- MLV
multilamellar vesicle
- DSC
differential scanning calorimetry
- Tm
phase transition temperature
- ΔH
calorimetric enthalpy
- MPER
the membrane-proximal external region.
Footnotes
This work was supported by the Canadian Institutes of Health Research (MA-7654), the “Ministère de la Région Wallonne” contract n°14540 (PROTMEM). We would also like to thank the “FRSM” (contract n°3.4505.02) for financial support. The work on mutagenesis and HIV-1 fusion was supported by grant R01 AI-33319 from the National Institutes of Health. R.B. is Research Director at the National Funds for Scientific Research of Belgium (FNRS). A.T. is Research Director at the “Institut National de la Santé et de la Recherche Médicale” (INSERM France). R.M.E. is a Senior Research Investigator of the Canadian Institutes for Health Research.
References
- 1.Epand RM. Do proteins facilitate the formation of cholesterol-rich domains? Biochim. Biophys. Acta. 2004;1666:227–238. doi: 10.1016/j.bbamem.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Hammond AT, Heberle FA, Baumgart T, Holowka D, Baird B, Feigenson GW. Crosslinking a lipid raft component triggers liquid ordered-liquid disordered phase separation in model plasma membranes. Proc. Natl. Acad. Sci. U. S. A. 2005;102:6320–6325. doi: 10.1073/pnas.0405654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Resh MD. Membrane targeting of lipid modified signal transduction proteins. Subcell. Biochem. 2004;37:217–232. doi: 10.1007/978-1-4757-5806-1_6. [DOI] [PubMed] [Google Scholar]
- 4.van Duyl BY, Meeldijk H, Verkleij AJ, Rijkers DT, Chupin V, De Kruijff B, Killian JA. A synergistic effect between cholesterol and tryptophan-flanked transmembrane helices modulates membrane curvature. Biochemistry. 2005;44:4526–4532. doi: 10.1021/bi047937n. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Papadopoulos V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology. 1998;139:4991–4997. doi: 10.1210/endo.139.12.6390. [DOI] [PubMed] [Google Scholar]
- 6.Vincent N, Genin C, Malvoisin E. Identification of a conserved domain of the HIV-1 transmembrane protein gp41 which interacts with cholesteryl groups. Biochim. Biophys. Acta. 2002;1567:157–164. doi: 10.1016/s0005-2736(02)00611-9. [DOI] [PubMed] [Google Scholar]
- 7.Epand RM, Sayer BG, Epand RF. Peptide-induced formation of cholesterol-rich domains. Biochemistry. 2003;42:14677–14689. doi: 10.1021/bi035587j. [DOI] [PubMed] [Google Scholar]
- 8.Salzwedel K, West JT, Hunter E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J. Virol. 1999;73:2469–2480. doi: 10.1128/jvi.73.3.2469-2480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saez-Cirion A, Nir S, Lorizate M, Agirre A, Cruz A, Perez-Gil J, Nieva JL. Sphingomyelin and cholesterol promote HIV-1 gp41 pretransmembrane sequence surface aggregation and membrane restructuring. J Biol. Chem. 2002;277:21776–21785. doi: 10.1074/jbc.M202255200. [DOI] [PubMed] [Google Scholar]
- 10.Saez-Cirion A, Arrondo JL, Gomara MJ, Lorizate M, Iloro I, Melikyan G, Nieva JL. Structural and functional roles of HIV-1 gp41 pretransmembrane sequence segmentation. Biophys. J. 2003;85:3769–3780. doi: 10.1016/S0006-3495(03)74792-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao Z, Cimakasky LM, Hampton R, Nguyen DH, Hildreth JE. Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS Res. Hum. Retroviruses. 2001;17:1009–1019. doi: 10.1089/088922201300343690. [DOI] [PubMed] [Google Scholar]
- 12.Sarin PS, Gallo RC, Scheer DI, Crews F, Lippa AS. Effects of a novel compound (AL 721) on HTLV-III infectivity in vitro. N. Engl. J Med. 1985;313:1289–1290. doi: 10.1056/NEJM198511143132011. [DOI] [PubMed] [Google Scholar]
- 13.Schaffner CP, Plescia OJ, Pontani D, Sun D, Thornton A, Pandey RC, Sarin PS. Anti-viral activity of amphotericin B methyl ester: inhibition of HTLV-III replication in cell culture. Biochem. Pharmacol. 1986;35:4110–4113. doi: 10.1016/0006-2952(86)90037-7. [DOI] [PubMed] [Google Scholar]
- 14.Liao Z, Graham DR, Hildreth JE. Lipid rafts and HIV pathogenesis: virion-associated cholesterol is required for fusion and infection of susceptible cells. AIDS Res. Hum. Retroviruses. 2003;19:675–687. doi: 10.1089/088922203322280900. [DOI] [PubMed] [Google Scholar]
- 15.Epand RF, Sayer BG, Epand RM. The tryptophan-rich region of HIV gp41 and the promotion of cholesterol-rich domains. Biochemistry. 2005;44:5525–5531. doi: 10.1021/bi0500224. [DOI] [PubMed] [Google Scholar]
- 16.Privalov G, Kavina V, Freire E, Privalov PL. Precise scanning calorimeter for studying thermal properties of biological macromolecules in dilute solution. Anal. Biochem. 1995;232:79–85. doi: 10.1006/abio.1995.9957. [DOI] [PubMed] [Google Scholar]
- 17.Sober HA, editor. Handbook of Biochemistry: Selected Data for Molecular Biology. The Chemical Rubber Co.; Cleveland, OH: 1970. pp. B-75–B-76. [Google Scholar]
- 18.Bruccoleri RE, Haber E, Novotny J. Structure of antibody hypervariable loops reproduced by a conformational search algorithm. Nature. 1988;335:564–568. doi: 10.1038/335564a0. [DOI] [PubMed] [Google Scholar]
- 19.Dudek MJ, Scheraga HA. Protein-Structure Prediction Using A Combination of Sequence Homology and Global Energy Minimization.1. Global Energy Minimization of Surface Loops. Journal of Computational Chemistry. 1990;11:121–151. [Google Scholar]
- 20.Moult J, James MN. An algorithm for determining the conformation of polypeptide segments in proteins by systematic search. Proteins. 1986;1:146–163. doi: 10.1002/prot.340010207. [DOI] [PubMed] [Google Scholar]
- 21.Lins L, Brasseur R, De Pauw M, Van Biervliet JP, Ruysschaert JM, Rosseneu M, Vanloo B. Helix-helix interactions in reconstituted high-density lipoproteins. Biochim. Biophys. Acta. 1995;1258:10–18. doi: 10.1016/0005-2760(95)00080-v. [DOI] [PubMed] [Google Scholar]
- 22.Leach AR. In: Molecular Modelling: Principles and Applications. Leach AR, editor. Longman Ltd., Harlow; England: 1996. pp. 171–177. [Google Scholar]
- 23.Thomas A, Milon A, Brasseur R. Partial atomic charges of amino acids in proteins. Proteins. 2004;56:102–109. doi: 10.1002/prot.20093. [DOI] [PubMed] [Google Scholar]
- 24.Brasseur R. Simulating the Folding of Small Proteins by Use of the Local Minimum Energy and the Free Solvation Energy Yields Native-Like Structures. Journal of Molecular Graphics. 1995;13:312–322. doi: 10.1016/0263-7855(95)00052-6. [DOI] [PubMed] [Google Scholar]
- 25.Ducarme P, Rahman M, Brasseur R. IMPALA: a simple restraint field to simulate the biological membrane in molecular structure studies. Proteins. 1998;30:357–371. [PubMed] [Google Scholar]
- 26.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents. Chemother. 2002;46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimpton J, Emerman M. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J. Virol. 1992;66:2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Epand RM, Bach D, Borochov N, Wachtel E. Cholesterol crystalline polymorphism and the solubility of cholesterol in phosphatidylserine. Biophys. J. 2000;78:866–873. doi: 10.1016/S0006-3495(00)76644-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loomis CR, Shipley GG, Small DM. The phase behavior of hydrated cholesterol. J. Lipid Res. 1979;20:525–535. [PubMed] [Google Scholar]
- 30.Palmer M. Cholesterol and the activity of bacterial toxins. FEMS Microbiol. Lett. 2004;238:281–289. doi: 10.1016/j.femsle.2004.07.059. [DOI] [PubMed] [Google Scholar]
- 31.Jamin N, Neumann JM, Ostuni MA, Vu TK, Yao ZX, Murail S, Robert JC, Giatzakis C, Papadopoulos V, Lacapere JJ. Characterization of the cholesterol recognition amino acid consensus sequence of the peripheral-type benzodiazepine receptor. Mol. Endocrinol. 2005;19:588–594. doi: 10.1210/me.2004-0308. [DOI] [PubMed] [Google Scholar]
- 32.Lins L, Charloteaux B, Heinen C, Thomas A, Brasseur R. De novo design of peptides with specific lipid-binding properties. Biophys. J. 2006;90:470–479. doi: 10.1529/biophysj.105.068213. [DOI] [PMC free article] [PubMed] [Google Scholar]